

Abstract
Tropomyosin (Tm) is a two-stranded α-helical coiled-coil protein, and when associated with troponin, it is responsible for the actin filament-based regulation of muscle contraction in vertebrate skeletal and cardiac muscles. It is widely believed that Tm adopts a flexible rod-like structure in which the flexibility must play a crucial role in its functions. To obtain more information about the flexibility of Tm, we solved and compared two crystal structures of the identical C-terminal segments, spanning ∼40% of the entire length. We also compared these structures with our previously reported crystal structure of an almost identical Tm segment in a distinct crystal form. The parameters specifying the local coiled-coil geometry, such as the separation between two helices and the local helical pitch, undulate along the length of Tm in the same way as among the three crystal structures, indicating that these parameters are defined by the amino acid sequence. In the region of increased separation, around Glu-218 and Gln-263, the hydrophobic core is disrupted by three holes. Moreover, for the first time to our knowledge, for Tm, water molecules have been identified in these holes. In some structures, the B-factors are higher around the holes than in the rest of the molecule. The Tm coiled-coil must be destabilized and therefore may be flexible, not only in the alanine clusters but also in the regions of the broken core. A closer look at the local staggering between the two chains and the local bending revealed that the strain accumulates at the alanine cluster and may be relaxed in the broken core region. Moreover, the strain is distributed over a long range, even when a deformation like bending may occur at a limited number of spots. Thus, Tm should not be regarded as a train of short rigid rods connected by flexible linkers, but rather as a seamless rubber rod patched with relatively more flexible regions.
INTRODUCTION
The Ca2+ regulation of vertebrate skeletal and cardiac muscle contraction is exerted by the actin-containing muscle thin filament, consisting of troponin (Tn), tropomyosin (Tm), and actin at a ratio of 1:1:7 (1). In these muscles, Tm is a dimer of homologous (αα-Tm) or highly homologous (αβ-Tm) α-helices 284-residues long, forming a parallel coiled-coil over almost the entire length of 400 Å. The coiled-coil forming peptide is characterized by the heptad repeat (a-b-c-d-e-f-g), where the residues at the a- and d-positions are usually hydrophobic (2,3). These hydrophobic residues interlock in a “knobs-into-holes” manner (4) and form the continuous hydrophobic core that stabilizes the coiled-coil structure.
Upon a transient increase in the myoplasmic Ca2+ concentration ([Ca2+]), Ca2+ binds to Tn, which must induce conformational changes in Tn. These conformation changes in Tn must be transmitted to Tm and then to the entire actin filament. It is widely believed that for this transmittance, the flexibility of the Tm strands on the actin filament must play an important role. Nevertheless, the dynamic properties of Tm have remained obscure.
To understand the dynamic properties of Tm, the atomic structure of the entire molecule is required. The whole Tm molecule formed crystals that diffracted x-rays poorly (5,6) and, at best, the structure has been obtained at 7 Å resolution (7). Recently, Tm segments have been crystallized with or without an extension at either end of the coiled-coil of the leucine-zipper sequence of GCN4, and the structures have been solved at an atomic resolution (Fig. 1 a). These include the N-terminal 81 residues of chicken skeletal muscle α-Tm (Tm81) at 2.0 Å resolution (8), the C-terminal 31 residues of rat skeletal muscle α-Tm at 2.7 Å resolution (9), and the midregion of rat striated muscle α-Tm (Mid-Tm), containing residues 89–208, at 2.3 Å resolution (10). In our previous article, we reported the crystal structure of a C-terminal fragment of rabbit skeletal muscle α-Tm, ZrsTm176, containing residues 176–284 at 2.6 Å resolution (11). That was the first atomic structure of the last missing segment of α-Tm, including the region interacting with Tn.
FIGURE 1.

(a) Segments of skeletal muscle Tm whose atomic structures have been reported (black boxes), together with locations of specific structural features (pink and green circles). The black boxes indicate the segment 1–81 of chicken skeletal muscle α-Tm (Tm81), with Protein Data Bank (PDB) accession number 1IC2 (8), the segment 254–284 of rat skeletal muscle α-Tm, 1KQL (9), and the segment 89–208 (the midregion) of rat striated muscle α-Tm (Mid-Tm), 2B9C (10). The red boxes indicate the segment 176–273 of rabbit skeletal muscle α-Tm (Z-rsTm(176–273)-Z), which are reported here, and the segment 176–284 of rabbit skeletal muscle α-Tm (ZrsTm176), 2D3E (11), which are compared with Z-rsTm(176–273)-Z. Specific structural features are the alanine cluster (pink circles) and the break in hydrophobic core (green circle) with the residue numbers at a- and d-positions indicated in the boxes. (b) Comparison of the two constructs and the three crystal forms reported and discussed in this report, with the PDB accession codes in parentheses. The amino acid sequences of segments of Tm and of fused GCN4 are indicated as pink and cyan bars, respectively. The numbers above and below the bars are numbers of amino acids and the end residues for each segments with positions in parentheses in the heptad repeat, respectively.
In the atomic structures of the Tm segments reported thus far, two types of structural patterns have been identified as regions that may destabilize the Tm structure (Fig. 1 a). The first pattern is the alanine cluster, where consecutive alanine or serine residues are clustered at the core, as described in the structure of Tm81. The alanine cluster is associated with decreased spacing together with a small, but significant axial stagger between the two α-helices. It was hypothesized that the alanine cluster would allow Tm to bend at either end of the cluster so that Tm could wrap around the actin filament. The second pattern is the broken core region, which is formed around the acidic core residues, Asn-137(d) and Glu-218(a), as observed in the structures of Mid-Tm and ZrsTm176, respectively. The broken core region is associated with an increase in the spacing between the two α-helices, leaving a hole in the middle of the core. Since the core residues in the hole are exposed to the solvent, this structure must substantially destabilize the entire molecule.
In this study, we obtained and compared two distinct crystal structures, type 1 and type 2 crystals, from the identical C-terminal segment, Z-rsTm(176–273)-Z, of rabbit skeletal muscle α-Tm, at 1.8 Å and 2.0 Å resolutions, respectively. Comparisons were also made with the previously reported crystal structure of the nearly identical Tm segment ZrsTm176 in a distinct crystal form (11). These two constructs, Z-rsTm(176–273)-Z and ZrsTm176, contain the Tm sequence 176–273 in common but differ in their GCN4 extensions: the former molecule has extensions at both ends, whereas the latter has an extension only at the N-terminus (Fig. 1 b). Our comparisons enabled us for the first time to our knowledge to distinguish between the properties intrinsic to the Tm sequence and those caused by the crystal packing. The structural aspects which are shared by the three crystal forms must be intrinsic to the Tm sequence. From the structural aspects uniquely associated with a particular crystal form, we can learn how the external forces imposed by the crystal lattice onto the Tm segment cause the deformation of the segment. In this article, by comparing these three structures, two questions will be addressed. First, do the broken core regions originate from the amino acid sequence of Tm, rather than from the force imposed by neighboring molecules, and what are the functional roles of these regions? Second, should Tm be regarded as a chain of rigid rods connected by flexible linkers or as a continuous soft rod?
A short preliminary description of the type 2 crystal structure of Z-rsTm(176–273)-Z was provided in our previous report, as a part of the meeting proceedings (11).
MATERIALS AND METHODS
Protein preparation
The recombinant protein used has the following amino acid sequence: M146-RMKQLEDKVEELLSKNYHLENEVARLKK-L175 (numbered 146–175: the sequence 147–175 corresponds to 249–277 of GCN4) followed by L176-ERAEERAEL SEGKSAELEE ELKTVTNNLK SLEAQAEKYS QKEDKYEEEI KVLSDKLKEA ETRAEFAERS VTKLEKSIDD LEDELYAQKL KYKAISE-E273 (176–273 of rabbit skeletal muscle α-Tm with the replacement C190S) and M274-KQLEDKVEELLSKNYHLENEVARLKK-L301 (numbered 274–301, which corresponds to 250–277 of GCN4).
This sequence differs from ZrsTm176 (11) in two points: the molecule in this study contains a Tm sequence 11 residues shorter than that in ZrsTm176, and it has a leucine-zipper sequence extension at both ends, compared to only one at the N-terminus in ZrsTm176.
Expression and purification of Z-rsTm(176–273)-Z
pETZ-rsTm(176–273)-Z was constructed as follows: the Z-rsTm(176–273)-Z fragment was polymerase chain reaction (PCR) amplified using oligo1: ggc atg cca tgg gca tga aac agc tgg aag ata aag tgg and oligo2: ggg cgg atc cta cag ttt ttt cag acg cgc cac ttc gtt ttc cag gtg gta gtt ttt gct cag cag ttc ttc cac. The template DNA plasmid was pETZrsTm(176–284)C190S (see below). The PCR fragment was digested by the restriction enzymes NcoI and BamHI and was ligated into pET3d, which was previously digested by NcoI and BamHI. This plasmid DNA was mutated to change the first ccatgg to ccatgc, which was used for cloning. The mutagenesis was carried out with a QuickChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) using the following oligos: oligo3: gga gat ata cca tgc gca tga aac agc tg and oligo4: cag ctg ttt cat gcg cat ggt ata tct cc.
pETZrsTm(176–284)C190S was constructed as follows: the ZrsTm(176–284) fragment was PCR amplified by using oligo5: cat gcc atg gaa gat aaa gtg gaa gaa ctg ctg agc aaa aac tac cac ctg gaa aac gaa gtg gcg cgt ctg aaa aaa ctg ctg gag cgt gca gag gag cgg gcc gag c and oligo6: ctc ggg atc cgt gaa gca aag aaa c. The template DNA plasmid was pB(2-1)Tm (full-length rabbit Tm clone) (12). The PCR fragment was digested by the restriction enzymes NcoI and BamHI and was ligated into pET3d, which was previously digested by NcoI and BamHI. The plasmid pETZrsTm(176–284) thus generated was mutated to change cysteine 190 to serine with a QuickChange Site-Directed Mutagenesis Kit, using oligo7: ctc tca gaa ggc aaa tct gcc gag ctt gaa gaa g and oligo8: ctt ctt caa gct cgg cag ctt tgc ctt ctg aga g, to make pETZrsTm(176–284)C190S. The protein was expressed in BL21(DE3) cells and was purified as described by Nitanai et al. (11), with minor modifications.
Crystallization and data collection
The Z-rsTm(176–273)-Z protein was crystallized by the sitting drop vapor diffusion method. In the case of the type 1 crystal, a drop containing 1 μl of protein solution (26 mg/ml of Z-rsTm(176–273)-Z, 10 mM Tris-HCl (pH = 8.0) and 1 mM dithiothreitol) and 1 μl of reservoir solution (0.2 M Mg(OAc)2, 2.2 M 1,6-hexanediol, and 0.1 M HEPES (pH = 7.5)) was equilibrated against 1 ml of reservoir solution at 20°C. The type 2 crystal was crystallized with the following reservoir solution (0.4 M Mg(OAc)2, 40% m-phenylenediamine, and 0.1 M Tris-HCl (pH = 8.5)). The crystals were flash-cooled in a cold nitrogen flow. The 1.8 Å resolution data set for the type 1 crystal and the 2.0 Å resolution data set for the type 2 crystal were collected at the RIKEN Structural Biology Beamline (BL44B2) of SPring-8 (13).
The type 1 crystal belonged to the space group P1, with unit cell dimensions a = 42.36 Å, b = 65.84 Å, c = 71.14 Å, α = 66.40°, β = 89.93°, γ = 90.04°. There were two dimeric molecules and 50% solvent content in the unit cell. The type 2 crystal belonged to the space group P1, with unit cell dimensions a = 43.20 Å, b = 63.72 Å, c = 73.26 Å, α = 66.64°, β = 89.99°, γ = 90.30° and had 51% solvent content.
The data of the type 1 crystal were processed using the programs DENZO and SCALEPACK from the HKL2000 package (14). The data of the type 2 crystal were processed with the program Crystal Clear (15).
Structure determination
The atomic structures were determined by the molecular replacement method with the ZrsTm176 coordinates, using Phaser (16) in the CCP4 program suite (17). Refinements were carried out by CNS (18) and ARP/wARP, and model building was accomplished with Coot (19). The data collection and refinement statistics are summarized in Table 1.
TABLE 1.
Data collection and refinement statistics
| Statistic | Type 1 | Type 2 | ||
|---|---|---|---|---|
| Data collection | ||||
| Space group | P1 | P1 | ||
| Unit cell dimensions (Å, degree) | a = 42.36, b = 65.84, c = 71.14 | a = 43.20, b = 63.72, c = 73.26 | ||
| α = 66.40, β = 89.93, γ = 90.04 | α = 66.64, β = 89.99, γ = 90.30 | |||
| Spacing (Å) | 20.0 − 1.80 | 1.86 − 1.80 | 29.25 − 2.00 | 2.07 − 2.00 |
| No. of measured reflections | 244,351 | 182,859 | ||
| No. of unique reflections | 62,779 | 47,224 | ||
| Average redundancy | 3.9 | 3.7 | 3.9 | 3.7 |
| Completeness (%) | 96.3 | 95.1 | 97.4 | 93.6 |
| Rmerge (I)* | 0.064 | 0.201 | 0.038 | 0.110 |
| Average I/σ (I) | 17.1 | 5.5 | 21.0 | 4.8 |
| Solvent content (%)/VM | 50.1/2.47 | 51.0/2.47 | ||
| Refinement | ||||
| No. of reflections (%) | 62,708 (96.0) | 47,200 (97.2) | ||
| No. of reflections in working set (%) | 59,547 (91.3) | 44,801 (92.4) | ||
| No. of reflections in test set (%) | 3161 (4.8) | 2399 (4.9) | ||
| R† | 0.237 | 0.249 | ||
| Free R‡ | 0.316 | 0.304 | ||
| No. of peptide chains | 4 | 4 | ||
| No. of protein/water atoms | 5118/1460 | 5096/361 | ||
| RMSD bond length (Å) | 0.008 | 0.008 | ||
| RMSD bond angle (degree) | 1.1 | 1 | ||
| Average B factor | 42.4 | 52.9 | ||
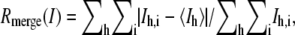 where Ih,i is the intensity of the ith observation of the reflection h.
where Ih,i is the intensity of the ith observation of the reflection h.
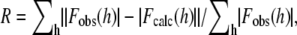 where Fobs(h) and Fobs(h) are the observed and calculated structure factors, respectively.
where Fobs(h) and Fobs(h) are the observed and calculated structure factors, respectively.
Free R is calculated for randomly selected 5% of the reflection data, which were not included in the refinement.
Analysis of the coiled-coil geometry
The program TWISTER (20) was used to determine the local coiled-coil parameters as a function of the residue number. In this program, the center of each α-helix, Chelix(n), at the position of residue n, the center of the coiled-coil Ccc (n), and the local pitch (LP) were obtained. From the coordinates of Chelix(n) and Ccc(n), the interhelical distance (HD), the local bending angle (LBA) and the local staggering angle (LSA) were calculated. The definitions of HD, LBA, and LSA were described previously by Nitanai et al. (11). These parameters help to clarify the local coiled-coil geometry.
Comparison of the conformations between the two molecules
The conformational differences between the two molecules were analyzed in two ways. On one hand, the Cα-Cα distances were plotted against the residue number n after the two molecules were aligned, so that the sum of the root mean-square differences (RMSD) over the entire lengths of the two molecules was minimized. On the other hand, the tendency of the Cα-Cα displacement, a “differentiation” of the displacement, was obtained as the RMSD as a function of the residue number n, after aligning each pair of 14-residue segments (from n − 6 to n + 7) for the minimal sum of the RMSD over a 14-residue window.
RESULTS AND DISCUSSION
Overall structure
The protein crystallized in this study is Z-rsTm(176–273)-Z, expressed in Escherichia coli. This protein consists of amino acid residues 176–273 of the rabbit skeletal muscle α-Tm sequence, with a 30-residue extension of the leucine-zipper sequence of GCN4 at the N-terminus, and another 28-residue extension of the leucine-zipper sequence at the C-terminus (for the amino acid sequence, see the Materials and Methods section). From two different batches of the same protein preparations of the protein, two forms of crystals, type 1 and type 2, were obtained under slightly different conditions of crystallization. The crystal structure of type 1 was solved at 1.8 Å, whereas that of type 2 was at 2.0 Å resolution. The crystal lattices are almost identical to each other; the percentage differences in the lattice constants between type 1 and type 2 are +1.9%, −3.3%, and +2.9% in a, b, and c, respectively, and +1.8% in the volume.
In common, each crystal form has two dimers in the asymmetric unit. Each dimer belongs to one of the two layers, the lower layer or the upper layer, as shown in Fig. 2. The direction of the molecules in each layer is inclined by ∼20° relative to each other. In this report, the two polypeptide chains forming the dimer molecule in the lower layer are designated chains A and B, whereas the two chains forming the molecule in the upper layer are designated chains C and D. Despite the nearly identical behavior of the two types of crystals, the conformations of the individual molecules are quite different. This is consistent with the distinct intensity distribution of each data set. Thus, the difference in the diffraction intensities between two scaled data sets is over 50%, which is substantially larger than 20% for the difference between crystals of the same type.
FIGURE 2.

The molecular arrangement of Z-rsTm(176–273)-Z in the crystal. (a) Two-dimer molecules (pink), one (dimer of chains A and B) in the lower layer and the other (CD dimer) in the upper layer, are included in the asymmetric unit. The N- and C-termini are marked. The white circle indicates a crossover point, and its side-chain arrangement is indicated in c. Type 1 crystal. (b) The same crystal as in a, viewed from the direction of the molecular axis of the lower layer. (c) Distinct manner of contacts at the crossover point between a molecule in the upper layer and another molecule in the lower layer for the type 1 crystal (upper panel) and for the type 2 crystal (lower panel). The residues contributing to the contacts are labeled, and possible hydrogen bonds are indicated by the dotted lines with the bond lengths. Residue numbers larger than 274 are from GCN4.
The distinct crystal packing between the type 1 and type 2 crystals must be due, at least in part, to the different mode of intermolecular interactions between the two layers at the crossover point (Fig. 2 c). The hydrogen-bond distance between Arg-244(f) of a dimer in the upper layer and Asn-293(f) of the interacting dimer in the lower layer, 3.1 Å, is shared by type 1 and type 2 crystals, whereas the distance between Lys-248(c) of a dimer in the upper layer and Glu-292(e) of the interacting dimer in the lower layer is 4.2 Å for type 1 and 2.8 Å for type 2. Arg-244(f) and Lys-248(c) are in the vicinity of an alanine cluster of core residues, A235(d)–A239(a)–A242(d), whereas Glu-292(e) and Asn-293(f) are in the GCN4 extension.
Holes in the hydrophobic core and water molecules
The most remarkable feature in the crystal structures here is that the hydrophobic core is not continuous, but is interrupted at three points by holes. In addition, for the first time to our knowledge for Tm, as many as three water molecules have been identified within each hole (Fig. 3, Table 2). This is in contrast to the widely accepted concept of the coiled-coil structure that is stabilized by a tightly packed hydrophobic core along the coiled-coil axis (2,3). The breaks in the hydrophobic core are associated with increased separation between the two chains, which was also observed in the previous crystal structure of ZrsTm176 at 2.6 Å resolution (11).
FIGURE 3.

(a) The electron density map around the hole (214–221), and (b) around the hole (263–267) of molecule AB of the type 1 crystal. The upper maps are viewed from a direction perpendicular to the coiled-coil axis, and the lower maps are viewed along the coiled-coil axis. The peaks assigned to water molecules are indicated as yellow arrows. The maps are indicated as 2Fo − Fc electron density maps contoured at 1.4 σ.
TABLE 2.
Water molecules in the holes
| Type 1 Crystal
|
Type 2 Crystal
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Holes (amino acid residues) | Molecule AB
|
Molecule CD
|
Molecule AB
|
Molecule CD
|
||||
| Number of water molecules | Hydrogen bonds to | Number of water molecules | Hydrogen bonds to | Number of water molecules | Hydrogen bonds to | Number of water molecules | Hydrogen bonds to | |
| 207–214 | 3 | Chain A, Glu-210, side chain C=O, 2.87 Å Chain A, Leu-207, main chain C=O, 2.99 Å | 2 | Chain C, Glu-210, side chain C=O, 2.85 Å Chain C, Leu-207, main chain C=O, 2.87 Å | 1 | Chain A, Glu-210, side chain C=O, 2.69 Å Chain A, Leu-207, main chain C=O, 2.91 Å | 2 | Chain C, Glu-210, side chain C=O, 2.77 Å Chain C, Leu-207, main chain C=O, 3.17 Å |
| Chain B, Glu-210, side chain C=O, 2.92 Å Chain B, Leu-207, main chain C=O, 2.91 Å | Chain D, Glu-210, side chain C=O, 2.83 Å Chain D, Leu-207, main chain C=O, 2.72 Å | Chain D, Glu-210, side chain C=O, 3.55 Å Chain D, Leu-207, main chain C=O, 2.66 Å | ||||||
| Chain A, Glu-210, side chain C=O, 2.87 Å Chain B, Leu-207, main chain C=O, 3.41 Å | ||||||||
| 214–221 | 2 | Chain B, Glu-218, side chain C=O, 2.93 Å Chain A, Tyr-214, main chain C=O, 3.05 Å | 2 | Chain D, Glu-218, side chain C=O, 2.86 Å Chain C, Tyr-214, main chain C=O, 3.09 Å | 0 | 0 | ||
| Chain B, Tyr-214, main chain C=O, 3.08 Å | Chain C, Glu-218, side chain C=O, 2.76 Å Chain D, Tyr 214, main chain C=O, 3.19 Å | |||||||
| 263–267 | 1 | Chain A, Gln-263, side chain N-H or C=O, 3.34 Å Chain B, Gln-263, side chain N-H or C=O, 3.14 Å Chain A, Gln-263, main chain C=O, 2.92 Å Chain B, Gln-263, main chain C=O, 3.66 Å | 1 | Chain C, Gln-263, side chain N-H or C=O, 3.02 Å Chain D, Gln-263, side chain N-H or C=O, 3.02 Å Chain C, Gln-263, main chain C=O, 3.16 Å Chain D, Gln-263, main chain C=O, 3.60 Å | 1 | Chain A, Gln-263, side chain N-H or C=O, 2.73 Å Chain B, Gln-263, side chain N-H or C=O, 3.12 Å Chain A, Gln-263, main chain C=O, 3.00 Å Chain B, Gln-263, main chain C=O, 3.74 Å | 1 | Chain C, Gln-263, side chain N-H or C=O, 2.90 Å Chain D, Gln-263, side chain N-H or C=O, 3.24 Å Chain C, Gln-263, main chain C=O, 2.98 Å Chain D, Gln-263, main chain C=O, 3.65 Å |
The number of water molecules localized in the holes in the hydrophobic core, together with possible hydrogen bonds and distances between water molecules and surrounding atoms.
The first hole (207–214) and the second hole (214–221) are adjacent to each other, and they may also be considered two sectors of a single hole. These holes are also associated with increased separation between the two chains (Fig. 4 a: HD = 11.0 Å, compared with the average value, 9.6 Å) and local unwinding of the coiled-coil (Fig. 4 a: LP = 242–258 Å, compared with the average value, 156 Å). The sequence of the core residues there is Leu-207(d)–Ala-211(a)–Tyr-214(d)–Glu-218(a)–Tyr-221(d). In particular, Glu-218(a) seems to play an important role in forming the holes. Systematic studies on the relationship between the stability and the amino acid residues at the core positions (a- and d-positions) in a homodimeric coiled-coil indicated that charged residues and, to a lesser extent, polar residues at the core positions destabilize the coiled-coil (21,22).
FIGURE 4.

(a and b) Distributions of the local geometrical parameters in molecule AB of the type 1 crystal. (a) The green signs connected by green lines are for the local HD, and the red signs are for the LP. (b) The black signs are for the LBA, and the blue signs are the LSA. (c) A comparison between the local geometrical parameters of the type 1 and type 2 crystals. The black and gray lines indicate the LBAs of type 1 and type 2, respectively. The blue and light blue lines indicate the LSAs of type 1 and type 2, respectively. (a–c) The yellow bars indicate the leucine-zipper sequence regions. The purple bars indicate the alanine clusters Ala-179(d)–Ala-183(a)–Ser-186(d)–Cys-190(a) and Ala-235(d)–Ala-239(a)–Ala-242(d). The green bars indicate Tyr-214(d)–Glu-218(a)–Tyr-221(d).
In the first hole (207–214), the water molecules form hydrogen bonds with the side chains of Glu-210(g), whereas in the second hole (214–221) of the type 1 crystal, each of the two water molecules forms a hydrogen bond with Glu-218(a) of each chain (Fig. 3 a, Table 2). In the second hole (214–221) of the type 2 crystal, however, no water molecule was found in molecule AB or molecule CD (Table 2) although the helical parameters in this region are almost identical between the two types of crystals. This difference between the type 1 and type 2 crystals is likely to be due to the high B-factors in the type 2 crystal. The average B-factor of this region in the type 2 crystal is ∼1.7 times higher than the corresponding value in the type 1 crystal (43.2 Å2). This must hinder the identification of water molecules. This is consistent with the observation that, in the type 2 crystal, the side chains including Tyr-214(d) at the core are poorly defined, whereas they are well defined in the type 1 crystal.
The third hole (263–267), formed between Gln-263(d) and Tyr-267(a), also coincides with the splaying point of the coiled-coil (9). This hole is also associated with a slight, but significant, expansion of the helical spacing HD to 10.2 Å (Fig. 4 a). In this hole, one water molecule interacts with Gln-263(d) in all of the molecules analyzed in this study (Fig. 3 b, Table 2). Gln-263(d) is a hydrophilic residue, which is unsuitable for the core d-position in the heptad repeat (21,22).
The interruption of the tight hydrophobic core as well as the exposure of the hydrophobic core residues to the solvent should greatly destabilize the Tm coiled-coil. Water molecules have been identified in each of the three holes. This means not only that the core residues at the holes are exposed to the solvent but also that the water molecules are structured and are localized at stable positions in the vicinities of the core residues.
As described above, the broken core region around Glu-218(a) is associated with a peak of the B-factors in the type 2 crystal. In the previous ZrsTm176 crystal, in which the molecule is remarkably bent at this broken core region, the average B-factor around Glu-218(a) was unusually high: 110.2 Å2 (11). These findings suggest that, in the broken core region, Tm is not only readily bent but also dynamically flexible.
The formation of core-breaking holes would be defined by the Tm sequence, not by external forces. This is because the hole formation is associated with increased chain separation (HD), and the HD profiles are independent of the crystal form (Fig. 4 a; 11). What aspects of the Tm sequence give rise to the holes in the core? The presence of a charged or a polar residue at the a- or d-position seems to play the major role in the formation of a hole, like Glu-218(a) for the extended hole (214–221) or Gln-263(d) for hole (263–267). Especially, the pair of negatively charged side chains of Glu-218(a) dramatically destabilizes the coiled-coil structure (22). Moreover, Glu-218(a) is well conserved from Caenorhabditis elegans to Homo sapiens, indicating that this residue must play an important role in the function of Tm. Second, the occurrence of bulky side chains, Tyr-214(d) and Tyr-221(d), at either side of Glu-218(a), must contribute to the larger chain separation. Third, the compact side chain of Ala-211(a), placed between Leu-207(d) and Tyr-214(d), could leave an empty space in the core.
Another broken core region at around Asp-137(d)?
In the amino acid sequence of rabbit skeletal muscle α-Tm, only two acidic residues are located at the core, Asp-137(d) and Glu-218(a). Therefore, the region around Asp-137(d) is another candidate for a broken core region. This residue is also evolutionally well conserved. In the recently reported crystal structure of Mid-Tm (10), the authors explained that the hole was in the core around Ala-134(a), between the large residues Met-127(a) and Met-141(a). No water molecule was identified in the hole, presumably because the high B-factor, 70.54 Å2 on average between Ala-134(a) and Met-141(a), obscured the water molecules. We suggest that the broken core region around Asp-137(d) would also be flexible. After submission of this manuscript, Sumida et al. (23) reported that the Asp-137 region is much more susceptible to tryptic digestion than any other part (including the Glu-218 region) in the middle of Tm. Although they demonstrated that the Asp-137 is flexible, the results do not necessarily deny the flexible nature of the Glu-218 region. It is important to know how the two regions are different in local dynamics, as they pointed out.
Functional importance of the flexibility around Gln-263(d)
The broken core region around Gln-263(d) has also been thought to be flexible. It was proposed (24) that Gln-263(d) is required to form a ternary complex between troponin T, the C-terminus, and the N-terminus of Tm, and for the complex formation, the flexibility at this point should be essential. Consistent with this idea, the replacement Q263L made the fragment 251–284 more stable but less capable of forming a ternary complex (24). Recently, the NMR structure of the overlapping complex between the N- and C-terminal Tm fragments indicated that the side chain of Gln-263(d) existed in a dynamic equilibrium between two (or more) conformations in both the free state and the bound state with the N-terminal Tm (25). The side-chain flexibility is well accounted for by the increased (and presumably fluctuating) separation of the two chains. Residue 263 may also be important in relation to the Tn binding. Thus, this position is occupied by Gln and Glu in the vertebrate and invertebrate skeletal muscle Tms, respectively, which function together with Tn. In contrast, in smooth muscle and nonmuscle tissues, which lack Tn, Ala is at position 263 of Tm.
Functional significance of the broken core regions around Glu-218(a) and Asp-137(d)
The functional significance of the broken core regions is not well understood. However, it is worth noting that the alanine clusters (consisting of three or more consecutive core residues with small side chains, like Ala or Ser) and the broken core region occur alternately (Figs. 1 a and 4 b). Moreover, the alanine clusters appear to accumulate the strain, whereas the broken core regions seem to relax the strain (see below).
Undulating coiled-coil
Our analyses of the distributions of the local helical parameters along the length of Tm revealed that the Tm molecule is an undulating coiled-coil. The manner of undulation, but not the bending, is specified by the amino acid sequence. The local helical parameters of the molecules were calculated using the program TWISTER (20). The HD, the LP of the twist of each α-helix about the axis of the coiled-coil, the LBA, and the LSA, were defined previously (11).
Interestingly, the profiles of HD and LP (Fig. 4 a) are almost identical, not only between the type 1 and type 2 crystals of this study but also to the previously reported ZrsTm176 crystal (11). In each crystal, the minima in HD coincide with two alanine clusters, A179(d)–A183(a)–S186(d) and A235(d)–A239(a)–A242(d), where HD is 8.0 Å at residue 238, compared with the averaged value of 9.6 Å. In the broken core region Y214(d)–E218(a)–Y221(d), HD yields the peak value of 11.0 Å at residue 217, in both crystal types in this study. This peak HD value is also comparable to the corresponding value of 11.3 Å, in the previous crystal of ZrsTm176, although the structural consequences between the case here and previous cases are substantially different: neither the type 1 nor type 2 molecule bends at this point, whereas the previous molecule ZrsTm176 had a remarkable bend at this position (11).
The LP profile (Fig. 4 a) also shows a maximum around the region of Tyr-214(d)–Glu-218(a)–Tyr-221(d). In the type 1 crystal, the maximum in the crossover distance is 242.0 Å at around residue 224, as compared with the averaged value of 152.2 Å. In the type 2 crystal, the maximum is 257.5 Å at residue 218, as compared with the averaged value of 159.7 Å. In the previously solved ZrsTm176 crystal, the corresponding value was 334.7 Å at residue 218. This extremely large value must be contributed by the extra bending of the molecule at this point.
In contrast to HD and LP, the profiles of the LBA and the LSA of the type 1 and type 2 crystals here differ substantially from each other (Fig. 4, b and c) as well as from the previous crystal of Zrs176 (11). In both the type 1 and type 2 crystals, the molecules bend at around Arg-244(f), proximal to the alanine cluster Ala-235(d)–Ala-239(a)–Ala-242(d). This bending is probably caused by the crystal packing, because the bending position coincides with the interaction locus between Arg-244(f) of the molecule in the upper layer and Asn-293(f) (of the GCN4 sequence) of the lower layer (Fig. 2 c). In the crystal of ZrsTm176 (11), the molecules were almost straight at the residue Arg-244(f).
The profiles of the LSA indicate that two peaks coincide with the positions of two alanine clusters, Ala-179(d)–Ala-183(a)–Ser-186(d) and Ala-235(d)–Ala-239(a)–Ala-242(d) (Fig. 4, b and c). In the vicinity of the first alanine cluster, the two chains stagger in the same direction in the type 1 and in type 2 crystals, whereas in the vicinity of the second alanine cluster, the two chains stagger in opposite directions between the two types of crystals (Fig. 4 c). The interchain staggering in opposite directions is probably caused by the distinct manner of the crystal packing around this region in the two crystals (Fig. 2 c). The distinct hydrogen-bond networks around the alanine cluster are likely to exert forces on this region in different directions, which give rise to staggering due to weak interactions between the small side chains of the core residues.
These results indicate that the separation of the two chains, HD, and the LP must be determined by the amino acid sequence and are not substantially influenced by the molecular packing in the crystal lattice. On the other hand, the LBA and the associated LSA are primarily caused by the molecular packing. Third, the regions that are bent in at least one crystal form, including either end of the alanine clusters Ala-179(d)–Ala-183(a)–Ser-186(d) and Ala-235(d)–Ala-239(a)–Ala-242(d) as well as the broken core region Tyr-214(d)–Glu-218(a)–Tyr-221(d) seemed to readily bend by an external force from other molecules in the crystal lattice. Consistent with this view, the B-factors are high in the region Tyr-214(d)–Glu-218(a)–Tyr-221(d) (see above).
The strain in Tm is not localized but is distributed throughout the molecule
The question arises whether the staggering (as well as the bending) is localized in a limited part of the molecule, leaving the remaining parts unaltered, or if the strain is distributed over a wide range of the molecule. To address this question, the conformations of two molecules, molecule AB of type 1 crystal versus molecule AB of type 2 crystal, were compared in two different ways. When the two molecules were aligned by minimizing the sum of the RMSD over the entire molecule, the profile of the Cα-Cα distances plotted against the residue number n revealed a broad peak around the second alanine cluster, with a peak displacement as large as 4 Å, which is roughly equivalent to the radius of an α-helix (Fig. 5 a, black line). Consistent with this, when the two molecules were superposed by aligning the middle parts of the molecule, the C-termini of the molecules were displaced substantially relative to each other (Fig. 5 b). The substantial differences in the overall conformations of the two molecules are expected from the distinct manners of crystal packing. On the other hand, when the two molecules are aligned in segment-to-segment bases, for the minimal sum of RMSD over each 14 residue segment (from n − 6 to n + 7), the profile of the Cα-Cα distances plotted against the residue number n is totally flat (Fig. 5 a, red line).
FIGURE 5.

(a) Comparison of the conformations between chain A of the type 1 crystal and chain A of the type 2 crystal. Black lines represent the conformational differences as the Cα- Cα distance plotted against the residue number n, after the two chains were aligned for the minimal sum of the RMSD over the entire chains. Red lines represent the same plot as above, but after aligning each pair of 14-residue chain segments (from n − 6 to n + 7) for the minimal sum of the RMSD over the window. If the conformational differences between the two chains (black line) were restricted to hinge-like, narrow regions, then the red profile would have sharp peaks. This graph indicates that this is not the case. (b) The superposition of the main chains of the two molecules, molecule AB of type 1 (white) and molecule AB of type 2 (cyan). The left panel is the side view, and the right panel is the end-on view from the C-terminus. The two molecules are placed by superposing the segment (169–225) of each molecule, in which the Cα- Cα distance is relatively small, as indicated in the box in a. The colored bars along the abscissa are the same as in Fig. 3.
The results described above are not consistent with the view that the Tm molecule consists of a limited number of regions with large strain and the remaining canonical and unaltered regions, which would give rise to a limited number of sharp peaks in the RMSD profile. The flat RMSD profile upon the alignment on segment-to-segment bases indicates that the strain is distributed over wide regions along the length. In other words, Tm should not be regarded as being composed of segments of rigid rods connected by flexible joints, but rather as a seamless rubber rod patched with more flexible regions, as compared to the remainder. Even if a Tm segment had ideal core residues for canonical coiled-coil formation, it may not always form a stable coiled-coil. We speculate that, in this way, all of the parts of Tm may communicate with one another.
It is worth noting that, although the strain is widely distributed along the length of Tm, there is a tendency in the distribution. The stagger (LSA) is maximal at the alanine clusters, whereas it is almost null in the broken core regions (Fig. 4, b and c). There is a possibility that the alanine clusters can accumulate the strains, and the broken core regions can relax the strain.
Possible influences of GCN4 sequence extensions
Finally, it is worth noting three points about possible influences of the GCN sequence extensions on the structures and properties of Tm. First, the GCN4 sequence extensions appear to facilitate good quality crystal formation, not only through the possible stabilizing effect of the coiled-coil of Tm but also (or more so) through providing suitable sites for intermolecular interactions for the crystal formation. Both in type 1 and type 2 crystals, Glu-292(e) and Asn-293(f) of the GCN4 extension contribute to the crystal formation. This was also true for the crystal of ZrsTm176 (11): the side chain contact between Tyr-261(b) of Tm and Tyr-163(b) of the GCN4 extension appeared to be crucial for the crystal formation.
Second, among the three crystals studied, the profiles of the chain separation (HD) and local helix pitch (LP) were almost identical, whereas the shapes (LBA and LSA) of the Tm segment were different to each other. Because the three crystal forms were distinct in the crystal lattice and because the lattice is formed through the GCN4 extension, the GCN4 extension does not influence the properties associated with HD and LP, although it does exert stress to change the shape of the Tm segment.
Third, it might be true that the GCN4 extension should make the Tm segments less flexible. Because all of the parts of Tm may communicate with one another (see above), Tm segments must be influenced by any extension at either end. Nevertheless we can learn much about the flexibility of Tm from these crystal structures with the GCN4 extensions. Flexibility may consist of two parts: one is intrinsic to the Tm sequence, which is supposed to be unaffected by the extensions, whereas the other is affected by the GCN4 extensions. We can learn about the former, leaving the latter for future study.
Acknowledgments
We thank T. Hikima for assistance with data collection.
This work was supported by the ERATO grant from the Japan Science and Technology Agency (JST).
The coordinate set has been deposited with the RCSB Protein Data Bank, with the accession code of 2EFR (type 1 crystal) and 2EFS (type 2 crystal).
Editor: Malcolm Irving.
References
- 1.Ebashi, S., and M. Endo. 1968. Calcium ion and muscle contraction. Prog. Biophys. Mol. Biol. 18:123–183. [DOI] [PubMed] [Google Scholar]
- 2.McLachlan, A. D., and M. Stewart. 1975. Tropomyosin coiled-coil interactions: evidence for an unstaggered structure. J. Mol. Biol. 98:293–304. [DOI] [PubMed] [Google Scholar]
- 3.Parry, D. A. 1975. Analysis of the primary sequence of alpha-tropomyosin from rabbit skeletal muscle. J. Mol. Biol. 98:519–535. [DOI] [PubMed] [Google Scholar]
- 4.Crick, F. 1953. The packing of α-helices: simple coiled-coils. Acta Crystallogr. 6:689–697. [Google Scholar]
- 5.Phillips, G. N. Jr., J. P. Fillers, and C. Cohen. 1986. Tropomyosin crystal structure and muscle regulation. J. Mol. Biol. 192:111–131. [DOI] [PubMed] [Google Scholar]
- 6.Whitby, F. G., H. Kent, F. Stewart, M. Stewart, X. Xie, V. Hatch, C. Cohen, and G. N. Phillips Jr. 1992. Structure of tropomyosin at 9 angstroms resolution. J. Mol. Biol. 227:441–452. [DOI] [PubMed] [Google Scholar]
- 7.Whitby, F. G., and G. N. Phillips Jr. 2000. Crystal structure of tropomyosin at 7 angstroms resolution. Proteins. 38:49–59. [PubMed] [Google Scholar]
- 8.Brown, J. H., K. H. Kim, G. Jun, N. J. Greenfield, R. Dominguez, N. Volkmann, S. E. Hitchcock-DeGregori, and C. Cohen. 2001. Deciphering the design of the tropomyosin molecule. Proc. Natl. Acad. Sci. USA. 98:8496–8501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li, Y., S. Mui, J. H. Brown, J. Strand, L. Reshetnikova, L. S. Tobacman, and C. Cohen. 2002. The crystal structure of the C-terminal fragment of striated-muscle α-tropomyosin reveals a key troponin T recognition site. Proc. Natl. Acad. Sci. USA. 99:7378–7383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown, J. H., Z. Zhou, L. Reshetnikova, H. Robinson, R. D. Yammani, L. S. Tobacman, and C. Cohen. 2005. Structure of the mid-region of tropomyosin: bending and binding sites for actin. Proc. Natl. Acad. Sci. USA. 102:18878–18883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nitanai, Y., S. Minakata, K. Maeda, N. Oda, and Y. Maeda. 2007. Crystal structures of tropomyosin: flexible coiled-coil. Adv. Exp. Med. Biol. 592:137–151. [DOI] [PubMed] [Google Scholar]
- 12.Kluwe, L., K. Maeda, A. Miegel, S. Fujita-Becker, Y. Maeda, G. Talbo, T. Houthaeve, and R. Kellner. 1995. Rabbit skeletal muscle α α-tropomyosin expressed in baculovirus-infected insect cells possesses. the authentic N-terminus structure and functions. J. Muscle Res. Cell Motil. 16:103–110. [DOI] [PubMed] [Google Scholar]
- 13.Adachi, S., T. Oguchi, H. Tanida, S. Y. Park, H. Shimizu, H. Miyatake, N. Kamiya, Y. Shiro, Y. Inoue, T. Ueki, and T. Iizuka. 2001. The RIKEN structural biology beamline II (BL44B2) at the SPring-8. Nucl. Instrum. Methods Phys. Res. A. 467:711–714. [Google Scholar]
- 14.Otwinowski, Z., and W. Minor. 1997. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276:307–326. [DOI] [PubMed] [Google Scholar]
- 15.Pflugrath, J. W. 1999. The finer things in x-ray diffraction data collection. Acta Crystallogr. D Biol. Crystallogr. 55:1718–1725. [DOI] [PubMed] [Google Scholar]
- 16.McCoy, A. J., R. W. Grosse-Kunstleve, L. C. Storoni, and R. J. Read. 2005. Likelihood-enhanced fast translation functions. Acta Crystallogr. D Biol. Crystallogr. 61:458–464. [DOI] [PubMed] [Google Scholar]
- 17.Collaborative Computational Project, Number 4. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50:760–763. [DOI] [PubMed] [Google Scholar]
- 18.Brunger, A. T., P. D. Adams, G. M. Clore, W. L. DeLano, P. Gros, R. W. Grosse-Kunstleve, J. S. Jiang, J. Kuszewski, M. Nilges, N. S. Pannu, R. J. Read, L. M. Rice, T. Simonson, and G. L. Warren. 1998. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54:905–921. [DOI] [PubMed] [Google Scholar]
- 19.Emsley, P., and K. Cowtan. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 20.Strelkov, S. V., and P. Burkhard. 2002. Analysis of alpha-helical coiled coils with the program TWISTER reveals a structural mechanism for stutter compensation. J. Struct. Biol. 137:54–64. [DOI] [PubMed] [Google Scholar]
- 21.Tripet, B., K. Wagschal, P. Lavigne, C. T. Mant, and R. S. Hodges. 2000. Effects of side-chain characteristics on stability and oligomerization state of a de novo-designed model coiled-coil: 20 amino acid substitutions in position “d”. J. Mol. Biol. 300:377–402. [DOI] [PubMed] [Google Scholar]
- 22.Wagschal, K., B. Tripet, P. Lavigne, C. Mant, and R. S. Hodges. 1999. The role of position a in determining the stability and oligomerization state of alpha-helical coiled coils: 20 amino acid stability coefficients in the hydrophobic core of proteins. Protein Sci. 8:2312–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sumida, J. P., E. Wu, and S. S. Lehrer. 2008. Conserved Asp137 imparts flexibility to tropomyosin and affects function. J. Biol. Chem. 283:6728–6734. [DOI] [PubMed] [Google Scholar]
- 24.Greenfield, N. J., T. Palm, and S. E. Hitchcock-DeGregori. 2002. Structure and interactions of the carboxyl terminus of striated muscle alpha-tropomyosin: it is important to be flexible. Biophys. J. 83:2754–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Greenfield, N. J., Y. J. Huang, G. V. Swapna, A. Bhattacharya, B. Rapp, A. Singh, G. T. Montelione, and S. E. Hitchcock-DeGregori. 2006. Solution NMR structure of the junction between tropomyosin molecules: implications for actin binding and regulation. J. Mol. Biol. 364:80–96. [DOI] [PubMed] [Google Scholar]