

Abstract
Photosynthetic light-harvesting proceeds by the collection and highly efficient transfer of energy through a network of pigment-protein complexes. Interchromophore electronic couplings and interactions between pigments and the surrounding protein determine energy levels of excitonic states, and dictate the mechanism of energy flow. The excitonic structure (orientation of excitonic transition dipoles) of pigment-protein complexes is generally deduced indirectly from x-ray crystallography, in combination with predictions of transition energies and couplings in the chromophore site basis. We demonstrate that coarse-grained, excitonic, structural information in the form of projection angles between transition dipole moments can be obtained from the polarization-dependent, two-dimensional electronic spectroscopy of an isotropic sample, particularly when the nonrephasing or free polarization decay signal, rather than the photon echo signal, is considered. This method provides an experimental link between atomic and electronic structure, and accesses dynamical information with femtosecond time resolution. In an investigation of the Fenna-Matthews-Olson complex from green sulfur bacteria, the energy transfer connecting two particular exciton states in the protein was isolated as the primary contributor to a crosspeak in the nonrephasing two-dimensional spectrum at 400 femtoseconds under a specific sequence of polarized excitation pulses. The results suggest the possibility of designing experiments using combinations of tailored polarization sequences to separate and monitor individual relaxation pathways.
INTRODUCTION
Photosynthesis begins with the harvesting of sunlight by antenna pigments that rapidly funnel energy to reaction centers. The spectral coverage and energy transfer characteristics of light-harvesting systems are determined by the structural arrangement of pigments and their interactions with the surrounding environment, which is often the interior of a protein. Even among organisms with chlorophyll as the primary light absorber, the architecture of antenna systems varies widely in nature (1). Investigation of these specialized light-harvesting structures, which evolved under different light environments to fuel photosynthesis with optimal efficiency, could have applications in solar-energy conversion devices, and such investigations become increasingly important as we search for clean energy alternatives.
The Fenna-Matthews-Olson (FMO) pigment-protein complex is found in low light-adapted green sulfur bacteria, which harvest light primarily in a large antenna structure called the chlorosome. The FMO complex is tasked with transporting energy collected in the chlorosome to the reaction center, thereby initiating the photochemistry that ultimately leads to the chemical storage of energy. The FMO complex was the first chlorophyll protein structure solved by x-ray crystallography, and comprises three identical subunits, each containing seven bacteriochlorophyll (BChl) pigments nested within beta sheets (2,3). The closest center-to-center distance between neighboring intrasubunit BChls is 11 Å, with the largest coupling energies between them estimated to be on the order of 100 cm−1. The closest approach of intersubunit BChls is ∼24 Å, with corresponding coupling energies of <20 cm−1 (4). For this reason, it has been assumed (and borne out by spectroscopic studies) that the exciton wave function is rapidly localized on individual subunits upon excitation (5). Compared to the highly symmetric ring structures of the light-harvesting apparatus in the similarly widely studied purple photosynthetic bacteria, the asymmetric arrangement of the seven pigments in FMO is more reminiscent of light harvesting in higher plants. Because of its lack of symmetry and the early availability of x-ray structure information in conjunction with its relatively small size, FMO has been considered a model system for the investigation of photosynthetic energy transfer.
A number of spectroscopic experiments and theoretical studies have contributed to a good understanding of the energetic landscape in FMO (5–7). The lack of symmetry within the FMO subunit has presented a challenge for researchers modeling experimental spectra, because each of the seven BChls experiences a different local environment due to, for example, the proximity of charged residues or bowing of the BChl macrocycle, resulting in variations of Qy site energies (the S0-S1 transition energy of the chromophore) by as much as 600 cm−1 (4). Adolphs and Renger performed calculations of site energies, using an electrostatic method that for the first time predicted absorption and linear and circular dichroism spectra (8). Müh et al. (9) further incorporated atomic details of the protein, including the backbone, into quantum chemical/electrostatic calculations of excitation energies, and discovered a pronounced effect of the alpha-helix dipole on BChl site energies. In this way, the protein itself can direct excitation flow through the complex by tuning the site energies.
Several time-resolved spectroscopic studies showed that energy transfer in FMO proceeds with time constants ranging from ∼100 fs to several ps, with faster transfer generally occurring on the blue side of the Qy band (10). Two-dimensional (2D) electronic spectroscopy is a particularly incisive tool for investigating photosynthetic energy transfer, yielding maps of coupling and dynamical processes of electronic transitions. Brixner et al. (11) identified two major pathways of energy flow in FMO, using 2D electronic spectroscopy. The nature of energy transfer within the protein was also investigated, and evidence of quantum coherence in fluorescence anisotropy (12) and 2D spectroscopy data (13) imply that quantum mechanical wavelike energy transfer rather than purely dissipative “hopping” may contribute to the efficiency of light-harvesting by FMO.
Excitation energy transfer in photosynthetic complexes is controlled by the orientation and spatial extent of exciton states, referred to here as the excitonic structure, in addition to the energetic landscape of the protein. In a multichromophore system, nonnegligible coupling energies give rise to exciton states, with transition dipole moments distinct from those of the individual chromophores. The Coulombic couplings redistribute electron transition density among chromophore sites, shifting transition energies and reorienting transition dipole moments. The excitonic structure of photosynthetic complexes is generally determined indirectly from estimates of site and coupling energies, with the latter often based on the dipole-dipole approximation despite the close packing of chromophores. An experimental method to probe excitonic structure directly for a further understanding of energy transfer dynamics is therefore desirable.
Here, we demonstrate that 2D electronic spectroscopy can be used to obtain excitonic structure information, in addition to elucidating energetic relaxation pathways. Based on the generation of a three-pulse photon echo, analogous to the spin echo in NMR, 2D spectroscopy is sensitive to the third-order optical polarization of the system. In effect, the technique connects absorption and emission frequencies by interrogating the sample with three time-delayed femtosecond laser pulses, and monitoring the emitted signal (14). The frequency-domain signal, collected as a function of the two scanned time delays called the “coherence” time, or τ (between the first two pulses), and the “population” time, or T (between the second two pulses), is Fourier- transformed over τ to yield 2D frequency-frequency plots as a function of T. The ability to overcome phase fluctuations has played a key role in making 2D spectroscopy feasible at visible wavelengths. To this end, passively stabilized 2D setups based on diffractive optics (15,16), or actively stabilized techniques using feedback-controlled optics, are employed (17–19). Grumstrup et al. demonstrated the phase stable measurement of absorptive 2D electronic spectra, using pulse shapers (20).
Although 2D spectra contain information often unavailable or obscured in other femtosecond laser experiments, broad line-widths can nevertheless complicate the interpretation of spectra. Two-dimensional spectra contain signals arising from many different energetic processes, and the separation of individual contributions is desirable for a better characterization of excited state dynamics. This can be achieved in part by rotating the polarization of the laser pulses to suppress or enhance particular signals, based on the relative orientations of transition dipoles in the system. Polarization conditions for 2D infrared (IR) spectroscopy of an isotropic sample were pioneered by Hochstrasser (21) and Zanni et al. (22), and were further explored theoretically by Dreyer et al. (23) Sequences have been identified that suppress diagonal peaks in 2D spectra, and it was shown that disentangling 2D spectra in this way is also feasible in the electronic regime (24,25). Using cross-polarized excitation pulses, angles between transition dipole moments were determined from 2D IR spectra (26,27). These polarization conditions do not require aligning the molecules in the sample cell, but rather take advantage of the set relative orientations of excited transitions within molecules. Here, we use polarization-dependent 2D femtosecond electronic spectroscopy to probe the excitonic structure and dynamics of FMO from Prosthecochloris aestuarii.
Separating rephasing (photon echo) and nonrephasing contributions to the signal is an additional tool for disentangling the complicated interference pattern that makes up a 2D spectrum. So-called phase-twisted lineshapes, with a mixed absorptive and dispersive character, were identified in 2D NMR and IR spectra when rephasing and nonrephasing signals were measured separately, and addition of the two spectra was used to obtain absorptive lineshapes (28,29). Ge et al. discussed the separate measurement of rephasing and nonrephasing signals for enhanced resolution of closely spaced peaks in 2D IR spectra (30). In rephasing contributions, the system evolves in conjugate frequencies during τ and t (the time delay between the last two pulses). This gives rise to a photon echo signal, which reveals the system's ability to effectively reverse inhomogeneous dephasing. For nonrephasing signals, the phase factors governing the evolution of coherence during τ and t have the same sign, resulting in free polarization decay during t. The measurement of rephasing and nonrephasing spectra is achieved experimentally by reversing the order of arrival of the first two pulses at the sample, and the relative amplitude of the two signals at long T is a measure of the degree of inhomogeneous broadening in the system (27). Here, we monitor both types of signals separately, and observe distinct spectral features in nonrephasing spectra. Combined with the use of specific polarization schemes, the nonrephasing spectra directly reveal information about the excitonic structure, or orientation of excitonic transition dipoles, of the protein complex, and give a more detailed view of energetic relaxation.
EXPERIMENTAL METHODS
The FMO from P. aestuarii was isolated as described in Li et al. (31). A Sephacryl (Pharmacia, Uppsala, Sweden ) S-300 high-resolution gel-filtration column was used as a final purification step. The optical density (OD) ratio of OD 267 nm/OD 371 nm was <0.55, indicative of a highly purified complex. The sample was dissolved in a buffer solution (at pH 8) of 20 mM Tris-HCl, and 0.1% of lauryldimethylamine oxide (LDAO) was added to prevent aggregation. The sample was then mixed with glycerol (30:70 by volume) for formation of a glass in a 200-μm-thick quartz cell for measurements at 77 K. The sample OD was 0.22 at 805 nm.
Details of the experimental apparatus were described elsewhere (24,32). Briefly, 45-fs pulses at 805 nm, with a spectral bandwidth of 31-nm output from a home-built Ti:Sapphire regenerative amplifier, are used to measure 2D spectra. The beam is split into four by a beam-splitter, followed by a diffractive optic optimized for the transmission of ±1st orders. The time delay between the first two pulses (coherence time, τ) is scanned by the movement of paired glass wedges controlled by a computerized stepper motor, and the time delay between the second and third pulses (population time, T) is controlled by a retroreflector delay stage. The beams are focused to a 70-μm spot-size on the sample in the boxcar geometry, with 4 nJ/pulse in pulses 1, 2, and 3. The fourth pulse is attenuated by four orders of magnitude to serve as a local oscillator for heterodyne detection, and passes through the sample before the other pulses. A mask selects the signal emitted from the sample in the phase-matched direction ks = −k1 + k2 + k3, collinear with beam 4. The signal is spectrally resolved with a spectrometer and charge-coupled device camera. The polarizations of pulses are controlled by true zero-order half-waveplates inserted in beams 1 and 2, and in this work the combinations 〈0°, 0°, 0°, 0°〉, 〈45°, −45°, 0°, 0°〉, and 〈75°, −75°, 0°, 0°〉 are used (with the bracket expressions denoting the linear polarizations of pulses 〈1, 2, 3, 4〉, where 4 is the local oscillator). The measurement is performed by scanning the coherence time from −300 to 300 fs, where negative τ corresponds to the arrival of pulse 2 before pulse 1, for each population time point. For a given T, Fourier transformation is performed, using all coherence time points to yield the total 2D spectra, whereas for rephasing and nonrephasing spectra, only positive and negative time points are Fourier-transformed, respectively.
Theoretical methods
Theoretical calculations of 2D spectra are performed based on Zigmantas et al. (33) In brief, a Frenkel exciton model of the FMO monomer is adopted in which 1-exciton and 2-exciton manifolds are built from site and coupling energies of Qy transitions of the seven BChls, with orientations of site transition dipole moments determined from the protein x-ray structure (34). Couplings between BChls in different monomeric subunits of the protein are neglected, because of their relatively weak strength. An overdamped Brownian oscillator model for nuclear motions resulting from interaction with the protein environment (electron-phonon coupling) is adopted (35), with no correlation between energetic fluctuations at different sites. Dynamics between exciton states are calculated, using a modified Redfield theory in the secular approximation (36,37). A Gaussian distribution of site energies represents inhomogeneous broadening because of slow fluctuations of the protein environment, as determined by the line widths in the spectra and the amplitude ratio of rephasing to nonrephasing spectra. All model parameters are published in the Supplementary Material (Data S1).
Two-dimensional spectra are calculated using the third-order nonlinear response function, R(3), described in detail by Mukamel (35). The response function describes a sum of Liouville energy level pathways contributing to the signal in the phase-matched direction, assuming the impulsive excitation limit and the rotating wave approximation. The 2D spectrum, S2D, is then proportional to the response function:
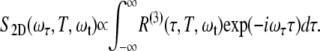 |
(1) |
The rephasing and nonrephasing spectra are calculated using response pathways
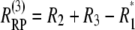 |
(2) |
and
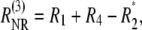 |
(3) |
respectively, in the conventional nomenclature. Each of these time-domain response pathways describes a sequence of promotions and demotions of electrons between states of the system stimulated by the laser pulse fields, and includes the evolution of the system during intervening time periods. The full form of the response functions can be found elsewhere (23,35), but here it is useful to note that the contribution to the 2D spectrum by each pathway is controlled by a prefactor of the form 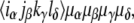 where α, β, γ, and δ represent transitions connecting pairs of states,
where α, β, γ, and δ represent transitions connecting pairs of states, 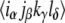 is the orientationally averaged projection of the laboratory-frame linear polarizations of the laser pulses onto the molecular frame transition dipole moments, and
is the orientationally averaged projection of the laboratory-frame linear polarizations of the laser pulses onto the molecular frame transition dipole moments, and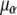 denotes the dipole moment magnitude of molecular transition α.
denotes the dipole moment magnitude of molecular transition α.
RESULTS AND DISCUSSION
Fig. 1 shows the real total, rephasing, and nonrephasing 2D spectra of FMO from P. aestuarii at 77 K, denoted as S(λτ,T,λt), S(λ+τ,T,λt), and S(λ−τ,T,λt), respectively. Features with a positive sign are shown in green to red, and correspond to stimulated emission or bleaching, whereas blue features are negative, indicating an excited state absorption. Peaks in the rephasing and nonrephasing parts tilt along and perpendicular to the diagonal, respectively, as is characteristic in 2D spectra from NMR through the optical regime. The total spectrum, which is equivalent to the summation of the two signals, is dominated by the rephasing part. The ratio of rephasing to nonrephasing signal amplitudes (all normalized in Fig. 1 for clarity) is 4.7 at T = 0 and 3.3 at T = 400 fs. The rephasing signal decays more quickly than the nonrephasing signal because of rapid homogeneous dephasing, or loss of memory, which diminishes the system's ability to emit a photon echo. As discussed previously by Ernst et al. (28), phase-twisted lineshapes in separated rephasing and nonrephasing signals give rise to distinctive interference effects in the spectra. Nonrephasing spectra have the advantage that closely spaced peaks are easier to distinguish than in rephasing spectra because of their antidiagonal tilt, i.e., the diagonal line-width of an individual transition is narrower in the nonrephasing spectrum than in the rephasing spectrum. In both types of spectra, interference can give rise to shifts in the center positions of peaks, obscuring the true transition energies. Adding the two types of signals to obtain the total spectrum serves to cancel the dispersive character of peaks, in favor of purely absorptive lines.
FIGURE 1.

Real 2D spectra at 77 K of FMO from P. aestuarii. Spectra shown from left to right are total, rephasing, and nonrephasing signals at T = 0 (top) and T = 400 fs (bottom). All spectra are normalized to unity. The T = 0 nonrephasing spectrum clearly shows distinctive diagonal peaks and crosspeaks not apparent in the rephasing spectrum, and energetic relaxation out of the upper band is visible in the nonrephasing spectrum at T = 400 fs.
In Fig. 1, the T = 0 total spectrum is characterized by a strong positive signal along the diagonal comprising several overlapping bands, weaker positive off-diagonal features that merge into the diagonal peaks, and excited state absorption above the diagonal. The rephasing and nonrephasing signals are strikingly different. The T = 0 rephasing spectrum contains almost no features aside from one wide, inhomogeneously broadened diagonal band, whereas the nonrephasing spectrum contains several distinct peaks along the diagonal, as well as strong off-diagonal features. The total spectrum, i.e., the sum of both signals, retains some of this structure. In particular, the two most prominent diagonal bands of the nonrephasing spectrum give rise to the strong double peak along the diagonal in the total spectrum (peaks at 814 nm and 804 nm). In the second row of Fig. 1, the T = 400 fs spectra show that the nonrephasing spectrum gives a clearer view of dynamics. The rephasing spectrum shows little evolution on this timescale, whereas the excitation energy migration out of the upper bands (most notably, the disappearance of the (λτ = 804 nm, λt = 804 nm) diagonal peak) is clearly visible in the nonrephasing spectrum.
The rephasing and nonrephasing parts of the third-order polarization are sensitive to the same excitation energy transfer dynamics. However, energy transfer is more apparent in the nonrephasing spectrum. This results from the narrower diagonal line widths of nonrephasing signals and from the photon echo effect that gives rise to dominant diagonal peaks in rephasing spectra, obscuring off-diagonal peaks that may reveal information about energy transfer. In addition, as discussed by Cheng and Fleming (38), Liouville pathways that cause excitonic quantum beating appear off and on the diagonal in rephasing and nonrephasing spectra, respectively. Thus a clearer view of energetic relaxation is obtained from the off-diagonal region of nonrephasing spectra. In Fig. 1, it is clear that splitting the total spectrum into its constituent +τ and −τ parts aids in analysis because, although the total spectrum has the advantage of absorptive lineshapes, the nonrephasing part shows more clearly the individual excitonic features.
Additional separation of signals can be achieved in 2D spectra by the use of specific polarization combinations of excitation pulses. The polarization conditions used in this experiment were chosen because it is theoretically possible to derive φ, the projection angle between a pair of exciton states, directly from the ratio of the 〈45°, −45°, 0°, 0°〉 and 〈75°, −75°, 0°, 0°〉 spectra (except that it is always impossible to distinguish between φ and 180° − φ). This scheme is similar to the cross-polarized 〈90°, 90°, 0°, 0°〉/〈0°, 0°, 0°, 0°〉 method used previously in 2D IR spectroscopy (26,27). The advantage of the sequences used here is that the diagonal peak amplitudes are more suppressed compared with those in the cross-polarized experiment, which is necessary for gleaning information from 2D electronic spectra with broad line widths and closely spaced states. Although it is still difficult to separate individual transitions, which prevents direct acquisition of φ, comparison of the spectra measured under these polarization conditions enables the deduction of excitonic structural information.
Fig. 2 shows the absolute magnitude 2D spectra of FMO from P. aestuarii measured under the two polarization schemes, 〈45°, −45°, 0°, 0°〉 and 〈75°, −75°, 0°, 0°〉, at T = 400 fs. The spectra in the top row are absolute magnitude rephasing, |S(λ+τ,T,λt)|, and those in the bottom row are absolute magnitude nonrephasing, |S(λ−τ,T,λt)|. In magnitude spectra, the diagonal and antidiagonal tilts of rephasing and nonrephasing peaks become difficult to distinguish. The striking aspect of the spectra is that large differences between these two different polarization experiments are only readily visible in the nonrephasing spectra. This again points to the utility of isolating the nonrephasing component to decongest spectra, and shows that polarization conditions become particularly powerful when combined with separation of the +τ and −τ parts. The disadvantage to the polarization schemes used here involves the inability to phase the data using a separate pump-probe experiment, as is performed for the real spectra shown in Fig. 1. Under experimental conditions of extremely good phase stability, it is possible to collect 2D spectra with reliable phase, without the need for an auxiliary experiment, as discussed by Read et al. (24) However, as indicated by these data, in the absence of sufficient phase stability, the absolute magnitude spectra are still valuable.
FIGURE 2.

Rephasing (top) and nonrephasing (bottom) absolute magnitude 2D spectra of FMO from P. aestuarii at T = 400 fs, measured with polarizations 〈45°, −45°, 0°, 0°〉 (left) and 〈75°, −75°, 0°, 0°〉 (right) of laser pulses 1, 2, 3, and 4, respectively. The nonrephasing spectra show distinct spectral features under the two polarization conditions that can be used to obtain excitonic structural information. The strong crosspeak at (λτ = 804 nm, λt = 814 nm) in the 〈75°, −75°, 0°, 0°〉 nonrephasing spectrum is indicative of excitation energy transfer.
To simulate the spectra, we begin with the full Hamiltonian of the FMO monomer published by Müh et al. (9), and we perform simultaneous fitting of the linear and 2D rephasing, nonrephasing, and polarized spectra. The parameters arrived at after fitting are similar to those in Müh et al. (9). Because the peaks overlap less, the parameters required to model polarized and nonrephasing spectra satisfactorily are more constrained. The similarity of our fit parameters to those arrived at by pure theory in Müh et al. (9) underscores the robustness of their method. Theoretical nonrephasing spectra are shown in the top row of Fig. 3. The general features of the experimental spectra at 400 fs are captured, and the model also shows excellent agreement with the experimental rephasing spectra (not shown). Fig. 3 (upper right) shows the experimental and theoretical linear absorption spectra, along with the spectrum of laser pulses used to measure the 2D spectra. The pulse spectrum is incorporated in 2D simulations by weighting each calculated dipole strength by the experimental laser spectrum, which has the small but noticeable effect on the calculated spectra of enhancing the higher-energy peaks.
FIGURE 3.

Theoretical and experimental nonrephasing spectra of FMO from P. aestuarii at T = 400 fs and 77 K. (Top row, left to right) Theoretical 〈0°, 0°, 0°, 0°〉, 〈45°, −45°, 0°, 0°〉, and 〈75°, −75°, 0°, 0°〉 2D spectra. (Top right) Experimental and theoretical linear absorption spectra in black and red, respectively; the dotted line indicates the laser spectrum of pulses used to measure 2D spectra. (Bottom row, left to right): Experimental 〈0°, 0°, 0°, 0°〉, 〈45°, −45°, 0°, 0°〉, and 〈75°, −75°, 0°, 0°〉 2D spectra.
The T = 400 fs 〈45°, −45°, 0°, 0°〉 and 〈75°, −75°, 0°, 0°〉 spectra in Fig. 3, simulated using the full FMO monomer Hamiltonian, reproduce the major aspect of the corresponding experimental spectra (bottom row): in the 〈45°, −45°, 0°, 0°〉 case at 400 fs, the spectrum is dominated by a diagonal peak at (λτ = 814 nm, λt = 814 nm), whereas in the 〈75°, −75°, 0°, 0°〉 spectrum, a pronounced crosspeak at (λτ = 804 nm, λt = 814 nm) is also visible. This crosspeak is due primarily to excitonic energy transfer during the population time, T. The simulated 〈75°, −75°, 0°, 0°〉 nonrephasing spectrum in Fig. 3 shows a complete disappearance of the diagonal peak at 804 nm by 400 fs, whereas the experimental spectrum shows that the upper diagonal peak persists in this timescale. This may result from an overestimation of the excitation energy transfer rate, or from an incomplete description of coherence dynamics (expected to contribute strongly to the diagonal in nonrephasing spectra) afforded by the modified Redfield theory. However, the generally similar experimental and theoretical spectra suggest that the model accurately accounts for the energetic and excitonic structure of the FMO complex and the major pathways of energy flow. The striking difference between the 〈45°, −45°, 0°, 0°〉 and 〈75°, −75°, 0°, 0°〉 spectra can be explained by the orientational factors 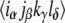 controlling the peak amplitudes for Liouville pathways, corresponding to energy transfer between 1-exciton states, as discussed above. Fig. 4 a shows the dependence of these orientational factors on the projection angle φ between a pair of exciton states for the two polarization schemes. Note that the orientational factors can be positive or negative, which we cannot distinguish in the current work because of the incompatibility of polarization schemes with the auxiliary phasing experiment. As seen in Fig. 4 a, the amplitude of an energy-transfer crosspeak in a 〈45°, −45°, 0°, 0°〉 spectrum is maximum at a projection angle of 0°, and vanishes at 52°, whereas in a 〈75°, −75°, 0°, 0°〉 spectrum, it increases (in absolute value) monotonically from 0° to 90°.
controlling the peak amplitudes for Liouville pathways, corresponding to energy transfer between 1-exciton states, as discussed above. Fig. 4 a shows the dependence of these orientational factors on the projection angle φ between a pair of exciton states for the two polarization schemes. Note that the orientational factors can be positive or negative, which we cannot distinguish in the current work because of the incompatibility of polarization schemes with the auxiliary phasing experiment. As seen in Fig. 4 a, the amplitude of an energy-transfer crosspeak in a 〈45°, −45°, 0°, 0°〉 spectrum is maximum at a projection angle of 0°, and vanishes at 52°, whereas in a 〈75°, −75°, 0°, 0°〉 spectrum, it increases (in absolute value) monotonically from 0° to 90°.
FIGURE 4.

Energy transfer pathway orientational factor dependence on projection angle between exciton states for 〈45°, −45°, 0°, 0°〉 and 〈75°, −75°, 0°, 0°〉 polarization sequences (a). Below, calculated 2D nonrephasing dimer spectra at 400 fs (top to bottom: all parallel, 〈45°, −45°, 0°, 0°〉 and 〈75°, −75°, 0°, 0°〉 polarizations) for a dimer with projection angle φ = 5° (b) and φ = 40° (c). The spectra in c reproduce more closely the behavior seen in the FMO polarization-dependent nonrephasing experiment because of the ∼40° relative orientation of the dominant transition dipoles.
The manifestation of the orientational factor dependence on φ is illustrated in Fig 4, b and c, by simulated nonrephasing 2D spectra of an excitonically coupled dimer with coupling energy at 40 cm−1, site energies at 810 and 802 nm, and an energy-transfer time constant from the upper to lower exciton states calculated from modified Redfield theory to be 350 fs. The dimer Hamiltonian parameters were chosen to reproduce roughly the two most prominent peaks in the FMO spectrum at 814 and 804 nm, to illustrate the effect of transition dipole orientation on polarization-dependent 2D nonrephasing spectra. Fig. 4 b shows the T = 400 fs spectra calculated for the dimer with φ = 5°, whereas Fig. 4 c shows the spectra calculated for the same dimer system, but with φ = 40° (the dipole strengths are adjusted to give the same relative amplitudes of diagonal peaks). The φ = 5° spectra under the 〈45°, −45°, 0°, 0°〉 and 〈75°, −75°, 0°, 0°〉 polarization schemes are nearly indistinguishable, whereas the φ = 40° spectra approximate the behavior we observed in the FMO experiment, with a crosspeak because of energy transfer appearing strongly in the 〈75°, −75°, 0°, 0°〉 spectrum and only weakly in the 〈45°, −45°, 0°, 0°〉 spectrum. Characterization of the observed energetic states and dynamics of FMO requires use of the full Hamiltonian, taking into account all seven of the BChls, as shown in Fig. 3. However, the simplified dimer model of Fig. 4 clearly illustrates that the origin of the contrast between the 〈45°,−45°, 0°, 0°〉 and 〈75°, −75°, 0°, 0°〉 nonrephasing 2D spectra lies in a relative orientation between prominent transitions near φ = 40°.
The exciton model from the full FMO monomer Hamiltonian, determined by fitting 2D spectra, is consistent with analysis of the φ = 40° dimer. According to the present FMO model, excitons 2 and 4 have the highest dipole strength, and are primarily responsible for the two strongest bands in the linear and 2D spectra at 814 and 804 nm, respectively. These states are characterized by a projection angle of 38° and a 4–2 energy-transfer time constant of 440 fs. Therefore, the φ = 40° dimer spectra at T = 400 fs generally reproduce the dominant features, resulting from excitons 2 and 4, of the corresponding FMO experimental spectra, and the (λτ = 804 nm, λt = 814 nm) crosspeak is mainly due to relaxation from excitons 4→2. As discussed above, the exciton transition dipoles contribute to the crosspeak through 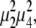 and the peak is enhanced in the 〈75°, −75°, 0°, 0°〉 spectrum by a favorable orientational factor. The positioning of excitonic transition dipoles is strongly dependent on the model Hamiltonian parameters, so that subtle changes in site and coupling energies have a pronounced effect on φ. In fitting the spectra, input parameters can be identified that produce good fits to rephasing and all-parallel spectra, but overestimate the amplitude of the crosspeak in the T = 400 fs 〈75°, −75°, 0°, 0°〉 nonrephasing spectrum, because of too large a projection angle (too close to 90°) between the dominant transitions.
and the peak is enhanced in the 〈75°, −75°, 0°, 0°〉 spectrum by a favorable orientational factor. The positioning of excitonic transition dipoles is strongly dependent on the model Hamiltonian parameters, so that subtle changes in site and coupling energies have a pronounced effect on φ. In fitting the spectra, input parameters can be identified that produce good fits to rephasing and all-parallel spectra, but overestimate the amplitude of the crosspeak in the T = 400 fs 〈75°, −75°, 0°, 0°〉 nonrephasing spectrum, because of too large a projection angle (too close to 90°) between the dominant transitions.
The simulations underscore the fact that the nonrephasing polarized spectra are uniquely sensitive to excitonic structure, and can be used to isolate individual energy-transfer pathways. The polarization conditions serve to identify particular relative orientations of coupled transitions, and a consideration of the nonrephasing spectra, in which fewer Liouville pathways contribute to the off-diagonal region, is useful for focusing on energy-transfer processes. Relative amplitudes of energy-transfer crosspeaks under different polarization conditions yield excitonic structural information directly. This point is demonstrated here, in that an estimate of the relative orientation of two transition dipoles, determined from the orientational factor dependence on φ (as illustrated by the dimer model), is borne out by the calculation based on the full FMO Hamiltonian. In other words, even without previously obtained crystallographic or spectroscopic data, a coarse-grained view of excitonic structure can be obtained using combined polarization sequences in 2D electronic spectroscopy. However, this is only feasible when crosspeaks can be sufficiently resolved, and therefore the use of nonrephasing spectra is particularly powerful in conjunction with polarization-dependent 2D experiments.
The site and exciton basis transition dipole moments predicted by the full FMO model are shown in Fig. 5. In the absence of interchromophore interaction, i.e., when the J coupling is 0, the transitions correspond to the Qy transition dipoles localized on each BChl, resulting from π→π* excitation within the conjugated plane of the molecule parallel to the axis connecting the NB and ND nitrogens (in the Protein Data Bank notation). The lengths of the transition dipole moments shown in Fig. 5 are proportional to their magnitudes, which are all assumed to be equal in the site basis (Fig. 5 a). In the absence of coupling, the site and exciton basis pictures are equivalent. When interchromophore electronic coupling is introduced (Fig. 5 b), linear combinations of the sites give rise to reoriented transition dipole moments with redistributed dipole strength. The excitons are numbered according to their energies, the site numbering used is in accordance with the Protein Data Bank crystal structure (34), and the color-coding in Fig. 5 matches the sites on the left with the exciton states on the right to which they contribute predominantly. However, this correspondence is only approximate, because some of the excitons are delocalized over multiple sites. Transition dipole moments are positioned in Fig. 5 b according to the site basis coefficients making up the exciton states, thereby taking into account the delocalization length of the excitons. In particular, exciton 5 is highly delocalized over sites 4, 5, 6, and 7, as reflected by the pronounced shift in position of the green arrow from site 6 to a position spanning sites 4, 5, 6, and 7 in the exciton basis. In contrast, excitons 1 and 7 are largely localized on sites 3 and 5, respectively, as evidenced by the similar positioning of the dipole moments in the site and exciton bases. The energetic ordering of states changes upon moving from the site to the exciton basis, reflecting the change in energetic landscape effected by the Coulombic coupling. The dominant transitions corresponding to excitons 2 and 4 are shown in red and cyan in Fig. 5 b, respectively, in the geometry supported by spectral fits as discussed above.
FIGURE 5.

Qy transition dipole moments in the site basis (a) and exciton basis (b). The site basis is the case in the limit of negligible interchromophore couplings, and the transitions are localized on individual BChls. Coupling between BChls redistributes dipole strength among sites, resulting in different positions and orientations of the excitonic transition dipoles. The dominant peaks in the short-time 2D spectra arise from excitons 2 and 4 in red and cyan, respectively.
The orientation of the FMO complex between the chlorosome and reaction center has not yet been verified by crystallography. However, as discussed by Adolphs and Renger (8), the funneling of energy by FMO suggests that the lower energy states are linked to the reaction center. Thus, the most probable conformation for the complex is with BChl sites 1 and 6 interfacing with the chlorosome, and sites 3 and 4 close to the reaction center. As with the results of Brixner et al. (11) for FMO from Chlorobium tepidum (a structurally similar but spectroscopically distinct complex from a different green sulfur bacterium) and the results of Adolphs and Renger (8), the model presented here supports two major energetic pathways through the P. aestuarii FMO complex. In the model presented here, these pathways correspond generally to migration from exciton 7 to 5 to 4 to 2 to 1, and from 6 to 3 to 2 to 1. As discussed by Brixner et al. (11), the spatial extent of the excitons determines the energy migration pathway, rather than the energy levels alone, so that relaxation can skip energetically intermediate states. When sites 1 and 6 are linked to the chlorosome, energy can couple into both of the dominant pathways, corresponding generally to the left-hand and right-hand sides of the protein in Fig. 5. The model suggests that at 77 K, energy equilibrates among the six highest exciton states within ∼1 ps, and further relaxation occurs only after 3 ps. That energy transfer among the higher states is so much more rapid than transfer to the lowest state is apparent in the short-time experimental spectra presented in Figs. 1–3, where pronounced relaxation into, but not out of, the 814-nm band (corresponding to excitons 2 and 3) is visible. The discrepancy in energy-transfer rates is a manifestation of the localization of exciton 1 on site 3 because of the pronounced red shift of its site energy relative to others, so that little spatial overlap occurs with other exciton states. The evolutionary advantage is most likely the resultant unidirectionality of energy flow, such that once the excitation migrates to site 3, it is essentially trapped and directed to the reaction center.
CONCLUSION
Because it has been so widely studied, FMO is a model photosynthetic light-harvesting protein. In this work, we demonstrate the use of polarization sequences for 2D electronic spectroscopy that, through comparison of nonrephasing spectra, generally corroborate the orientation of exciton transitions predicted for FMO previously, and allow further refinement of Hamiltonian parameters. In addition, the experiment allows the identification of a crosspeak dominated by an energy transfer coupling two particular states in a crowded spectrum. Rapid energy transfer from exciton 4 to exciton 2 is observed within 400 fs, and the projection angle between the excitons is estimated at ∼40° by comparison with theoretical spectra. The technique can be used to obtain a coarse-grained view of excitonic structure directly, without previous knowledge of a system's structure or energetics, and without alignment of the sample. When prior knowledge is available, specific polarization sequences can be chosen to isolate particular energetic pathways. For example, identifying a polarization scheme under which relaxation between states with a known φ disappears allows that process to be essentially subtracted out of the spectrum, enabling a clearer visualization of other processes. More complex experiments using combinations of polarization sequences, similar to the work presented here, could be designed by choosing two polarization schemes with the maximum difference between orientational factors for a predicted crosspeak. In this way, the subtraction of one type of spectrum from the other would highlight peaks arising only from the process of interest. Extending these techniques using oriented samples, one could further suppress unwanted signals and probe individual relaxation pathways. This method promises to be especially useful for studies of photosynthesis, allowing researchers to tease out the underlying states and their dynamics disguised by the spectral overlap typical of large multichromophore aggregates. On the complex energetic landscape of the photosynthetic light-harvesting apparatus, existing dynamical theories are often less than satisfactory, owing to high disorder, a wide range of coupling energies, and poorly understood interactions with the protein environment. It is therefore necessary to use experimental tools that can probe specific relaxation processes and thereby obtain rate constants directly from experiments to be used for the calibration of dynamical theories.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
Simulations were performed using the NOSE simulation package, originally developed by Tomáš Mančal, now at Charles University, Prague, Czech Republic. G.S.E. thanks the Miller Institute for Basic Research in Science for its support.
This work was supported by grant DE-AC03-76SF000098 and grant DE-FG02-07ER15902 to G.R.F. and R.E.B., respectively, from the Chemical Sciences, Geosciences and Biosciences Division, Office of Basic Energy Sciences, Office of Science, U.S. Department of Energy, and by a grant from the National Science Foundation to G.R.F.
Gregory S. Engel's present address is the Dept. of Chemistry, University of Chicago, Chicago, IL 60637.
Editor: Klaus Schulten.
References
- 1.Blankenship, R. E. 2002. Molecular Mechanisms of Photosynthesis. Blackwell Science, Malden, MA.
- 2.Fenna, R. E., and B. W. Matthews. 1975. Chlorophyll arrangement in a bacteriochlorophyll protein from Chlorobium-limicola. Nature 258:573–577. [Google Scholar]
- 3.Camara-Artigas, A., R. E. Blankenship, and J. P. Allen. 2003. The structure of the FMO protein from Chlorobium tepidum at 2.2 angstrom resolution. Photosynth. Res. 75:49–55. [DOI] [PubMed] [Google Scholar]
- 4.van Amerongen, H., L. Valkunas, and R. van Grondelle. 2000. Photosynthetic excitons. World Scientific, River Edge, NJ.
- 5.Louwe, R. J. W., J. Vrieze, A. J. Hoff, and T. J. Aartsma. 1997. Toward an integral interpretation of the optical steady-state spectra of the FMO-complex of Prosthecochloris aestuarii. 2. Exciton simulations. J. Phys. Chem. B. 101:11280–11287. [Google Scholar]
- 6.Vulto, S. I. E., M. A. de Baat, R. J. W. Louwe, H. P. Permentier, T. Neef, M. Miller, H. van Amerongen, and T. J. Aartsma. 1998. Exciton simulations of optical spectra of the FMO complex from the green sulfur bacterium Chlorobium tepidum at 6 K. J. Phys. Chem. B. 102:9577–9582. [Google Scholar]
- 7.Wendling, M., M. A. Przyjalgowski, D. Gulen, S. I. E. Vulto, T. J. Aartsma, R. van Grondelle, and H. van Amerongen. 2002. The quantitative relationship between structure and polarized spectroscopy in the FMO complex of Prosthecochloris aestuarii: refining experiments and simulations. Photosynth. Res. 71:99–123. [DOI] [PubMed] [Google Scholar]
- 8.Adolphs, J., and T. Renger. 2006. How proteins trigger excitation energy transfer in the FMO complex of green sulfur bacteria. Biophys. J. 91:2778–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Müh, F., M. E. A. Madjet, J. Adolphs, A. Abdurahman, B. Rabenstein, H. Ishikita, E. W. Knapp, and T. Renger. 2007. Alpha-helices direct excitation energy flow in the Fenna-Matthews-Olson protein. Proc. Natl. Acad. Sci. USA 104:16862–16867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Savikhin, S., D. R. Buck, and W. S. Struve. 1998. Toward level-to-level energy transfers in photosynthesis: the Fenna-Matthews-Olson protein. J. Phys. Chem. B. 102:5556–5565. [Google Scholar]
- 11.Brixner, T., J. Stenger, H. M. Vaswani, M. Cho, R. E. Blankenship, and G. R. Fleming. 2005. Two-dimensional spectroscopy of electronic couplings in photosynthesis. Nature 434:625–628. [DOI] [PubMed] [Google Scholar]
- 12.Savikhin, S., D. R. Buck, and W. S. Struve. 1997. Oscillating anisotropies in a bacteriochlorophyll protein: evidence for quantum beating between exciton levels. Chem. Phys. 223:303–312. [Google Scholar]
- 13.Engel G. S., T. R. Calhoun, E. L. Read, T.-K. Ahn, T. Mancal, Y.-C. Chung, R. E. Blankenship, and G. R. Fleming. 2007. Evidence for wavelike energy transfer through quantum coherence in photosynthetic systems. Nature 446:782–786. [DOI] [PubMed] [Google Scholar]
- 14.Jonas, D. M. 2003. Two-dimensional femtosecond spectroscopy. Annu. Rev. Phys. Chem. 54:425–463. [DOI] [PubMed] [Google Scholar]
- 15.Cowan, M. L., J. P. Ogilvie, and R. J. D. Miller. 2004. Two-dimensional spectroscopy using diffractive optics based phased-locked photon echoes. Chem. Phys. Lett. 386:184–189. [Google Scholar]
- 16.Brixner, T., T. Manĉal, I. V. Stiopkin, and G. R. Fleming. 2004. Phase-stabilized two-dimensional electronic spectroscopy. J. Chem. Phys. 121:4221–4236. [DOI] [PubMed] [Google Scholar]
- 17.Hybl, J. D., A. W. Albrecht, S. M. G. Faeder, and D. M. Jonas. 1998. Two-dimensional electronic spectroscopy. Chem. Phys. Lett. 297:307–313. [Google Scholar]
- 18.Tian, P. F., D. Keusters, Y. Suzaki, and W. S. Warren. 2003. Femtosecond phase-coherent two-dimensional spectroscopy. Science 300:1553–1555. [DOI] [PubMed] [Google Scholar]
- 19.Zhang, T. H., C. N. Borca, X. Q. Li, and S. T. Cundiff. 2005. Optical two-dimensional Fourier transform spectroscopy with active interferometric stabilization. Opt. Express. 13:7432–7441. [DOI] [PubMed] [Google Scholar]
- 20.Grumstrup, E. M., S. H. Shim, M. A. Montgomery, N. H. Damrauer, and M. T. Zanni. 2007. Facile collection of two-dimensional electronic spectra using femtosecond pulse-shaping technology. Opt. Express. 15:16681–16689. [DOI] [PubMed] [Google Scholar]
- 21.Hochstrasser, R. M. 2001. Two-dimensional IR-spectroscopy: polarization anisotropy effects. Chem. Phys. 266:273–284. [Google Scholar]
- 22.Zanni, M. T., N. H. Ge, Y. S. Kim, and R. M. Hochstrasser. 2001. Two-dimensional IR spectroscopy can be designed to eliminate the diagonal peaks and expose only the crosspeaks needed for structure determination. Proc. Natl. Acad. Sci. USA 98:11265–11270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dreyer, J., A. M. Moran, and S. Mukamel. 2003. Tensor components in three pulse vibrational echoes of a rigid dipeptide. Bull. Korean Chem. Soc. 24:1091–1096. [Google Scholar]
- 24.Read, E. L., G. S. Engel, T. R. Calhoun, T. Mancal, T. K. Ahn, R. E. Blankenship, and G. R. Fleming. 2007. Cross-peak-specific two-dimensional electronic spectroscopy. Proc. Natl. Acad. Sci. USA 104:14203–14208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang, T. H., I. Kuznetsova, T. Meier, X. C. Li, R. P. Mirin, P. Thomas, and S. T. Cundiff. 2007. Polarization-dependent optical 2D Fourier transform spectroscopy of semiconductors. Proc. Natl. Acad. Sci. USA 104:14227–14232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Woutersen, S., and P. Hamm. 2000. Structure determination of trialanine in water using polarization sensitive two-dimensional vibrational spectroscopy. J. Phys. Chem. B. 104:11316–11320. [Google Scholar]
- 27.Khalil, M., N. Demirdoven, and A. Tokmakoff. 2003. Coherent 2D IR spectroscopy: molecular structure and dynamics in solution. J. Phys. Chem. A. 107:5258–5279. [Google Scholar]
- 28.Ernst, R. R., G. Bodenhausen, and A. Wokaun. 1987. Two-dimensional Fourier spectroscopy. In Principles of Nuclear Magnetic Resonance in One and Two Dimensions. J. S. Rowlinson, editor. Oxford Science Publications, Oxford. 283–357.
- 29.Khalil, M., N. Demirdoven, and A. Tokmakoff. 2003. Obtaining absorptive line shapes in two-dimensional infrared vibrational correlation spectra. Phys. Rev. Lett. 90:047401. [DOI] [PubMed] [Google Scholar]
- 30.Ge, N. H., M. T. Zanni, and R. M. Hochstrasser. 2002. Effects of vibrational frequency correlations on two-dimensional infrared spectra. J. Phys. Chem. A. 106:962–972. [Google Scholar]
- 31.Li, Y. F., W. L. Zhou, R. E. Blankenship, and J. P. Allen. 1997. Crystal structure of the bacteriochlorophyll: a protein from Chlorobium tepidum. J. Mol. Biol. 271:456–471. [DOI] [PubMed] [Google Scholar]
- 32.Brixner, T., I. V. Stiopkin, and G. R. Fleming. 2004. Tunable two-dimensional femtosecond spectroscopy. Opt. Lett. 29:884–886. [DOI] [PubMed] [Google Scholar]
- 33.Zigmantas, D., E. L. Read, T. Mancal, T. Brixner, A. T. Gardiner, R. J. Cogdell, and G. R. Fleming. 2006. Two-dimensional electronic spectroscopy of the B800–B820 light-harvesting complex. Proc. Natl. Acad. Sci. USA 103:12672–12677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tronrud, D. E., M. F. Schmid, and B. W. Matthews. 1986. Structure and x-ray amino-acid-sequence of a bacteriochlorophyll—a protein from Prosthecochloris-aestuarii refined at 1.9 Å resolution. J. Mol. Biol. 188:443–454. [DOI] [PubMed] [Google Scholar]
- 35.Mukamel, S. 1995. Principles of Nonlinear Optical Spectroscopy. Oxford University Press, New York.
- 36.Zhang, W. M., T. Meier, V. Chernyak, and S. Mukamel. 1998. Exciton-migration and three-pulse femtosecond optical spectroscopies of photosynthetic antenna complexes. J. Chem. Phys. 108:7763–7774. [Google Scholar]
- 37.Yang, M. N., and G. R. Fleming. 2002. Influence of phonons on exciton transfer dynamics: comparison of the Redfield, Forster, and modified Redfield equations. Chem. Phys. 275:355–372. [Google Scholar]
- 38.Cheng, Y. C., and G. R. Fleming. 2008. Coherence quantum beats in the two-dimensional electronic spectroscopy. J. Phys. Chem. A. 112:4254–4260. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.