

Abstract
Single-stranded junctions/loops are frequently occurring structural motifs in nucleic acid structures. Due to the polyanionic nature of the nucleic acid backbone, metal ions play a crucial role in the loop stability. Here we use the tightly bound ion theory, which can account for the possible ion correlation and ensemble (fluctuation) effects, to predict the ion-dependence of loop and stem-loop (hairpin) free energies. The predicted loop free energy is a function of the loop length, the loop end-to-end distance, and the ion (Na+ and Mg2+ in this study) concentrations. Based on the statistical mechanical calculations, we derive a set of empirical formulas for the loop thermodynamic parameters as functions of Na+ and Mg2+ concentrations. For three specific types of loops, namely, hairpin, bulge, and internal loops, the predicted free energies agree with the experimental data. Further applications of these empirical formulas to RNA and DNA hairpin stability lead to good agreements with the available experimental data. Our results indicate that the ion-dependent loop stability makes significant contribution to the overall ion-dependence of the hairpin stability.
INTRODUCTION
Nucleic acids (RNAs and DNAs) are highly charged polyanionic molecules. The metal ions in the solution, such as Na+ and Mg2+ ions, play an essential role in stabilizing the folded structure through electrostatic screening (1–23). Single-stranded junctions and loops between helices are important structural and functional components of nucleic acids (24–42). Thermodynamic properties of loops and junctions, such as the ion-dependence of loop stability, play an important role in the overall stability of the nucleic acid structures. This study addresses the ion-dependent loop stability and its effect on the ion-dependence of hairpin-folding stability.
The thermodynamic parameters for different types of (hairpin, bulge, and internal) RNA and DNA loops have been measured at standard 1 M NaCl condition (24–42). These parameters form the basis for predicting nucleic-acid folding stability (24–42). However, different ionic conditions can lead to different thermodynamic behaviors of the molecules (43–58), especially in the presence of multivalent ions, such as Mg2+ (59–68). For DNA and RNA helices, salt-dependent extensions of the thermodynamic parameters have been derived from experimental data (30,36,56–58) and from statistical mechanical modeling (67,68). However, no such relationship is available for loops. In this article, we develop a statistical mechanical model for the ion-dependent loop stability.
For treating the ion-nucleic acid interactions, there have been mainly two types of classic polyelectrolyte theories: the counterion condensation (CC) theory (69,70) and the Poisson-Boltzmann (PB) theory (71–77). Both (CC and PB) theories are successful in predicting thermodynamics for a broad range of systems with nucleic acids in ionic solutions (69–77). The CC theory is based on the simplified nucleic acid structural model and is a double-limit law, i.e., it is developed for dilute salt solution and nucleic acids of infinite length. The PB theory is a mean-field theory and ignores ion correlation and fluctuation effects which can be important for multivalent ions (e.g., Mg2+) solutions (78–82). Recently, we developed a statistical mechanical theory, namely, tightly-bound ion (TBI) theory, to account for correlations and fluctuations for strongly correlated ions (79–82). The theory has been validated through extensive comparisons with experiments on the stability of DNA and RNA helices in pure/mixed Na+/Mg2+ solutions (67,68) and on the ion-mediated DNA helix assembly and bending (80–82).
Here we will use the TBI theory to investigate the folding thermodynamics of loops and RNA hairpins in Na+ and Mg2+ solutions. Specifically, we will use the virtual-bond polymer model (Vfold model; (83–85)) to produce loop conformational ensembles, and for each conformation, we use the TBI theory to treat ion-chain interactions. The ensemble average over all the possible conformations gives the ion-dependent loop free energy. We will calculate the loop thermodynamics for different chain length, end-to-end distance, and Na+ and Mg2+ concentrations. Furthermore, based on the computed loop free energies, we will derive a set of simple empirical formulas for the ion-dependent loop thermodynamic parameters. We will also present extensive experimental tests for these empirical formulas and apply the theory to predict the ion-dependent nucleic acid hairpin folding stabilities.
METHODS
Chain conformational ensemble
For a given length of polynucleotide chain, we apply the Vfold model (83–85) to generate the conformational ensemble of loop conformations. The basic idea is to represent a nucleotide by two virtual bonds: C4-P and P-C4 (83–87), where P and C4 stand for the phosphate and carbon (C4) atoms, respectively; see Fig. 1 A. The conformation of an N-nt chain is described by the three-dimensional configuration of the 2N virtual bonds. The conformational ensemble of the loop can be generated through self-avoiding walks of the 2N virtual bonds in the three-dimensional space. A survey for the virtual bond configurations in the known RNA structures suggests that diamond lattice bonds can well describe the virtual bonds in realistic structures (88). Therefore, we can generate the loop conformational ensemble through an exhaustive self-avoiding walk of the virtual bonds in a diamond lattice.
FIGURE 1.

Illustrations of the structural model for single-stranded nucleotide chain used in the work. (A) The virtual-bond model for nucleotide chain (83–85). (B) The coarse-grained chain model where P atoms are represented by a series of spheres with charge −e (electronic charge) at the centers (79,89). (C) A three-dimensional chain conformation for the coarse-grained chain model where coordinates of P atoms are produced from the virtual bond model on diamond lattice (83–85). (D) The RNA and DNA hairpin sequences used in our calculations. The thermodynamic parameters at 1 M NaCl, and experimental references for these hairpins are listed in Table 2. The values i and j are the numbers of nucleotides of U's and T's in RH6 and DH1 loops, respectively.
To model the electrostatic properties of the chain, we represent each phosphate group, which carries one electronic charge (−e), by a sphere of radius 2.1 Å (79,89) and place a point charge −e at the center of the sphere (see Fig. 1, B and C). The radius of the sphere is equal to 2.1 Å, which is adopted from the groove-primitive model (79,89). For each chain conformation, we use the TBI theory (79–82) to calculate the electrostatic free energy.
Tightly-bound ion theory
The TBI theory has been described in detail in the literature (67,68,79–82). Here, we only give a brief overview of the theory.
Tightly-bound ions and tightly-bound region
In the TBI theory (67,68,79–82), the monovalent ions are treated as the ionic background as described by the mean-field Poisson-Boltzmann equation (PB) (68,82). For multivalent ions, to account for the possible ion-ion correlation effect, we classify them into two types (79–82) according to the correlation strength: the (strongly correlated) tightly bound ions; and the (weakly correlated) diffusive ions. Correspondingly, the regions where the two types of ions are distributed are denoted as the tightly-bound region and the diffusive region, respectively. For the diffusive ions, we use PB, and for the tightly-bound ions, we use a separate treatment by considering the ion-ion correlations and fluctuations (ensemble) of ion distributions.
Electrostatic free energy
For a single-stranded RNA (or DNA) chain with N phosphate spheres, we divide the whole tightly-bound region into N cells, each around a phosphate. We define a mode of the tightly-bound ion distribution by a set of numbers m1, m2, …, mN, where mi is the number of the tightly-bound ions in the ith cell. For z-valent (z ≥ 2) multivalent ions, a cell with mi = 1 multivalent cation would become positively charged, which makes further addition of ions less likely. Therefore, in practice, we assume that mi = 0 or 1.
The total electrostatic partition function Z for a given polynucleotide chain conformation is given by the summation over all the possible binding modes M for z-valent ions,
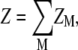 |
(1) |
where ZM is the partition function for a given ion-binding mode M,
 |
(2) |
where Z(id) is the partition function for the uniform ion solution (without the polyelectrolyte). Nz/V is the bulk concentration of the z-valent ion for a 1:z ionic solution (Nz is the total number of the z-valent ions and V is the volume). Nb is the total number of the tightly-bound ions for mode M. 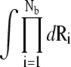 is the volume integral for the tightly-bound ions. ΔGb in Eq. 2 is the mean Coulombic interaction energy between all the charge-charge pairs (including the phosphate groups and the tightly-bound ions) in the tightly-bound region; ΔGd in Eq. 2 includes the free energy for the electrostatic interactions between the diffusive ions and between the diffusive ions and the charges in the tightly-bound region, as well as the entropic free energy of the diffusive ions.
is the volume integral for the tightly-bound ions. ΔGb in Eq. 2 is the mean Coulombic interaction energy between all the charge-charge pairs (including the phosphate groups and the tightly-bound ions) in the tightly-bound region; ΔGd in Eq. 2 includes the free energy for the electrostatic interactions between the diffusive ions and between the diffusive ions and the charges in the tightly-bound region, as well as the entropic free energy of the diffusive ions. 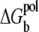 is the (Born) polarization energy for the charges in the tightly-bound region (82). The calculations of ΔGb, ΔGd, and
is the (Born) polarization energy for the charges in the tightly-bound region (82). The calculations of ΔGb, ΔGd, and 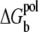 have been described in detail in the literature (67,68,79–82).
have been described in detail in the literature (67,68,79–82).
From Eqs. 1 and 2, the electrostatic free energy GE for a polynucleotide chain conformation is equal to
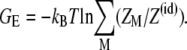 |
(3) |
Parameter sets and numerical details in TBI calculations
In this study, the ions are assumed to be hydrated, and the radii of hydrated Na+ and Mg2+ ions are taken as 3.5 Å and 4.5 Å (67,68,79–82,90), respectively. The dielectric constant ε of molecular interior is set to be 20 (82), and ε of solvent is set as the value of bulk water (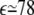 at 25°C). A thin layer of thickness equal to one cation radius is added to the molecular surface to account for the excluded volume layer of the cations (9,79–82). Moreover, we use the three-step focusing process to obtain the detailed ion distribution near the molecules (71,79–82). For each run, the electrostatic potentials are iterated to a convergence of <10−4 kBT/e. The grid size of the first run depends on the salt concentration used. Generally, we keep it larger than four times of the Debye length, and the resolution of the first run varies with the grid size to make the iterative process computationally feasible (67,68,79–82). The grid size (Lx, Ly, Lz) for the second and the third runs are kept at (204 Å, 204 Å, 204 Å) and (102 Å, 102 Å, 102 Å), respectively, and the resolutions are kept at 1.36 Å per grid and 0.68 Å per grid, respectively. Correspondingly, the number of the grid points is 151 × 151 × 151 in the second and the third runs. Our results are tested against different grid sizes, and the results are stable.
at 25°C). A thin layer of thickness equal to one cation radius is added to the molecular surface to account for the excluded volume layer of the cations (9,79–82). Moreover, we use the three-step focusing process to obtain the detailed ion distribution near the molecules (71,79–82). For each run, the electrostatic potentials are iterated to a convergence of <10−4 kBT/e. The grid size of the first run depends on the salt concentration used. Generally, we keep it larger than four times of the Debye length, and the resolution of the first run varies with the grid size to make the iterative process computationally feasible (67,68,79–82). The grid size (Lx, Ly, Lz) for the second and the third runs are kept at (204 Å, 204 Å, 204 Å) and (102 Å, 102 Å, 102 Å), respectively, and the resolutions are kept at 1.36 Å per grid and 0.68 Å per grid, respectively. Correspondingly, the number of the grid points is 151 × 151 × 151 in the second and the third runs. Our results are tested against different grid sizes, and the results are stable.
Loop free energy
As we discussed above, we use exhaustive enumeration of the self-avoiding walk trajectories (on diamond lattice) (83–85) to generate the ensemble of the loop conformations. We use the end-to-end distance x to describe the conformation of the loop, where x is the distance between the phosphates at the 5′ end and the phosphate at the 3′ end of the loop. In general, there are a large number of chain conformations for a given x. We use Ω(N, x) to denote the number of conformations for an N-nt chain with end-to-end distance x.
Loop conformations in the context of RNA structures are often described by the coordinates/configurations of the (two) terminal nucleotides. A given end-to-end distance x, which is defined through the positions of the phosphates, can correspond to multiple possible configurations of the terminal nucleotides of the loop. The average number of loop conformations for given set of configurations of the terminal nucleotides is given by 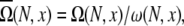 where ω (N, x) is the number of different configurations of the terminal nucleotides for a given x. The number of coil conformations is given by the summation over all possible x values:
where ω (N, x) is the number of different configurations of the terminal nucleotides for a given x. The number of coil conformations is given by the summation over all possible x values: 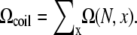
A rigorous treatment for the electrostatic contribution to the loop and coil free energies requires the computation of the electrostatic free energy for each conformational state. However, the number of chain conformations is huge. For instance, for a 13-nt chain, the number of coil conformations is ∼15 × 1010. Therefore, it is practically impossible to calculate the electrostatic free energy for every chain conformation. In our computational procedure, we randomly select Ω0(N, x) conformations from the conformational ensemble. During the enumeration of chain conformations, we select conformations through a pseudo random number generator, and simultaneously store the coordinates of P atoms for each selected conformation. In our practice, we choose 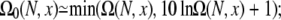 see Table 1. For each selected conformation, we calculate the electrostatic free energy GE (through Eq. 3). The average over all loop conformations gives the free energy ΔG(N, x) for an N-nt chain with end-to-end distance x,
see Table 1. For each selected conformation, we calculate the electrostatic free energy GE (through Eq. 3). The average over all loop conformations gives the free energy ΔG(N, x) for an N-nt chain with end-to-end distance x,
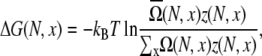 |
(4) |
where z(N, x) is the (electrostatic) partition function averaged over loop conformations,
 |
(5) |
Here 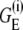 is the electrostatic free energy of the ith loop conformation.
is the electrostatic free energy of the ith loop conformation.  is the electrostatic free energy of the reference state, which is chosen as the fully stretched conformation: x = Nd with
is the electrostatic free energy of the reference state, which is chosen as the fully stretched conformation: x = Nd with  equal to the distance between adjacent nucleotides. If ignoring the electrostatic interactions, then
equal to the distance between adjacent nucleotides. If ignoring the electrostatic interactions, then 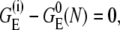 and Eq. 4 is reduced to the following form:
and Eq. 4 is reduced to the following form:
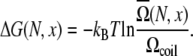 |
(6) |
TABLE 1.
The number of randomly selected loop conformations Ω0 used in calculations
|
x/d
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N (−nt)* | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 |
| 13 | 232 | 237 | 232 | 239 | 257 | 225 | 236 | 233 | 216 | 180 | 150 | 99 | 1 |
| 11 | 178 | 194 | 192 | 188 | 176 | 170 | 175 | 175 | 153 | 93 | 1 | − | − |
| 9 | 115 | 144 | 139 | 175 | 158 | 144 | 102 | 73 | 1 | − | − | − | − |
| 7 | 89 | 111 | 117 | 112 | 112 | 76 | 1 | − | − | − | − | − | − |
| 5 | 71 | 85 | 60 | 58 | 1 | − | − | − | − | − | − | − | − |
| 3† | 12 | 26 | 1 | − | − | − | − | − | − | − | − | − | − |
| 2† | 2 | 1 | − | − | − | − | − | − | − | − | − | − | − |
In the calculations, we use these number of single-stranded chain conformations for calculating electrostatic free energy, and the partition function for the whole conformation ensemble can be calculated through a mean-field approach; see Eqs. 4 and 5.
N is the chain length and the loop size is N – 2.
For these N values, we make calculations for more values of x/d than listed here.
RESULTS AND DISCUSSIONS
In the following, by considering the electrostatic interactions, we calculate the loop free energies for different chain length, end-to-end distance x, and a broad range of Na+ and Mg2+ concentrations: [Na+] ∈ [0.001 M,1 M], [Mg2+] ∈ [0.0001 M,0.1 M]. Based on the calculations, we will derive empirical formulas for the loop free energy for three specific types of loops, namely, hairpin, bulge, and internal loops. The derived empirical relations will be validated through experimental comparisons for the salt-dependent RNA and DNA hairpin stability in Na+ and Mg2+ solutions.
Conformational ensemble
Fig. 2, A–D, show the values of Ω(N, x), ω(N, x), 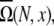 and
and 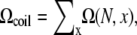 respectively. From Ω(N, x), ω(N, x),
respectively. From Ω(N, x), ω(N, x),  we calculate the loop free energy in the absence of the electrostatic interactions (Eq. 6) by assuming the chain backbone is electrically neutral. Fig. 3 shows the chain free energy as a function of end-to-end distance. The predicted loop free energy ΔG(N, x) (Eq. 6) is positive because the loop formation is entropically unfavorable. As compared with experimental data for DNA hairpin loop free energy, the predicted electrostatics-free ΔG(N, x) from Eq. 6 overestimates the loop stability (N = 13 nt and
we calculate the loop free energy in the absence of the electrostatic interactions (Eq. 6) by assuming the chain backbone is electrically neutral. Fig. 3 shows the chain free energy as a function of end-to-end distance. The predicted loop free energy ΔG(N, x) (Eq. 6) is positive because the loop formation is entropically unfavorable. As compared with experimental data for DNA hairpin loop free energy, the predicted electrostatics-free ΔG(N, x) from Eq. 6 overestimates the loop stability (N = 13 nt and  for the phosphate-phosphate distance for a basepair in an B-form DNA helix, and d = 6.4 Å is the phosphate-phosphate distance for the adjacent nucleotides along the sequence). The discrepancy between the electrostatics-free predictions and the experimental data may be partially attributed to the neglect of electrostatic interactions. Nucleotide chain is negatively charged and the Coulombic repulsion between monomers on backbone would make loop formation more unfavorable than an electrically neutral chain. In the following, we investigate how the electrostatic interactions (in addition to the conformational entropy) determine the loop stability.
for the phosphate-phosphate distance for a basepair in an B-form DNA helix, and d = 6.4 Å is the phosphate-phosphate distance for the adjacent nucleotides along the sequence). The discrepancy between the electrostatics-free predictions and the experimental data may be partially attributed to the neglect of electrostatic interactions. Nucleotide chain is negatively charged and the Coulombic repulsion between monomers on backbone would make loop formation more unfavorable than an electrically neutral chain. In the following, we investigate how the electrostatic interactions (in addition to the conformational entropy) determine the loop stability.
FIGURE 2.

The chain conformational ensemble produced from the virtual bond model on diamond lattice (83–85). (A) The number Ω(N, x) of all conformations with end-to-end distance x; (B) the number ω(N, x) of lattice nodes visited by the flexible chain end with end-to-end distance x; (C) the number of chain conformations with x averaged to one direction 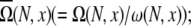 and (D) the number Ωcoil of chain coil states, which includes all the conformations produced by the self-avoiding walk on a diamond lattice.
and (D) the number Ωcoil of chain coil states, which includes all the conformations produced by the self-avoiding walk on a diamond lattice.
FIGURE 3.

The calculated loop formation free energy ΔG as functions of end-to-end distance x for different chain-lengths N for the electrostatics-free case (calculated through Eq. 6), i.e., by assuming chain backbone is neutral. The dotted line denotes the DNA hairpin loop free energy at 1 M NaCl for a 13-nt chain (loop size = 11-nt) (36).
Ion-dependent loop free energy
First, we calculate the electrostatic partition function z(N, x) (Eq. 5) for each given end-to-end distance x and chain-length N. The predicted z(N, x) (Fig. 4) decreases with the decrease of end-to-end distance x, and such effect is more pronounced for lower salt concentration and longer chain. This means that the small-x states are electrostatically unfavorable. Physically, this is because the electrostatic repulsion between the nucleotides is stronger for more compact chain conformations (smaller x). Therefore, z(N, x) decreases for smaller x.
FIGURE 4.

The partition function z(N, x) averaged for one chain conformation as a function of end-to-end distance x for different Na+ (A–C) and Mg2+ (D–F) concentrations. The chain lengths are 5-nt (A and D), 9-nt (B and E), and 13-nt (C and F). The averaging is over Ω0 (lines) and Ω0/2 (symbols) randomly selected chain conformations except for the fully stretched conformation. The agreement between lines and symbols show that the calculations are rather stable for different (large) numbers of conformations sampled. The Ω0 values used for calculations are listed in Table 1. For the comparisons, we also show z(N, x) calculated from the PB theory (dotted lines).
Due to ion-polynucleotide electrostatic interaction, counterions in solution tend to be distributed around the polynucleotide chain (often termed diffusive-binding) to partially neutralize the backbone charge. At a lower ion concentration, such ion binding would accompany a large decrease in the ion's translational entropy and thus lead to a weak ion binding. As a result, fewer ions become bound for low ion concentration, causing weaker screening/neutralization and stronger polynucleotide charge-charge repulsion effect. Thus, the decrease of z(N, x) with x is more pronounced at low ion concentration.
For a longer loop (larger N), the electrostatic repulsion would be stronger, and the compaction of longer chain with small x would cause more massive charge repulsion, thus z(N, x) decreases with decreasing x more strongly for longer chain than for shorter chain.
In addition, Fig. 4 shows that Mg2+ gives much more effective neutralization than Na+ even at the same ionic strength. For example, z(N, x) at 0.1 M [Mg2+] is larger than that at 1 M [Na+]. This is due to the higher valency of Mg2+ than Na+ and hence stronger ion-polynucleotide interaction. Also shown in Fig. 4 are the z(N, x) results calculated with the PB theory. It is obvious that the PB theory underestimates the ability of Mg2+ in neutralizing the polyanionic nucleotide chain, i.e., z(N, x) from the PB decreases more sharply than that from the TBI. Physically, due to the higher charge of Mg2+, the interion correlations can be strong, while the PB theory ignores such interion correlation with the mean-field approximation. One of the effects of ion correlation is that the bound Mg2+ ions can self-organize to form low-energy state beyond the mean-field state, causing stronger Mg2+ ion effect. Ignoring this effect would cause underestimation for the effect of Mg2+ ion binding (79,91). Our TBI model takes into account the interion correlation and ion-binding ensemble (fluctuation) effects and gives improved predictions on Mg2+-binding and electrostatic free energy (79–82).
From the partition function z(N, x), we compute the loop stability ΔG(N, x) through Eq. 4 (shown in Fig. 5). The loop free energy is more positive, and the loop is less stable for longer loops and low salt concentration. In RNA and DNA folding, the unfavorable loop formation is often compensated by the favorable formation of helices and other noncanonical basepairing and base stacking. In this model, two factors determine the loop stability: the conformational entropy of the nucleotide chain and the electrostatic interactions. Both factors oppose the loop formation due to the decreased chain entropy and the increased charge-charge repulsion, respectively. For example, for longer loop, the random coil state has many more conformations, thus the loop formation would bring a larger decrease in chain entropy. The contribution of intrachain Coulombic repulsion can be modulated by the ions in solutions, which will be discussed in the following.
FIGURE 5.

The three-dimensional loop free-energy landscapes ΔG/kBT as functions of end-to-end distance x for different chain lengths N and different [Na+] (A–D) and [Mg2+] (E–H). For the comparisons, we also show the loop free energy for different [Mg2+] computed from PB theory (I–L). The three-dimensional plots are produced from the empirical formulas for Na+ (Eq. 9), Mg2+ (Eq. 12), and for Mg2+ with the PB treatment (Data S1, Eq. S1). The red and blue colors represent the low and high loop free energies, respectively. In Data S1, Figs. S11–S13, we shows the comparisons between the calculated results and the respective empirical formulas.
In Na+ solutions
For loops with small/moderate end-to-end distance x (x/d ≲ N/2) (d is the distance between two adjacent nucleotides and Nd is the length of the fully stretched chain), the loop formation is less unfavorable for higher [Na+] (see Fig. 5, A–D; also shown in Supplementary Material, Data S1, Fig. S11). Such [Na+]-dependence of the loop stability is stronger for larger loops. This is because closing a loop with small/moderate end-to-end distance is opposed by the repulsion between the closely approached backbone negative charges. A higher [Na+] would reduce such repulsive force due to stronger charge neutralization/screening and thus improve the loop stability. For larger loops, more charges are involved and the charge repulsion effect is more significant. Therefore, the [Na+]-induced loop stability change is more pronounced.
In Mg2+ solutions
The ion concentration-dependence of the loop free energy in a Mg2+ solution (see Fig. 5, E–H; also see Data S1, Fig. S11) shows qualitatively similar behavior as in a Na+ solution. The loop stability increases with the increase of [Mg2+] due to the stronger Mg2+ binding. Compared with Na+ (Fig. 5, A–D), 1), Mg2+ is more effective in neutralizing the negative backbone charges, thus the loop formation in Mg2+ is apparently less unfavorable than in Na+ (even at the same ionic strength); and 2), the dependence of loop stability on [Mg2+] is obviously weaker than on [Na+]. Such difference between Mg2+ and Na+ comes from the higher ionic charge of Mg2+. Due to the stronger Mg2+-phosphate attraction, Mg2+-binding is more enthalpically favorable and (effectively) less entropically unfavorable (than Na+). As a result, the polynucleotide chain reaches stronger charge neutralization (screening) in Mg2+ (than Na+) solution. Therefore, Mg2+ is much more efficient in charge neutralization than Na+ and the loop formation free energy in Mg2+ solution exhibits the weaker dependence on [Mg2+].
For comparison, we use the PB theory to calculate the free energy of loop formation in Mg2+ solutions; see Fig. 5, I–L (also see Data S1, Fig. S12). Fig. 5, E–L, shows that the PB theory underestimates the role of Mg2+ in stabilizing loop, especially for large loop at high [Mg2+]. This may arise from the ignored interion correlations in the PB theory and the consequent underestimation of Mg2+ ion binding (79,91). Our results indicate that the PB theory underestimates the loop stability in Mg2+, especially for larger loop and higher [Mg2+], which may involve stronger ion-ion correlations. By contrast, the TBI model, which accounts for the ion correlation and ion-binding ensemble effects, gives improved free energy predictions (67,68,79–82).
Na+ versus Mg2+
To quantitatively compare the loop free energies in Na+ and Mg2+ solutions, we choose two typical ionic conditions: 1 M [Na+] and 0.01 M [Mg2+]. 1 M [Na+] and 0.01 M [Mg2+] have been previously shown to be approximately equivalent in stabilizing short DNA (and RNA) helices (60,67,68). For the two cases, Fig. 6 shows that the loops approximately have the same free energies, suggesting that 1 M [Na+] and 0.01 M [Mg2+] are approximately equivalent in stabilizing loops. Such TBI-predicted equivalence is in accordance with the experiment (60) and is beyond the mean-field description (e.g., ionic strength effect) due to the effects of ion correlation and ion-binding ensemble (fluctuation) for Mg2+ ions.
FIGURE 6.

The comparisons between the calculated loop free energy ΔG for two typical ionic conditions: 0.01 M MgCl2 and 1 M NaCl. The chain lengths are N = 13, 9, and 5 nucleotides, respectively.
In mixed Na+/Mg2+ solutions
Since the mixed ion solution is of biological significance, we also make the calculations for mixed Na+/Mg2+ solutions. As shown in Data S1, Fig. S13, the free energy for loop formation depends on the competition between [Na+] and [Mg2+]. In analogy to the DNA and RNA helix stability in mixed [Na+]/[Mg2+] solutions (68), there are three regimes: for high [Mg2+] (relatively to [Na+]), the system is dominated by Mg2+, and the loop free energy is close to that in pure Mg2+ solution; for high [Na+] (relatively to [Mg2+]), Na+ ion effect dominates the system, and the loop free energy is close to the values in pure Na+ solution; for the intermediate regime, loop free energy is determined by the competition between [Mg2+] and [Na+]. When [Mg2+] is high (relative to [Na+]), Mg2+-binding is dominating due to strong Mg2+-phosphate attraction and low ion-binding entropy penalty, and Na+-binding is fully suppressed. Thus, Mg2+ dominates the loop stability. With the addition of Na+, Mg2+ ion-binding would be suppressed and hence the efficient roles of Mg2+ in stabilizing loop conformation is weakened. When [Na+] becomes very high, Mg2+ would be completely pushed away from the molecular surface, and the (high-concentration) Na+ ions dominate the loop stability.
Thermodynamic parameters for ion-dependent loop free energy
In this section, following the previous works (30,36,67,68), we will fit empirical formulas for loop free energies as functions of chain length N, end-to-end distance x, and Na+ and Mg2+ concentrations. Such empirical formulas for loop thermodynamic parameters are potentially useful for predicting secondary structure stability in an arbitrary Na+/Mg2+ solution.
In Na+ solutions
Based on our calculations, we obtain the following empirical relations for the partition functions of loop ( see Eq. 4) and coil (
see Eq. 4) and coil (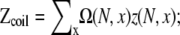 see Eq. 4) in pure Na+ solutions:
see Eq. 4) in pure Na+ solutions:
 |
(7) |
The coefficients a1, b1, c1, and d1 are given by
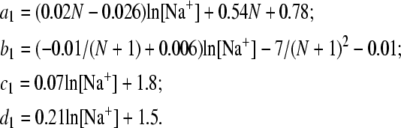 |
(8) |
Here, Zloop(N, x) and Zcoil are the partition functions for the loop and the coil, respectively. Then the free energy for a loop formation in a Na+ solution is given by Eq. 4:
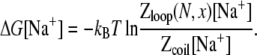 |
(9) |
As shown in Data S1, Fig. S11, A–D, the above empirical relations fit the TBI calculations very well for loop formation free energy in Na+ solutions.
In Mg2+ solutions
In analogy to Na+ solutions, for Mg2+ solutions, we obtain the following similar empirical relations:
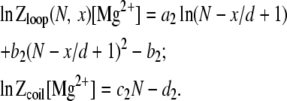 |
(10) |
The coefficients a2, b2, c2, and d2 are given by
 |
(11) |
Then the free energy for a loop formation in Mg2+ solution is calculated by
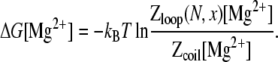 |
(12) |
As shown in Data S1, Fig. S11, E–H, the above empirical relations give good fit for loop formation free energy in Mg2+ solutions, as compared with the TBI calculations.
In mixed Na+/Mg2+ solutions
For a mixed Na+/Mg2+ solution, the free energy for a loop formation is given by
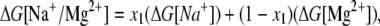 |
(13) |
where ΔG[Na+] and ΔG[Mg2+] are given by the above formulas (Eqs. 9 and 12). The expressions x1 and 1 − x1 describe the fractional contributions from Na+ and Mg2+, respectively, and x1 is given by
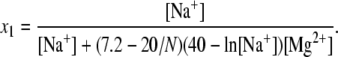 |
(14) |
As shown in Data S1, Fig. S13, the formulas give very good fits to the TBI calculations for mixed Na+/Mg2+ solutions. These formulas for mixed Na+/Mg2+ may also be used to estimate loop formation free energy in mixed K+/Mg2+ solutions, since K+ and Na+ have similar electrostatic properties (36).
For comparison, based on the calculations with the PB theory for the pure Mg2+ and mixed Na+/Mg2+ solutions, we also fit the empirical formulas for the loop free energies from the PB calculations (see Data S1, Fig. S12).
Ion-dependent hairpin, bulge, and internal loop free energies
After obtaining the above general empirical relations, we can conveniently calculate the ion-dependent free energies for the three types of specific loops, namely, hairpin, bulge, and internal loops.
Hairpin loop
For RNA and DNA, the end-end distance of the hairpin loop is  (83,92). Using xhairpin for x in Eqs. 9, 12, and 13 gives the free energy for an N-nt hairpin loop:
(83,92). Using xhairpin for x in Eqs. 9, 12, and 13 gives the free energy for an N-nt hairpin loop:
 |
(15) |
The fluctuation of xhairpin at ∼17 Å only brings very slight fluctuation in the estimated hairpin loop free energy (see Fig. 5, and Data S1, Fig. S11).
Fig. 7, A and D, shows the hairpin loop free energies for different [Na+] and [Mg2+]. Also shown in the figures are the available experimental data (1 M NaCl). The predicted free energy ΔGhairpin loop at 1 M NaCl is slightly higher than the experimental data for DNA loop (28), and slightly lower than the data for RNA loop (27). With the decrease of ion concentration, the predicted loop free energy increases and the loop becomes less stable. Such ion effect is stronger for Na+ and for longer loops, which is in accordance with the recent experimental measurements (93).
FIGURE 7.

The calculated loop free energies ΔG as functions of loop size (N – 2) for different [Na+] and [Mg2+]. (A and D) Hairpin loop; (B and E) bulge loop; and (C and F) internal loop. The symbols are experimental data for RNA (♦) (27) and DNA (□) (36) at 1 M NaCl, and the lines are calculated from Eqs. 15–17, respectively. From the bottom to top, ion concentrations are: (A–C) [Na+] = 1 M, 0.1 M, 0.01 M, and 0.001 M; (D–F) [Mg2+] = 0.1 M, 0.01 M, 0.001 M, and 0.0001 M, respectively.
Bulge loop
For a bulge loop, considering that the two helical arms connected by the bulge loop can fluctuate or bend, we allow the end-to-end distance x of the loop to fluctuate in a certain range. Due to the possible stacking between the two helix stems (83,85), very large end-end distance x for a bulge loop may be unfavorable. We select a fluctuation range of x ∈ [d, 2d], where d is the distance between two adjacent nucleotides. Our control test for the different fluctuation ranges of x indicates that the predicted loop free energy is not very sensitive to the choice of the fluctuation limits of x. For instance, using [d, 3d] instead of [d, 2d] causes an decrease in loop free energy by 10% at 1 M NaCl and 8% at 0.001 M NaCl. We compute the folding free energy for an N-nt bulge loop as
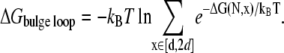 |
(16) |
Fig. 7, B and E, shows the folding free energy for bulge loops as a function of loop size for Na+ and Mg2+ solutions. Compared with experimental data at 1 M NaCl, our predicted free energy ΔG is slightly larger than that of DNA loop (36) and slightly smaller than that of RNA loop (27). As [Na+] or [Mg2+] is decreased, the loop free energy ΔG increases. Such effect is more pronounced for larger loops.
Internal loop
An internal loop contains two single-stranded chains. For a given N-nt internal loop, the two single-stranded chains can have different lengths (N1-nt and N2-nt, respectively). To compute the loop free energy as a function of the loop size N, we average over different N1 and N2 values with the constraint N1 + N2 = N,
 |
(17) |
where 〈…〉 denotes averaging over different N1 and N2 values and xmax(= min(N1d, N2d)) is the length of the shorter chain in the internal loop. The value xmax is the fully stretched distance of the chain and is thus the maximum end-to-end distance for the single-stranded chains for given N1 and N2.
Fig. 7, C and F, shows the free energy for the formation of internal loop in Na+ and Mg2+ solutions. The comparisons between the predicted free energy and the available experimental data at 1 M NaCl show the same trend (Fig. 7, A, B, D, and E). Our predictions slightly underestimate the internal loop free energy ΔG, as compared with the experimental data for RNA loop (27), and slightly overestimate ΔG, as compared with the data for DNA loop (36). The decrease of ion (Na+ and Mg2+) concentration and the increase of loop size both increase the free energy penalty for loop formation. Fig. 7, C and F, show that internal loop has a weaker ion-dependence of the free energy than hairpin and bulge loops of the same size (N). Such weaker ion-dependence is due to the shorter single-stranded chains (of lengths N1 and N2) in an internal loop than in a hairpin or bulge loop (of length N = N1 + N2).
As shown in Fig. 7, for the three types of loops, our predicted free energies for 1 M NaCl generally lie between the experimental values for RNA and DNA loops, except for very small loops. One possible reason is that our used value of d, distance between two adjacent nucleotide, is ∼6.4 Å, which lies between the values for RNA (∼6 Å) and DNA (∼6.6 Å) chains. A smaller d would result in a stronger intrachain Coulombic repulsion and consequently make the loop formation more unfavorable. Thus, with the use of the intermediate d, the predicted loop free energies are larger than the values of DNA loops and smaller than those of RNA loops. For very short loops (e.g., 1-nt bulge loop), the predictions obviously deviate from the experimental measurements, which may be due to the neglected intraloop and loop-helix interactions. In Fig. 7, we only present the predictions for pure Na+ and Mg2+ solutions. For mixed Na+/Mg2+ solutions, the loop free energy parameters can be conveniently calculated by using the general formulas for loop formation parameter (Eq. 13) and the specific formulas for the three types of loops (Eqs. 15–17).
Ion-dependent RNA and DNA hairpin stability
To validate the above empirical formulas for ion-dependent loop free energy, we compute the ion-dependent RNA and DNA hairpin stability by combining the above analytical formulas (derived from our TBI model) for loop free energy and the previously obtained analytical formulas (derived also from TBI) for RNA and DNA helix thermodynamic parameters (67,68). Based on the assumption of additive nearest-neighbor model (27,36), the enthalpy, entropy, and free energy for a hairpin can be calculated as
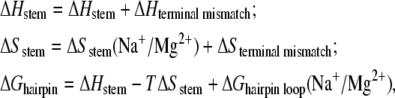 |
(18) |
where ΔHstem, ΔHterminal mismatch, and ΔSterminal mismatch can be obtained from the nearest-neighbor model with the measured thermodynamic parameters (Turner rules) (27,31,36,42). In our calculations, the parameters for base stacks in helix stem are from Xia et al. (31) for RNA hairpin and from SantaLucia (30) for DNA hairpin. The RNA terminal mismatch parameters are from Serra and Turner (27). For DNA hairpin, the terminal mismatch parameters, which are not directly available (36), are approximated by the dangling end parameters (94). ΔGhairpin loop(Na+/Mg2+) are calculated through the above analytical formula (Eq. 15), and ΔSstem(Na+/Mg2+) can be calculated from the previously developed analytical formulas for ion-dependent RNA and DNA helix stability (67,68). After obtaining ΔGhairpin, the melting temperature can also be calculated from the condition ΔGhairpin = 0.
Based on the above formulas (Eq. 18), we investigate the salt-dependent stability for RNA and DNA hairpins which are shown in Fig. 1. The thermodynamic parameters for these hairpins are listed in Table 2, along with the experimental references (20,49,93,95–97). We calculate the free energy and melting temperature over wide ranges of [Na+] and [Mg2+], and make quantitative comparisons with the available experimental data. In the calculations, for mixed K+/Mg2+ solution, we also use our formulas for mixed Na+/Mg2+, since Na+ and K+ have similar electrostatic properties (36).
TABLE 2.
The thermodynamic parameters for nucleic acid hairpins used in the calculations
| Label* | Ref. | Helix length (bp) | Loop size | −ΔH° (kcal/mol)† | −ΔS° (cal/mol K)† | ΔG°loop (kBT)‡ | −ΔG°37 (kcal/mol)§ |
|---|---|---|---|---|---|---|---|
| RH1 | (49) | 6 | 10 | 63.6 | 161.7 | 8.4 | 8.3 |
| RH2 | (49) | 6 | 8 | 63.6 | 161.7 | 7.9 | 8.6 |
| RH3 | (20,95) | 5 | 6 | 60.0 | 150.3 | 7.2 | 8.9 |
| RH4 | (96) | 7 | 9 | 68.8 | 179.3 | 8.2 | 8.1 |
| RH5 | (97) | 7 | 12 | 77.9 | 203.8 | 8.7 | 9.3 |
| RH6 | (93) | 5 | j+2 | 50.2 | 131.0 | ¶ | ¶ |
| DH1 | (93) | 7 | i | 53‖ | 143‖ | ¶ | ¶ |
In our calculations for salt-dependent nucleic acid hairpin thermodynamics, we use the values listed in the table at 1 M NaCl.
The sequences of the hairpins are shown in Fig. 1.
The listed thermodynamic parameters are calculated for helix stem at standard salt (1 M NaCl), through the nearest-neighbor model. The parameters for base stacking are taken from Xia et al. (31), and the parameters for terminal mismatches are taken from Serra et al. (27).
The loop free energy is obtained from our calculations for loops (Eq. 15), rather than the experimentally derived values.
Calculated for the whole RNA hairpin at standard salt (1 M NaCl).
The loop and hairpin free energies depend on the loop sizes j+2 and i.
In Na+ solutions
Fig. 8 shows the folding free energy ΔG°37 and melting temperature Tm as functions of [Na+] for three hairpins: RH1, RH2, and RH3; see Fig. 1 and Table 2. The increase of [Na+] enhances the RNA hairpin folding stability (decrease in the folding free energy and increase in the melting temperature) (20,49,95,98). Our predictions agree with the available experimental data very well (20,49,95). Physically, at higher [Na+], the entropic cost for Na+-binding is lower, and consequently more Na+ ions bind near the phosphate groups to neutralize the negatively charged backbone. Thus, hairpins have higher stability (lower ΔG°37 and higher Tm) at higher [Na+].
To test the importance of the ion-dependence of the loop stability, instead of using the above [Na+]-independent loop free energies, we use loop free energies at fixed 1 M NaCl for all other [Na+] values. Our test results (Fig. 8) show that ignoring the ion-dependence of the loop free energy would lead to notable inaccuracy, especially for large loop (e.g., see the curves for RH1 in Fig. 8). Therefore, the salt-dependence of loop free energy cannot be ignored, though the helix stem may contribute predominantly to the overall salt dependence of the hairpin stability due to its higher charge density.
FIGURE 8.

The RNA hairpin folding free energy ΔG°37 (A) and melting temperature Tm (B) as functions of [Na+] for three sequences RH1, RH2, and RH3. (Symbols) Experimental data: ⋄, RH3 in Na+ solution (20); ▴, RH3 in Na+ solution (95); ♦, RH2 in Na+ solution (49); and □, RH1 in Na+ solution (49). (Solid lines) Predictions with salt-dependent loop free energy (ΔG[Na+]); (dotted lines) predictions with invariable loop free energy at 1 M NaCl (ΔG[1 M Na+]). Hairpins RH1, RH2, and RH3 are shown in Fig. 1 and Table 2.
In mixed Na+/Mg2+ solutions
Fig. 9 shows the folding free energy ΔG°25 and melting temperature Tm in mixed Na+ (or K+)/Mg2+ solutions for three RNA hairpins: RH3, RH4, and RH5 (as shown in Fig. 1, and Table 2). Comparisons with the experimental data indicate that our model gives good predictions for RNA hairpin stability in mixed Na+ (or K+)/Mg2+ solutions. As shown in Fig. 9, similar to the DNA and RNA helix stability in mixed Na+/Mg2+ solutions (68), the ion-dependence of the hairpin stability can be classified into three of the aforementioned [Na+]/[Mg2+] regimes (see In Mixed Na+/Mg2+ Solutions). When Na+ (or K+) and Mg2+ compete with each other, adding Na+ (or K+) ions can weaken Mg2+ ion binding and thus destabilize the hairpin.
The predictions with invariable loop free energy at 1 M NaCl (ΔG[1 M Na+]) are also shown in Fig. 9. The comparisons with experimental data show that the Na+ (or K+)/Mg2+-dependence of loop free energy plays an important role in the overall ion-dependence of hairpin stability, especially for large loop; e.g., see the curves for RH5 in Fig. 9.
FIGURE 9.

The RNA hairpin folding free energy ΔG°25 (A–C) and melting temperature Tm (D–F) as functions of [Mg2+] for different [Na+]. Three sequences used are RH3, RH4, and RH5, which are shown in Fig. 1 and Table 2. (Symbols) Experimental data: (A) ♦, RH3 in 0.054 M [Na+] (20); (E) ♦, RH4 in 0.1 M [K+] (96); (F) ♦, RH5 in 0.1 M [K+] (97). (Solid lines) Predictions with salt (Na+/Mg2+) dependent loop free energy ΔG[Na+/Mg2+]; (dotted lines) predictions with invariable loop free energy at 1 M NaCl (ΔG[1 M Na+]). In panels A–C, we calculate ΔG°25 instead of ΔG°37 because of the available experimental data at 25°C. In the calculations, we use the same salt empirical formula for K+ as that for Na+, since Na+ and K+ have the similar electrostatic properties (36). For the comparisons, we also show the predictions with the loop free energy from the PB empirical formulas (Data S1, Eqs. S1 and S3) (dashed lines).
Also shown in Fig. 9 are the curves from the PB calculations on loop free energy. The comparisons between the predictions of the TBI model, the PB theory, and the experimental data suggest that the PB theory underestimates the ability of Mg2+ in stabilizing loop, especially for large loops at high [Mg2+]. As discussed above, the PB theory neglects the ion-ion correlation and ion fluctuation, and consequently predicts less-bound Mg2+ ions (79,91), causing the overestimation of (positive) loop free energy in Mg2+. Our TBI model explicitly accounts for these effects (79–82) and can give improved predictions for hairpin stability in Mg2+ solutions.
Loop-size dependence of hairpin stability
Fig. 10 shows the melting temperatures Tm for RH6 and DH1 (Fig. 1) as a function of the loop size. In general, the theoretical results agree well with the experimental data (93) except for 33 mM [Mg2+] in Fig. 10 B, where the theory slightly overestimates the Tm. This theory-experiment difference may come from neglecting loop-stem interactions, which can cause overestimation on the number of loop conformations and hence the loop stability. As shown in Fig. 10, larger loop gives lower hairpin stability. Such effect is more pronounced for lower ion concentration and for monovalent ion solution due to weaker charge neutralization and thus stronger charge repulsion upon loop closure.
FIGURE 10.

The melting temperatures Tm of DNA (DH1, A and B) and RNA (RH6, C) hairpins as a function of loop size for different ionic conditions: 0.1 M Na+ (A), 2.5 mM and 33 mM Mg2+ (B), and 2.5 mM Mg2+ (C). (Lines) Predictions from our theory; (symbols) experimental data (93).
CONCLUSIONS AND DISCUSSIONS
In this article, the tightly-bound ion (TBI) theory (67,68,79–82) and the Vfold model (83–85) are combined together to quantify the ionic dependence of nucleic acid loop thermodynamics. Based on the TBI calculations, we obtain fitted analytical formulas for loop free energies as functions of end-to-end distance, chain length, and Na+/Mg2+ concentrations. The analytical formulas are validated by quantitative comparisons with the extensive experimental data for ion-dependent RNA and DNA hairpin stability. The following are the major conclusions:
Loop formation is unfavorable due to backbone charge-charge repulsion. Na+ and Mg2+ can increase the loop flexibility by neutralizing the phosphate charges, causing the loop formation to be less unfavorable. Therefore, the increase of [Na+] and [Mg2+] leads to the decrease of the free energy cost for loop formation.
Mg2+ is more effective than Na+ in neutralizing backbone charges, and ion concentration-dependence of loop free energy for Mg2+ is weaker than that for Na+. Specifically, 1 M [Na+] and 0.01 M [Mg2+] are approximately equivalent in stabilizing loops.
For the loop formation in Mg2+ solutions, the TBI model makes better predictions for loop free energy than the PB theory, which tends to overestimate the (unfavorable) loop free energy, especially for large loops at high Mg2+ concentration.
The ion-dependence of loop free energy plays an important role in the overall salt-dependence of hairpin stability, especially for large loops.
Based on the TBI calculations, we obtain a set of fitted analytical formulas for loop free energy as function of chain length, end-to-end distance, and Na+/Mg2+ concentrations. These formulas are validated through comparisons with experimental data. These formulas, combined with the previously derived analytical formulas for the ion-dependent helix stabilities, can give good predictions for RNA and DNA hairpins at arbitrary Na+/Mg2+ concentrations.
Although our calculations can give quantitative predictions that are validated by the extensive experimental data, we have made several important simplifications and approximations in our theory. First, we use a coarse-grained chain model to represent the polynucleotide chain. As a result, the model cannot treat atomic details, which can be important for more accurate and detailed description of ion-binding. Second, the theory does not treat bases and possible intraloop basepairing and stacking (92), which can be important for sequence-specific interactions that help to stabilize the triloops and tetraloops (35,36,99). Thus this theory is unable to predict the sequence-specific tetraloop stability. Third, in our calculation for the loop stability, we ignored the influence from the helices that are connected to the loop. The loop-helix interactions can affect the loop conformational distribution (83). Fourth, in the TBI theory, we use hydrated ions and the current model cannot treat ion dehydration effect and possible specific ion-binding (9,100). In addition, we ignore the contributions from the dangling tails to the overall hairpin stability. Single-strand stacking in dangling tails can contribute sequence-dependent stability. For example, at room temperature, while poly(U) forms a random coil, poly(A) is largely stacked (1). Finally, our present computation is based on a randomly sampled loop conformational ensemble because the electrostatic calculations for the complete conformational ensemble is computationally not viable. Nevertheless, the agreements with available experimental data suggest that our predictions and the obtained analytical formulas are able to provide reliable ion-dependent thermodynamic stabilities for loops and hairpins. Further development of the theory might enable us to treat RNA (DNA) secondary and even simple tertiary structures at different ionic conditions.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Acknowledgments
We are grateful to Dr. Song Cao and Liang Liu for valuable discussions on the virtual bond model, and Dr. Irina A. Shkel for helpful communications on modeling nucleic acid hairpin stability. Parts of the computation were performed on the HPC resources at the University of Missouri Bioinformatics Consortium.
This research was supported by National Institutes of Health/National Institute of General Medical Sciences through grant No. GM063732 (to S.-J.C).
Editor: Angel E. Garcia.
References
- 1.Bloomfield, V. A., D. M. Crothers, and I. Tinoco, Jr. 2000. Nucleic Acids: Structure, Properties and Functions. University Science Books, Sausalito, CA.
- 2.Tinoco, I., and C. Bustamante. 1999. How RNA folds. J. Mol. Biol. 293:271–281. [DOI] [PubMed] [Google Scholar]
- 3.Anderson, C. F., and T. M. Record. 1995. Salt-nucleic acid interactions. Annu. Rev. Phys. Chem. 46:657–700. [DOI] [PubMed] [Google Scholar]
- 4.Bloomfield, V. A. 1997. DNA condensation by multivalent cations. Biopolymers. 44:269–282. [DOI] [PubMed] [Google Scholar]
- 5.Brion, P., and E. Westhof. 1997. Hierarchy and dynamics of RNA folding. Annu. Rev. Biophys. Biomol. Struct. 26:113–137. [DOI] [PubMed] [Google Scholar]
- 6.Pyle, A. M. 2002. Metal ions in the structure and function of RNA. J. Biol. Inorg. Chem. 7:679–690. [DOI] [PubMed] [Google Scholar]
- 7.Sosnick, T. R., and T. Pan. 2003. RNA folding: models and perspectives. Curr. Opin. Struct. Biol. 13:309–316. [DOI] [PubMed] [Google Scholar]
- 8.Woodson, S. A. 2005. Metal ions and RNA folding: a highly charged topic with a dynamic future. Curr. Opin. Chem. Biol. 9:104–109. [DOI] [PubMed] [Google Scholar]
- 9.Draper, D. E., D. Grilley, and A. M. Soto. 2005. Ions and RNA folding. Annu. Rev. Biophys. Biomol. Struct. 34:221–243. [DOI] [PubMed] [Google Scholar]
- 10.Thirumalai, D., and C. Hyeon. 2005. RNA and protein folding: common themes and variations. Biochemistry. 44:4957–4970. [DOI] [PubMed] [Google Scholar]
- 11.Chen, S. J. 2008. RNA Folding: conformational statistics, folding kinetics, and ion electrostatics. Annu. Rev. Biophys. 37:197–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rook, M. S., D. K. Treiber, and J. R. Williamson. 1999. An optimal Mg2+ concentration for kinetic folding of the Tetrahymena ribozyme. Proc. Natl. Acad. Sci. USA. 96:12471–12476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heilman-Miller, S. L., D. Thirumalai, and S. A. Woodson. 2001. Role of counterion condensation in folding of the Tetrahymena ribozyme. I. equilibrium stabilization by cations. J. Mol. Biol. 306:1157–1166. [DOI] [PubMed] [Google Scholar]
- 14.Takamoto, K., Q. He, S. Morris, M. R. Chance, and M. Brenowitz. 2002. Monovalent cations mediate formation of native tertiary structure of the Tetrahymena thermophila ribozyme. Nat. Struct. Biol. 9:928–933. [DOI] [PubMed] [Google Scholar]
- 15.Das, R., L. W. Kwok, I. S. Millett, Y. Bai, T. T. Mills, J. Jacob, G. S. Maskel, S. Seifert, S. G. J. Mochrie, P. Thiyagarajan, S. Doniach, L. Pollack, and D. Herschlag. 2003. The fastest global events in RNA folding: electrostatic relaxation and tertiary collapse of the Tetrahymena ribozyme. J. Mol. Biol. 332:311–319. [DOI] [PubMed] [Google Scholar]
- 16.Misra, V. K., R. Shiman, and D. E. Draper. 2003. A thermodynamic framework for the magnesium-dependent folding of RNA. Biopolymers. 69:118–136. [DOI] [PubMed] [Google Scholar]
- 17.Kwok, L. W., I. Shcherbakova, J. S. Lamb, H. Y. Park, K. Andresen, H. Smith, M. Brenowitz, and L. Pollack. 2006. Concordant exploration of the kinetics of RNA folding from global and local perspectives. J. Mol. Biol. 355:282–293. [DOI] [PubMed] [Google Scholar]
- 18.Waldsich, C., and A. M. Pyle. 2007. A folding control element for tertiary collapse of a group II intron ribozyme. Nat. Struct. Mol. Biol. 14:37–44. [DOI] [PubMed] [Google Scholar]
- 19.Koculi, E., C. Hyeon, D. Thirumalai, and S. A. Woodson. 2007. Charge density of divalent metal cations determines RNA stability. J. Am. Chem. Soc. 129:2676–2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soto, A. M., V. Misra, and D. E. Draper. 2007. Tertiary structure of an RNA pseudoknot is stabilized by “diffuse” Mg2+ ions. Biochemistry. 46:2973–2983. [DOI] [PubMed] [Google Scholar]
- 21.Grilley, D., V. Misra, G. Caliskan, and D. E. Draper. 2007. Importance of partially unfolded conformations for Mg2+-induced folding of RNA tertiary structure: structural models and free energies of Mg2+ interactions. Biochemistry. 46:10266–10278. [DOI] [PubMed] [Google Scholar]
- 22.Chauhan, S., and S. A. Woodson. 2008. Tertiary interactions determine the accuracy of RNA folding. J. Am. Chem. Soc. 130:1296–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garcia, A. E., and D. Paschek. 2008. Simulation of the pressure and temperature folding/unfolding equilibrium of a small RNA hairpin. J. Am. Chem. Soc. 130:815–817. [DOI] [PubMed] [Google Scholar]
- 24.Freier, S. M., R. Kierzek, J. A. Jaeger, N. Sugimoto, M. H. Caruthers, T. Neilson, and D. H. Turner. 1986. Improved free-energy parameters for predictions of RNA duplex stability. Proc. Natl. Acad. Sci. USA. 83:9373–9377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Breslauer, K. J., R. Frank, H. Blocker, and L. A. Marky. 1986. Predicting DNA duplex stability from the base sequence. Proc. Natl. Acad. Sci. USA. 83:3746–3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hall, K. B., and L. W. McLaughlin. 1991. Thermodynamic and structural properties of pentamer DNA.DNA, RNA.RNA, and DNA.RNA duplexes of identical sequence. Biochemistry. 30:10606–10613. [DOI] [PubMed] [Google Scholar]
- 27.Serra, M. J., and D. H. Turner. 1995. Predicting thermodynamic properties of RNA. Methods Enzymol. 259:242–261. [DOI] [PubMed] [Google Scholar]
- 28.SantaLucia, J., H. T. Allawi, and P. A. Seneviratne. 1996. Improved nearest-neighbor parameters for predicting DNA duplex stability. Biochemistry. 35:3555–3562. [DOI] [PubMed] [Google Scholar]
- 29.Sugimoto, N., S. I. Nakano, M. Yoneyama, and K. I. Honda. 1996. Improved thermodynamic parameters and helix initiation factor to predict stability of DNA duplexes. Nucleic Acids Res. 24:4501–4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.SantaLucia, J., Jr. 1998. A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl. Acad. Sci. USA. 95:1460–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xia, T., J. SantaLucia, M. E. Burkard, R. Kierzek, S. J. Schroeder, X. Jiao, C. Cox, and D. H. Turner. 1998. Thermodynamic parameters for an expanded nearest-neighbor model for formation of RNA duplexes with Watson-Crick base pairs. Biochemistry. 37:14719–14735. [DOI] [PubMed] [Google Scholar]
- 32.Mathews, D. H., J. Sabina, M. Zuker, and D. H. Turner. 1999. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 288:911–940. [DOI] [PubMed] [Google Scholar]
- 33.Chen, S. J., and K. A. Dill. 2000. RNA folding energy landscapes. Proc. Natl. Acad. Sci. USA. 97:646–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang, W. B., and S. J. Chen. 2002. RNA hairpin-folding kinetics. Proc. Natl. Acad. Sci. USA. 99:1931–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dimitrov, R. A., and M. Zuker. 2004. Prediction of hybridization and melting for double-stranded nucleic acids. Biophys. J. 87:215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.SantaLucia, J., Jr., and D. Hicks. 2004. The thermodynamics of DNA structural motifs. Annu. Rev. Biophys. Biomol. Struct. 33:415–440. [DOI] [PubMed] [Google Scholar]
- 37.Yildirim, I., and D. H. Turner. 2005. RNA challenges for computational chemists. Biochemistry. 44:13225–13234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang, W. B., and S. J. Chen. 2006. Exploring the complex folding kinetics of RNA hairpins: I. General folding kinetics analysis. Biophys. J. 90:765–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang, W. B., and S. J. Chen. 2006. Exploring the complex folding kinetics of RNA hairpins: II. Effect of sequence, length, and misfolded states. Biophys. J. 90:778–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mathews, D. H., and D. H. Turner. 2006. Prediction of RNA secondary structure by free energy minimization. Curr. Opin. Struct. Biol. 16:270–278. [DOI] [PubMed] [Google Scholar]
- 41.Ma, H., C. Wan, A. Wu, and A. H. Zewail. 2007. DNA folding and melting observed in real time redefine the energy landscape. Proc. Natl. Acad. Sci. USA. 104:712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu, Z. J., D. H. Turner, and D. H. Mathews. 2007. A set of nearest neighbor parameters for predicting the enthalpy change of RNA secondary structure formation. Nucleic Acids Res. 34:4912–4924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Elson, E. L., I. E. Scheffler, and R. L. Baldwin. 1970. Helix formation by (TA) oligomers. III. Electrostatic effects. J. Mol. Biol. 54:401–415. [DOI] [PubMed] [Google Scholar]
- 44.Hickey, D. R., and D. H. Turner. 1985. Solvent effects on the stability of A7U7p. Biochemistry. 24:2086–2094. [DOI] [PubMed] [Google Scholar]
- 45.Blommers, M. J., J. A. Walters, C. A. Haasnoot, J. M. Aelen, G. A. van der Marel, J. H. van Boom, and C. W. Hilbers. 1989. Effects of base sequence on the loop folding in DNA hairpins. Biochemistry. 28:7491–7498. [DOI] [PubMed] [Google Scholar]
- 46.Braunlin, W. H., and V. A. Bloomfield. 1991. 1H NMR study of the base pairing reaction of the d(GGAATTCC): salt effects on the equilibria and kinetics of strand association. Biochemistry. 30:754–758. [DOI] [PubMed] [Google Scholar]
- 47.Delcourt, S. G., and R. D. Blake. 1991. Stacking energies in DNA. J. Biol. Chem. 266:15160–15169. [PubMed] [Google Scholar]
- 48.Bond, J. P., C. F. Anderson, and M. T. Record, Jr. 1994. Conformational transitions of duplex and triplex nucleic acid helices: thermodynamic analysis of effects of salt concentration on stability using preferential interaction coefficients. Biophys. J. 67:825–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Williams, D. J., and K. B. Hall. 1996. Thermodynamic comparison of the salt dependence of natural RNA hairpins and RNA hairpins with non-nucleotide spacers. Biochemistry. 35:14665–14670. [DOI] [PubMed] [Google Scholar]
- 50.Soto, A. M., W. H. Gmeiner, and L. A. Marky. 2002. Energetic and conformational contributions to the stability of Okazaki fragments. Biochemistry. 41:6842–6849. [DOI] [PubMed] [Google Scholar]
- 51.Shkel, I., and M. T. Record, Jr. 2004. Effect of the number of nucleic acid oligomer charges on the salt dependence of stability (ΔG°37) and melting temperature (Tm): NLPB analysis of experimental data. Biochemistry. 43:7090–7101. [DOI] [PubMed] [Google Scholar]
- 52.You, Y., B. G. Moreira, M. A. Behlke, and R. Owczarzy. 2006. Design of LNA probes that improve mismatch discrimination. Nucleic Acids Res. 34:e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Siegfried, N. A., S. L. Metzger, and P. C. Bevilacqua. 2007. Folding cooperativity in RNA and DNA is dependent on position in the helix. Biochemistry. 46:172–181. [DOI] [PubMed] [Google Scholar]
- 54.Stellwagen, E., A. Abdulla, Q. Dong, and N. C. Stellwagen. 2007. Electrophoretic mobility is a reporter of hairpin structure in single-stranded DNA oligomers. Biochemistry. 46:10931–10941. [DOI] [PubMed] [Google Scholar]
- 55.Orden, A. V., and J. Jung. 2008. Review fluorescence correlation spectroscopy for probing the kinetics and mechanisms of DNA hairpin formation. Biopolymers. 89:1–16. [DOI] [PubMed] [Google Scholar]
- 56.Schildkraut, C., and S. Lifson. 1965. Dependence of the melting temperature of DNA on salt concentration. Biopolymers. 3:195–208. [DOI] [PubMed] [Google Scholar]
- 57.Blake, R. D., and S. G. Delcourt. 1998. Thermal stability of DNA. Nucleic Acids Res. 26:3323–3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Owczarzy, R., Y. You, B. G. Moreira, J. A. Manthey, L. Huang, M. A. Behlke, and J. A. Walder. 2004. Effects of sodium ions on DNA duplex oligomers: improved predictions of melting temperatures. Biochemistry. 43:3537–3554. [DOI] [PubMed] [Google Scholar]
- 59.Record, M. T., Jr. 1975. Effects of Na+ and Mg2+ ions on the helix-coil transition of DNA. Biopolymers. 14:2137–2158. [Google Scholar]
- 60.Williams, A. P., C. E. Longfellow, S. M. Freier, R. Kierzek, and D. H. Turner. 1989. Laser temperature-jump, spectroscopic, and thermodynamic study of salt effects on duplex formation by dGCATGC. Biochemistry. 28:4283–4291. [DOI] [PubMed] [Google Scholar]
- 61.Rippe, K., N. B. Ramsing, and T. M. Jovin. 1989. Spectroscopic properties and helical stabilities of 25-nt parallel-stranded linear DNA duplexes. Biochemistry. 28:9536–9541. [DOI] [PubMed] [Google Scholar]
- 62.Duguid, J. G., V. A. Bloomfield, J. M. Benevides, and G. J. Thomas, Jr. 1995. Raman spectroscopy of DNA-metal complexes. II. The thermal denaturation of DNA in the presence of Sr2+, Ba2+, Mg2+, Ca2+, Mn2+, Co2+, Ni2+, and Cd2+. Biophys. J. 69:2623–2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nakano, S., M. Fujimoto, H. Hara, and N. Sugimoto. 1999. Nucleic acid duplex stability: influence of base composition on cation effects. Nucleic Acids Res. 27:2957–2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Serra, M. J., J. D. Baird, T. Dale, B. L. Fey, K. Retatagos, and E. Westhof. 2002. Effects of magnesium ions on the stabilization of RNA oligomers of defined structures. RNA. 8:307–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weixlbaumer, A., A. Werner, C. Flamm, E. Westhof, and R. Schroeder. 2004. Determination of thermodynamic parameters for HIV DIS type loop-loop kissing complexes. Nucleic Acids Res. 32:5126–5133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lorenz, C., N. Piganeau, and R. Schroeder. 2006. Stabilities of HIV-1 DIS type RNA loop-loop interactions in vitro and in vivo. Nucleic Acids Res. 34:334–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tan, Z. J., and S. J. Chen. 2006. Nucleic acid helix stability: effects of salt concentration, cation valency and size, and chain length. Biophys. J. 90:1175–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tan, Z. J., and S. J. Chen. 2007. RNA helix stability in mixed Na+/Mg2+ solution. Biophys. J. 92:3615–3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Manning, G. S. 1978. The molecular theory of polyelectrolyte solutions with applications to the electrostatic properties of polynucleotides. Q. Rev. Biophys. 11:179–246. [DOI] [PubMed] [Google Scholar]
- 70.Manning, G. S. 2006. The contribution of transient counterion imbalances to DNA bending fluctuations. Biophys. J. 90:3208–3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gilson, M. K., K. A. Sharp, and B. Honig. 1987. Calculating the electrostatic potential of molecules in solution: method and error assessment. J. Comput. Chem. 9:327–335. [Google Scholar]
- 72.Sharp, K. A., and B. Honig. 1990. Calculating total electrostatic energies with the nonlinear Poisson-Boltzmann equation. J. Phys. Chem. 94:7684–7692. [Google Scholar]
- 73.You, T. J., and S. C. Harvey. 1993. Finite element approach to the electrostatics of macromolecules with arbitrary geometries. J. Comput. Chem. 14:484–501. [Google Scholar]
- 74.Baker, N. A., D. Sept, S. Joseph, M. J. Holst, and J. A. McCammon. 2000. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA. 98:10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Grant, J. A., B. T. Pickup, and A. Nicholls. 2001. A smooth permittivity function for Poisson-Boltzmann solvation methods. J. Comput. Chem. 22:608–640. [Google Scholar]
- 76.Sept, D., N. A. Baker, and J. A. McCammon. 2003. The physical basis of microtubule structure and stability. Protein Sci. 12:2257–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Boschitsch, A. H., and M. O. Fenley. 2007. A new outer boundary formulation and energy corrections for the nonlinear Poisson-Boltzmann equation. J. Comput. Chem. 28:909–921. [DOI] [PubMed] [Google Scholar]
- 78.Grochowski, P., and J. Trylska. 2008. Continuum molecular electrostatics, salt effects, and counterion binding—a review of the Poisson-Boltzmann theory and its modifications. Biopolymers. 89:93–113. [DOI] [PubMed] [Google Scholar]
- 79.Tan, Z. J., and S. J. Chen. 2005. Electrostatic correlations and fluctuations for ion binding to a finite length polyelectrolyte. J. Chem. Phys. 122:044903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tan, Z. J., and S. J. Chen. 2006. Ion-mediated nucleic acid helix-helix interactions. Biophys. J. 91:518–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tan, Z. J., and S. J. Chen. 2006. Electrostatic free energy landscape for nucleic acid helix assembly. Nucleic Acids Res. 34:6629–6639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tan, Z. J., and S. J. Chen. 2008. Electrostatic free energy landscapes for DNA helix bending. Biophys. J. 94:3137–3149. [DOI] [PMC free article] [PubMed]
- 83.Cao, S., and S. J. Chen. 2005. Predicting RNA folding thermodynamics with a reduced chain representation model. RNA. 11:1884–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cao, S., and S. J. Chen. 2006. Free energy landscapes of RNA/RNA complexes: with applications to snRNA complexes in spliceosomes. J. Mol. Biol. 357:292–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cao, S., and S. J. Chen. 2006. Predicting RNA pseudoknot folding thermodynamics. Nucleic Acids Res. 34:2634–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Olson, W. K., and P. J. Flory. 1972. Spatial configurations of polynucleotide chains. I. Steric interactions in polyribonucleotides: a virtual bond model. Biopolymers. 11:1–23. [DOI] [PubMed] [Google Scholar]
- 87.Olson, W. K. 1975. Configurational statistics of polynucleotide chains. A single virtual bond treatment. Macromolecules. 8:272–275. [DOI] [PubMed] [Google Scholar]
- 88.Wadley, L. M., K. S. Keating, C. M. Duarte, and A. M. Pyle. 2007. Evaluating and learning from RNA pseudotorsional space: quantitative validation of a reduced representation for RNA structure. J. Mol. Biol. 372:942–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Montoro, J. C. G., and J. L. F. Abascal. 1995. Ionic distribution around simple DNA models. I. Cylindrically averaged properties. J. Chem. Phys. 103:8273–8284. [Google Scholar]
- 90.Marcus, Y. 1985. Ion Solvation. John Wiley & Sons, Great Britain.
- 91.Ni, H. H., C. F. Anderson, and M. T. Record. 1999. Quantifying the thermodynamic consequences of cation (M2+, M+) accumulation and anion (X-) exclusion in mixed salt solutions of polyanionic DNA using Monte Carlo and Poisson-Boltzmann calculations of ion-polyion preferential interaction coefficients. J. Phys. Chem. B. 103:3489–3504. [Google Scholar]
- 92.Saenger, W. 1984. Principles of Nucleic Acid Structure. Springer-Verlag, New York.
- 93.Kuznetsov, S. V., C. C. Ren, S. A. Woodson, and A. Ansari. 2008. Loop dependence of the stability and dynamics of nucleic acid hairpins. Nucleic Acids Res. 36:1098–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bommarito, S., N. Peyret, and J. SantaLucia, Jr. Thermodynamic parameters for DNA sequences with dangling ends. Nucleic Acids Res. 28:1929–1934. [DOI] [PMC free article] [PubMed]
- 95.Nixon, P. L., and D. P. Giedroc. 2000. Energetics of a strongly pH dependent RNA tertiary structure in a frameshifting pseudoknot. J. Mol. Biol. 296:659–671. [DOI] [PubMed] [Google Scholar]
- 96.Nixon, P. L., C. A. Theimer, and D. P. Giedroc. 1999. Thermodynamics of stabilization of RNA pseudoknots by cobaltIII hexamine. Biopolymers. 50:443–458. [DOI] [PubMed] [Google Scholar]
- 97.Nixon, P. L., and D. P. Giedroc. 1998. Equilibrium unfolding (folding) pathway of a model H-type pseudoknotted RNA: the role of magnesium ions in stability. Biochemistry. 37:16116–16129. [DOI] [PubMed] [Google Scholar]
- 98.Vieregg, J., W. Cheng, C. Bustamante, and I. Tinoco, Jr. 2007. Measurement of the effect of monovalent cations on RNA hairpin stability. J. Am. Chem. Soc. 129:14966–14973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Williams, D. J., and K. B. Hall. 1999. Unrestrained stochastic dynamics simulations of the UUCG tetraloop using an implicit solvation model. Biophys. J. 76:3192–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Draper, D. E. 2004. A guide to ions and RNA structure. RNA. 10:335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]