

Abstract
As one of the best studied members of the pharmaceutically relevant family of G-protein-coupled receptors, rhodopsin serves as a prototype for understanding the mechanism of G-protein-coupled receptor activation. Here, we aim at exploring functionally relevant conformational changes and signal transmission mechanisms involved in its photoactivation brought about through a cis-trans photoisomerization of retinal. For this exploration, we propose a molecular dynamics simulation protocol that utilizes normal modes derived from the anisotropic network model for proteins. Deformations along multiple low-frequency modes of motion are used to efficiently sample collective conformational changes in the presence of explicit membrane and water environment, consistent with interresidue interactions. We identify two highly stable regions in rhodopsin, one clustered near the chromophore, the other near the cytoplasmic ends of transmembrane helices H1, H2, and H7. Due to redistribution of interactions in the neighborhood of retinal upon stabilization of the trans form, local structural rearrangements in the adjoining H3–H6 residues are efficiently propagated to the cytoplasmic end of these particular helices. In the structures obtained by our simulations, all-trans retinal interacts with Cys167 on H4 and Phe203 on H5, which were not accessible in the dark state, and exhibits stronger interactions with H5, while some of the contacts made (in the cis form) with H6 are lost.
INTRODUCTION
G-protein-coupled receptors (GPCRs) are a large family of integral membrane proteins that, due to their involvement in a wide variety of physiological processes, have been serving as key targets for pharmacological intervention against many diseases (1–3). Structural information is available for only two GPCRs: rhodopsin (4–6) and β2-adrenergic receptor (7–9). Being highly abundant in nature, rhodopsin has been the most widely studied member of the GPCR family. It is located in the outer segments of rod photoreceptor cells in the retina and responds to photons by initiating a series of intracellular events that result in an electrical signal processed by the visual system (10). The structure of rhodopsin can be divided into three regions, cytoplasmic (CP), transmembrane (TM), and extracellular (EC) (Fig. 1 A). The CP region includes three CP loops (CL1–CL3), a soluble helix (H8), and the C-terminus. Seven helices (H1–H7) span the TM region. The EC region consists of three loops (EL1–EL3) and the N-terminus (NT). EL2, connecting H5 and H6, forms a β-strand folded into the TM section underlying the EC side of the chromophore binding pocket (5). Light absorption by rhodopsin isomerizes its chromophore, 11-cis retinal, to the all-trans conformation on a subpicosecond timescale. The chromophore, composed of a β-ionone ring and a polyene chain covalently linked to Lys296 in H7 (Fig. 1 B), acts as an inverse agonist: its photoisomerization triggers the formation of a series of intermediates arising on picosecond to millisecond timescales, leading to the formation of metarhodopsin II (Meta II); the latter intermediate corresponds to the active conformation of rhodopsin (11).
FIGURE 1.

(A) Rhodopsin (red) and surrounding lipid (green) and water (cyan) molecules. Rhodopsin contains seven TM helices (H1–H7). The TM bundle encloses the chromophore, 11-cis retinal (purple). The simulation system consists of rhodopsin embedded in a lipid bilayer with 108 palmitoyloleoylphosphatidylcholine molecules, 6596 water molecules, and 27 ions. Cytoplasmic and extracellular regions are labeled for clarification. (B) 11-cis retinal. It consists of a β-ionone ring and a polyene chain and is covalently bound to Lys296. Carbon atoms are indexed from 1 to 20. The arrow shows the bond that isomerizes from cis to trans form upon light activation. (C) Closer view of the CP region encircled in A, near the ERY motif (E134, R135, and Y136). Helices H2–H8 are labeled.
A wide range of biophysical and biochemical experiments including site-directed mutagenesis, photoactivity, NMR, and engineered disulfide bonds have been used to elucidate the activation mechanism of rhodopsin and other GPCRs (10,12,13). These studies have shown that conformational changes in the CP region of rhodopsin are critical for activation and G-protein binding. Movements of the helical segments near this region presumably involve the disruption of specific contacts and release a number of constraints instrumental for transmitting signals. These constraints cluster in several conserved “microdomains”: 1), an electrostatic interaction site formed by the retinal Schiff base and the Glu113 counterion (14,15); 2), the highly conserved D(E)RY motif at the CP end of H3 (Fig. 1 C) and the XBBXXB motif at the CP end of H6 (B:basic) (16–24); 3), the Asn55-Asp83 pair in H1 and H2, respectively, and the NPXXY motif in H7 (4); and 4), the aromatic cluster surrounding the ligand binding pocket (25–27).
Recently, a photoactivated intermediate of rhodopsin has been structurally resolved at 4.15 Å resolution (6). However, the conformational changes observed were smaller than those indicated by biophysical and biochemical experiments (10,28,29), and the atomic coordinates of most side chains and the chromophore, as well as some backbone atoms, could not be determined (6). Further studies were therefore suggested to clarify the relationship between this photoactivated intermediate and the Meta II state (6).
Determination of the dark state structure of rhodopsin prompted many computational studies in recent years. In particular, the chromophore isomerization and conformational changes of rhodopsin at the early stages of photoactivation (30–35) and photointermediates (36,37) have been investigated by molecular dynamics (MD) simulations. In other computational studies, distance constraints derived from NMR and electron paramagnetic resonance spectroscopy and/or cysteine cross-linking experiments have been used to model the conformations of rhodopsin (38–40). The energy calculations have been used to model the active state of rhodopsin (41) and to determine the network of interacting residues (42). The effect of unsaturated lipids on rhodopsin stability and kinetics was studied by a series of independent 100-ns-long MD runs, performed for the dark-adapted rhodopsin, showing that the tightly packed unsaturated lipids weakened the interhelical packing by making specific interactions with the protein (43).
Even with these important contributions and extensive data on rhodopsin activation, details about how the conformational changes for activation are triggered and the molecular mechanisms explaining the experimental data on the Meta II state remain unknown. Although MD simulations provide valuable information on atomic motions and interactions, careful analysis of MD results reveals that they suffer from convergence problems, even when long trajectories of the order of 0.1 μs are generated (44). The timescale explored in MD simulations has been shown for many proteins to be too short to achieve adequate convergence properties, especially for “long” timescale motions (45,46). Likewise, the conformational sampling of the regions exposed to the environment in membrane proteins is usually less accurate compared to TM regions (47).
Various methods have been developed to accelerate MD simulations and increase sampling efficiency. Steered molecular dynamics (SMD) has been successfully used to observe large conformational changes by applying external forces (48,49). A similar method, targeted molecular dynamics, uses time-dependent, geometrical constraints to approach a known target conformation at ordinary temperature (50). Additionally, guiding MD trajectories along collective coordinates has proven useful for efficient sampling and expansion of the accessible conformational space (51). Berendsen and co-workers introduced the essential dynamics (ED) analysis, which is a principle-component analysis (PCA) of snapshots at fixed time intervals during MD trajectories (52). These collective modes have been used as constraints in MD simulations to achieve a more efficient sampling of the conformational space (52–54). Collective coordinates have been combined with ensemble sampling by Abheser and Nilges, by implementing a biasing potential that restrains the motions to those along the collective modes in a set of independent MD trajectories (55,56). However, the difficulties of using PCA to obtain essential modes describing the majority of the fluctuations due to sampling inefficiencies and the shortness of achievable MD simulation times have been demonstrated previously (57).
In our previous work (58), we adopted a different, purely analytical approach, and examined the collective motions predicted by two elastic network models (ENMs), the Gaussian network model (59) and the anisotropic network model (ANM) (60,61), followed by energy minimization to optimize side-chain conformations. We combined this analysis with systematic Cys→Ser replacement experiments to develop a model for providing insights into the potential reconfiguration of rhodopsin accompanying its activation (58).
ENMs have been widely used in recent years to explore the biologically relevant, long-timescale motions of large structures while avoiding expensive computations (for reviews, see (62–64)). An important extension has been the introduction of a perturbational variation of the elastic network (65–67), which allows for a quantitative analysis of allosteric effects. The comparative analysis of the results from this perturbational variation of the elastic network, and those from MD simulations biased with ED sampling, have recently proven to elucidate the coupling between nucleotide binding and TM domains in the ABC transporter BtuCD (68). Another technique, referred to as amplified collective motions, has been utilized for coupling low-frequency modes derived from the ANM to a higher temperature by using a weak coupling method (69). The basic ingredient in ENM-based models is the topology of interresidue contacts in the native structure, which turns out to be a major factor defining equilibrium dynamics. The interactions between residues in close proximity are represented by harmonic potentials with a uniform spring constant, and network junctions are usually identified by the Cα atoms (59,60,70). Low-frequency motions, also referred to as “global” modes, are insensitive to the details of the models and energy parameters used in normal-mode analyses at least as long as interresidue contacts are maintained (see for example (71,72)).
Despite their numerous insightful applications, ENM methods have limitations. They lack information on residue specificities, atomic details, or side-chain motions. Furthermore, the contribution of different modes to biologically relevant motions is usually unknown. We also note that although many functional mechanisms appear to be intrinsically defined by the 3-dimensional (3-D) structure of the protein itself (62,73,74), the effects of the interactions with the environment, such as the lipids and water molecules, are also crucial for membrane proteins.
Here, our aim is to find, in atomic detail, the biologically relevant conformations of rhodopsin that couple retinal isomerization to conformational changes in both the TM domain and the critical G-protein binding sites on the CP surface. We seek to explore the global dynamics while incorporating the effects of explicit residues and interactions with lipid and water in atomic detail. We propose, for this purpose, an algorithm, referred to as ANM-restrained MD, which uses the deformations derived from ANM analysis as restraints in MD trajectories. This permits us to sample the collective motions that are otherwise beyond the range of conventional MD simulations. With this new approach, we seek to incorporate the realism and accuracy of MD into ENM analysis while taking advantage of ENM to accelerate MD simulations. It is noteworthy that, using this method, we identify a hinge site that does not change with several applications of normal modes as restraints. This hinge site includes residues that are directly affected by the isomerization of retinal, as well as those stabilizing the resulting all-trans conformation of the chromophore. The CP ends of helices H3, H4, H5, and H6, and the connecting loops are found to enjoy an enhanced mobility facilitated by this hinge site. Several new interactions are observed to contribute to the mechanism of signal propagation from the retinal binding pocket to the G-protein-binding sites in the CP domain.
METHODS
The key idea for our approach is to use an ensemble of ANM modes in an iterative scheme, as described in Fig. 2. Essentially, our proposed algorithm consists of two loops. A first (inner) loop generates a succession of conformations using ANM modes as harmonic restraints in MD runs, succeeded in each case by a short energy minimization to allow the molecule to settle in a local energy minimum. For this purpose, we select from a pool of ANM modes the subsets of most distinctive (lowest-frequency modes whose eigenvalues are sufficiently separate from those of other modes) and cooperative (based on collectivity κ(k); see Eq. 3) modes. For each mode, we define two target conformations (see Eq. 4) and we run short MD simulations (of 20 ps) in the presence of harmonic restraints that favor these target structures. Because the restraints may lead to unrealistic strains in the structure, we perform a short energy minimization succeeding each run and choose among the two alternative structures the one that is energetically favored. After screening all selected modes, a new cycle (outer loop) is initiated, with the updated ANM modes corresponding to the final structure of the first cycle. This procedure was repeated until the final structure deviated from the starting one by a root mean-squared deviation (RMSD) of 3.5 Å in backbone coordinates. Two independent runs have been performed here, each composed of two cycles (the outer loop in Fig. 2), which yielded similar patterns (RMSDs and energy profiles) and redistributions of interresidue interactions comparable to experimental data (see Results and Discussion). The underlying assumption in this protocol is that ANM-derived restraints drive the excursion of the molecule toward a direction that would otherwise be naturally selected at much longer times. We explain below the steps involved in the actual calculations.
FIGURE 2.

ANM-restrained MD protocol. See text for details.
Generating the normal modes
We use the ANM for generating the modes of interest. ANM, described in detail previously (60), is a normal mode analysis that uses as potential
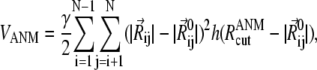 |
(1) |
where 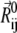 and
and  denote the original and instantaneous distance vectors between residues i and j, represented by their Cα-atom positions, N is the residue number, h(x) is the Heaviside step function equal to 1 if x is positive, and zero otherwise,
denote the original and instantaneous distance vectors between residues i and j, represented by their Cα-atom positions, N is the residue number, h(x) is the Heaviside step function equal to 1 if x is positive, and zero otherwise, 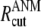 is the cutoff distance for interresidue interactions, taken as 13 Å (75), and γ is the force constant of the order of 1 kcal/(mol Å2), usually found by requiring the sum of 〈(ΔRi)2〉 over all residues (1 ≤ i ≤ N) to match the sum of the experimental B-factors. The mode shapes are insensitive to the absolute value of γ.The cross-correlation 〈ΔRi · ΔRj〉 between the fluctuations of residues i and j can be expressed as a sum
is the cutoff distance for interresidue interactions, taken as 13 Å (75), and γ is the force constant of the order of 1 kcal/(mol Å2), usually found by requiring the sum of 〈(ΔRi)2〉 over all residues (1 ≤ i ≤ N) to match the sum of the experimental B-factors. The mode shapes are insensitive to the absolute value of γ.The cross-correlation 〈ΔRi · ΔRj〉 between the fluctuations of residues i and j can be expressed as a sum
 |
(2) |
over the contributions of all modes (k), using the eigenvectors uk and eigenvalues λk of the Hessian H, evaluated from the second derivatives of VANM. Here kB is the Boltzmann constant, T is the absolute temperature, and a total of 1 ≤ k ≤ 3N − 6 nonzero modes contribute to 〈ΔRi · ΔRj〉. uk describes the displacements of residues induced by mode k,  scales with its frequency.
scales with its frequency.
Selection of distinctive and cooperative modes
In principle, we select the lowest-frequency modes. However, two additional criteria were considered: mode frequency dispersion (or eigenvalue distribution) and degree of collectivity. The former is examined to identify a subset of modes whose frequencies (eigenvalues) are sufficiently separate from those of the other modes. The degree of collectivity for the kth mode, on the other hand, is calculated using
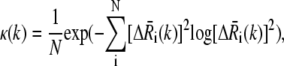 |
(3) |
where 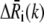 is the displacement of the ith residue driven by mode k.
is the displacement of the ith residue driven by mode k. 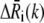 is normalized such that
is normalized such that 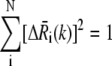 (72) to ascertain that the selected modes are cooperative enough. This second criterion helps in eliminating the cases where the low-frequency modes induce a motion in a loosely coupled chain segment only (e.g., the N- or C-terminus). In the case of rhodopsin, the lowest-frequency modes were also observed to be the most cooperative ones, and the frequency distribution indicated that the subset of the first three, or the first seven, modes were separable.
(72) to ascertain that the selected modes are cooperative enough. This second criterion helps in eliminating the cases where the low-frequency modes induce a motion in a loosely coupled chain segment only (e.g., the N- or C-terminus). In the case of rhodopsin, the lowest-frequency modes were also observed to be the most cooperative ones, and the frequency distribution indicated that the subset of the first three, or the first seven, modes were separable.
Generating new conformations
Since each mode corresponds to a fluctuation between two oppositely directed motions, both directions equally probable, two sets of deformations are considered for each mode, referred to as “plus” or “minus” displacements along the particular mode axis. The corresponding “target” conformations, represented by the 3N-dimensional position vectors  and
and 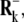 are evaluated from
are evaluated from
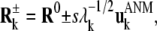 |
(4) |
where R0 is the conformation before the application of restraints, and s is a scaling parameter set by requiring the RMSD in Cα-atom positions to remain close to 1.5 Å after reconfiguration along the selected mode for mode 1, and 1.5(λi/λ1)1/2 Å for mode i. Note that  Each Cα atom is harmonically restrained to approach the target conformations, using a uniform spring constant of 1 kcal/(mol·Å2) for all residues in ANM-restrained MD simulations. After reaching the two target structures
Each Cα atom is harmonically restrained to approach the target conformations, using a uniform spring constant of 1 kcal/(mol·Å2) for all residues in ANM-restrained MD simulations. After reaching the two target structures  and
and  for a given mode, both are subjected to energy minimization (1000–3000 steps of steepest descent) without any restraints to relieve possible unrealistic distortions and to select the lower energy conformer among the two (see profiles in Fig. 3).
for a given mode, both are subjected to energy minimization (1000–3000 steps of steepest descent) without any restraints to relieve possible unrealistic distortions and to select the lower energy conformer among the two (see profiles in Fig. 3).
FIGURE 3.

Time evolution of the overall RMSD in the α-carbon coordinates compared to the starting conformation (A) and its energy (B) during a first inner cycle of the ANM-restrained MD protocol described in Fig. 2. The two curves refer to the trajectories sampled with ANM restraints along opposite directions, each succeeded by a short energy minimization that selects the lower energy conformation for the next run (with another mode).
The initial structure
Previously, Saam et al. (35) performed MD simulations to study the photoisomerization of retinal from 11-cis to all-trans and the relaxation of rhodopsin succeeding this isomerization. The system consists of rhodopsin embedded in a lipid bilayer with 108 palmitoyloleoylphosphatidylcholine molecules, 6596 water molecules, and 27 ions (Fig. 1). The size of this system is 39,964 atoms. All titratable groups in the protein were considered to be charged, with the exception of Glu122 and Asp83 (76). After minimization and equilibration of the system, 11-cis retinal was isomerized to all-trans retinal by transiently switching the dihedral potential energy function of the corresponding bond. We adopted the conformation at 495 ps of MD simulation after isomerization of retinal and the flip of β-ionone ring as our initial structure, because experimental evidence shows that rhodopsin undergoes conformational changes to reach the active Meta II state after the cis-trans isomerization of retinal and the flip of the β-ionone ring. We refer to the structure energy-minimized and equilibrated in the presence of water and membrane molecules right before isomerization of 11-cis retinal, as the “dark state”. To investigate the effect of the orientation of the β-ionone ring, we also repeated the simulations using an alternative initial conformation of the β-ionone ring in the absence of the ring flip taken at 212 ps of MD simulation after isomerization of retinal.
The results presented below were confirmed to be reproducible in two independent runs. In particular, the critical interactions in the chromophore binding pocket between retinal and neighboring helices, as well as the highly fluctuating sites on H6 and CP important for G-protein binding, were found to be robust features of the simulated structural dynamics.
RESULTS AND DISCUSSION
RMSD profiles and accompanying change in energy
Fig. 3 illustrates the time evolution of RMSD in Cα positions from the initial structure (upper), and the accompanying change in energy (lower) in a given cycle of our protocol. The two curves (black and red) refer to the opposite direction deformations along each normal mode used as targets in the MD simulations. The seven slowest modes are used in this cycle, each being succeeded by a short energy minimization. The conformer with the lower energy is chosen as the starting structure for the implementation of the successive modes. By the end of this cycle, the conformation departs from the original one by an RMSD of ∼2 Å (see Fig. 1S in the Supplementary Material, Data S1 for the corresponding energy and RMSD profiles of the second cycle). Two such cycles (the outer loop in Fig. 2) were performed, to reach an RMSD of 3.5 Å, and two independent runs of two cycles each were performed to verify the reproducibility of the results.
Two stable regions, retinal binding pocket and CP ends of H1, H2, and H7
Fig. 4 A shows the RMSD profiles of residues from the initial structure obtained by the end of cycle 1 (red curve) and cycle 2 (black curve). TM helices H1–H8 are indicated by gray bands. The two profiles in Fig. 4 A share similar features; the minima (labeled) maintained by the end of the two cycles point to residues participating in highly stable hinge sites. Except for Pro23, all such residues are clustered in two regions of rhodopsin, the chromophore binding pocket and the CP ends of H1, H2, and H7. The two sites are also clearly distinguished in Fig. 4 B, showing the rhodopsin ribbon diagram color coded by the RMSDs observed by the end of the second cycle, from red (least mobile) to blue (most mobile). It is interesting that, located separately from these two regions, one minimum is at Pro23 near the soluble N-terminus. The Pro23His mutation is associated with the most frequently occurring form of autosomal dominant retinitis pigmentosa, a hereditary progressive blinding disease (77). The pathogenicity of human mutant Pro23His causing retinal degeneration was confirmed by transgenic mice strain experiments (78,79). The structural disruption in the EC domain of rhodopsin caused by the Pro23His mutation results in severe misfolding of the entire protein, made irreversible by the formation of a wrong disulfide bond (80).
FIGURE 4.

RMSD profile as a function of residue index, reached by the end of cycles 1 and 2 of the ANM-restrained MD protocol. (A) The RMSDs exhibit an overall increase between cycles 1 (red) and 2 (black), although the overall profile tends to maintain similar features. The helices (H1–H8) are indicated by gray bands. Residues that lie at the minima are labeled. Non-TM regions exhibit higher mobility in general, especially CL2 (between H3 and H4) and CL3 (between H5 and H6). (B) Ribbon diagram of rhodopsin color-coded according to the RMSD profile reached at the end of cycle 2. The color code is orange (smallest motions), yellow, green, blue (largest motions). The all-trans retinal is colored brown. Residues that exhibit the lowest RMSDs are clustered in two regions: around the chromophore, and in the CP portion of helices 1, 2, and 7. The CP ends of H3–H6, including loops CP2 and CP3, exhibit high RMSDs.
Hinge site near retinal participates in rhodopsin activation
The hinge site includes Ala124 and Leu125 on H3; Trp161 and Cys167 on H4; Pro180, Glu181, Ser186, Cys187, and Gly188 on the β-strands at EL2; Met207, His211, and Phe212 on H5; Trp265 and Tyr268 on H6; and Tyr292 and Ala295 on H7 (Fig. 5). This cluster includes residues located in the retinal binding pocket in the dark state, such as Trp265 and Tyr268, as well as residues that form new interactions to stabilize all-trans retinal in the structure reached by the end of our simulations (e.g., Cys167; see below). Evidently, they would be very sensitive to conformational changes of retinal and hence critically important to coordinate the conformational changes necessary for the activation of rhodopsin.
FIGURE 5.

Hinge residues in the vicinity of the chromophore viewed from the CP regions. These are residues distinguished by their high stability (low mobility) in Fig. 4 A. They include Cys167 on H4, Ser186 and Cys187 on the β-sheet, Met207, His211, and Phe212 on H5, and Trp265 on H6. All-trans retinal is shown in orange.
Meta II stability of rhodopsin mutants is characterized by quantifying Meta II decay rates (81). This method has been useful to estimate the role of a given amino acid in the structure and function of the protein. We used our previously compiled extensive list for Meta II decay rates of rhodopsin mutants (58) to determine the effect of the hinge residues on Meta II stability. Meta II decay rates were investigated for 11 of the hinge residues, and it is noteworthy that all of them were found to affect the stability of Meta II. Five of the hinge residues remain to be tested: Trp161 on H4, Pro180 on EC2, Met207 and Phe212 on H5, and Tyr268 on H6. Along with the validated hinges, the residues are proposed to be critical for Meta II stability and good candidates for Meta II decay experiments.
Two members of this cluster, Cys187 and Cys110, form a disulfide bridge, critical for folding and stability of rhodopsin (82). Mutation of both residues was also found to be associated with autosomal dominant retinitis pigmentosa (83,84), most probably caused by the destabilization of the opsin structure near the chromophore binding site in Meta II and the dark state of rhodopsin. Cys187 forms the deepest minimum in the second-cycle curve (Fig. 4, black curve). Its RMSD and position do not change between the first and second cycle. Cys110 is in the deepest minimum of the first-cycle curve. Although it is still one of the minima in the second cycle, its RMSD is slightly higher than that of the first cycle.
Water molecules hydrogen-bonded to highly conserved residues
There exists an extensive interhelical hydrogen-bond network between H1, H2, and H7. This network includes highly conserved residues in the GPCR family, including the interhelical N-D pair (Asn55 on H1 and Asp83 on H2). We also note that the NPXXY motif between Asn302 and Tyr306 on H7 participates in this network of hydrogen bonds. These residues participate in the low RMSD cluster (CP region) that we have presently identified. The interior of rhodopsin contains water molecules that mediate interhelical interactions, and it has been noted that these interactions could play critical roles in regulating the activity of GPCRs (85) and the spectral sensitivity in visual pigment (4). Present simulations reveal that two residues belonging to the NPXXY motif on H7, Asn302 and Tyr306, are connected to H1 and H2 through water molecules located in the cavity between helices H1, H2, and H7. It is noteworthy that in the resulting conformation, Asn55 (H1), Asp83 (H2), and Asn302 (H7) are hydrogen bonded to a central water molecule (Fig. 6 A); a second water molecule interacts closely with Thr62 (H1), Asn73 (H2), and Tyr306 (H7) (Fig. 6 B). This second water molecule, already in contact with these residues in the x-ray structure (4), remains at the same position despite the implementation of 10 cycles of ANM restraints in MD simulations. Overall, ∼20 water molecules are observed to span the helical bundle from the EC to the CP region and some exchange neighbors during the simulations; they mostly interact with H1–H4 and H7 residues.
FIGURE 6.

Coordination of water molecules that take part in the hydrogen-bond network between H1, H2, and H7. (A and B) Two water molecules were found to be connected to highly conserved residues throughout the simulations.
Comparison of the initial and final structures
To visualize conformational changes of the helices and retinal, we have superimposed the initial and final structures in Fig. 7 A. The transparent and opaque ribbon diagrams correspond to initial and final conformations, respectively. The largest conformational changes occur at loops CL2 and CL3 and at the C-terminal end (see the analysis of the CP ends of rhodopsin below). In contrast, loops connecting the helices at the EC region exhibit smaller displacements. Although the NT end does not display a regular secondary structure, the fingerlike shape of the projecting loop is maintained throughout the simulations. The lower mobilities of the EC loops and the NT end help to maintain the important interactions between the residues at these sites, such as those involving Tyr102 at EL1 and Pro23 at the NT end. In both the initial and final structures, the retinal is in its all-trans conformation since the initial conformations are taken from the MD simulation right after the isomerization of the C11=C12 bond. Yet in the resulting structure, the chromophore assumes a more extended form (Fig. 7 B, red).
FIGURE 7.

Comparison of the dark state (initial) and the activated state (final) found from the simulations. (A) The resulting conformation (opaque) is superimposed on the initial conformation (transparent). (B) Redistribution of interactions in the neighborhood of the retinal. To clarify the change in the position of the β-ionone ring between the cis and trans conformations, retinal atoms 16 and 17 are labeled in both conformations. Initially, 11-cis retinal was in close contact with H6 residues such as Phe261, Trp265, Tyr268, and Ala269. At the end of the simulations, the all-trans retinal made new atomic contacts including Cys167 on H4 and Phe203 on H5. The number of atom-atom contacts with Met207 and His211 increased, in agreement with cross-linking and NMR experiments (22,73). See text for more details.
Rearrangements in chromophore binding pocket to accommodate all-trans retinal
The chromophore binding pocket is a densely packed region. Even small conformational changes in retinal would be sufficient to significantly affect the interactions in this region and violate the van der Waals volumes of interacting atoms. The isomerization of retinal and the accompanying flip of the β-ionone ring indeed cause steric clashes between the all-trans retinal and surrounding residues (58). These clashes are relieved upon rearrangements in the positions and orientations of helices, which in turn induce a redistribution of contacts in the chromophore binding pocket. We note in particular that the number of atom-atom contacts between Trp265 and all-trans retinal (calculated using a distance cutoff of 4.5 Å) is significantly lower than those made in the cis form. The β-ionone ring of 11-cis retinal in the dark state is approximately parallel to the aromatic ring of Trp265, such that many interatomic contacts are possible. In contrast, only one atom, C16, of all-trans retinal is in the vicinity of Trp265 (Fig. 7 B). Furthermore, Phe261, Tyr268, and Ala269 on H6 make contacts with the β-ionone ring to stabilize 11-cis retinal in the dark state (not shown), whereas these contacts are lost in the new structure. Instead, a new amino acid, Cys167 on H4, lines the chromophore binding pocket and interacts with atoms C3–C5 and C18 of the β-ionone ring of all-trans retinal. In addition, two residues on H5, Phe203 and His211, form new contacts with atoms C2–C4 and C18 of retinal. The number of contacts made by Met207 and Thr118 is larger in the resulting structure compared to those in the dark state. Changes in contacts of the polyene chain mostly occur at C20. In the dark state, C20 interacts with Trp265, Tyr268, and Ala292. In the newly obtained structure, on the other hand, it interacts with Glu181, Ser186, and Cys187 on the β-strands, and with Tyr268 (H6) and Ala295 (H7).
Some of these results are confirmed by cross-linking experiments that used photoactivatable anologs of 11-cis retinal (25,86) and NMR experiments with 13C-labeled 11-cis retinal (87). In the dark state, 11-cis retinal cross-links to Trp265, whereas the all-trans retinal cross-links to Ala169 instead of Trp265, consistent with the redistribution of contacts observed here. Further investigation by high-resolution solid-state NMR measurements also showed that Trp126 and Trp265 interact more weakly with retinal in the active state (87). In addition, the NMR data showed that both the side chain of Glu122 and the backbone carbonyl of His211 are disrupted by the orientation of the β-ionone ring of all-trans retinal in Meta II (87). Patel and co-workers (87) further proposed that the contact of the ionone ring with H5 near His211 moves H5 to an active-state orientation.
The orientation and stereochemistry of retinal have been explored in several other computational and experimental studies (35–37,88–90). Solid-state 2H NMR spectroscopy has been used to study conformational changes of retinal after isomerization. Although these studies are carried out before helical rearrangements accompanying the activation of rhodopsin, they give information on the conformations of retinal in the chromophore binding pocket. They suggested the following values for the dihedral angle C5-C6-C7-C8: ±15, ±28, ±32, ±57, ±96, ±116, ±155, and ±158°. Addition of further constraints showed that the most probable dihedral angle is −32 ± 7° (90,91). We have examined the distribution of the dihedral angle C5-C6-C7-C8 observed in this study. During the simulations starting from the structure taken at 212 ps after isomerization (all-trans conformer before the flip of the β-ionone ring (see Methods)), the dihedral angle shows a distribution centered around −40° with peaks around −42° and −34°. During the simulations starting from the structure taken at 495 ps after the isomerization, this dihedral was −170° initially and stabilized around −155° later in the simulations. It is interesting that the same pattern of structural changes was observed in all simulations of ANM-restrained MD. The interaction of the β-ionone ring with H6, which plays an important role in keeping rhodopsin in the inactive conformation, was lost and similar new interactions with H4 and H5 were established in both simulation systems. In particular, most of the interactions with Trp265 on H6 were lost. Cys167 on H4 was in the vicinity of the β-ionone ring, interacting with the C3–C5 and C18 atoms, whereas Phe203 and His211 on H5 interacted with atoms C2–C4 and C18. Most differences between the simulations with two different conformations of retinal resided in the neighborhood of C16–C18 of the β-ionone ring. These atoms underwent the largest changes in their spatial positions as a function of the β-ionone-ring rotational angle. Although C18 was stabilized by Thr118, Cys167, and Met207 in all simulations, the interatomic interactions were redistributed. In particular, C16 and C17 exchanged their interacting residues. During the simulations with the 495-ps starting structure, C16 was the only atom interacting with Trp265. In contrast, C17 was interacting with Trp265, and C16 is further stabilized by Met207 and Phe208 during the simulation with the 212-ps starting structure.
Glu181 stabilizes both the dark and active states of rhodopsin
Various counterion switch models have been proposed in which new roles for Glu181 in the chromophore binding pocket have been introduced. First, based on ultraviolet-visible and resonance Raman spectroscopic studies of rhodopsin mutants of Glu181, it was proposed that Glu181 is protonated (neutral) in the dark state and that the Schiff-base counterion would switch from Glu113 to Glu181 by a proton transfer (92). Hence, Glu181 would serve as the primary counterion until rhodopsin reached the active-state Meta II (before the deprotonation of the Schiff base). In this model, it was also suggested that Ser186 and two water molecules could stabilize the molecule during the switch. Later, Fourier-transform infrared (FTIR) studies were used to detect the protonation states of the acidic residues Glu113 and Glu181 (93). In contrast to previous measurements, FTIR spectroscopic data were more consistent with a deprotonated (charged) Glu181 in the dark state, which did not change during structural transitions. In light of these findings, a refinement of the counterion switch model, in which both Glu113 and Glu181 participate in a complex counterion to the protonated Schiff base in Meta I, was proposed (93). Using FTIR spectroscopy, Vogel et al. (93) also suggested that the region around the protonated Schiff base and the adjacent H-bonded network including Glu113 and Glu181 could serve as an interface that allosterically links the conformational changes in one part of the protein to those in other parts. The authors also proposed that this hydrogen-bond network could be coupled to that between the H1, H2, and H7 helices. The release of the constraint between the Schiff base and the protein counterion allows the interaction between H1, H2, and H7 to decouple from others in the networks of hydrogen bonds. Our model strongly supports the allosteric role of the hinge residues (including Glu181) located at the chromophore binding site in communicating the local conformational changes to the CP region of rhodopsin.
We also note that during the simulations, Glu181 makes new interactions with retinal's polyene chain atoms closer to the Schiff base. The most flexible atom of the polyene chain, C20, which initially interacts with Trp265 and Tyr268, switches to a closer interaction with Glu181. In both our previous study (58) and the simulations described here, Glu181 is found to occupy hinge positions along with Ser186, consistent with its key role in mediating the conformational changes required for activation. The mutants of Glu181, investigated by Meta II decay rate experiments (94), were shown in 15 of 20 cases to affect the Meta II decay rate, lending further support to the central role of Glu181.
Role of H3–H6 CP ends and loops CL2 and CL3 for G-protein binding and activation
To understand the conformational changes accompanying the activation of rhodopsin, extensive Cys scanning mutagenesis experiments were conducted in combination with site-directed spin labeling followed by electron paramagnetic resonance analysis of mobility, accessibility, spin-spin interactions, sulfhydryl reactivity, and disulfide cross-linking rates (10,13,95–102). Upon isomerization, the mobility of spin-labeled side chains at the buried surfaces of H1–H3, H6, and H7 were found to increase. Furthermore, experiments suggest that the CP ends of helices, especially H3, H5, and H6, need to be highly flexible to bind and activate the G-protein (10). Fig. 8 A shows the top view of rhodopsin, and Fig. 8 B the side view of helices close to the ERY motif. The final conformation reached (opaque) in the simulations described here is superimposed on the dark state (transparent). The motions of helices are indicated by arrows. The CP ends of H3–H6, and the connecting loops CL2 and CL3 at the CP region, are highly mobile, which is evident from their high RMSD values, in agreement with spin labeling and cross-linking experiments (10,25,86,100,103). The surface accessibility of the G-protein contact site near the ERY motif is found to increase during the simulations. The surface accessibilities of Glu134, Arg135, and Tyr136 forming this motif increase from 41.9 Å2 to 54 Å2, from 23.5Å2 to 33.3 Å2, and from 57.9 Å2 to 63.5 Å2, respectively. The increased accessibility of Val250 and Thr251 upon activation (95) and the changes in the spin-spin distances with respect to Val139Cys (100) were attributed to the movement of H6 away from the helical bundle such that its interaction with H3 and H7 was weakened. We have previously shown that the first two global modes of ANM drive the relative rearrangements of helices H3–H6 (58); the simulations described here closely reproduce these modes at the CP ends of these helices. On the other hand, the CP ends of H1, H2, and H7 are at present found to closely maintain their dark-state structure (Fig. 8 A), consistent with the relatively smaller conformational changes observed experimentally in this region (96,97,101,104–107).
FIGURE 8.

Comparison of the dark state (initial) and the resulting conformation of the CP end of the helical bundle (A) near the ERY motif (shown as sticks) on H3 (B). The transparent and opaque ribbon diagrams correspond to the initial and final conformations, respectively. Arrows indicate the displacements of the helices from the initial to the final conformation. Arrangements of H3–H6, along with loops CL2 and CL3, result in the exposure of the ERY motif on H3. The surface accessibility of the ERY motif residues Glu134, Arg135, and Tyr136 increases from 41.9 Å2 to 54 Å2, from 23.5Å2 to 33.3 Å2, and from 57.9 Å2 to 63.5 Å2, respectively, between the initial and final conformations. Compared to the rest of the helical bundle, H1, H2, and H7 undergo relatively small fluctuations at the CP region.
The largest RMSDs from the starting structure are found for the CP loop and the C-terminus, in both the simulations presented here and in our earlier ANM analysis (58), in agreement with the results from Grossfield et al. (44). The EC loops, on the other hand, show lower mobility, along with the N-terminus that maintains its fold throughout the simulations (Fig. 4). This suggests that retinal isomerization induces a larger conformational change in the CP region than the EC region. Note that the CP loops bind and activate the G-protein, and this higher mobility is consistent with the function of rhodopsin. The effect of retinal isomerization on the C-terminus is also consistent with the fact that the C-terminus contains two residues that are phosphorylated by rhodopsin kinase in Meta II, where arrestin binds to terminate the signal. On the other hand, the N-terminus should be less mobile for its role in the stability of the molecule (80). Considering the high fluctuations in the CP region and the associated nonlinear effects, we chose to take very conservative steps while perturbing the structure in each SMD run, and let the highly flexible loops and the C-terminus in this region relax during the energy minimization cycles. We note that the C-terminal conformation shows poor convergence in the 100-ns simulations conducted for the dark state of rhodopsin (44).
Refinements in the previous model inferred from simulations in this study
In our previous work (58), the side-chain conformations were generated by assuming that they accompany the backbone motion, and then, to correct for possible unrealistic distortions in bond length and bond angles, the conformations were subjected to a short energy minimization in vacuum (58). In this study, using ANM modes in SMD, we were able to generate conformations favored by a detailed atomic force field. Some residues identified in our previous work to act as global hinges maintained the same character here. These are all residues close to the chromophore: Ala124 and Ile125 on H3; Trp161 on H4; Phe212 on H5; and Pro180 and Cys187 on the β-sheet of the EC2. We also observed that the hinge site broadened in two directions, to include Cys167 on H4; Glu181 and Ser186 on the β-sheet of the EC2; Met207 and His211 on H5; Trp265 and Tyr268 on H6; and Tyr292 and Ala295 on H7. Previously, we detected steric clashes between Cys167 and the C1, C2, and C16 atoms of all-trans retinal in the dark state. Here, this residue is in close contact with retinal and is positioned at the hinge region. The hinge residues at H1, H2, and H7 are relatively closer to the CP region, where two water molecules are found to further stabilize the hinge site. Inclusion of explicit water in our model thus contributed to refine the precise location of the global hinge region, as well as to identify a hydrogen-bond network consolidated by water molecules. The conserved N-D pair (Asn55 and Asp83) also takes part in this network and is connected to Asn302 through a bridging water molecule.
Stability of the conformations obtained by simulations
To test the stability of the final rhodopsin conformation attained by our simulations, the final structure was subjected to two independent 5-ns unrestrained MD simulations. The protein exhibited an RMSD of only 1.75 Å from the starting conformation (i.e., the end structure obtained by our ANM-restrained MD). We did not include the C-terminus in these RMSD calculations, since this end of the molecule is highly flexible, as also pointed out previously (44). The critical interactions derived from the ANM-restrained MD simulations were confirmed to be stable, as illustrated in Fig. 9 (see Figs. 2S and 3S in the Supplementary Material, Data S1, for the relevant RMSD profiles). By the end of the unrestrained MD, Cys167 on H4 and Phe203 on H5 continued to be part of the chromophore binding site. The stronger interactions with H5 observed during ANM-restrained MD remained unchanged. NMR data shows that both the side chain of Glu122 and the backbone carbonyl of His211 are disrupted by the reorientation of the β-ionone ring of all-trans retinal in the Meta II state (87). These residues exhibited new interactions during the ANM-restrained MD simulations, which were retained during the unrestrained MD simulations. There were a number of interactions between the residues that stabilize the chromophore binding pocket in the dark state, which were lost during the ANM-restrained MD simulations and not restored in the unrestrained MD runs. These interactions mainly involved residues from H6, such as Trp265 and Ala269, consistent with previous cross-linking (25,86) and NMR experiments (87). These analyses suggest that the structure reached by the end of ANM-restrained MD is stable, especially near the chromophore binding pocket, whereas the C-terminus and the CP loops are very flexible and sample different conformations during the unrestrained MD.
FIGURE 9.

Stability of the chromophore binding pocket. The figure compares the end structure of the ANM-restrained MD (green) and that after an additional 5 ns unrestrained MD (cyan). The critical residues whose interactions with the chromophore were lost during the ANM-restrained MD simulations are labeled in red. Those residues that started to stabilize the chromophore during the ANM-restrained MD simulations are labeled black. These critical interactions are all maintained during the 5-ns unrestrained MD simulations.
CONCLUSION
In this study, we took advantage of the normal modes generated by the ANM to sample global conformational changes during MD simulations of rhodopsin. A major advantage of adopting such an ANM-restrained MD protocol is the ability to observe the global cooperative changes in full atomic detail in the presence of explicit water and membrane. SMD implements a force, or acceleration, along predefined directions. In this study, we take advantage of normal modes calculated for rhodopsin to identify the directions along which SMD forces will act, i.e., we “bias” the system along its inherent motions as calculated by the ANM analysis. Not knowing the conformation of the end point (activated state), we limited the structural changes to within 3.5 Å RMSD in backbone coordinates from the original state, using the most robust modes of motion, which have been updated as the structure was reconfigured.
We note that our simulations were performed at physiological temperature, i.e., there was no need to activate transitions using high temperatures, which is another advantage of adopting ANM restraints. Furthermore, ANM does not necessitate the generation of a trajectory, in contrast to ED analysis, or optimal superimposition of appropriate domains before PCA (108,109). Consequently, ANM restraints are not affected by convergence or inadequate sampling problems that may affect PCA-based analyses of MD trajectories (57). Knowledge of one (equilibrium) structure is sufficient to predict the most likely (lowest-energy ascent) directions of deformations (as the slowest modes) away from that local minimum. However, a limitation is the lack of knowledge of the size of motions, which are now being defined by the force fields that control the MD simulations and energy minimization steps. We note that our ANM-steered MD simulations can be readily extended to explore a larger portion of the conformational energy space in the neighborhood of the starting conformation, and sample multiple local minima. We have terminated here our runs after 10 cycles in view of the favorable comparison with experimental data on site-directed spin labeling, sulfhydryl reactivity, and disulfide cross-linking (10,12,13) observed at this stage.
Finally, the method explored here is easily implemented using the ANM modes in Nanoscale Molecular Dynamics (NAMD) (110). We have recently developed a fully automated server that releases these modes for any Protein Data Bank structure with known Cα coordinates (or any model submitted as query in Protein Data Bank format) (75,111). The incorporation of ANM restraints in MD packages such as NAMD is straightforward and flexible. In future work, ANM-restrained MD can be implemented as an option in NAMD or relevant MD software packages.
Although a wide range of information on both the dark and active states of rhodopsin exists, the details of the activation mechanism remain unclear. Here, we gained insights about the global motions that cooperatively propagate the local motions in the neighborhood of the isomerizing chromophore to the G-protein binding sites on the CP surface. Two global hinge centers have been identified that ensure this functional communication between distant positions. The first contains specific residues on H3–H6 that directly sense the local conformational changes induced by retinal isomerization. Some of these residues stabilize 11-cis retinal in the dark state (e.g., Trp265 and Tyr268 on H6), whereas others interact with all-trans retinal in the resulting structure (e.g., Cys167, Met207, and His211). In particular, the new contact found between Cys167 on H4 and all-trans retinal support findings from our previous study showing the significant effect of the Cys167Ser mutation on the Meta II decay rate (58). The second hinge, on the other hand, is located close to the CP end of helices H1, H2, and H7, and includes the NPXXY motif.
In the dark state, H6 is highly constrained by 11-cis retinal. The aromatic ring of Trp265 makes several contacts with the β-ionone ring. Isomerization to the trans form releases the constraints imposed on H6. In contrast, H4 and H5 form new contacts, and those made with H3 residues are redistributed. To accommodate these alterations in the chromophore binding site, H3–H6 undergo conformational changes that manifest themselves mainly at the CP ends of the helices. The local rearrangements of these helices near the chromophore binding packet efficiently propagate to these distal points due to the tight packing at the retinal-binding pocket, and the internal rigidity (like a moment arm) of helices. The enhanced ability of secondary structural elements—including, in particular, helices—to efficiently transmit signals to distant loci was pointed out in a recent study (112). It is interesting that some residues in the chromophore binding area (e.g., a disulfide bridge, Cys110-Cys187) are particularly rigid, as evidenced by their minimal RMSD by the end of our runs. The motions are even accentuated at the CP loops connecting helices H3–H6, leading to the exposure of the ERY motif crucial for G-protein binding (Fig. 4). It is worthy of note that in the activated structure, water molecules span the TM domain from the EC to the CP region. In addition to those detected by x-ray crystallography, new water molecules are located in the vicinity of highly conserved residues such as those in the cavity between Asn55, Asp83, and Asn302 (Fig. 6 A).
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
Part of this work was completed during the visit of Dr. Isin to the Theoretical and Computational Biophysics (TCB) Group, University of Illinois at Urbana-Champaign. Dr. Isin thanks all members of TCB group, especially J. C. Gumbart, Barry Isralewitz, and Chakra Chennubhotla (University of Pittsburgh) for useful discussions. We thank Judith Klein-Seetharaman (Department of Structural Biology, University of Pittsburgh) for helpful discussions throughout the course of this work and for critical reading of the manuscript.
Support from National Institutes of Health grants 5R33GM068400 (I.B.) and P41RR05969 and R01GM067887 (K.S. and E.T.) is gratefully acknowledged.
Editor: Peter Tieleman.
References
- 1.Gether, U. 2000. Uncovering molecular mechanisms involved in activation of G protein-coupled receptors. Endocr. Rev. 21:90–113. [DOI] [PubMed] [Google Scholar]
- 2.Müller, G. 2000. Towards 3D structures of G protein-coupled receptors: a multidisciplinary approach. Curr. Med. Chem. 7:861–888. [DOI] [PubMed] [Google Scholar]
- 3.Sakmar, T. P. 1998. Rhodopsin: a prototypical G protein-coupled receptor. Prog. Nucleic Acid Res. Mol. Biol. 5:1–34. [DOI] [PubMed] [Google Scholar]
- 4.Okada, T., Y. Fujiyoshi, M. Silow, J. Navarro, E. M. Landau, and Y. Shichida. 2002. Functional role of internal water molecules in rhodopsin revealed by X-ray crystallography. Proc. Natl. Acad. Sci. USA. 99:5982–5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Palczewski, K., T. Kumasaka, T. Hori, C. A. Behnke, H. Motoshima, B. A. Fox, I. Le Trong, D. C. Teller, T. Okada, R. E. Stenkamp, M. Yamamoto, and M. Miyano. 2000. Crystal structure of rhodopsin: a G protein-coupled receptor. Science. 289:739–745. [DOI] [PubMed] [Google Scholar]
- 6.Salom, D., D. T. Lodowski, R. E. Stenkamp, I. Le Trong, M. Golczak, B. Jastrzebska, T. Harris, J. A. Ballesteros, and K. Palczewski. 2006. Crystal structure of a photoactivated deprotonated intermediate of rhodopsin. Proc. Natl. Acad. Sci. USA. 103:16123–16128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cherezov, V., D. M. Rosenbaum, M. A. Hanson, S. G. Rasmussen, F. S. Thian, T. S. Kobilka, H. J. Choi, P. Kuhn, W. I. Weis, B. K. Kobilka, and R. C. Stevens. 2007. High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science. 318:1258–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosenbaum, D. M., V. Cherezov, M. A. Hanson, S. G. Rasmussen, F. S. Thian, T. S. Kobilka, H. J. Choi, X. J. Yao, W. I. Weis, R. C. Stevens, and B. K. Kobilka. 2007. GPCR engineering yields high-resolution structural insights into β2-adrenergic receptor function. Science. 318:1266–1273. [DOI] [PubMed] [Google Scholar]
- 9.Rasmussen, S. G., H. J. Choi, D. M. Rosenbaum, T. S. Kobilka, F. S. Thian, P. C. Edwards, M. Burghammer, V. R. Ratnala, R. Sanishvili, R. F. Fischetti, G. F. Schertler, W. I. Weis, and B. K. Kobilka. 2007. Crystal structure of the human β2 adrenergic G-protein-coupled receptor. Nature. 450:383–387. [DOI] [PubMed] [Google Scholar]
- 10.Hubbell, W. L., C. Altenbach, C. M. Hubbell, and H. G. Khorana. 2003. Rhodopsin structure, dynamics, and activation: a perspective from crystallography, site-directed spin labeling, sulfhydryl reactivity, and disulfide cross-linking. Adv. Protein Chem. 63:243–290. [DOI] [PubMed] [Google Scholar]
- 11.Palczewski, K. 2006. G protein-coupled receptor rhodopsin. Annu. Rev. Biochem. 75:743–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klein-Seetharaman, J. 2002. Dynamics in rhodopsin. ChemBioChem. 3:981–986. [DOI] [PubMed] [Google Scholar]
- 13.Meng, E. C., and H. R. Bourne. 2001. Receptor activation: what does the rhodopsin structure tell us? Trends Pharmacol. Sci. 22:587–593. [DOI] [PubMed] [Google Scholar]
- 14.Cohen, G. B., T. Yang, P. R. Robinson, and D. D. Oprian. 1993. Constitutive activation of opsin: influence of charge at position 134 and size at position 296. Biochemistry. 32:6111–6115. [DOI] [PubMed] [Google Scholar]
- 15.Sakmar, T. P., R. R. Franke, and H. G. Khorana. 1991. The role of the retinylidene Schiff base counterion in rhodopsin in determining wavelength absorbance and Schiff base pKa. Proc. Natl. Acad. Sci. USA. 88:3079–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alewijnse, A. E., H. Timmerman, E. H. Jacobs, M. J. Smit, E. Roovers, S. Cotecchia, and R. Leurs. 2000. The effect of mutations in the DRY motif on the constitutive activity and structural instability of the histamine H(2) receptor. Mol. Pharmacol. 57:890–898. [PubMed] [Google Scholar]
- 17.Ballesteros, J., S. Kitanovic, F. Guarnieri, P. Davies, B. J. Fromme, K. Konvicka, L. Chi, R. P. Millar, J. S. Davidson, H. Weinstein, and S. C. Sealfon. 1998. Functional microdomains in G-protein-coupled receptors. The conserved arginine-cage motif in the gonadotropin-releasing hormone receptor. J. Biol. Chem. 273:10445–10453. [DOI] [PubMed] [Google Scholar]
- 18.Ballesteros, J. A., A. D. Jensen, G. Liapakis, S. G. Rasmussen, L. Shi, U. Gether, and J. A. Javitch. 2001. Activation of the β2-adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6. J. Biol. Chem. 276:29171–29177. [DOI] [PubMed] [Google Scholar]
- 19.Greasley, P. J., F. Fanelli, O. Rossier, L. Abuin, and S. Cotecchia. 2002. Mutagenesis and modelling of the a1β-adrenergic receptor highlight the role of the helix 3/helix 6 interface in receptor activation. Mol. Pharmacol. 61:1025–1032. [DOI] [PubMed] [Google Scholar]
- 20.Hogger, P., M. S. Shockley, J. Lameh, and W. Sadee. 1995. Activating and inactivating mutations in N- and C-terminal i3 loop junctions of muscarinic acetylcholine Hm1 receptors. J. Biol. Chem. 270:7405–7410. [DOI] [PubMed] [Google Scholar]
- 21.Rasmussen, S. G., A. D. Jensen, G. Liapakis, P. Ghanouni, J. A. Javitch, and U. Gether. 1999. Mutation of a highly conserved aspartic acid in the β2 adrenergic receptor: constitutive activation, structural instability, and conformational rearrangement of transmembrane segment 6. Mol. Pharmacol. 56:175–184. [DOI] [PubMed] [Google Scholar]
- 22.Samama, P., S. Cotecchia, T. Costa, and R. J. Lefkowitz. 1993. A mutation-induced activated state of the β2-adrenergic receptor. Extending the ternary complex model. J. Biol. Chem. 268:4625–4636. [PubMed] [Google Scholar]
- 23.Scheer, A., F. Fanelli, T. Costa, P. G. De Benedetti, and S. Cotecchia. 1996. Constitutively active mutants of the a1β-adrenergic receptor: role of highly conserved polar amino acids in receptor activation. EMBO J. 15:3566–3578. [PMC free article] [PubMed] [Google Scholar]
- 24.Shi, L., G. Liapakis, R. Xu, F. Guarnieri, J. A. Ballesteros, and J. A. Javitch. 2002. β2 adrenergic receptor activation. Modulation of the proline kink in transmembrane 6 by a rotamer toggle switch. J. Biol. Chem. 277:40989–40996. [DOI] [PubMed] [Google Scholar]
- 25.Borhan, B., M. L. Souto, H. Imai, Y. Shichida, and K. Nakanishi. 2000. Movement of retinal along the visual transduction path. Science. 288:2209–2212. [DOI] [PubMed] [Google Scholar]
- 26.Nakayama, T. A., and H. G. Khorana. 1991. Mapping of the amino acids in membrane-embedded helices that interact with the retinal chromophore in bovine rhodopsin. J. Biol. Chem. 266:4269–4275. [PubMed] [Google Scholar]
- 27.Swaminath, G., X. Deupi, T. W. Lee, W. Zhu, F. S. Thian, T. S. Kobilka, and B. Kobilka. 2005. Probing the β2 adrenoceptor binding site with catechol reveals differences in binding and activation by agonists and partial agonists. J. Biol. Chem. 280:22165–22171. [DOI] [PubMed] [Google Scholar]
- 28.Menon, S. T., M. Han, and T. P. Sakmar. 2001. Rhodopsin: structural basis of molecular physiology. Physiol. Rev. 81:1659–1688. [DOI] [PubMed] [Google Scholar]
- 29.Sakmar, T. P., S. T. Menon, E. P. Marin, and E. S. Awad. 2002. Rhodopsin: insights from recent structural studies. Annu. Rev. Biophys. Biomol. Struct. 31:443–484. [DOI] [PubMed] [Google Scholar]
- 30.Crozier, P. S., M. J. Stevens, L. R. Forrest, and T. B. Woolf. 2003. Molecular dynamics simulation of dark-adapted rhodopsin in an explicit membrane bilayer: coupling between local retinal and larger scale conformational change. J. Mol. Biol. 333:493–514. [DOI] [PubMed] [Google Scholar]
- 31.Crozier, P. S., M. J. Stevens, and T. B. Woolf. 2007. How a small change in retinal leads to G-protein activation: initial events suggested by molecular dynamics calculations. Proteins. 66:559–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huber, T., A. V. Botelho, K. Beyer, and M. F. Brown. 2004. Membrane model for the G-protein-coupled receptor rhodopsin: hydrophobic interface and dynamical structure. Biophys. J. 86:2078–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lemaitre, V., P. Yeagle, and A. Watts. 2005. Molecular dynamics simulations of retinal in rhodopsin: from the dark-adapted state towards lumirhodopsin. Biochemistry. 44:12667–12680. [DOI] [PubMed] [Google Scholar]
- 34.Rohrig, U. F., L. Guidoni, and U. Rothlisberger. 2002. Early steps of the intramolecular signal transduction in rhodopsin explored by molecular dynamics simulations. Biochemistry. 41:10799–10809. [DOI] [PubMed] [Google Scholar]
- 35.Saam, J., E. Tajkhorshid, S. Hayashi, and K. Schulten. 2002. Molecular dynamics investigation of primary photoinduced events in the activation of rhodopsin. Biophys. J. 83:3097–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ishiguro, M., T. Hirano, and Y. Oyama. 2003. Modelling of photointermediates suggests a mechanism of the flip of the β-ionone moiety of the retinylidene chromophore in the rhodopsin photocascade. ChemBioChem. 4:228–231. [DOI] [PubMed] [Google Scholar]
- 37.Ishiguro, M., Y. Oyama, and T. Hirano. 2004. Structural models of the photointermediates in the rhodopsin photocascade, lumirhodopsin, metarhodopsin I, and metarhodopsin II. ChemBioChem. 5:298–310. [DOI] [PubMed] [Google Scholar]
- 38.Choi, G., J. Landin, J. F. Galan, R. R. Birge, A. D. Albert, and P. L. Yeagle. 2002. Structural studies of metarhodopsin II, the activated form of the G-protein coupled receptor, rhodopsin. Biochemistry. 41:7318–7324. [DOI] [PubMed] [Google Scholar]
- 39.Gouldson, P. R., N. J. Kidley, R. P. Bywater, G. Psaroudakis, H. D. Brooks, C. Diaz, D. Shire, and C. A. Reynolds. 2004. Toward the active conformations of rhodopsin and the β2-adrenergic receptor. Proteins. 56:67–84. [DOI] [PubMed] [Google Scholar]
- 40.Niv, M. Y., L. Skrabanek, M. Filizola, and H. Weinstein. 2006. Modeling activated states of GPCRs: the rhodopsin template. J. Comput. Aided Mol. Des. 20:437–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nikiforovich, G. V., and G. R. Marshall. 2005. Modeling flexible loops in the dark-adapted and activated states of rhodopsin, a prototypical G-protein-coupled receptor. Biophys. J. 89:3780–3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kong, Y., and M. Karplus. 2007. The signaling pathway of rhodopsin. Structure. 15:611–623. [DOI] [PubMed] [Google Scholar]
- 43.Grossfield, A., S. E. Feller, and M. C. Pitman. 2006. A role for direct interactions in the modulation of rhodopsin by omega-3 polyunsaturated lipids. Proc. Natl. Acad. Sci. USA. 103:4888–4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grossfield, A., S. E. Feller, and M. C. Pitman. 2007. Convergence of molecular dynamics simulations of membrane proteins. Proteins. 67:31–40. [DOI] [PubMed] [Google Scholar]
- 45.Hess, B. 2000. Similarities between principal components of protein dynamics and random diffusion. Phys. Rev. E Stat. Phys. Plasmas Fluids Relat. Interdiscip. Topics. 62:8438–8448. [DOI] [PubMed] [Google Scholar]
- 46.Hess, B. 2002. Convergence of sampling in protein simulations. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 65:031910. [DOI] [PubMed] [Google Scholar]
- 47.Faraldo-Gomez, J. D., L. R. Forrest, M. Baaden, P. J. Bond, C. Domene, G. Patargias, J. Cuthbertson, and M. S. Sansom. 2004. Conformational sampling and dynamics of membrane proteins from 10-nanosecond computer simulations. Proteins. 57:783–791. [DOI] [PubMed] [Google Scholar]
- 48.Isralewitz, B., J. Baudry, J. Gullingsrud, D. Kosztin, and K. Schulten. 2001. Steered molecular dynamics investigations of protein function. J. Mol. Graph. Model. 19:13–25. [DOI] [PubMed] [Google Scholar]
- 49.Sotomayor, M., and K. Schulten. 2007. Single-molecule experiments in vitro and in silico. Science. 316:1144–1148. [DOI] [PubMed] [Google Scholar]
- 50.Schlitter, J., M. Engels, and P. Kruger. 1994. Targeted molecular dynamics: a new approach for searching pathways of conformational transitions. J. Mol. Graph. 12:84–89. [DOI] [PubMed] [Google Scholar]
- 51.Kitao, A., and N. Go. 1999. Investigating protein dynamics in collective coordinate space. Curr. Opin. Struct. Biol. 9:164–169. [DOI] [PubMed] [Google Scholar]
- 52.Amadei, A., A. B. Linssen, and H. J. Berendsen. 1993. Essential dynamics of proteins. Proteins. 17:412–425. [DOI] [PubMed] [Google Scholar]
- 53.Amadei, A., A. B. Linssen, B. L. de Groot, D. M. van Aalten, and H. J. Berendsen. 1996. An efficient method for sampling the essential subspace of proteins. J. Biomol. Struct. Dyn. 13:615–625. [DOI] [PubMed] [Google Scholar]
- 54.de Groot, B. L., A. Amadei, R. M. Scheek, N. A. van Nuland, and H. J. Berendsen. 1996. An extended sampling of the configurational space of HPr from E. coli. Proteins. 26:314–322. [DOI] [PubMed] [Google Scholar]
- 55.Cui, Q., and I. Bahar. 2006. Normal Mode Analysis. Theory and Applications to Biological and Chemical Systems. CRC Press, Boca Raton, FL.
- 56.Abseher, R., and M. Nilges. 2000. Efficient sampling in collective coordinate space. Proteins. 39:82–88. [DOI] [PubMed] [Google Scholar]
- 57.Balsera, M. A., W. Wriggers, Y. Oono, and K. Schulten. 1996. Principal component analysis and long time protein dynamics. J. Phys. Chem. 100:2567–2572. [Google Scholar]
- 58.Isin, B., A. J. Rader, H. K. Dhiman, J. Klein-Seetharaman, and I. Bahar. 2006. Predisposition of the dark state of rhodopsin to functional changes in structure. Proteins. 65:970–983. [DOI] [PubMed] [Google Scholar]
- 59.Bahar, I., A. R. Atilgan, and B. Erman. 1997. Direct evaluation of thermal fluctuations in proteins using a single- parameter harmonic potential. Fold. Des. 2:173–181. [DOI] [PubMed] [Google Scholar]
- 60.Atilgan, A. R., S. R. Durell, R. L. Jernigan, M. C. Demirel, O. Keskin, and I. Bahar. 2001. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 80:505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Doruker, P., A. R. Atilgan, and I. Bahar. 2000. Dynamics of proteins predicted by molecular dynamics simulations and analytical approaches: application to a-amylase inhibitor. Proteins. 40:512–524. [PubMed] [Google Scholar]
- 62.Bahar, I., and A. J. Rader. 2005. Coarse-grained normal mode analysis in structural biology. Curr. Opin. Struct. Biol. 15:586–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ma, J. 2005. Usefulness and limitations of normal mode analysis in modeling dynamics of biomolecular complexes. Structure. 13:373–380. [DOI] [PubMed] [Google Scholar]
- 64.Tama, F., and C. L. Brooks. 2006. Symmetry, form, and shape: guiding principles for robustness in macromolecular machines. Annu. Rev. Biophys. Biomol. Struct. 35:115–133. [DOI] [PubMed] [Google Scholar]
- 65.Zheng, W., and B. R. Brooks. 2005. Normal-modes-based prediction of protein conformational changes guided by distance constraints. Biophys. J. 88:3109–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ming, D., and M. E. Wall. 2006. Interactions in native binding sites cause a large change in protein dynamics. J. Mol. Biol. 358:213–223. [DOI] [PubMed] [Google Scholar]
- 67.Ming, D., and M. E. Wall. 2005. Allostery in a coarse-grained model of protein dynamics. Phys. Rev. Lett. 95:198103. [DOI] [PubMed] [Google Scholar]
- 68.Sonne, J., C. Kandt, G. H. Peters, F. Y. Hansen, M. O. Jensen, and D. P. Tieleman. 2007. Simulation of the coupling between nucleotide binding and transmembrane domains in the ATP binding cassette transporter BtuCD. Biophys. J. 92:2727–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang, Z., Y. Shi, and H. Liu. 2003. Molecular dynamics simulations of peptides and proteins with amplified collective motions. Biophys. J. 84:3583–3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hinsen, K. 1998. Analysis of domain motions by approximate normal mode calculations. Proteins. 33:417–429. [DOI] [PubMed] [Google Scholar]
- 71.Lu, M., and J. Ma. 2005. The role of shape in determining molecular motions. Biophys. J. 89:2395–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tama, F., and Y. H. Sanejouand. 2001. Conformational change of proteins arising from normal mode calculations. Protein Eng. 14:1–6. [DOI] [PubMed] [Google Scholar]
- 73.Changeux, J. P., and S. J. Edelstein. 2005. Allosteric mechanisms of signal transduction. Science. 308:1424–1428. [DOI] [PubMed] [Google Scholar]
- 74.Eisenmesser, E. Z., O. Millet, W. Labeikovsky, D. M. Korzhnev, M. Wolf-Watz, D. A. Bosco, J. J. Skalicky, L. E. Kay, and D. Kern. 2005. Intrinsic dynamics of an enzyme underlies catalysis. Nature. 438:117–121. [DOI] [PubMed] [Google Scholar]
- 75.Eyal, E., C. Chennubhotla, L. W. Yang, and I. Bahar. 2007. Anisotropic fluctuations of amino acids in protein structures: insights from x-ray crystallography and elastic network models. Bioinformatics. In press. [DOI] [PubMed]
- 76.Fahmy, K., F. Jager, M. Beck, T. A. Zvyaga, T. P. Sakmar, and F. Siebert. 1993. Protonation states of membrane-embedded carboxylic acid groups in rhodopsin and metarhodopsin II: a Fourier-transform infrared spectroscopy study of site-directed mutants. Proc. Natl. Acad. Sci. USA. 90:10206–10210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Berson, E. L. 1993. Retinitis pigmentosa. The Friedenwald Lecture. Invest. Ophthalmol. Vis. Sci. 34:1659–1676. [PubMed] [Google Scholar]
- 78.Olsson, J. E., J. W. Gordon, B. S. Pawlyk, D. Roof, A. Hayes, R. S. Molday, S. Mukai, G. S. Cowley, E. L. Berson, and T. P. Dryja. 1992. Transgenic mice with a rhodopsin mutation (Pro23His): a mouse model of autosomal dominant retinitis pigmentosa. Neuron. 9:815–830. [DOI] [PubMed] [Google Scholar]
- 79.Wang, M., T. T. Lam, M. O. Tso, and M. I. Naash. 1997. Expression of a mutant opsin gene increases the susceptibility of the retina to light damage. Vis. Neurosci. 14:55–62. [DOI] [PubMed] [Google Scholar]
- 80.Hwa, J., J. Klein-Seetharaman, and H. G. Khorana. 2001. Structure and function in rhodopsin: mass spectrometric identification of the abnormal intradiscal disulfide bond in misfolded retinitis pigmentosa mutants. Proc. Natl. Acad. Sci. USA. 98:4872–4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Farrens, D. L., and H. G. Khorana. 1995. Structure and function in rhodopsin. Measurement of the rate of metarhodopsin II decay by fluorescence spectroscopy. J. Biol. Chem. 270:5073–5076. [DOI] [PubMed] [Google Scholar]
- 82.Hwa, J., P. J. Reeves, J. Klein-Seetharaman, F. Davidson, and H. G. Khorana. 1999. Structure and function in rhodopsin: further elucidation of the role of the intradiscal cysteines, Cys-110, -185, and -187, in rhodopsin folding and function. Proc. Natl. Acad. Sci. USA. 96:1932–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Richards, J. E., K. M. Scott, and P. A. Sieving. 1995. Disruption of conserved rhodopsin disulfide bond by Cys187Tyr mutation causes early and severe autosomal dominant retinitis pigmentosa. Ophthalmology. 102:669–677. [DOI] [PubMed] [Google Scholar]
- 84.Vaithinathan, R., E. L. Berson, and T. P. Dryja. 1994. Further screening of the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa. Genomics. 21:461–463. [DOI] [PubMed] [Google Scholar]
- 85.Lehmann, N., U. Alexiev, and K. Fahmy. 2007. Linkage between the intramembrane H-bond network around aspartic acid 83 and the cytosolic environment of helix 8 in photoactivated rhodopsin. J. Mol. Biol. 366:1129–1141. [DOI] [PubMed] [Google Scholar]
- 86.Nakayama, T. A., and H. G. Khorana. 1990. Orientation of retinal in bovine rhodopsin determined by cross-linking using a photoactivatable analog of 11-cis-retinal. J. Biol. Chem. 265:15762–15769. [PubMed] [Google Scholar]
- 87.Patel, A. B., E. Crocker, M. Eilers, A. Hirshfeld, M. Sheves, and S. O. Smith. 2004. Coupling of retinal isomerization to the activation of rhodopsin. Proc. Natl. Acad. Sci. USA. 101:10048–10053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lau, P. W., A. Grossfield, S. E. Feller, M. C. Pitman, and M. F. Brown. 2007. Dynamic structure of retinylidene ligand of rhodopsin probed by molecular simulations. J. Mol. Biol. 372:906–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Salgado, G. F., A. V. Struts, K. Tanaka, N. Fujioka, K. Nakanishi, and M. F. Brown. 2004. Deuterium NMR structure of retinal in the ground state of rhodopsin. Biochemistry. 43:12819–12828. [DOI] [PubMed] [Google Scholar]
- 90.Salgado, G. F., A. V. Struts, K. Tanaka, S. Krane, K. Nakanishi, and M. F. Brown. 2006. Solid-state 2H NMR structure of retinal in metarhodopsin I. J. Am. Chem. Soc. 128:11067–11071. [DOI] [PubMed] [Google Scholar]
- 91.Struts, A. V., G. F. Salgado, K. Tanaka, S. Krane, K. Nakanishi, and M. F. Brown. 2007. Structural analysis and dynamics of retinal chromophore in dark and meta I states of rhodopsin from 2H NMR of aligned membranes. J. Mol. Biol. 372:50–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yan, E. C., M. A. Kazmi, Z. Ganim, J. M. Hou, D. Pan, B. S. Chang, T. P. Sakmar, and R. A. Mathies. 2003. Retinal counterion switch in the photoactivation of the G protein-coupled receptor rhodopsin. Proc. Natl. Acad. Sci. USA. 100:9262–9267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ludeke, S., M. Beck, E. C. Yan, T. P. Sakmar, F. Siebert, and R. Vogel. 2005. The role of Glu181 in the photoactivation of rhodopsin. J. Mol. Biol. 353:345–356. [DOI] [PubMed] [Google Scholar]
- 94.Yan, E. C., M. A. Kazmi, S. De, B. S. Chang, C. Seibert, E. P. Marin, R. A. Mathies, and T. P. Sakmar. 2002. Function of extracellular loop 2 in rhodopsin: glutamic acid 181 modulates stability and absorption wavelength of metarhodopsin II. Biochemistry. 41:3620–3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Altenbach, C., K. Yang, D. L. Farrens, Z. T. Farahbakhsh, H. G. Khorana, and W. L. Hubbell. 1996. Structural features and light-dependent changes in the cytoplasmic interhelical E-F loop region of rhodopsin: a site-directed spin-labeling study. Biochemistry. 35:12470–12478. [DOI] [PubMed] [Google Scholar]
- 96.Altenbach, C., J. Klein-Seetharaman, J. Hwa, H. G. Khorana, and W. L. Hubbell. 1999. Structural features and light-dependent changes in the sequence 59–75 connecting helices I and II in rhodopsin: a site-directed spin-labeling study. Biochemistry. 38:7945–7949. [DOI] [PubMed] [Google Scholar]
- 97.Altenbach, C., K. Cai, H. G. Khorana, and W. L. Hubbell. 1999. Structural features and light-dependent changes in the sequence 306–322 extending from helix VII to the palmitoylation sites in rhodopsin: a site-directed spin-labeling study. Biochemistry. 38:7931–7937. [DOI] [PubMed] [Google Scholar]
- 98.Cai, K., R. Langen, W. L. Hubbell, and H. G. Khorana. 1997. Structure and function in rhodopsin: topology of the C-terminal polypeptide chain in relation to the cytoplasmic loops. Proc. Natl. Acad. Sci. USA. 94:14267–14272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Farahbakhsh, Z. T., K. D. Ridge, H. G. Khorana, and W. L. Hubbell. 1995. Mapping light-dependent structural changes in the cytoplasmic loop connecting helices C and D in rhodopsin: a site-directed spin labeling study. Biochemistry. 34:8812–8819. [DOI] [PubMed] [Google Scholar]
- 100.Farrens, D. L., C. Altenbach, K. Yang, W. L. Hubbell, and H. G. Khorana. 1996. Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science. 274:768–770. [DOI] [PubMed] [Google Scholar]
- 101.Klein-Seetharaman, J., J. Hwa, K. Cai, C. Altenbach, W. L. Hubbell, and H. G. Khorana. 2001. Probing the dark state tertiary structure in the cytoplasmic domain of rhodopsin: proximities between amino acids deduced from spontaneous disulfide bond formation between Cys316 and engineered cysteines in cytoplasmic loop 1. Biochemistry. 40:12472–12478. [DOI] [PubMed] [Google Scholar]
- 102.Sheikh, S. P., T. A. Zvyaga, O. Lichtarge, T. P. Sakmar, and H. R. Bourne. 1996. Rhodopsin activation blocked by metal-ion-binding sites linking transmembrane helices C and F. Nature. 383:347–350. [DOI] [PubMed] [Google Scholar]
- 103.Bourne, H. R. 1997. How receptors talk to trimeric G proteins. Curr. Opin. Cell Biol. 9:134–142. [DOI] [PubMed] [Google Scholar]
- 104.Altenbach, C., K. Cai, J. Klein-Seetharaman, H. G. Khorana, and W. L. Hubbell. 2001. Structure and function in rhodopsin: mapping light-dependent changes in distance between residue 65 in helix TM1 and residues in the sequence 306–319 at the cytoplasmic end of helix TM7 and in helix H8. Biochemistry. 40:15483–15492. [DOI] [PubMed] [Google Scholar]
- 105.Cai, K., J. Klein-Seetharaman, D. Farrens, C. Zhang, C. Altenbach, W. L. Hubbell, and H. G. Khorana. 1999. Single-cysteine substitution mutants at amino acid positions 306–321 in rhodopsin, the sequence between the cytoplasmic end of helix VII and the palmitoylation sites: sulfhydryl reactivity and transducin activation reveal a tertiary structure. Biochemistry. 38:7925–7930. [DOI] [PubMed] [Google Scholar]
- 106.Cai, K., J. Klein-Seetharaman, C. Altenbach, W. L. Hubbell, and H. G. Khorana. 2001. Probing the dark state tertiary structure in the cytoplasmic domain of rhodopsin: proximities between amino acids deduced from spontaneous disulfide bond formation betwen cysteine pairs engineered in cytoplasmic loops 1, 3, and 4. Biochemistry. 40:12479–12485. [DOI] [PubMed] [Google Scholar]
- 107.Klein-Seetharaman, J., J. Hwa, K. Cai, C. Altenbach, W. L. Hubbell, and H. G. Khorana. 1999. Single-cysteine substitution mutants at amino acid positions 55–75, the sequence connecting the cytoplasmic ends of helices I and II in rhodopsin: reactivity of the sulfhydryl groups and their derivatives identifies a tertiary structure that changes upon light-activation. Biochemistry. 38:7938–7944. [DOI] [PubMed] [Google Scholar]
- 108.Hayward, S., and N. Go. 1995. Collective variable description of native protein dynamics. Annu. Rev. Phys. Chem. 46:223–250. [DOI] [PubMed] [Google Scholar]
- 109.Hayward, S., A. Kitao, and H. J. Berendsen. 1997. Model-free methods of analyzing domain motions in proteins from simulation: a comparison of normal mode analysis and molecular dynamics simulation of lysozyme. Proteins. 27:425–437. [DOI] [PubMed] [Google Scholar]
- 110.Phillips, J. C., R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale, and K. Schulten. 2005. Scalable molecular dynamics with NAMD. J. Comput. Chem. 26:1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Eyal, E., L. W. Yang, and I. Bahar. 2006. Anisotropic network model: systematic evaluation and a new web interface. Bioinformatics. 22:2619–2627. [DOI] [PubMed] [Google Scholar]
- 112.Chennubhotla, C., and I. Bahar. 2007. Signal propagation in proteins and relation to equilibrium fluctuations. PLoS Comput. Biol. 3:1716–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.