

Abstract
Toll-like receptors (TLRs) are a family of pattern-recognition receptors expressed on cells of the innate immune system that allow for the recognition of conserved structural motifs on a wide array of pathogens, referred to as pathogen-associated molecular patterns, as well as some endogenous molecules. The recent emergence of studies examining TLRs in the central nervous system (CNS) indicates that these receptors not only play a role in innate immunity in response to infectious diseases but may also participate in CNS autoimmunity, neurode-generation, and tissue injury. This review summarizes the experimental evidence demonstrating a role for TLRs in the context of CNS inflammation in both infectious and noninfectious conditions.
Keywords: Toll-like receptor (TLR), pattern recognition receptor (PRR), pathogen-associated molecular pattern (PAMP), central nervous system
INNATE IMMUNITY IN THE CNS
The immune system is subdivided into two interactive branches, namely, the innate (cellular) and adaptive (humoral) immune systems. Both systems have been shown to participate in infectious and autoimmune responses in the CNS. In general, innate immunity represents the first line of defense against pathogens and does not require prior exposure to foreign antigens to be triggered. Cell types that make up the innate immune system include macrophages, neutrophils, dendritic cells, natural killer cells, and in the CNS microglia and perivascular macrophages. In contrast, the induction of adaptive immunity requires signals provided by the innate immune system to facilitate the expansion of antigen-specific T and B lymphocytes, which are important for antibody production and the formation of long-lived memory cells. Unlike adaptive immunity, in which an infinite number of potential antigens can be recognized by T and B cells because of random gene rearrangements of their specific antigen receptors, cells of the innate immune system must recognize their cognate antigens by virtue of a predetermined subset of germline-encoded receptors. As a result of this limited receptor expression, cells of the innate immune system may not be able to recognize every possible antigen; rather, they focus on a few highly conserved structures expressed by large groups of microorganisms. These conserved structural motifs are referred to as pathogen-associated molecular patterns (PAMPs), and the receptors of the innate immune system that recognize these structures are called pattern recognition receptors (PRRs).
The CNS was once considered to be an immune-privileged site; however, it is now well accepted that immune surveillance does occur in the normal CNS and that inflammatory responses can and do take place in the context of disease (Hickey, 1999). The CNS launches an organized series of innate immune responses during both localized and systemic infections (Nguyen et al., 2002; Rivest, 2003). In addition, recent evidence suggests a role of innate immunity in response to injury (Owens et al., 2005; Tanga et al., 2005).
Microglia are the resident mononuclear phagocytes of the CNS parenchyma and participate in innate and adaptive immune responses, which include the induction of neuroinflammation through the release of proinflammatory cytokines and chemokines, phagocytosis, cytotoxicity, and regulation of T lymphocyte responses through antigen presentation (Aloisi, 2001; Hanisch, 2002). As such, microglia are uniquely poised to provide an initial line of defense against invading pathogens in the CNS prior to leukocyte infiltration. Astrocytes also play a role dictating the type and extent of CNS inflammation. These cells likely participate in the initial recruitment and activation of peripheral immune cells into the CNS during neuroinflammation through the production of several cytokines and chemokines (Dong and Benveniste, 2001). As detailed in the following sections, both microglia and astrocytes express numerous Toll-like receptors (TLRs), which allow the recognition of diverse PAMPs and potentially endogenous TLR agonists. The immediate activation of resident glia via TLRs likely serves as an amplification pathway to maximize proinflammatory responses within the CNS compartment.
OVERVIEW OF TLR AND SIGNALING PATHWAYS
The identification of TLRs in the human and mouse was based on their high degree of homology with the Toll family of proteins in Drosophila. The Drosophila Toll receptor was originally described for its role in regulating dorsoventral patterning during development (Anderson et al., 1985). Interestingly, Toll mutant flies were found to be more susceptible to fungal infections, implicating Toll in antifungal host defense (Lemaitre et al., 1996). Likewise, another Drosophila Toll family member, 18-Wheeler, was found to be pivotal in mediating antibacterial responses (Williams et al., 1997). Sequencing of these Drosophila receptors led to the discovery that their cytoplasmic domains shared a high degree of homology with the signaling domain of the mammalian interleukin-1 (IL-1) receptor (IL-1R; Anderson, 2000). Based on the relatedness between these Drosophila and mammalian proteins that play pivotal roles in innate immunity, the search for mammalian Toll-like homologues was initiated.
TLRs are a family of PRR expressed on cells of the innate immune system that allow for the recognition of invariant molecular motifs of bacteria, fungi, and viruses that are essential for pathogen survival and are conserved across broad subclasses of pathogens (Medzhitov and Janeway, 2000; Kopp and Medzhitov, 2003; Kaisho and Akira, 2004). In the human and mouse, 11 TLRs have been identified to date, and at least one agonist has been identified for each TLR, with the exception of TLR10 (Kopp and Medzhitov, 2003; Takeda et al., 2003). TLR1 recognizes triacylated lipoproteins in association with TLR2 that are cell wall constituents of various bacterial pathogens, including Borrelia burgorferi. TLR2 recognizes the widest array of PAMPs, including lipoproteins and peptidoglycan (PGN) that are components of all bacteria possessing cell walls and lipoteichoic acid (LTA) from the cell wall of gram-positive bacteria, as well as zymosan that is present in the yeast cell wall. TLR3 recognizes double-stranded RNA (dsRNA) which is generated during the replication of RNA viruses in host cells. TLR4 is the major receptor responsible for mediating effects to lipopolysaccharide (LPS) from the outer cell wall of gram-negative bacteria. TLR5 recognizes flagellin, a monomer of bacterial flagella that extends from the outer cell wall of gram-negative bacteria and propels organisms through aqueous environments. TLR6 forms functional heterodimers with TLR2 to recognize diacylated lipoproteins from Mycobacterium species that are unique in that they do not possess an enzyme required for the triacylation of lipoproteins (TLR1 agonist; Omueti et al., 2005). TLR7 and TLR8 mediate responses to GU-rich single-stranded RNA (ssRNA) that is present in virus-infected cells (Diebold et al., 2004; Heil et al., 2004); however, mammalian RNA also contains GU-rich sequences, suggesting that it may serve as a trigger for autoimmunity, which is supported by the finding that patients with systemic lupus erythrematosus (SLE) have autoantibodies against RNA (Lau et al., 2005). TLR9 recognizes bacterial and viral DNAs that contain high levels of unmethylated CpG motifs (Takeda et al., 2003); although these sequences also occur in mammalian DNA, they are typically methylated and thus do not trigger TLR9-mediated signaling. Finally, TLR11 recognizes pathogenic bacteria commonly associated with urinary tract infections, such as uropathogenic Escherichia coli, as well as a profilin-like protein from the parasite Toxoplasma gondii (Zhang et al., 2004; Lauw et al., 2005; Yarovinsky et al., 2005). Functional inactivation of many TLR genes has been accomplished by knockout (KO) strategies (Takeuchi et al., 1999, 2000a, 2002; Alexopoulou et al., 2001) as well as natural TLR4 mutations arising in various mouse strains (Poltorak et al., 1998a,b; Hoshino et al., 1999; Qureshi et al., 1999). All of these model systems provide excellent opportunities to examine the roles of TLRs on microglia and astrocytes both in vitro and in vivo as well as the functional importance of TLRs in CNS responses to injury, infection, and homeostasis.
As previously mentioned, the cytoplasmic domains of the TLRs share a high degree of homology with the intracellular portion of the IL-1R (Anderson, 2000; Takeda et al., 2003). Indeed, signaling via both the IL-1R and the TLRs culminates in activation of nuclear factor-κB (NF-κB), a transcription factor that regulates the expression of a wide array of genes involved in immune responses. A diagram depicting the major steps involved in TLR2 and TLR4 signaling pathways is presented in Figure 1. TLR activation results in the recruitment of the adaptor protein MyD88, which is associated with the serine/threonine kinase IL-1R-associated kinase (IRAK). IRAK then interacts with the tumor necrosis factor (TNF) receptor-associated factor (TRAF) adaptor protein TRAF6, which provides a bridge to the protein kinase NF-κB-inducing kinase (NIK). NIK then phosphorylates IKK (IκB kinase), resulting in the phosphorylation of IκB. IκB phosphorylation targets the protein for ubiquitination and proteasome-mediated degradation, resulting in the release and nuclear translocation of NF-κB, whereupon it can influence the expression of numerous immune response genes. Several groups have demonstrated the importance of MyD88 (Medzhitov et al., 1998) in signaling through numerous TLRs either in MyD88 KO mice (Adachi et al., 1998; Kawai et al., 1999; Takeuchi et al., 2000a,b) or through the expression of a mutant MyD88 protein that no longer is capable of transducing an activation signal (Underhill et al., 1999a,b). However, recent evidence has unveiled the existence of alternative adaptor molecules that transduce signals from TLRs via a MyD88-independent pathway (Akira and Takeda, 2004). These adaptors include TIRAP and TRIF, which are pivotal for the expression of interferon (IFN)-inducible genes following TLR4 activation (Akira and Takeda, 2004; Yamamoto et al., 2004). A distinction between adaptor usage for TLR4-dependent signaling is seen when comparing the kinetics of inflammatory mediator release in response to the TLR4 agonist lipopolysaccharide (LPS). For example, after LPS exposure, proinflammatory cytokine release induced by the MyD88-dependent pathway is rapid, whereas the production of IFN-inducible genes (MyD88-independent pathway) occurs much later (Akira and Takeda, 2004; Yamamoto et al., 2004).
Fig. 1.
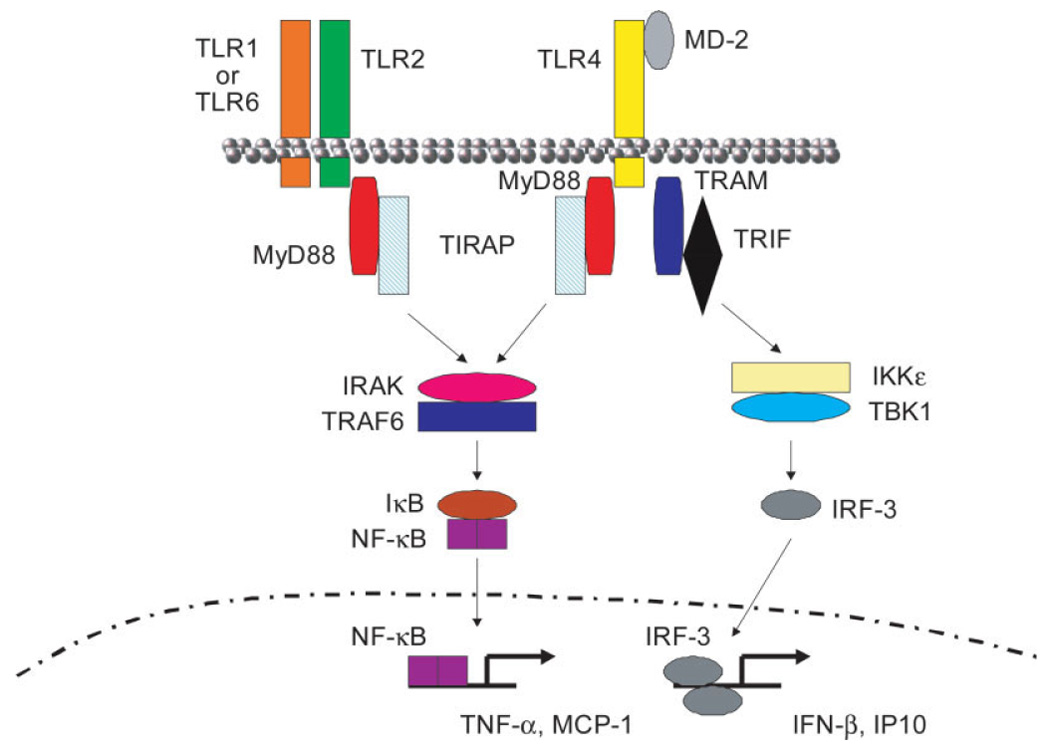
Signaling cascades initiated via TLR2- and TLR4-dependent activation. Engagement of TLR2 on the cell surface as a heterodimer with either TLR1 or TLR6 leads to the recruitment of the adaptor protein MyD88 and interaction with TIR-domain-containing adaptor protein (TIRAP) via death-domain interactions. Subsequently, IL-1-receptor-associated kinase (IRAK) and tumor necrosis factor-associated factor 6 (TRAF6) are recruited, leading to the phosphorylation and degradation of inhibitory-κB (IκB), allowing the nuclear translocation of NF-κB and subsequent induction of target genes such as tumor necrosis factor-α (TNF-α) and monocyte chemoattractant protein-1 (MCP-1/CCL2). The adaptor molecule MD-2 is involved in TLR4-mediated activation, which can lead to the recruitment of either of two adaptor proteins: the MyD88-dependent pathway occurs as described for TLR2; however, an alternative pathway has recently been identified. Upon TLR4 activation, the adaptor protein TRIF-related adaptor molecule (TRAM) can be activated, which associates with TIR-domain-containing adaptor-inducing IFN-β (TRIF). This complex activates IKKε and interacts with TBK-1 to induce translocation of the IFN-inducible transcription factor interferon response factor-3 (IRF-3) to the nucleus, where it transcriptionally activates IFN-inducible genes, such as IFN-β and IFN-inducible protein of 10 kDa (IP-10). Figure adapted from Microbes and Infection, Vol. 6, Kaisho T. and Akira S., Pleiotropic function of Toll-like receptors, p. 1388–1394, 2004, with permission from Elsevier.
TLRS IN GLIA: EXPRESSION AND FUNCTIONAL ROLES IN VITRO
In general, studies investigating TLR expression in CNS glia have emerged over the past 5 years; therefore, there is limited information available regarding the functional importance of many of these PRRs. However, the field is rapidly expanding, with publications related to the expression and function of TLRs in glia emerging at a rapid rate. A brief introduction of those TLRs that have been well defined will be presented initially, followed by a discussion of less-well-known TLRs, along with the available information related to their expression/function in microglia and astrocytes.
TLR2
Recent studies have established that microglia constitutively express TLR2 (Laflamme et al., 2001, 2003; Bsibsi et al., 2002; Kielian et al., 2002, 2005a; Rasley et al., 2002; Zekki et al., 2002; Olson and Miller, 2004), with elevated receptor expression observed following exposure to the gram-negative cell wall component LPS in vivo (Laflamme et al., 2001, 2003). With regard to classical TLR2 antigens, as defined by studies investigating the functional importance of TLR2 in macrophage activation, microglia are capable of recognizing numerous TLR2 ligands, including PGN, LTA, and tripalmitoyl-S-glyceryl-cysteine (Pam3Cys; Kielian et al., 2002; Olson and Miller, 2004; Chien et al., 2005; Ebert et al., 2005; Jung et al., 2005).
With regard to the functional importance of TLR2 in microglial activation, our group was the first to report that this PRR plays an essential role in enabling PGN recognition, using primary microglia isolated from TLR2 KO and wild-type (WT) mice (Kielian et al., 2005a). Interestingly, microglial responses to the gram-positive pathogen Staphylococcus aureus were found to be largely TLR2-independent, with the majority of proininflammatory mediators examined in response to intact bacteria equivalently expressed in both TLR2 KO and WT cells, suggesting that microglia utilize alternative PRRs for S. aureus recognition (Kielian et al., 2005a). PGN is released during normal bacterial growth as well as from dying organisms within infected tissues. In addition, many antibiotics that are used to treat CNS gram-positive infections enhance PGN release from the bacterial cell wall (van der Flier et al., 2003; Weber et al., 2003b), liberating additional antigen to engage PRRs, such as TLR2. CD14 is another PRR involved in PAMP recognition and is expressed on cells of the myeloid lineage, including microglia and macrophages (Becher et al., 1996; Nadeau and Rivest, 2000b; Saito et al., 2000; Kielian et al., 2002, 2005a). It has been well established that, because CD14 is a glycosylphosphatidyl inositol (GPI)-anchored receptor, it requires interaction with TLR4 to transduce activation signals in response to LPS (Haziot et al., 1988; Dobrovolskaia and Vogel, 2002; Fitzgerald et al., 2004; Palsson-McDermott and O’Neill, 2004). However, recent evidence also supports a role for CD14 in the recognition of gram-positive PAMPs, such as PGN and LTA, through its ability to interact with TLR2/TLR1 and/or TLR2/TLR6 heterodimers (Cleveland et al., 1996; Gupta et al., 1996; Dziarski et al., 2000; Henneke et al., 2001; Schroder et al., 2003; Weber et al., 2003a; Manukyan et al., 2005). Recent studies from our laboratory have revealed that, similarly to TLR2, CD14 participates in PGN-dependent microglial activation, whereas responses to intact gram-positive S. aureus are primarily CD14 independent (Esen and Kielian, 2005). The similarity in responses observed between TLR2 and CD14 KO microglia are highly suggestive that these two receptors function cooperatively to influence maximal PGN recognition. Indeed, evidence to support the collaborative actions of TLR2 and CD14 in the recognition of gram-positive bacteria is provided by recent studies demonstrating that macrophage activation in response to group B streptococci (GBS) involves the coordinated efforts of both receptors (Medvedev et al., 1998; Yoshimura et al., 1999). In terms of CNS disease pathogenesis, GBS are a major cause of neonatal meningitis and sepsis (Koedel et al., 2002; Nau and Bruck, 2002). Studies are underway to assess directly the cooperation between TLR2 and CD14 in regulating PGN-dependent microglial activation by examining primary microglia from TLR2/CD14 double KO mice. However, many important questions remain with regard to microglial TLR expression. First, recent studies have established microglial heterogeneity in various CNS compartments (Schmid et al., 2002; Carson et al., 2004; Wirenfeldt et al., 2005). Although a few reports have demonstrated constitutive expression of TLR2 and TLR4 in regions of the CNS that possess a leaky blood–brain barrier (BBB; i.e., circumventricular organs and choroid plexus), these studies did not allow for the unequivocal identification of TLR-expressing cells as microglia or macrophages (Laflamme et al., 2001, 2003; Chakravarty and Herkenham, 2005). Therefore, the issue of TLR heterogeneity in various microglial subsets remains an open question. In addition, numerous CNS diseases and trauma are associated with a mononuclear cell infiltrate consisting of macrophages and/or dendritic cells, both of which express TLRs. It remains to be demonstrated whether these peripheral mononuclear cell infiltrates express a different TLR repertoire compared with parenchymal microglia. Definitive proof of differential receptor levels between these populations awaits analysis by flow cytometric-based methods, with which individual cell types can be identified based on the expression of well-defined surface markers (Ford et al., 1995; Carson et al., 1998).
Several studies have demonstrated TLR2 expression in primary mouse astrocytes that can be enhanced following the treatment of cells with various PAMPs (Bowman et al., 2003; Esen et al., 2004; Carpentier et al., 2005). Recently, by using primary astrocytes isolated from TLR2 KO and WT mice, our laboratory has demonstrated that TLR2 is pivotal for enabling astrocytes to recognize both intact S. aureus and its cell wall product PGN. It is intriguing that this relationship was not observed in primary microglia (Kielian et al., 2005a), where cells utilized TLR2 primarily for PGN recognition but not for intact bacteria. This finding may be explained by the fact that, unlike astrocytes, microglia are considered to be potent bactericidal effector cells and likely possess a more extensive receptor repertoire to respond to infectious pathogens.
Although studies examining TLR2 expression in cultured mouse astrocytes have been in agreement (Bowman et al., 2003; Esen et al., 2004; Carpentier et al., 2005), studies examining TLR2 expression in other systems have produced some conflicting results. For example, in two reports examining TLR2 expression in human astrocytes, Bsibsi et al. (2002) reported that TLR2 mRNA was present, whereas Farina et al. (2005) were unable to detect this receptor. In addition, in situ hybridization studies of LPS- or cytokine-activated mouse brain have led to the identification of TLR2 mRNA expression in microglia but not astrocytes (Rivest, 2003; Owens, 2005). Collectively, the discrepancies between these studies may stem from species differences, route of PAMP administration in in vivo studies, and/or length of time for which astrocytes are co-cultured with microglia prior to purification in in vitro studies (Owens, 2005). The latter possibility seems plausible, in that human fetal astrocytes are relatively devoid of microglia without any additional purification steps (Farina et al., 2005), whereas adult human astrocytes, as utilized by Bsibsi et al. (2002), initially contain relatively high numbers of microglia prior to purification. It could be argued that the coculture of astrocytes with microglia prior to astrocyte purification is more reminiscent of the interactions between these cells in vivo, although the artificial conditions under which in vitro studies are performed cannot be denied. Finally, primary human oligodendrocytes have been shown to express TLR2 (Bsibsi et al., 2002); however, the functional significance of TLR2 in this cell type remains unknown.
Nonetheless, examination of purified glial cells provides a direct means of evaluating the roles of various TLRs on glial activation in a setting that is not confounded by the multiple influences encountered during in vivo studies. It will be important to evaluate further TLR2 expression on astrocytes in vivo to resolve these discrepancies; however, progress on this front has been hampered to date by the lack of commercially available antibodies for the reliable detection of TLR2 by immuno fluorescence staining techniques. Another approach that our laboratory is currently using to address astrocytic expression of TLR2 in vivo is laser capture microdissection to recover astrocytes from the inflamed and normal CNS to evaluate TLR2 mRNA expression by qRT-PCR. This approach will allow for the identification of TLR2-expressing astrocytes in their natural microenvironment. Finally, it is important to acknowledge that the context in which the CNS encounters PAMPs may be vitally important for detecting TLR expression on astrocytes. For example, the systemic injection of a PAMP may provide subthreshold signals to astrocytes in the brain parenchyma, which may prevent visualization of TLR2 expression by ISH techniques. In contrast, direct CNS parenchymal infection with a pathogen (i.e., bacteria, virus) might lead to a dramatic increase in receptor expression that may become detectable via this approach. In addition, the biochemical nature of the PAMPs used to examine systemic activation of CNS innate immunity should be considered, because PGN represents a particulate, nonsoluble polymer that exhibits a high degree of cross-linking (Dziarski, 2003; Weber et al., 2003b), and its bioavailability following i.p. administration may be quite distinct from a soluble PAMP, such as LPS. However, these possibilities remain speculative at the present time and warrant additional studies.
TLR3
TLR3 recognizes dsRNA, which is an intermediate produced during viral replication in cells (Alexopoulou et al., 2001). Studies investigating the potential role of TLR3-mediated signaling commonly utilize the synthetic TLR3 agonist polyinosine:cytosine [poly(I:C)]. Microglia have been reported to express TLR3 (Bsibsi et al., 2002; Olson and Miller, 2004) and respond to poly(I:C) and Theiler’s murine encephalomyelitis virus (TMEV) with elevated immune receptor expression as well as the production of select cytokines, including IFN-β, IL-1β, and IL-6 (Olson and Miller, 2004). Interestingly, TLR3 expression does not appear to be regulated by poly(I:C) in microglia, which differs from some of the other TLRs where receptor levels are augmented following exposure to their natural agonist(s) (Olson and Miller, 2004; Kielian et al., 2005a). However, exposure of microglia to TMEV did augment TLR3 mRNA expression (Olson and Miller, 2004), which may be attributed to the complex antigenic milieu provided by intact virus that likely stimulates additional, as yet unidentified TLRs.
Several recent studies have reported TLR3 expression in astrocytes (Bsibsi et al., 2002; Carpentier et al., 2005; Farina et al., 2005; Scumpia et al., 2005). A central role for astrocytes in sensing viral infections in the CNS is supported by the finding that cells are responsive to the TLR3 agonist poly(I:C), as evident from the production of several proinflammatory mediators. In addition, two studies have reported that both oligodendrocytes (Bsibsi et al., 2002) and neurons (Prehaud et al., 2005) express TLR3. The physiological implications of TLR3 in these cell populations remain to be defined but likely are involved in the host antiviral IFN response to dsRNA. The direct demonstration that TLR3 regulates glial and/or neuronal responses to dsRNA awaits the examination of primary cells isolated from TLR3-deficient mice. In addition, the use of primary glia and neurons from TLR3 KO mice will allow for a thorough investigation into the signal transduction pathways elicited upon receptor engagement to determine whether they differ from those already described in other immune cell populations that utilize TRIF, rather than MyD88, as a central adaptor (Akira and Takeda, 2004; Kaisho and Akira, 2004).
TLR4
As mentioned above, TLR4 is a central PRR responsible for the recognition of the gram-negative cell wall component LPS by cells of the mononuclear phagocyte lineage. Many of the early studies on human TLR2 and TLR4 evaluated the responses of HEK293 and Chinese hamster ovary (CHO) cell lines stably transfected with these expression constructs. From this initial work, it appeared that both TLR4 and TLR2 conferred sensitivity to LPS (Yang et al., 1998, 1999). However, subsequent studies established that TLR4, but not TLR2, was the receptor responsible for LPS-dependent signaling (Heine et al., 1999; Hirschfeld et al., 2000; Lien et al., 2000; Tapping et al., 2000). This original discrepancy regarding the role of TLR2 in LPS activation was attributed to contaminating lipoproteins (TLR2 agonists) in commercially available LPS preparations, because repurification of LPS to remove trace amounts of lipoproteins resulted in the inability of LPS to signal via TLR2 (Hirschfeld et al., 2000; Tapping et al., 2000). The consensus that TLR4 mediates responses to gram-negative bacteria is supported by findings in several mouse models. Targeted disruption of TLR4 has revealed that this receptor mediates macrophage activation in response to LPS (Takeuchi et al., 1999). In addition, after years of remaining elusive, the LPS hyporesponsive phenotypes of the C3H/HeJ and 10ScCr mouse strains were found to map to a point mutation and premature stop codon in the TLR4 gene, respectively, both resulting in the functional inactivation of TLR4-dependent signaling (Hoshino et al., 1999; Poltorak et al., 1998a,b; Qureshi et al., 1999). In contrast to other TLRs, the molecular pathways involved in LPS-mediated activation of TLR4 are relatively well-defined (Fig. 1). The lipid A portion of LPS represents the biologically active component and binds directly to a host-derived soluble molecule termed MD-2, which is essential for LPS recognition and signaling (Shimazu et al., 1999; Viriyakosol et al., 2000). MD-2 forms a receptor complex with TLR4, which represents the active signal-transducing component of the LPS receptor complex. In contrast to the known direct interactions between MD-2 and LPS, the question of whether LPS physically associates with TLR4 remains a subject of contention (Visintin et al., 2005).
With respect to CNS glia, it has long been acknowledged that LPS serves as a potent stimulus for microglial activation typified by the robust production of numerous proinflammatory mediators. It follows that numerous studies have reported that microglia express TLR4 (Laflamme and Rivest, 2001; Bsibsi et al., 2002; Lehnardt et al., 2002, 2003; Laflamme et al., 2003; Rivest, 2003; Olson and Miller, 2004; Chakravarty and Herkenham, 2005; Jung et al., 2005). The use of microglia from TLR4-deficient mice has allowed for the direct demonstration that this receptor is responsible, in part, for microglial activation in response LPS (Kitamura et al., 2001; Qin et al., 2005). Importantly, LPS-dependent microglial activation can occur in a TLR4-independent manner when cells are stimulated with high-dose LPS (i.e., >1 µg/ml; Kitamura et al., 2001; Qin et al., 2005), which has been previously reported in macrophages and may be a consequence of engaging lower affinity receptors under conditions of excess antigen (Lynn et al., 1993; Perera et al., 1997; Haziot et al., 1999). This is further supported by recent studies from our laboratory using primary microglia isolated from MyD88 KO mice, in which microglial responses to high-dose LPS were found to occur in a MyD88-dependent manner (Kielian, submitted). The identity of the receptor(s) that may be responsible for mediating TLR4-independent microglial activation in response to LPS is currently unknown.
As previously mentioned, CD14 interacts with TLR4 to induce maximal responses to LPS in macrophages (Dobrovolskaia and Vogel, 2002; O’Neill, 2004; Palsson-McDermott and O’Neill, 2004). Although microglia have been shown to express CD14 (Becher et al., 1996; Nadeau and Rivest, 2000a; Kielian et al., 2002), the functional significance of this receptor in mediating LPS-dependent microglial activation had not yet been demonstrated. By using primary microglia isolated from CD14 KO mice, we have recently demonstrated that CD14 is essential for microglial activation in response to low-dose LPS (Esen and Kielian, 2005). Similar to studies with TLR4-deficient microphages, we found that high doses of LPS (i.e., >1 µg/ml) were capable of activating microglia via a CD14-independent manner (Esen and Kielian, 2005). Future studies examining the signal transduction intermediates that are elicited in microglia upon TLR4 activation as well as the identification of additional PRRs that are involved in LPS recognition by microglia are warranted.
A recent report has described prolonged exposure of microglia to LPS (i.e., 72 hr) induces apoptosis that is mediated by TLR4 and involves caspase-11 and -3 activation (Jung et al., 2005). Interestingly, the autocrine production of IFN-β by LPS-activated microglia was found to be essential for driving microglial apoptosis in a TLR4-dependent manner. Indeed, TLRs have been shown under certain conditions to mediate apoptosis in other cell types, including monocytes (Aliprantis et al., 1999, 2000). However, it should be noted that the apoptosis- inducing signal(s) originating from TLR activation is expected to be complex and likely cell type dependent. In addition to the autocrine induction of apoptosis-inducing factors in LPS-activated microglia, TLR4-dependent activation of microglia elicits the production of mediators such as nitric oxide (NO), superoxide, and cytokines, which are capable of inducing apoptotic cell death in susceptible neurons and oligodendrocytes in coculture paradigms. For example, stimulation of oligodendrocyte and microglia co-cultures from TLR4-deficient mice with LPS failed to induce oligodendrocyte cell death, whereas cytotoxicity was significant when microglia from TLR4 WT animals were used in the co-culture paradigm (Lehnardt et al., 2002). A similar requirement for TLR4 has been demonstrated for mediating neuronal cell death caused by LPS-activated microglia (Lehnardt et al., 2003). Evidence also exists that TLR4 plays a role in mediating microglial activation to heat shock proteins (HSP), which may represent endogenous TLR4 agonists in the context of cellular damage. Specifically, TNF-α and IL-6 release as well as β-amyloid phagocytosis in response to human HSP90 and −70 and rat HSP32 was nearly completely suppressed in TLR4-deficient primary microglia (Kakimura et al., 2002). The authors state that the reported involvement of TLR4 in microglial activation by HSPs was not a result of LPS contamination, because heat treatment of HSP preparations completely destroyed their ability to induce proinflammatory cytokine release in microglia (Kakimura et al., 2002). However, as discussed below, the use of heat sensitivity as a sole indicator of LPS contamination can be misleading, in that LPS may also exhibit heat-labile properties, which appears to be an underappreciated concept (Vikstrom, 2002; Gao and Tsan, 2003a,b; Tsan and Gao, 2004).
In contrast to the case for microglia, it appears more controversial whether astrocytes express TLR4. Several groups have been unable to demonstrate astrocytic TLR4 expression in vitro (Farina et al., 2005; Kielian, unpublished observations) or in vivo (Laflamme and Rivest, 2001; Lehnardt et al., 2002, 2003); however, others have been able to detect low, constitutive expression of TLR4 in astrocytes that is increased upon cell activation (Bsibsi et al., 2002; Bowman et al., 2003; Carpentier et al., 2005). These discrepancies are reminiscent of the data obtained to date with TLR2 and, again, may be explained by species differences and/or the length of coculture with microglia prior to purification.
TLR9
TLR9 mediates responses to bacterial DNA, viral DNA, and synthetic oligodeoxynucleotides (ODN), all of which contain unmethylated CpG motifs (Takeda et al., 2003). Although CpG sequences also occur in mammalian DNA, they are typically methylated and thus do not trigger TLR9-mediated signaling. Synthetic unmethylated CpG-containing ODN are commonly used to simulate bacterial/viral DNA, because they can be manufactured in a highly purified form, which minimizes concerns about contaminating PAMPs such as endotoxin. Several reports have demonstrated that microglia express TLR9 and respond to CpG DNA with the robust production of numerous proinflammatory mediators as well as increased expression of immune receptor molecules (Takeshita et al., 2001; Dalpke et al., 2002; Iliev et al., 2004; Olson and Miller, 2004; Zhang et al., 2005). Related to its ability to induce proinflammatory mediator release in microglia, CpG ODN-stimulated microglia were found to induce neuron cell death in a neuron–microglia co-culture paradigm (Iliev et al., 2004). This microglial-mediated neuronal toxicity following CpG ODN treatment is reminiscent of what is observed following stimulation with the TLR4 agonist LPS (Lehnardt et al., 2003), suggesting, in part, the conservation of microglial responses to diverse PAMPs and a link between TLRs and neurodegeneration. Finally, a recent report by Lotz et al. (2005) has demonstrated that CpG ODN is capable of attenuating β-amyloid-dependent production of NO and TNF-α in microglia. In contrast, coadministration of the TLR2 or TLR4 agonists (Pam3-Cys and LPS, respectively) with β-amyloid augmented microglial proinflammatory mediator release (Lotz et al., 2005). This finding may account, in part, for the finding that Alzheimer’s disease patients often suffer clinical deterioration during infections (Perry et al., 2003) and reveals that not all TLR ligands modulate responses to β-amyloid equally.
Astrocytes are also activated upon exposure to CpG ODN (Takeshita et al., 2001; Bowman et al., 2003; Hosoi et al., 2004; S. Lee et al., 2004) and, as such, express TLR9 (Bowman et al., 2003; Hosoi et al., 2004; Carpentier et al., 2005). CpG ODN treatment of astrocytes induced p38 MAPK activation and subsequent inducible NO synthase (iNOS) expression, which was found to be MyD88-dependent, in that these effects were not observed in primary astrocytes isolated from MyD88 KO mice (Hosoi et al., 2004). Further investigation into the signal transduction pathways triggered in astrocytes following CpG ODN exposure revealed the activation of IKK and c-Jun N-terminal kinase (JNK), the latter of which was shown to be pivotal for inducing cytokine and chemokine expression (S. Lee et al., 2004). Despite these reports documenting that astrocytes express TLR9 and are activated in response to CpG DNA, one group has not been able to detect TLR9 in primary human astrocytes (Farina et al., 2005). This discrepancy could be the result of species differences, insofar as the studies in which TLR9 was detected in astrocytes were performed with primary rodent astrocytes (Bowman et al., 2003; Hosoi et al., 2004; Carpentier et al., 2005), whereas Farina et al. (2005) utilized primary human fetal astrocytes. Although studies have not yet been conducted with primary microglia and astrocytes from TLR9 KO mice, extrapolating from the evidence to date establishing that TLR9 is the receptor for prokaryotic DNA in macrophages coupled with the finding that astrocytes from MyD88 KO mice do not respond to CpG ODN (Hosoi et al., 2004) provides strong evidence for an important role of TLR9 in mediating glial responses to CpG DNA.
Other TLRs (TLR1, TLR5, TLR6, TLR7, TLR8, TLR10, TLR11)
As part of the emergence of reports examining TLR biology in the field of neuroimmunology, a few initial studies have been performed characterizing the array of additional TLRs expressed by microglia and astrocytes. Because no functional studies have been performed to assess the functional significance of these receptors to date using cells from KO mice, only a brief description of the known agonists for these TLRs along with their expression pattern in CNS glia, if known, will be presented.
TLR1 and TLR6 form functional heterodimers with TLR2 to mediate signals in response to gram-positive PAMPs (Ozinsky et al., 2000; Takeuchi et al., 2001, 2002). This functional cooperativity may facilitate the customization of immune responses elicited by diverse TLR2 agonists. Both TLR1 and TLR6 have been shown to be expressed by microglia (Bsibsi et al., 2002; Kielian et al., 2002; Olson and Miller, 2004) and astrocytes (Carpentier et al., 2005). However, experiments designed to investigate the functional interactions between TLR2/TLR1 and TLR2/TLR6 in dictating glial responses to diverse TLR2 agonists are lacking; these could be examined with the use of double-receptor-KO mice once they are developed.
TLR5 serves as an agonist for flagellin, a monomer of bacterial flagella, which extends from the outer cell wall of gram-negative bacteria and propels organisms through aqueous environments (Hayashi et al., 2001). Microglia (Bsibsi et al., 2002; Olson and Miller, 2004) and astrocytes (Bowman et al., 2003; Carpentier et al., 2005) have been reported to express TLR5; however, only one study has examined the responses of astrocytes to the TLR5 ligand flagellin from S. typhimurium, which led to an increase in TLR2, TLR4, and TLR5 expression along with an induction in IL-6 production (Bowman et al., 2003). Additional studies are needed to characterize responses of microglia to flagellin as well as to delineate the functional significance of TLR5 in both astrocytes and microglia using cells isolated from TLR5-deficient mice.
TLR7 and TLR8 are highly homologous to each other and are important for immune responses elicited by GU-rich ssRNA as well as synthetic chemicals, including the imidazoquinoline compounds (i.e., imiquimod and resiquimod) and guanosine analogs (Hemmi et al., 2002; Diebold et al., 2004; Heil et al., 2004). The latter compounds were initially described for their ability to activate TLR7 and TLR8 and are potent immune response modifiers leading to the production of cytokines (i.e., IFNs) that exert important antiviral and antitumor activities (Hemmi et al., 2002). Both imiquimod and resiquimod are used clinically for the localized treatment of herpesvirus infections of the skin, so systemic engagement of TLR7/8 would not be expected to occur. The structural similarities of these compounds to nucleic acids led to the identification of ssRNA as a natural agonist for TLR7/8 (Diebold et al., 2004; Heil et al., 2004). Interestingly, mammalian RNA also contains GU-rich sequences, suggesting that it may serve as an autoimmune trigger, which is supported by the finding that patients with systemic lupus erythrematosus (SLE) have autoantibodies against RNA (Lau et al., 2005). Although there is evidence that TLR7 and TLR8 are expressed in microglia (Bsibsi et al., 2002; Olson and Miller, 2004) and astrocytes (Carpentier et al., 2005), there are no available studies investigating either the consequences of TLR7/8 agonist treatment or the responses in TLR7 or TLR8-deficient glia. A recent report has identified a critical role for TLR8 in regulating the activity of CD4+ regulatory T cells, which may play an important role in controlling immune responses to cancer and autoimmune diseases (Peng et al., 2005).
TLR10 is an orphan member of the TLR family and has recently been described to form homodimers as well as heterodimerize with TLR1 and TLR2 (Hasan et al., 2005). The expression of TLR10 appears to be rather restricted, with only B cells and specialized dendritic cell subsets demonstrating expression (Hasan et al., 2005). Mouse TLR11 is involved in the recognition of uropathogenic bacteria (such as E. coli) as well as a profilin-like molecule of T. gondii, whereas the human receptor is nonfunctional because of the presence of a stop codon (Zhang et al., 2004; Lauw et al., 2005; Yarovinsky et al., 2005). Based on the limited and unique expression patterns of these receptors, it seems unlikely that these receptors will be found in CNS glia. However, based on accumulating evidence demonstrating that immune molecules can often assume different functions in the context of the CNS, it remains possible that a new role for TLR10 and/or TLR11 may emerge relevant to CNS homeostasis and/or disease.
EXPRESSION AND FUNCTIONAL ROLES OF TLRS IN VIVO
Responses to Purified PAMPs in the CNS
Initial studies examining the localization of TLRs in the CNS in vivo characterized expression patterns in the brain following systemic exposure to the TLR2 and TLR4 ligands PGN and LPS, respectively (Laflamme et al., 2001, 2003; Laflamme and Rivest, 2001). Under physiological conditions, in situ hybridization approaches have localized constitutive expression of TLR2 and TLR4 to regions of the CNS that possess a leaky BBB, namely, the circumventricular organs (CVO) and choroid plexus (Laflamme et al., 2001, 2003; Laflamme and Rivest, 2001; Chakravarty and Herkenham, 2005). However, a recent study has revealed that constitutive TLR4 expression may be more extensive than previously reported, with detectable signals observed across the brain parenchyma, including the meninges and fenestrated capillaries (Chakravarty and Herkenham, 2005). It is thought that the constitutive expression of TLR2 and TLR4 in these specialized areas that lack an intact BBB serves as an initial sensor of systemic infection. Subsequently, the engagement of these PRRs leads to the initiation of proinflammatory signaling cascades to propagate activation signals to immunocompetent cells within the CNS parenchyma (Nguyen et al., 2002; Rivest, 2003). Indeed, studies examining the induction of proinflammatory molecules in the CNS following systemic administration of bacterial PAMPs support this concept, as described below.
Systemic LPS administration to rats and mice induces a robust increase in TLR2 expression in the CNS originating from the CVO and choroid plexus emanating into deeper parenchymal regions, whereas TLR4 levels are reduced (Laflamme et al., 2001, 2003; Laflamme and Rivest, 2001). It is intriguing that TLR2 levels are augmented within the CNS during systemic LPS challenge in the absence of any obvious ligand. This response has been proposed potentially to serve three purposes. 1) The first is to arm the cerebral innate immune response against a potential invading pathogen that utilizes TLR2. 2) Chronic expression of TLR2 in the CNS may be detrimental through augmenting proinflammatory bystander damage to normal parenchyma. 3) Prolonged elevations in TLR2 may induce microglial apoptosis to prevent bystander damage to surrounding neurons resulting from inappropriate proinflammatory mediator release (Laflamme et al., 2001; Rivest, 2003).
Subsequent studies have directly examined the functional importance of TLR2 and TLR4 in the CNS response to systemic PAMPs using receptor-KO mice. Systemic infection is associated with the worsening of numerous CNS diseases, such as multiple sclerosis (Sibley et al., 1985; Rapp et al., 1995; Buljevac et al., 2002), and microglial activation is a hallmark of several CNS disorders, including multiple sclerosis, Alzheimer’s disease, HIV encephalitis and dementia, and ischemia (Eikelenboom et al., 2002; Griffin and Mrak, 2002; Koedel et al., 2002; McGeer and McGeer, 2002; Nau and Bruck, 2002; Kielian, 2004b). These diseases are associated, to variable extents, with oligodendrocyte and/or neuron damage. Products of activated microglia have been implicated in mediating cell death of both CNS populations via bystander damage; numerous studies using co-culture paradigms have documented that LPS-activated microglia induce oligodendrocyte and neuron cytotoxicity (Hewett et al., 1999; Pang et al., 2000; Arai et al., 2004; Qin et al., 2004; Shie et al., 2005); however, whether this effect was mediated by TLR4 had not yet been demonstrated. In vivo evidence for a role of TLR4 in LPS-dependent CNS injury was revealed by using a model of neurodegeneration in which a systemic injection of LPS converts a subthreshold hypoxic-ischemic insult that normally does not induce CNS injury into one with severe axonal and neuronal loss (Lehnardt et al., 2003). Exposure of TLR4 mutant mice to this treatment did not induce any detectable neurodegeneration, whereas WT animals displayed significant axonal and neuronal loss, typified by a reduction in NeuN+ and neurofilament+ cells, establishing a direct link between innate immunity and neuronal injury in the CNS (Fig. 2; Lehnardt et al., 2003).
Fig. 2.

Potential roles of TLRs in the CNS response to infection and injury. Microglia and astrocytes respond to numerous PAMPs, including peptidoglycan (PGN; TLR2 agonist), double-stranded RNA (dsRNA; TLR3 agonist), lipopolysaccharide (LPS; TLR4 agonist), and unmethylated CpG oligodeoxynucleotides and/or bacterial DNA (CpG DNA; TLR9 agonist). The resultant effect of glial PAMP stimulation is the elaboration of a wide array of proinflammatory cytokines (including TNF-α, IL-1β, and IL-12), chemokines [including macrophage inflammatory protein-2 (MIP-2/CXCL2) and monocyte chemoattractant protein-1 (MCP-1)], and reactive oxygen/nitrogen species [ROI/RNI; including superoxide () and nitric oxide (NO)]. These proinflammatory mediators contribute to enhanced blood–brain barrier (BBB) permeability and the resultant influx of peripheral immune cells into the CNS parenchyma, the extent of which is dictated by the nature of the proinflammatory milieu. Endogenous TLR ligands may be released from injured cells within the CNS parenchyma that may serve to augment neuroinflammation further; however, their role in TLR-dependent glial activation remains to be determined. The outcome of this neuroinflammatory response (i.e., beneficial vs. detrimental) is unclear and likely depends on the context of the insult and duration of inflammation. (Figure adapted from Kielian, 2004a.)
An intriguing relationship was found to exist between TLR2 and TLR4 in regulating CNS innate immunity that was suggestive of receptor cooperativity (Laflamme et al., 2003). Previous studies had demonstrated that a systemic bolus of LPS led to a dramatic increase in TLR2 in expression the CNS (Laflamme et al., 2001), so it was hypothesized that a subsequent exposure to the TLR2 agonist PGN would result in heightened levels of proinflammatory mediator expression. On the contrary, PGN delivered subsequent to a systemic LPS administration largely abolished the LPS-induced expression of inflammatory mediator expression in parenchymal microglia (Laflamme et al., 2003). This PGN-induced effect was mediated by TLR2, in that PGN was unable to inhibit ongoing CNS inflammation following an initial systemic bolus of LPS in TLR2 KO mice. The cause(s) of this phenomenon is not clearly understood, but it could result from the induction of a negative regulatory factor of TLR-dependent signaling, such as Toll-interacting protein (TOLLIP), which inhibits TLR2 and TLR4 activation by preventing the phosphorylation of IRAK1, or alternatively MyD88 short, a splice variant of MyD88 that cannot interact with TRAF6, culminating in signal transduction inhibition (Burns et al., 2000, 2003; Zhang and Ghosh, 2002; Akira and Takeda, 2004). Collectively, these studies revealed that, in response to systemic LPS, TLR4 is required to trigger expression of TLR2 and other inflammatory genes in the CNS, whereas TLR2 is involved in the second wave of proinflammatory mediator expression in the context of severe endotoxemia.
Although the studies described above have assigned an essential role for TLR4 in mediating the expression of proinflammatory molecules in the CNS in response to systemic LPS, the origin of LPS-responding cells remained controversial. Specifically, it was not clear whether the induction of proinflammatory mediators in the CNS following endotoxemia was due to the action of cytokines released by activation of TLR4-expressing hematopoietic cells or whether LPS was directly capable of activating signal transduction in CNS cells. This question was recently addressed in a series of elegant studies by Chakravarty and Herkenham (2005), in which the contribution of TLR4 in the CNS vs. hematopoietic cells was assessed by using radiation bone marrow chimeric mice. With this approach, the authors demonstrated that, after systemic LPS administration, the early induction of proinflammatory mediators within the CNS compartment is due to both hematopoietic and resident CNS cells, whereas the late inflammatory response in the CNS requires parenchymal signaling via TLR4 (Chakravarty and Herkenham, 2005). Collectively, this study provided direct evidence that TLR4 functions in LPS-dependent signaling in resident CNS parenchymal cells, leading to the subsequent release of proinflammatory mediators. Reiterating the importance of reagent purity, this study confirmed previous reports that commercial LPS preparations contain contaminants that signal via TLR4-independent pathways, insofar as LPS repurification eliminated the induction of proinflammatory mediator expression in TLR4 mutant mice that was observed with the nonpurified preparation.
During bacterial infections, bacterial DNA is released from dying organisms either as a consequence of the host immune response or through the actions of antibiotic therapy. Bacterial DNA is extremely immunogenic owing to its high content of unmethylated CpG motifs, whereas eukaryotic DNA is predominantly methylated. Therefore, the release of bacterial DNA during CNS infections may serve as a major stimulus for the propagation and/or persistence of innate immune responses. These responses may be beneficial and/or detrimental depending on the context and duration of proinflammatory mediator release in the CNS. Injection of the TLR9 agonist CpG ODN into the CNS induces meningitis and macrophage/microglial activation (Deng et al., 2001; Schluesener et al., 2001; Dalpke et al., 2002). Interestingly, CpG ODN was found to synergize with suboptimal doses of either LPS or PGN to induce meningitis, which more accurately reflects the complex antigenic milieu encountered during CNS bacterial infections (Deng et al., 2001). In addition, it has been demonstrated that CpG DNA can exacerbate Theiler’s murine encephalomyelitis infection (Tsunoda et al., 1999) and relapsing-remitting EAE as well as disease induction in a myelin basic protein model of EAE (Segal et al., 1997, 2000). Collectively, these results suggest that bacterial DNA may serve as a potent activator of CNS innate immune responses. However, the continual presence of this PAMP could exacerbate CNS damage through persistent engagement of TLR9-mediated glial activation. The context and functional importance of TLR9-dependent signals in CNS infectious disease and potential adjuvant effects of CpG DNA await examination in TLR9-deficient mice.
Role of TLR2 in CNS Gram-Positive Bacterial Infections
TLR2 plays an important role in innate immune recognition of conserved structural motifs on a wide array of pathogens, including various gram-positive bacteria, fungi, and protozoa (Kopp and Medzhitov, 2003). An important role for TLR2 in recognition of the gram-positive CNS pathogens Strep. pneumoniae and Staph. aureus was recently demonstrated by using mouse models of bacterial meningitis (Echchannaoui et al., 2002; Koedel et al., 2003) and brain abscess (Kielian et al., 2005b). Although these studies did reveal roles for TLR2 in CNS antibacterial immune responses, collectively the evidence strongly suggests the involvement of additional, as yet unidentified receptors in bacterial recognition and elimination in the CNS.
The expression of TLR2 is increased during pneumococcal meningitis (Bottcher et al., 2003; Koedel et al., 2003). This finding, coupled with the fact that TLR2 is pivotal for the containment of other gram-positive infections (Takeuchi et al., 2000a; Echchannaoui et al., 2002; Drennan et al., 2004; Wieland et al., 2004), led to studies investigating the functional importance of TLR2 in the pathogenesis of Strep. pneumoniae meningitis by using TLR2 KO mice, with minor differences noted in each of the experimental models (Echchannaoui et al., 2002; Koedel et al., 2003). Both studies demonstrated enhanced disease severity in TLR2-deficient mice and elevated bacterial burdens within the CNS; however, only Koedel et al. (2003) found that systemic bacterial titers were significantly elevated in TLR2 KO mice, whereas Echchannaoui et al. (2002) reported that Strep. pneumoniae levels in the blood were not significantly different between TLR2 KO and WT animals. There were also differences regarding the relative role of TLR2 in regulating CNS cytokine expression between the two studies. For example, Echchannaoui et al. reported that TNF-α levels were significantly elevated in the cerebrospinal fluid (CSF) of TLR2 KO mice compared with WT animals, which correlated with enhanced BBB disruption in the former. In contrast, Koedel et al. did not find any evidence of elevated TNF-α expression in the brain parenchyma of TLR2 KO mice; however, CSF levels were not examined in this study. These discrepancies could be due to the route of bacterial administration (intracerebroventricular vs. intracranial injection) or the different compartments in which cytokine expression was examined (i.e., parenchyma vs. CSF).
Our laboratory has recently completed studies investigating the functional importance of TLR2 in the context of CNS parenchymal infection by evaluating the pathogenesis of Staph. aureus-induced experimental brain abscess in TLR2 KO and WT mice, since it may differ from meningitis based on the highly focal nature of lesions in the former (Kielian et al., 2005b). The expression of several proinflammatory mediators, including iNOS, TNF-α, and macrophage inflammatory protein-2/CXCL2, was significantly attenuated in brain abscesses of TLR2 KO compared with WT mice during the acute phase of infection. Conversely, IL-17, a cytokine produced by activated and memory T cells (Aggarwal and Gurney, 2002; Witowski et al., 2004), was significantly elevated in lesions of TLR2 KO mice, suggesting an association between innate and adaptive immunity in brain abscess (Kielian et al., 2005b). Despite these differences, brain abscess severity was similar between TLR2 KO and WT animals, with comparable mortality rates, bacterial titers, and BBB permeability, implying a role for alternative PRRs. The relative importance of TLR2 in brain abscess pathogenesis was not as dramatic compared with recent reports examining the role of this receptor in Strep. pneumoniae meningitis (Echchannaoui et al., 2002; Koedel et al., 2003). For example, both meningitis studies revealed that TLR2 regulated bacterial burdens in the CNS, whereas we did not observe a critical role for TLR2 in pathogen containment in brain abscess. This finding could be explained by the differential extent of infection in both models, where bacteremia occurs in conjunction with CNS infection during meningitis, whereas brain abscess is typified by a focal infection confined to the CNS parenchyma. Indeed, studies documenting a critical role for TLR2 in the pathogenesis of gram-positive infections have been conducted with disseminated systemic infectious disease models (Takeuchi et al., 2000a; Echchannaoui et al., 2002; Drennan et al., 2004; Wieland et al., 2004). Nonetheless, it appears that TLR2 plays a role in the host antibacterial immune response in both bacterial meningitis and brain abscess, although it is apparent that additional, as yet undefined PRRs contribute to pathogen recognition.
To assess the involvement of alternative TLRs in bacterial meningitis, Koedel et al. (2004) have examined the host response to Strep. pneumoniae by using MyD88 KO mice. The authors observed that MyD88 KO mice displayed elevated bacterial burdens in the CNS; however, there was less evidence for active CNS inflammation as shown by a significant decrease in the expression of numerous proinflammatory mediators and cellular influx into the CNS, reduced BBB permeability, and less edema, suggesting a key role for MyD88-dependent signal transduction pathways in the recognition of Strep. pneumoniae (Koedel et al., 2004). However, it is important to acknowledge that MyD88 is a key adaptor molecule in signaling via the IL-1 and IL-18 receptors; these pathways have individually been implicated in modulating host innate immune responses during bacterial meningitis (Koedel et al., 2002; Zwijnenburg et al., 2003a,b), suggesting that the phenotype observed in MyD88 KO mice might result from the collective inactivation of multiple TLRs in addition to IL-1- and IL-18-dependent signals. Collectively, these studies have illuminated an important point, namely, that the development of antibacterial immune responses in the CNS parenchyma cannot be accounted for by the activity of a single receptor, a concept that has emerged in recent years (Henneke et al., 2001, 2002; Koedel et al., 2003; Mukhopadhyay et al., 2004). Indeed, recent evidence in other models of systemic infectious disease support the concept that multiple PRRs act in concert to induce protective antibacterial immune responses (Henneke et al., 2002; Reiling et al., 2002; Koedel et al., 2003; Nicolle et al., 2004; Knapp et al., 2004). This concept of receptor redundancy is not unexpected; bacterial pathogens such as Staph. aureus have the potential to elicit devastating consequences in a tissue that has limited regenerative capacity such as the CNS (Kielian, 2004a). Therefore, the host repertoire of available PRRs should be substantial, ensuring that an effective antibacterial immune response will be rapidly elicited upon infection of the CNS parenchyma.
Role of TLRs in Parasitic and Viral CNS Infections
To date, there are only a limited number of studies directly investigating the roles of TLRs in nonbacterial CNS infectious diseases. With a mouse model of T. gondii infection, several groups have demonstrated that MyD88-dependent pathways are critical for systemic host resistance to parasites (Scanga et al., 2002; Mun et al., 2003; Hitziger et al., 2005); however, only one study has directly examined parasite burdens in the CNS, in which pathogen loads were elevated in MyD88 KO mice in response to an avirulent strain of T. gondii (Mun et al., 2003). In contrast, the functional importance of TLR2 in T. gondii host defense in the CNS appears more controversial. For example, Mun et al. (2003) demonstrated that TLR2 KO mice displayed elevated CNS parasite burdens and mortality compared with WT animals; however, this effect was dose-dependent, in that a role for TLR2 was not observed with a lower parasite inoculum. In contrast, Hitziger et al. (2005) did not find any evidence for a role of TLR2, TLR1, TLR4, TLR6, or TLR9 in controlling T. gondii replication in the CNS. The discrepancy regarding the functional importance of TLR2 in the host response to T. gondii infection in the CNS may be attributed to the strain of T. gondii used, dose of parasite injected, and/or sensitivity of methods used to determine parasite burdens in the infected CNS (i.e., standard plaquing assays vs. quantitative competitive PCR). Nonetheless, the requirement for MyD88 in host resistance to T. gondii suggests the involvement of numerous members of the TLR family as well as potentially additional TIR signaling pathways.
Two recent studies have investigated the role of TLRs in viral infections that affect the CNS. Although it has been described as a major receptor for gram-positive bacterial and fungal PAMPs, TLR2 has been shown to be important for mediating the host immune response during herpes simplex virus 1 (HSV-1)-induced encephalitis (Kurt-Jones et al., 2004). Another recent study has examined the functional role of TLR3 in response to West Nile virus (WNV) encephalitis (Wang et al., 2004). Systemic infection of TLR3 KO mice with WNV revealed an intriguing compartmental requirement for TLR3 in the host response to infection. Specifically, TLR3 KO mice were more resistant to lethal WNV infection and displayed reduced TNF-α levels systemically; however, viral burdens were elevated in KO mice compared with WT animals (Wang et al., 2004). In contrast, in the brain, viral load, cytokine expression, and cellular infiltrates were significantly attenuated in TLR3 KO mice compared with WT animals. The diminished penetration of WNV into the CNS of TLR3 KO mice was attributed to attenuated BBB compromise in the context of reduced TNF-α production in the periphery. Interestingly, there were no significant differences in neuropathogenesis between TLR3 KO and WT mice following i.c.v. administration of WNV, highlighting the importance of the infection route and BBB permeability in TLR3-mediated responses (Wang et al., 2004). Additional evidence to support a role for TLR3 in CNS inflammation is the finding that the chronic infusion of poly(I:C) into the lateral ventricle induced chronic neurodegeneration that was morphologically similar to Alzheimer’s disease (Melton et al., 2003). It will be interesting to determine whether TLR3 KO mice display similar differences upon chronic infusion of dsRNA, which would establish a functional role for TLR3 in this neurodegenerative process.
Multiple Sclerosis
It has long been acknowledged that infections precipitate clinical relapses in MS patients (Sibley et al., 1985; Rapp et al., 1995; Buljevac et al., 2002); however, the molecular mechanisms responsible for this phenomenon are unclear. Recent experimental evidence suggests that various bacterial PAMPs [i.e., PGN, pertussis toxin (PTX), and CpG DNA] act to break tolerance (i.e., enable the inappropriate immune-mediated recognition of self antigens) to CNS myelin antigens through effects on antigen-presenting cells (APCs), such as dendritic cells and macrophages (Segal et al., 2000; Ichikawa et al., 2002; Kerfoot et al., 2004; Waldner et al., 2004; Visser et al., 2005). These effects have been attributed, in part, to engagement of TLRs on the APC surface.
In the majority of EAE animal models, PTX is required to induce disease, which was thought to occur by its ability to modulate BBB permeability and subsequent immune cell entry into the CNS (Racke et al., 2005). However, a recent study by Kerfoot et al. (2004) demonstrated that BBB compromise is a secondary occurrence following PTX administration, with its primary effect modulating the adhesion molecule (P-selectin)-dependent rolling of leukocytes on the cerebral vascular endothelium. Furthermore, Kerfoot et al. revealed that PTX-induced cellular recruitment in the brain is attenuated in TLR4-deficient mice, suggesting that TLR4-mediated signals may be required for EAE induction. However, EAE studies in TLR4 KO mice were not consistent among individual experiments (Kerfoot et al., 2004), suggesting that additional factors influence the establishment of clinical disease in addition to TLR4/PTX interactions. Kerfoot et al. proposed that unknown environmental factors may contribute to the establishment of EAE in this model, leading to the observed variability in their studies with TLR4-deficient mice. This is supported by previous studies demonstrating that autoimmune diseases, such as EAE, are more readily induced in animals that are housed in conventional quarters compared with those bred and maintained in a specific pathogen-free environment (Goverman et al., 1993). A separate study by Waldner et al. (2004) also demonstrated that PTX was capable of enabling T cells to recognize inappropriately the self myelin proteolipid protein (PLP) peptide 139–151, although it was less effective at inducing disease compared with CpG ODN. Nonetheless, it appears that PTX is capable of acting as a substitute for environmental conditions to drive the induction of CNS autoimmunity (Kerfoot et al., 2004; Racke et al., 2005).
With regard to the potential involvement of TLR4 in MS, studies have been performed to examine whether genetic variants of TLR4 are related to MS development (Reindl et al., 2003; Kroner et al., 2005). Two mutations of TLR4 have been observed at a relatively high frequency in the human population, namely, Asp299Gly and Thr399Ile (Arbour et al., 2000; Lorenz et al., 2001), where the former substitution alters the extracellular domain of TLR4, resulting in defective TLR4-mediated signaling. Examination of MS patients revealed that the frequency of the Asp299Gly mutation was not significantly different compared with age-matched controls, indicating that TLR4 polymorphism has no influence on the incidence and progression of MS (Reindl et al., 2003; Kroner et al., 2005). However, a functional role of TLR4 cannot be excluded based on the apparent interactions between infectious agents and disease exacerbation, some of which may be mediated by TLR4-dependent signals.
In addition to TLR4, it is also possible that TLR2 participates in the CNS innate immune response during EAE; TLR2 expression was found to increase progressively during the course of MOG35–55-induced EAE in distinct CNS regions (Zekki et al., 2002). This is supported by the finding that the TLR2 agonist PGN is capable of inducing clinical disease in a MOG model of EAE when emulsified in incomplete Freund’s adjuvant (IFA), whereas MOG in IFA alone is incapable of inducing disease (Visser et al., 2005). Additional studies are needed to ascertain the context in which TLR2 may participate in MS pathology.
Recent evidence has also revealed a role for TLR9 in the adjuvant effect exhibited by CpG ODN on EAE induction. Using a MBP87–106 model of EAE, Segal et al. (2000) demonstrated that the adjuvant activity of complete Freund’s adjuvant (CFA) is duplicated by CpG ODN in IFA. Interestingly, to induce disease, CpG ODN and IFA required co-administration in a single emulsion, whereas, if these entities were delivered at physically separate sites, animals remained asymptomatic. This finding suggests that either the same APC must present MBP and produce CpG-induced factors or neighboring APCs must release these factors to act in a paracrine fashion (Segal et al., 2000). A subsequent report from the same group revealed that stimulation of APCs with CpG ODN provides the requisite signal(s) to induce the activation of autoreactive T cells specific for MBP and the development of active EAE (Ichikawa et al., 2002). In addition, another recent study demonstrated that CpG ODN was also capable of breaking tolerance in an EAE-resistant transgenic mouse model (Waldner et al., 2004). Collectively, these studies provide insights into mechanisms linking autoimmune exacerbations with infectious diseases, which routinely occur in MS patients (Sibley et al., 1985; Rapp et al., 1995; Buljevac et al., 2002). Detailed studies investigating the functional importance of various TLRs in the adjuvant effects of various PAMPs using TLR-deficient mice are needed to establish further the involvement of these pathways in disease induction and/or exacerbation. However, based on the available evidence obtained from other studies investigating the roles of TLRs in infectious diseases, it is likely that the multiple TLRs (not to mention additional, as yet unidentified PRRs) will contribute to autoimmune disease.
Other CNS Inflammatory Conditions
Because of the ability of TLRs to recognize a wide array of PAMPs in addition to endogenous molecules, studies examining TLRs have begun to emerge in other noninfectious CNS disorders. For example, cerebral ischemia elicits acute inflammation that, in turn, exacerbates primary brain damage (Danton and Dietrich, 2003). Activation of the innate immune system (i.e., neutrophils, macrophages, and microglia) is an important component of the inflammatory response observed following an ischemic episode. A phenomenon referred to as LPS preconditioning, in which a subthreshold dose of systemic LPS affords protection against a subsequent ischemic attack, has been demonstrated in several models of CNS ischemia (Puisieux et al., 2000; Bordet et al., 2000; Bastide et al., 2003; Kariko et al., 2004b; Rosenzweig et al., 2004). The molecular mechanisms responsible for LPS preconditioning of stroke are poorly understood, and, although to date no studies have directly examined the role of TLR4 in LPS-dependent ischemic preconditioning, it is likely that this PRR plays an important role in reprogramming the host immune response to a subsequent ischemic attack. The relative role of TLR4 as well as endogenous ligands that may be released from damaged CNS tissue following an ischemic episode in modulating the LPS preconditioning response to stroke await further study. Similarly to recent studies in MS patients, there is no evidence to suggest that natural TLR4 mutations predispose individuals to cerebral ischemia (Reismann et al., 2004).
Involvement of TLRs in Response to Injury
Evidence is emerging that TLRs not only participate in CNS innate immunity in response to pathogens but also regulate the subsequent host reactions to injury (Owens et al., 2005; Tanga et al., 2005). This is an intriguing concept, in that traditional TLR agonists (i.e., PAMPs of infectious agents) are normally not present in an injury paradigm. By using a mouse model of painful neuropathy elicited by an L5 nerve transaction, Tanga et al. (2005) demonstrated a critical role for TLR4 in the induction phase of behavioral hypersensitivity. Specifically, the evaluation of two independent TLR4 mutant mouse strains revealed that mechanical allodynia and thermal hypersensitivity that are normally observed in L5 transected mice were significantly attenuated in TLR4 mutant animals. This dampening of behavioral hypersensitivity was associated with the decreased expression of markers of microglial activation and proinflammatory mediator release within the lumbar spinal cord, which are normally increased in transected animals (Tanga et al., 2004, 2005).
Another group has also begun to examine CNS innate immune responses in the absence of pathogenic insults. Owens et al. (2005) have investigated the relative importance of TLR signaling pathways in glial responses to axonal degeneration in the dentate gyrus of the hippocampus using MyD88 and TLR2 KO mice. In this model, axonal terminals located in the entorhinal cortex are transected, leading to a reproducible pattern of axonal degeneration that is associated with an early glial response and subsequent peripheral immune cell infiltrates (Jensen et al., 1997, 1999; Finsen et al., 1999). Relevant to TLR signaling, axotomy in MyD88 KO mice resulted in reduced numbers of both macrophages and lymphocytes infiltrating the hippocampus (Owens et al., 2005). In addition, microglial numbers and T cell infiltrates that are normally increased at specific times post-lesion were reduced in hippocampi of TLR2 KO mice, suggesting an important role for TLR2 in dictating the resulting inflammatory response that ensues following injury (Owens et al., 2005).
One intriguing observation was made in both of these noninfectious injury paradigms; namely, that a complete inhibition of behavioral hypersensitivity or axonal inflammation was not observed in TLR4 or MyD88 mutant mice, respectively, indicating that additional receptors/ factors are involved in eliciting maximal neuroinflammatory responses (Owens et al., 2005; Tanga et al., 2005). These findings raise an important point of similarity between studies performed with infectious agents and those performed in noninfectious injury models; namely, the experimental evidence in both systems reveals that CNS innate immune responses, whether they are directed against infectious pathogens or endogenous molecules, are likely mediated by a combination of receptors that may include members of the TLR family.
There have been numerous reports in recent years suggesting that, in addition to PAMPs, several endogenous molecules are capable of activating the innate immune system in a TLR-dependent manner (Kariko et al., 2004b; Tsan and Gao, 2004). These molecules have been referred to as endogenous TLR agonists to distinguish them from PAMPs. The list of endogenous molecules reported to activate cells via TLR-dependent pathways includes fibrinogen (TLR4; Smiley et al., 2001), fibronectin (TLR4; Okamura et al., 2001), heparan sulfate (TLR4; Johnson et al., 2002, 2004), β-defensin (TLR4; Biragyn et al., 2002), hyaluronan (TLR4; Termeer et al., 2002), mRNA (Kariko et al., 2004a), HSPs (TLR2 and TLR4; Ohashi et al., 2000; Asea et al., 2002; Vabulas et al., 2002a,b), and saturated fatty acids (TLR2; J.Y. Lee et al., 2001, 2004; Weatherill et al., 2005). To date, it is unclear what exact role these suggested endogenous TLR agonists play in homeostasis and/or disease, although it has been suggested that they may contribute to the pathogenesis of various autoimmune diseases or represent a physiological response to tissue injury (Beg, 2002; Kariko et al., 2004b).
A word of caution must be mentioned with regard to the potential contamination of endogenous TLR agonists with exogenous molecules of microbial origin. This point was recently emphasized by several studies demonstrating that the cytokine-stimulatory effects of various HSP preparations could be attributed to contaminating endotoxin (Bausinger et al., 2002; Gao and Tsan, 2003a,b; Reed et al., 2003), suggesting that they may not serve as bona fide TLR agonists. In addition to LPS, non-LPS bacterial cell wall components, such as lipoproteins, may also contribute to the TLR-dependent effects of some reported endogenous agonists; however, this possibility remains to be examined. The contamination of many of these reagents may be explained by the production of recombinant products produced by genetically engineered E. coli, which can introduce trace amounts of LPS and/or alternative PAMPs that are difficult to detect but nonetheless are capable of initiating cytokine release by highly sensitive cells of the mononuclear phagocyte lineage. Studies investigating whether any of the reported endogenous TLR agonists described above are capable of directly modulating glial activation in a TLR-dependent manner have not yet been performed, but the available evidence suggests that stimulation of TLRs by an as yet unidentified endogenous molecule(s) is pivotal for regulating CNS inflammation in response to injury when classical microbial PAMPs are absent (Owens et al., 2005; Tanga et al., 2005).
CONCLUSIONS, PERSPECTIVES, AND CHALLENGES
Reports investigating the expression and roles of various TLRs in the CNS are increasing at a rapid rate; however, much work remains to be performed. Specifically, additional studies are needed to examine the functional importance of various TLRs in the context of both infectious and noninfectious inflammatory diseases of the CNS, which can be accomplished with the use of newly generated TLR KO mice. In addition, insofar as recent evidence suggests that certain TLRs function in a cooperative manner, studies investigating disease pathogenesis in TLR double KO mice will be particularly interesting and may reveal novel insights that might be masked by the deletion of each receptor individually. Detailed studies investigating cell types in the CNS expressing TLR proteins in homeostatic and disease states have been hampered by the lack of high-quality commercially available antibodies for immunofluorescence staining. However, new antibody reagents have recently been developed that allow for the accurate detection of TLR2 and TLR4 on mouse cells by flow cytometry, allowing the analysis of TLR expression on CNS cells immediately ex vivo.
As studies investigating the role of TLRs in the CNS continue, care should be taken to ensure that TLR agonists are of high quality and are not contaminated by other biologically active PAMPs. For example, non-LPS reagents should be screened for endotoxin contamination using Limulus amebocyte lysate assays, and examination of phosphate levels in samples can be used to evaluate contamination with lipoproteins and bacterial DNA (Gao et al., 2001). This is particularly important in that the purity of TLR agonists has been a recurring theme that has plagued the field of TLR biology over the years (Gao et al., 2001; Lee et al., 2002; Travassos et al., 2004; Tsan and Gao, 2004). Although much information remains to be revealed regarding the roles of TLRs in nonbacterial CNS infectious diseases, the evidence to date supports what has been noted in bacterial infections, namely, that multiple PRRs are involved in mounting an effective host innate immune response in the CNS.
ACKNOWLEDGMENT
The author thanks Dr. Joyce DeLeo for critical review of this manuscript.
Contract grant sponsor: NIH; Contract grant number: NS-40730; Contract grant number: MH-65297; Contract grant sponsor: Arkansas Biosciences Institute.
REFERENCES
- Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, Akira S. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9:143–150. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- Aggarwal S, Gurney AL. IL-17: prototype member of an emerging cytokine family. J Leukoc Biol. 2002;71:1–8. [PubMed] [Google Scholar]
- Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- Aliprantis AO, Yang RB, Mark MR, Suggett S, Devaux B, Radolf JD, Klimpel GR, Godowski P, Zychlinsky A. Cell activation and apoptosis by bacterial lipoproteins through Toll-like receptor-2. Science. 1999;285:736–739. doi: 10.1126/science.285.5428.736. [DOI] [PubMed] [Google Scholar]
- Aliprantis AO, Yang RB, Weiss DS, Godowski P, Zychlinsky A. The apoptotic signaling pathway activated by Toll-like receptor-2. EMBO J. 2000;19:3325–3336. doi: 10.1093/emboj/19.13.3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aloisi F. Immune function of microglia. Glia. 2001;36:165–179. doi: 10.1002/glia.1106. [DOI] [PubMed] [Google Scholar]
- Anderson KV. Toll signaling pathways in the innate immune response. Curr Opin Immunol. 2000;12:13–19. doi: 10.1016/s0952-7915(99)00045-x. [DOI] [PubMed] [Google Scholar]
- Anderson KV, Jurgens G, Nusslein-Volhard C. Establishment of dorsal-ventral polarity in the Drosophila embryo: genetic studies on the role of the Toll gene product. Cell. 1985;42:779–789. doi: 10.1016/0092-8674(85)90274-0. [DOI] [PubMed] [Google Scholar]
- Arai H, Furuya T, Yasuda T, Miura M, Mizuno Y, Mochizuki H. Neurotoxic effects of lipopolysaccharide on nigral dopaminergic neurons are mediated by microglial activation, interleukin-1β, and expression of caspase-11 in mice. J Biol Chem. 2004;279:51647–51653. doi: 10.1074/jbc.M407328200. [DOI] [PubMed] [Google Scholar]
- Arbour NC, Lorenz E, Schutte BC, Zabner J, Kline JN, Jones M, Frees K, Watt JL, Schwartz DA. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet. 2000;25:187–191. doi: 10.1038/76048. [DOI] [PubMed] [Google Scholar]
- Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, Stevenson MA, Calderwood SK. Novel signal transduction pathway utilized by extracellular HSP70: role of Toll-like receptor (TLR) 2 and TLR4. J Biol Chem. 2002;277:15028–15034. doi: 10.1074/jbc.M200497200. [DOI] [PubMed] [Google Scholar]
- Bastide M, Gele P, Petrault O, Pu Q, Caliez A, Robin E, Deplanque D, Duriez P, Bordet R. Delayed cerebrovascular protective effect of lipopolysaccharide in parallel to brain ischemic tolerance. J Cereb Blood Flow Metab. 2003;23:399–405. doi: 10.1097/01.WCB.0000050064.57184.F2. [DOI] [PubMed] [Google Scholar]
- Bausinger H, Lipsker D, Ziylan U, Manie S, Briand JP, Cazenave JP, Muller S, Haeuw JF, Ravanat C, de la Salle H, Hanau D. Endotoxin-free heat-shock protein 70 fails to induce APC activation. Eur J Immunol. 2002;32:3708–3713. doi: 10.1002/1521-4141(200212)32:12<3708::AID-IMMU3708>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Becher B, Fedorowicz V, Antel JP. Regulation of CD14 expression on human adult central nervous system-derived microglia. J Neurosci Res. 1996;45:375–381. doi: 10.1002/(SICI)1097-4547(19960815)45:4<375::AID-JNR6>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Beg AA. Endogenous ligands of Toll-like receptors: implications for regulating inflammatory and immune responses. Trends Immunol. 2002;23:509–512. doi: 10.1016/s1471-4906(02)02317-7. [DOI] [PubMed] [Google Scholar]
- Biragyn A, Ruffini PA, Leifer CA, Klyushnenkova E, Shakhov A, Chertov O, Shirakawa AK, Farber JM, Segal DM, Oppenheim JJ, Kwak LW. Toll-like receptor 4-dependent activation of dendritic cells by β-defensin 2. Science. 2002;298:1025–1029. doi: 10.1126/science.1075565. [DOI] [PubMed] [Google Scholar]
- Bordet R, Deplanque D, Maboudou P, Puisieux F, Pu Q, Robin E, Martin A, Bastide M, Leys D, Lhermitte M, Dupuis B. Increase in endogenous brain superoxide dismutase as a potential mechanism of lipopolysaccharide-induced brain ischemic tolerance. J Cereb Blood Flow Metab. 2000;20:1190–1196. doi: 10.1097/00004647-200008000-00004. [DOI] [PubMed] [Google Scholar]
- Bottcher T, von Mering M, Ebert S, Meyding-Lamade U, Kuhnt U, Gerber J, Nau R. Differential regulation of Toll-like receptor mRNAs in experimental murine central nervous system infections. Neurosci Lett. 2003;344:17–20. doi: 10.1016/s0304-3940(03)00404-x. [DOI] [PubMed] [Google Scholar]
- Bowman CC, Rasley A, Tranguch SL, Marriott I. Cultured astrocytes express Toll-like receptors for bacterial products. Glia. 2003;43:281–291. doi: 10.1002/glia.10256. [DOI] [PubMed] [Google Scholar]
- Bsibsi M, Ravid R, Gveric D, van Noort JM. Broad expression of Toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol. 2002;61:1013–1021. doi: 10.1093/jnen/61.11.1013. [DOI] [PubMed] [Google Scholar]
- Buljevac D, Flach HZ, Hop WC, Hijdra D, Laman JD, Savelkoul HF, van Der Meche FG, van Doorn PA, Hintzen RQ. Prospective study on the relationship between infections and multiple sclerosis exacerbations. Brain. 2002;125:952–960. doi: 10.1093/brain/awf098. [DOI] [PubMed] [Google Scholar]
- Burns K, Clatworthy J, Martin L, Martinon F, Plumpton C, Maschera B, Lewis A, Ray K, Tschopp J, Volpe F. Tollip, a new component of the IL-1RI pathway, links IRAK to the IL-1 receptor. Nat Cell Biol. 2000;2:346–351. doi: 10.1038/35014038. [DOI] [PubMed] [Google Scholar]
- Burns K, Janssens S, Brissoni B, Olivos N, Beyaert R, Tschopp J. Inhibition of interleukin 1 receptor/Toll-like receptor signaling through the alternatively spliced, short form of MyD88 is due to its failure to recruit IRAK-4. J Exp Med. 2003;197:263–268. doi: 10.1084/jem.20021790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpentier PA, Begolka WS, Olson JK, Elhofy A, Karpus WJ, Miller SD. Differential activation of astrocytes by innate and adaptive immune stimuli. Glia. 2005;49:360–374. doi: 10.1002/glia.20117. [DOI] [PubMed] [Google Scholar]
- Carson MJ, Reilly CR, Sutcliffe JG, Lo D. Mature microglia resemble immature antigen-presenting cells. Glia. 1998;22:72–85. doi: 10.1002/(sici)1098-1136(199801)22:1<72::aid-glia7>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Carson MJ, Thrash JC, Lo D. Analysis of microglial gene expression: identifying targets for CNS neurodegenerative and autoimmune disease. Am J Pharmacogenom. 2004;4:321–330. doi: 10.2165/00129785-200404050-00005. [DOI] [PubMed] [Google Scholar]
- Chakravarty S, Herkenham M. Toll-like receptor 4 on nonhematopoietic cells sustains CNS inflammation during endotoxemia, independent of systemic cytokines. J Neurosci. 2005;25:1788–1796. doi: 10.1523/JNEUROSCI.4268-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien HF, Yeh KY, Jiang-Shieh YF, Wei IH, Chang CY, Chang ML, Wu CH. Signal transduction pathways of nitric oxide release in primary microglial culture challenged with gram-positive bacterial constituent, lipoteichoic acid. Neuroscience. 2005;133:423–436. doi: 10.1016/j.neuroscience.2004.09.067. [DOI] [PubMed] [Google Scholar]
- Cleveland MG, Gorham JD, Murphy TL, Tuomanen E, Murphy KM. Lipoteichoic acid preparations of gram-positive bacteria induce interleukin-12 through a CD14-dependent pathway. Infect Immun. 1996;64:1906–1912. doi: 10.1128/iai.64.6.1906-1912.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalpke AH, Schafer MK, Frey M, Zimmermann S, Tebbe J, Weihe E, Heeg K. Immunostimulatory CpG-DNA activates murine microglia. J Immunol. 2002;168:4854–4863. doi: 10.4049/jimmunol.168.10.4854. [DOI] [PubMed] [Google Scholar]
- Danton GH, Dietrich WD. Inflammatory mechanisms after ischemia and stroke. J Neuropathol Exp Neurol. 2003;62:127–136. doi: 10.1093/jnen/62.2.127. [DOI] [PubMed] [Google Scholar]
- Deng GM, Liu ZQ, Tarkowski A. Intracisternally localized bacterial DNA containing CpG motifs induces meningitis. J Immunol. 2001;167:4616–4626. doi: 10.4049/jimmunol.167.8.4616. [DOI] [PubMed] [Google Scholar]
- Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- Dobrovolskaia MA, Vogel SN. Toll receptors, CD14, and macrophage activation and deactivation by LPS. Microbes Infect. 2002;4:903–914. doi: 10.1016/s1286-4579(02)01613-1. [DOI] [PubMed] [Google Scholar]
- Dong Y, Benveniste EN. Immune function of astrocytes. Glia. 2001;36:180–190. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- Drennan MB, Nicolle D, Quesniaux VJ, Jacobs M, Allie N, Mpagi J, Fremond C, Wagner H, Kirschning C, Ryffel B. Toll-like receptor 2-deficient mice succumb to Mycobacterium tuberculosis infection. Am J Pathol. 2004;164:49–57. doi: 10.1016/S0002-9440(10)63095-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziarski R. Recognition of bacterial peptidoglycan by the innate immune system. Cell Mol Life Sci. 2003;60:1793–1804. doi: 10.1007/s00018-003-3019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziarski R, Ulmer AJ, Gupta D. Interactions of CD14 with components of gram-positive bacteria. Chem Immunol. 2000;74:83–107. doi: 10.1159/000058761. [DOI] [PubMed] [Google Scholar]
- Ebert S, Gerber J, Bader S, Muhlhauser F, Brechtel K, Mitchell TJ, Nau R. Dose-dependent activation of microglial cells by Toll-like receptor agonists alone and in combination. J Neuroimmunol. 2005;159:87–96. doi: 10.1016/j.jneuroim.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Echchannaoui H, Frei K, Schnell C, Leib SL, Zimmerli W, Landmann R. Toll-like receptor 2-deficient mice are highly susceptible to Streptococcus pneumoniae meningitis because of reduced bacterial clearing and enhanced inflammation. J Infect Dis. 2002;186:798–806. doi: 10.1086/342845. [DOI] [PubMed] [Google Scholar]
- Eikelenboom P, Bate C, Van Gool WA, Hoozemans JJ, Rozemuller JM, Veerhuis R, Williams A. Neuroinflammation in Alzheimer’s disease and prion disease. Glia. 2002;40:232–239. doi: 10.1002/glia.10146. [DOI] [PubMed] [Google Scholar]
- Esen N, Tanga FY, DeLeo JA, Kielian T. Toll-like receptor 2 (TLR2) mediates astrocyte activation in response to the gram-positive bacterium Staphylococcus aureus. J Neurochem. 2004;88:746–758. doi: 10.1046/j.1471-4159.2003.02202.x. [DOI] [PubMed] [Google Scholar]
- Esen N, Kielian T. Recognition of staphylococcus aureus-derived peptidoglycan (PGN) but not intact bacteria is mediated by CD14 in microglia. J Neuroimmunol. 2005;170:93–104. doi: 10.1016/j.jneuroim.2005.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina C, Krumbholz M, Giese T, Hartmann G, Aloisi F, Meinl E. Preferential expression and function of Toll-like receptor 3 in human astrocytes. J Neuroimmunol. 2005;159:12–19. doi: 10.1016/j.jneuroim.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Finsen B, Jensen MB, Lomholt ND, Hegelund IV, Poulsen FR, Owens T. Axotomy-induced glial reactions in normal and cytokine transgenic mice. Adv Exp Med Biol. 1999;468:157–171. doi: 10.1007/978-1-4615-4685-6_13. [DOI] [PubMed] [Google Scholar]
- Fitzgerald KA, Rowe DC, Golenbock DT. Endotoxin recognition and signal transduction by the TLR4/MD2-complex. Microbes Infect. 2004;6:1361–1367. doi: 10.1016/j.micinf.2004.08.015. [DOI] [PubMed] [Google Scholar]
- Ford AL, Goodsall AL, Hickey WF, Sedgwick JD. Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. J Immunol. 1995;154:4309–4321. [PubMed] [Google Scholar]
- Gao B, Tsan MF. Endotoxin contamination in recombinant human heat shock protein 70 (Hsp70) preparation is responsible for the induction of tumor necrosis factor alpha release by murine macrophages. J Biol Chem. 2003a;278:174–179. doi: 10.1074/jbc.M208742200. [DOI] [PubMed] [Google Scholar]
- Gao B, Tsan MF. Recombinant human heat shock protein 60 does not induce the release of tumor necrosis factor alpha from murine macrophages. J Biol Chem. 2003b;278:22523–22529. doi: 10.1074/jbc.M303161200. [DOI] [PubMed] [Google Scholar]
- Gao JJ, Xue Q, Zuvanich EG, Haghi KR, Morrison DC. Commercial preparations of lipoteichoic acid contain endotoxin that contributes to activation of mouse macrophages in vitro. Infect Immun. 2001;69:751–757. doi: 10.1128/IAI.69.2.751-757.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goverman J, Woods A, Larson L, Weiner LP, Hood L, Zaller DM. Transgenic mice that express a myelin basic protein-specific T cell receptor develop spontaneous autoimmunity. Cell. 1993;72:551–560. doi: 10.1016/0092-8674(93)90074-z. [DOI] [PubMed] [Google Scholar]
- Griffin WS, Mrak RE. Interleukin-1 in the genesis and progression of and risk for development of neuronal degeneration in Alzheimer’s disease. J Leukoc Biol. 2002;72:233–238. [PMC free article] [PubMed] [Google Scholar]
- Gupta D, Kirkland TN, Viriyakosol S, Dziarski R. CD14 is a cell-activating receptor for bacterial peptidoglycan. J Biol Chem. 1996;271:23310–23316. doi: 10.1074/jbc.271.38.23310. [DOI] [PubMed] [Google Scholar]
- Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40:140–155. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- Hasan U, Chaffois C, Gaillard C, Saulnier V, Merck E, Tancredi S, Guiet C, Briere F, Vlach J, Lebecque S, Trinchieri G, Bates EE. Human TLR10 is a functional receptor, expressed by B cells and plasmacytoid dendritic cells, which activates gene transcription through MyD88. J Immunol. 2005;174:2942–2950. doi: 10.4049/jimmunol.174.5.2942. [DOI] [PubMed] [Google Scholar]
- Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, Eng JK, Akira S, Underhill DM, Aderem A. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature. 2001;410:1099–1103. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- Haziot A, Chen S, Ferrero E, Low MG, Silber R, Goyert SM. The monocyte differentiation antigen, CD14, is anchored to the cell membrane by a phosphatidylinositol linkage. J Immunol. 1988;141:547–552. [PubMed] [Google Scholar]
- Haziot A, Hijiya N, Schultz K, Zhang F, Gangloff SC, Goyert SM. CD14 plays no major role in shock induced by Staphylococcus aureus but down-regulates TNF-alpha production. J Immunol. 1999;162:4801–4805. [PubMed] [Google Scholar]
- Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S. Species-specific recognition of single-stranded RNA via Toll-like receptor 7 and 8. Science. 2004;303:1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- Heine H, Kirschning CJ, Lien E, Monks BG, Rothe M, Golenbock DT. Cutting edge: cells that carry A null allele for Toll-like receptor 2 are capable of responding to endotoxin. J Immunol. 1999;162:6971–6975. [PubMed] [Google Scholar]
- Hemmi H, Kaisho T, Takeuchi O, Sato S, Sanjo H, Hoshino K, Horiuchi T, Tomizawa H, Takeda K, Akira S. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol. 2002;3:196–200. doi: 10.1038/ni758. [DOI] [PubMed] [Google Scholar]
- Henneke P, Takeuchi O, van Strijp JA, Guttormsen HK, Smith JA, Schromm AB, Espevik TA, Akira S, Nizet V, Kasper DL, Golenbock DT. Novel engagement of CD14 and multiple Toll-like receptors by group B streptococci. J Immunol. 2001;167:7069–7076. doi: 10.4049/jimmunol.167.12.7069. [DOI] [PubMed] [Google Scholar]
- Henneke P, Takeuchi O, Malley R, Lien E, Ingalls RR, Freeman MW, Mayadas T, Nizet V, Akira S, Kasper DL, Golenbock DT. Cellular activation, phagocytosis, and bactericidal activity against group B streptococcus involve parallel myeloid differentiation factor 88-dependent and independent signaling pathways. J Immunol. 2002;169:3970–3977. doi: 10.4049/jimmunol.169.7.3970. [DOI] [PubMed] [Google Scholar]
- Hewett JA, Hewett SJ, Winkler S, Pfeiffer SE. Inducible nitric oxide synthase expression in cultures enriched for mature oligodendrocytes is due to microglia. J Neurosci Res. 1999;56:189–198. doi: 10.1002/(sici)1097-4547(19990415)56:2<189::aid-jnr8>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Hickey WF. Leukocyte traffic in the central nervous system: the participants and their roles. Semin Immunol. 1999;11:125–137. doi: 10.1006/smim.1999.0168. [DOI] [PubMed] [Google Scholar]
- Hirschfeld M, Ma Y, Weis JH, Vogel SN, Weis JJ. Cutting edge: repurification of lipopolysaccharide eliminates signaling through both human and murine Toll-like receptor 2. J Immunol. 2000;165:618–622. doi: 10.4049/jimmunol.165.2.618. [DOI] [PubMed] [Google Scholar]
- Hitziger N, Dellacasa I, Albiger B, Barragan A. Dissemination of Toxoplasma gondii to immunoprivileged organs and role of Toll/interleukin-1 receptor signalling for host resistance assessed by in vivo bioluminescence imaging. Cell Microbiol. 2005;7:837–848. doi: 10.1111/j.1462-5822.2005.00517.x. [DOI] [PubMed] [Google Scholar]
- Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the LPS gene product. J Immunol. 1999;162:3749–3752. [PubMed] [Google Scholar]
- Hosoi T, Suzuki S, Nomura J, Ono A, Okuma Y, Akira S, Nomura Y. Bacterial DNA induced iNOS expression through MyD88-p38 MAP kinase in mouse primary cultured glial cells. Brain Res Mol Brain Res. 2004;124:159–164. doi: 10.1016/j.molbrainres.2004.02.014. [DOI] [PubMed] [Google Scholar]
- Ichikawa HT, Williams LP, Segal BM. Activation of APCs through CD40 or Toll-like receptor 9 overcomes tolerance and precipitates autoimmune disease. J Immunol. 2002;169:2781–2787. doi: 10.4049/jimmunol.169.5.2781. [DOI] [PubMed] [Google Scholar]
- Iliev AI, Stringaris AK, Nau R, Neumann H. Neuronal injury mediated via stimulation of microglial Toll-like receptor-9 (TLR9) FASEB J. 2004;18:412–414. doi: 10.1096/fj.03-0670fje. [DOI] [PubMed] [Google Scholar]
- Jensen MB, Finsen B, Zimmer J. Morphological and immunophenotypic microglial changes in the denervated fascia dentata of adult rats: correlation with blood-brain barrier damage and astroglial reactions. Exp Neurol. 1997;143:103–116. doi: 10.1006/exnr.1996.6337. [DOI] [PubMed] [Google Scholar]
- Jensen MB, Hegelund IV, Poulsen FR, Owens T, Zimmer J, Finsen B. Microglial reactivity correlates to the density and the myelination of the anterogradely degenerating axons and terminals following perforant path denervation of the mouse fascia dentata. Neuroscience. 1999;93:507–518. doi: 10.1016/s0306-4522(99)00139-6. [DOI] [PubMed] [Google Scholar]
- Johnson GB, Brunn GJ, Kodaira Y, Platt JL. Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by Toll-like receptor 4. J Immunol. 2002;168:5233–5239. doi: 10.4049/jimmunol.168.10.5233. [DOI] [PubMed] [Google Scholar]
- Johnson GB, Brunn GJ, Platt JL. Cutting edge: an endogenous pathway to systemic inflammatory response syndrome (SIRS)-like reactions through Toll-like receptor 4. J Immunol. 2004;172:20–24. doi: 10.4049/jimmunol.172.1.20. [DOI] [PubMed] [Google Scholar]
- Jung DY, Lee H, Jung BY, Ock J, Lee MS, Lee WH, Suk K. TLR4, but not TLR2, signals autoregulatory apoptosis of cultured microglia: a critical role of IFN-β as a decision maker. J Immunol. 2005;174:6467–6476. doi: 10.4049/jimmunol.174.10.6467. [DOI] [PubMed] [Google Scholar]
- Kaisho T, Akira S. Pleiotropic function of Toll-like receptors. Microbes Infect. 2004;6:1388–1394. doi: 10.1016/j.micinf.2004.08.019. [DOI] [PubMed] [Google Scholar]
- Kakimura J, Kitamura Y, Takata K, Umeki M, Suzuki S, Shibagaki K, Taniguchi T, Nomura Y, Gebicke-Haerter PJ, Smith MA, Perry G, Shimohama S. Microglial activation and amyloid-β clearance induced by exogenous heat-shock proteins. FASEB J. 2002;16:601–603. doi: 10.1096/fj.01-0530fje. [DOI] [PubMed] [Google Scholar]
- Kariko K, Ni H, Capodici J, Lamphier M, Weissman D. mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem. 2004a;279:12542–12550. doi: 10.1074/jbc.M310175200. [DOI] [PubMed] [Google Scholar]
- Kariko K, Weissman D, Welsh FA. Inhibition of Toll-like receptor and cytokine signaling—a unifying theme in ischemic tolerance. J Cereb Blood Flow Metab. 2004b;24:1288–1304. doi: 10.1097/01.WCB.0000145666.68576.71. [DOI] [PubMed] [Google Scholar]
- Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11:115–122. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- Kerfoot SM, Long EM, Hickey MJ, Andonegui G, Lapointe BM, Zanardo RC, Bonder C, James WG, Robbins SM, Kubes P. TLR4 contributes to disease-inducing mechanisms resulting in central nervous system autoimmune disease. J Immunol. 2004;173:7070–7077. doi: 10.4049/jimmunol.173.11.7070. [DOI] [PubMed] [Google Scholar]
- Kielian T. Immunopathogenesis of brain abscess. J Neuroinflamm. 2004a;1:16. doi: 10.1186/1742-2094-1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T. Microglia and chemokines in infectious diseases of the nervous system: views and reviews. Front Biosci. 2004b;9:732–750. doi: 10.2741/1266. [DOI] [PubMed] [Google Scholar]
- Kielian T, Mayes P, Kielian M. Characterization of microglial responses to Staphylococcus aureus: effects on cytokine, costimulatory molecule, and Toll-like receptor expression. J Neuroimmunol. 2002;130:86–99. doi: 10.1016/s0165-5728(02)00216-3. [DOI] [PubMed] [Google Scholar]
- Kielian T, Esen N, Bearden ED. Toll-like receptor 2 (TLR2) is pivotal for recognition of S. aureus peptidoglycan but not intact bacteria by microglia. Glia. 2005a;49:567–576. doi: 10.1002/glia.20144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T, Haney A, Mayes PM, Garg S, Esen N. Toll-like receptor 2 modulates the proinflammatory milieu in Staphylococcus aureus-induced brain abscess. Infect Immun. 2005b;73:7428–7435. doi: 10.1128/IAI.73.11.7428-7435.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura Y, Kakimura J, Koike H, Umeki M, Gebicke-Haerter PJ, Nomura Y, Taniguchi T. Effects of 15-deoxy-delta(12,14) prostaglandin J and interleukin-4 in Toll-like receptor-4-mutant glial cells. Eur J Pharmacol. 2001;411:223–230. doi: 10.1016/s0014-2999(00)00910-9. [DOI] [PubMed] [Google Scholar]
- Knapp S, Wieland CW, van ’t Veer C, Takeuchi O, Akira S, Florquin S, van der Poll T. Toll-like receptor 2 plays a role in the early inflammatory response to murine pneumococcal pneumonia but does not contribute to antibacterial defense. J Immunol. 2004;172:3132–3138. doi: 10.4049/jimmunol.172.5.3132. [DOI] [PubMed] [Google Scholar]
- Koedel U, Scheld WM, Pfister HW. Pathogenesis and pathophysiology of pneumococcal meningitis. Lancet Infect Dis. 2002;2:721–736. doi: 10.1016/s1473-3099(02)00450-4. [DOI] [PubMed] [Google Scholar]
- Koedel U, Angele B, Rupprecht T, Wagner H, Roggenkamp A, Pfister HW, Kirschning CJ. Toll-like receptor 2 participates in mediation of immune response in experimental pneumococcal meningitis. J Immunol. 2003;170:438–444. doi: 10.4049/jimmunol.170.1.438. [DOI] [PubMed] [Google Scholar]
- Koedel U, Rupprecht T, Angele B, Heesemann J, Wagner H, Pfister HW, Kirschning CJ. MyD88 is required for mounting a robust host immune response to Streptococcus pneumoniae in the CNS. Brain. 2004;127:1437–1445. doi: 10.1093/brain/awh171. [DOI] [PubMed] [Google Scholar]
- Kopp E, Medzhitov R. Recognition of microbial infection by Toll-like receptors. Curr Opin Immunol. 2003;15:396–401. doi: 10.1016/s0952-7915(03)00080-3. [DOI] [PubMed] [Google Scholar]
- Kroner A, Vogel F, Kolb-Maurer A, Kruse N, Toyka KV, Hemmer B, Rieckmann P, Maurer M. Impact of the Asp299Gly polymorphism in the Toll-like receptor 4 (tlr-4) gene on disease course of multiple sclerosis. J Neuroimmunol. 2005;165:161–165. doi: 10.1016/j.jneuroim.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Kurt-Jones EA, Chan M, Zhou S, Wang J, Reed G, Bronson R, Arnold MM, Knipe DM, Finberg RW. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc Natl Acad Sci U S A. 2004;101:1315–1320. doi: 10.1073/pnas.0308057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laflamme N, Rivest S. Toll-like receptor 4: the missing link of the cerebral innate immune response triggered by circulating gram-negative bacterial cell wall components. FASEB J. 2001;15:155–163. doi: 10.1096/fj.00-0339com. [DOI] [PubMed] [Google Scholar]
- Laflamme N, Soucy G, Rivest S. Circulating cell wall components derived from gram-negative, not gram-positive, bacteria cause a profound induction of the gene-encoding Toll-like receptor 2 in the CNS. J Neurochem. 2001;79:648–657. doi: 10.1046/j.1471-4159.2001.00603.x. [DOI] [PubMed] [Google Scholar]
- Laflamme N, Echchannaoui H, Landmann R, Rivest S. Cooperation between Toll-like receptor 2 and 4 in the brain of mice challenged with cell wall components derived from gram-negative and gram-positive bacteria. Eur J Immunol. 2003;33:1127–1138. doi: 10.1002/eji.200323821. [DOI] [PubMed] [Google Scholar]
- Lau CM, Broughton C, Tabor AS, Akira S, Flavell RA, Mamula MJ, Christensen SR, Shlomchik MJ, Viglianti GA, Rifkin IR, Marshak-Rothstein A. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J Exp Med. 2005;202:1171–1177. doi: 10.1084/jem.20050630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauw FN, Caffrey DR, Golenbock DT. Of mice and man: TLR11 (finally) finds profilin. Trends Immunol. 2005;26:509–511. doi: 10.1016/j.it.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Lee HK, Lee J, Tobias PS. Two lipoproteins extracted from Escherichia coli K-12 LCD25 lipopolysaccharide are the major components responsible for Toll-like receptor 2-mediated signaling. J Immunol. 2002;168:4012–4017. doi: 10.4049/jimmunol.168.8.4012. [DOI] [PubMed] [Google Scholar]
- Lee JY, Sohn KH, Rhee SH, Hwang D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem. 2001;276:16683–16689. doi: 10.1074/jbc.M011695200. [DOI] [PubMed] [Google Scholar]
- Lee JY, Zhao L, Youn HS, Weatherill AR, Tapping R, Feng L, Lee WH, Fitzgerald KA, Hwang DH. Saturated fatty acid activates but polyunsaturated fatty acid inhibits Toll-like receptor 2 dimerized with Toll-like receptor 6 or 1. J Biol Chem. 2004;279:16971–16979. doi: 10.1074/jbc.M312990200. [DOI] [PubMed] [Google Scholar]
- Lee S, Hong J, Choi SY, Oh SB, Park K, Kim JS, Karin M, Lee SJ. CpG oligodeoxynucleotides induce expression of proinflammatory cytokines and chemokines in astrocytes: the role of c-Jun N-terminal kinase in CpG ODN-mediated NF-kappaB activation. J Neuroimmunol. 2004;153:50–63. doi: 10.1016/j.jneuroim.2004.04.013. [DOI] [PubMed] [Google Scholar]
- Lehnardt S, Lachance C, Patrizi S, Lefebvre S, Follett PL, Jensen FE, Rosenberg PA, Volpe JJ, Vartanian T. The Toll-like receptor TLR4 is necessary for lipopolysaccharide-induced oligodendrocyte injury in the CNS. J Neurosci. 2002;22:2478–2486. doi: 10.1523/JNEUROSCI.22-07-02478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnardt S, Massillon L, Follett P, Jensen FE, Ratan R, Rosenberg PA, Volpe JJ, Vartanian T. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc Natl Acad Sci U S A. 2003;100:8514–8519. doi: 10.1073/pnas.1432609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell. 1996;86:973–983. doi: 10.1016/s0092-8674(00)80172-5. [DOI] [PubMed] [Google Scholar]
- Lien E, Means TK, Heine H, Yoshimura A, Kusumoto S, Fukase K, Fenton MJ, Oikawa M, Qureshi N, Monks B, Finberg RW, Ingalls RR, Golenbock DT. Toll-like receptor 4 imparts ligand-specific recognition of bacterial lipopolysaccharide. J Clin Invest. 2000;105:497–504. doi: 10.1172/JCI8541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz E, Frees KL, Schwartz DA. Determination of the TLR4 genotype using allele-specific PCR. Biotechniques. 2001;31:22–24. doi: 10.2144/01311bm01. [DOI] [PubMed] [Google Scholar]
- Lotz M, Ebert S, Esselmann H, Iliev AI, Prinz M, Wiazewicz N, Wiltfang J, Gerber J, Nau R. Amyloid β peptide 1–40 enhances the action of Toll-like receptor-2 and -4 agonists but antagonizes Toll-like eceptor-9-induced inflammation in primary mouse microglial cell cultures. J Neurochem. 2005;94:289–298. doi: 10.1111/j.1471-4159.2005.03188.x. [DOI] [PubMed] [Google Scholar]
- Lynn WA, Liu Y, Golenbock DT. Neither CD14 nor serum is absolutely necessary for activation of mononuclear phagocytes by bacterial lipopolysaccharide. Infect Immun. 1993;61:4452–4461. doi: 10.1128/iai.61.10.4452-4461.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manukyan M, Triantafilou K, Triantafilou M, Mackie A, Nilsen N, Espevik T, Wiesmuller KH, Ulmer AJ, Heine H. Binding of lipopeptide to CD14 induces physical proximity of CD14, TLR2 and TLR1. Eur J Immunol. 2005;35:911–921. doi: 10.1002/eji.200425336. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. Local neuroinflammation and the progression of Alzheimer’s disease. J Neurovirol. 2002;8:529–538. doi: 10.1080/13550280290100969. [DOI] [PubMed] [Google Scholar]
- Medvedev AE, Flo T, Ingalls RR, Golenbock DT, Teti G, Vogel SN, Espevik T. Involvement of CD14 and complement receptors CR3 and CR4 in nuclear factor-kappaB activation and TNF production induced by lipopolysaccharide and group B streptococcal cell walls. J Immunol. 1998;160:4535–4542. [PubMed] [Google Scholar]
- Medzhitov R, Janeway C., Jr Innate immunity. N Engl J Med. 2000;343:338–344. doi: 10.1056/NEJM200008033430506. [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, Janeway CA., Jr MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol Cell. 1998;2:253–258. doi: 10.1016/s1097-2765(00)80136-7. [DOI] [PubMed] [Google Scholar]
- Melton LM, Keith AB, Davis S, Oakley AE, Edwardson JA, Morris CM. Chronic glial activation, neurodegeneration, and APP immunoreactive deposits following acute administration of double-stranded RNA. Glia. 2003;44:1–12. doi: 10.1002/glia.10276. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S, Herre J, Brown GD, Gordon S. The potential for Toll-like receptors to collaborate with other innate immune receptors. Immunology. 2004;112:521–530. doi: 10.1111/j.1365-2567.2004.01941.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mun HS, Aosai F, Norose K, Chen M, Piao LX, Takeuchi O, Akira S, Ishikura H, Yano A. TLR2 as an essential molecule for protective immunity against Toxoplasma gondii infection. Int Immunol. 2003;15:1081–1087. doi: 10.1093/intimm/dxg108. [DOI] [PubMed] [Google Scholar]
- Nadeau S, Rivest S. Role of microglial-derived tumor necrosis factor in mediating CD14 transcription and nuclear factor kappa B activity in the brain during endotoxemia. J Neurosci. 2000a;20:3456–3468. doi: 10.1523/JNEUROSCI.20-09-03456.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau S, Rivest S. Role of microglial-derived tumor necrosis factor in mediating CD14 transcription and nuclear factor kappa B activity in the brain during endotoxemia. J Neurosci. 2000b;20:3456–3468. doi: 10.1523/JNEUROSCI.20-09-03456.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nau R, Bruck W. Neuronal injury in bacterial meningitis: mechanisms and implications for therapy. Trends Neurosci. 2002;25:38–45. doi: 10.1016/s0166-2236(00)02024-5. [DOI] [PubMed] [Google Scholar]
- Nguyen MD, Julien JP, Rivest S. Innate immunity: the missing link in neuroprotection and neurodegeneration? Nat Rev Neurosci. 2002;3:216–227. doi: 10.1038/nrn752. [DOI] [PubMed] [Google Scholar]
- Nicolle D, Fremond C, Pichon X, Bouchot A, Maillet I, Ryffel B, Quesniaux VJ. Long-term control of Mycobacterium bovis BCG infection in the absence of Toll-like receptors (TLRs): investigation of TLR2-, TLR6-, or TLR2-TLR4-deficient mice. Infect Immun. 2004;72:6994–7004. doi: 10.1128/IAI.72.12.6994-7004.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi K, Burkart V, Flohe S, Kolb H. Cutting edge: heat shock protein 60 is a putative endogenous ligand of the Toll-like receptor-4 complex. J Immunol. 2000;164:558–561. doi: 10.4049/jimmunol.164.2.558. [DOI] [PubMed] [Google Scholar]
- Okamura Y, Watari M, Jerud ES, Young DW, Ishizaka ST, Rose J, Chow JC, Strauss JF., 3rd The extra domain A of fibronectin activates Toll-like receptor 4. J Biol Chem. 2001;276:10229–10233. doi: 10.1074/jbc.M100099200. [DOI] [PubMed] [Google Scholar]
- Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173:3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- Omueti KO, Beyer JM, Johnson CM, Lyle EA, Tapping RI. Domain exchange between human Toll-like receptors 1 and 6 reveals a region required for lipopeptide discrimination. J Biol Chem. 2005;280:36616–36625. doi: 10.1074/jbc.M504320200. [DOI] [PubMed] [Google Scholar]
- O’Neill LA. TLRs: Professor Mechnikov, sit on your hat. Trends Immunol. 2004;25:687–693. doi: 10.1016/j.it.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Owens T. Toll-like receptors on astrocytes: patterning for immunity. J Neuroimmunol. 2005;159:1–2. doi: 10.1016/j.jneuroim.2004.10.015. [DOI] [PubMed] [Google Scholar]
- Owens T, Babcock AA, Millward JM, Toft-Hansen H. Cytokine and chemokine inter-regulation in the inflamed or injured CNS. Brain Res Brain Res Rev. 2005;48:178–184. doi: 10.1016/j.brainresrev.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Ozinsky A, Underhill DM, Fontenot JD, Hajjar AM, Smith KD, Wilson CB, Schroeder L, Aderem A. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between Toll-like receptors. Proc Natl Acad Sci U S A. 2000;97:13766–13771. doi: 10.1073/pnas.250476497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palsson-McDermott EM, O’Neill LA. Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology. 2004;113:153–162. doi: 10.1111/j.1365-2567.2004.01976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang Y, Cai Z, Rhodes PG. Effects of lipopolysaccharide on oligodendrocyte progenitor cells are mediated by astrocytes and microglia. J Neurosci Res. 2000;62:510–520. doi: 10.1002/1097-4547(20001115)62:4<510::AID-JNR5>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Peng G, Guo Z, Kiniwa Y, Voo KS, Peng W, Fu T, Wang DY, Li Y, Wang HY, Wang RF. Toll-like receptor 8-mediated reversal of CD4+ regulatory T cell function. Science. 2005;309:1380–1384. doi: 10.1126/science.1113401. [DOI] [PubMed] [Google Scholar]
- Perera PY, Vogel SN, Detore GR, Haziot A, Goyert SM. CD14-dependent and CD14-independent signaling pathways in murine macrophages from normal and CD14 knockout mice stimulated with lipopolysaccharide or taxol. J Immunol. 1997;158:4422–4429. [PubMed] [Google Scholar]
- Perry VH, Newman TA, Cunningham C. The impact of systemic infection on the progression of neurodegenerative disease. Nat Rev Neurosci. 2003;4:103–112. doi: 10.1038/nrn1032. [DOI] [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998a;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- Poltorak A, Smirnova I, He X, Liu MY, Van Huffel C, McNally O, Birdwell D, Alejos E, Silva M, Du X, Thompson P, Chan EK, Ledesma J, Roe B, Clifton S, Vogel SN, Beutler B. Genetic and physical mapping of the Lps locus: identification of the Toll-4 receptor as a candidate gene in the critical region. Blood Cells Mol Dis. 1998b;24:340–355. doi: 10.1006/bcmd.1998.0201. [DOI] [PubMed] [Google Scholar]
- Prehaud C, Megret F, Lafage M, Lafon M. Virus infection switches TLR-3-positive human neurons to become strong producers of β interferon. J Virol. 2005;79:12893–12904. doi: 10.1128/JVI.79.20.12893-12904.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puisieux F, Deplanque D, Pu Q, Souil E, Bastide M, Bordet R. Differential role of nitric oxide pathway and heat shock protein in preconditioning and lipopolysaccharide-induced brain ischemic tolerance. Eur J Pharmacol. 2000;389:71–78. doi: 10.1016/s0014-2999(99)00893-6. [DOI] [PubMed] [Google Scholar]
- Qin L, Liu Y, Wang T, Wei SJ, Block ML, Wilson B, Liu B, Hong JS. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem. 2004;279:1415–1421. doi: 10.1074/jbc.M307657200. [DOI] [PubMed] [Google Scholar]
- Qin L, Li G, Qian X, Liu Y, Wu X, Liu B, Hong JS, Block ML. Interactive role of the Toll-like receptor 4 and reactive oxygen species in LPS-induced microglia activation. Glia. 2005;52:78–84. doi: 10.1002/glia.20225. [DOI] [PubMed] [Google Scholar]
- Qureshi ST, Lariviere L, Leveque G, Clermont S, Moore KJ, Gros P, Malo D. Endotoxin-tolerant mice have mutations in Toll-like receptor 4 (Tlr4) J Exp Med. 1999;189:615–625. doi: 10.1084/jem.189.4.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racke MK, Hu W, Lovett-Racke AE. PTX cruiser: driving autoimmunity via TLR4. Trends Immunol. 2005;26:289–291. doi: 10.1016/j.it.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Rapp NS, Gilroy J, Lerner AM. Role of bacterial infection in exacerbation of multiple sclerosis. Am J Phys Med Rehabil. 1995;74:415–418. doi: 10.1097/00002060-199511000-00004. [DOI] [PubMed] [Google Scholar]
- Rasley A, Anguita J, Marriott I. Borrelia burgdorferi induces inflammatory mediator production by murine microglia. J Neuroimmunol. 2002;130:22–31. doi: 10.1016/s0165-5728(02)00187-x. [DOI] [PubMed] [Google Scholar]
- Reed RC, Berwin B, Baker JP, Nicchitta CV. GRP94/gp96 elicits ERK activation in murine macrophages. A role for endotoxin contamination in NF-kappa B activation and nitric oxide production. J Biol Chem. 2003;278:31853–31860. doi: 10.1074/jbc.M305480200. [DOI] [PubMed] [Google Scholar]
- Reiling N, Holscher C, Fehrenbach A, Kroger S, Kirschning CJ, Goyert S, Ehlers S. Cutting edge: Toll-like receptor (TLR)2- and TLR4-mediated pathogen recognition in resistance to airborne infection with Mycobacterium tuberculosis. J Immunol. 2002;169:3480–3484. doi: 10.4049/jimmunol.169.7.3480. [DOI] [PubMed] [Google Scholar]
- Reindl M, Lutterotti A, Ingram J, Schanda K, Gassner C, Deisenhammer F, Berger T, Lorenz E. Mutations in the gene for Toll-like receptor 4 and multiple sclerosis. Tissue Antigens. 2003;61:85–88. doi: 10.1034/j.1399-0039.2003.610108.x. [DOI] [PubMed] [Google Scholar]
- Reismann P, Lichy C, Rudofsky G, Humpert PM, Genius J, Si TD, Dorfer C, Grau AJ, Hamann A, Hacke W, Nawroth PP, Bierhaus A. Lack of association between polymorphisms of the Toll-like receptor 4 gene and cerebral ischemia. J Neurol. 2004;251:853–858. doi: 10.1007/s00415-004-0447-7. [DOI] [PubMed] [Google Scholar]
- Rivest S. Molecular insights on the cerebral innate immune system. Brain Behav Immun. 2003;17:13–19. doi: 10.1016/s0889-1591(02)00055-7. [DOI] [PubMed] [Google Scholar]
- Rosenzweig HL, Lessov NS, Henshall DC, Minami M, Simon RP, Stenzel-Poore MP. Endotoxin preconditioning prevents cellular inflammatory response during ischemic neuroprotection in mice. Stroke. 2004;35:2576–2581. doi: 10.1161/01.STR.0000143450.04438.ae. [DOI] [PubMed] [Google Scholar]
- Saito S, Matsuura M, Tominaga K, Kirikae T, Nakano M. Important role of membrane-associated CD14 in the induction of IFN-β and subsequent nitric oxide production by murine macrophages in response to bacterial lipopolysaccharide. Eur J Biochem. 2000;267:37–45. doi: 10.1046/j.1432-1327.2000.00956.x. [DOI] [PubMed] [Google Scholar]
- Scanga CA, Aliberti J, Jankovic D, Tilloy F, Bennouna S, Denkers EY, Medzhitov R, Sher A. Cutting edge: MyD88 is required for resistance to Toxoplasma gondii infection and regulates parasite-induced IL-12 production by dendritic cells. J Immunol. 2002;168:5997–6001. doi: 10.4049/jimmunol.168.12.5997. [DOI] [PubMed] [Google Scholar]
- Schluesener HJ, Seid K, Deininger M, Schwab J. Transient in vivo activation of rat brain macrophages/microglial cells and astrocytes by immunostimulatory multiple CpG oligonucleotides. J Neuroimmunol. 2001;113:89–94. doi: 10.1016/s0165-5728(00)00428-8. [DOI] [PubMed] [Google Scholar]
- Schmid CD, Sautkulis LN, Danielson PE, Cooper J, Hasel KW, Hilbush BS, Sutcliffe JG, Carson MJ. Heterogeneous expression of the triggering receptor expressed on myeloid cells-2 on adult murine microglia. J Neurochem. 2002;83:1309–1320. doi: 10.1046/j.1471-4159.2002.01243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder NW, Morath S, Alexander C, Hamann L, Hartung T, Zahringer U, Gobel UB, Weber JR, Schumann RR. Lipoteichoic acid (LTA) of Streptococcus pneumoniae and Staphylococcus aureus activates immune cells via Toll-like receptor (TLR)-2, lipopolysaccharide-binding protein (LBP), and CD14, whereas TLR-4 and MD-2 are not involved. J Biol Chem. 2003;278:15587–15594. doi: 10.1074/jbc.M212829200. [DOI] [PubMed] [Google Scholar]
- Scumpia PO, Kelly KM, Reeves WH, Stevens BR. Double-stranded RNA signals antiviral and inflammatory programs and dysfunctional glutamate transport in TLR3-expressing astrocytes. Glia. 2005;52:153–162. doi: 10.1002/glia.20234. [DOI] [PubMed] [Google Scholar]
- Segal BM, Klinman DM, Shevach EM. Microbial products induce autoimmune disease by an IL-12-dependent pathway. J Immunol. 1997;158:5087–5090. [PubMed] [Google Scholar]
- Segal BM, Chang JT, Shevach EM. CpG oligonucleotides are potent adjuvants for the activation of autoreactive encephalitogenic T cells in vivo. J Immunol. 2000;164:5683–5688. doi: 10.4049/jimmunol.164.11.5683. [DOI] [PubMed] [Google Scholar]
- Shie FS, Montine KS, Breyer RM, Montine TJ. Microglial EP2 is critical to neurotoxicity from activated cerebral innate immunity. Glia. 2005;52:70–77. doi: 10.1002/glia.20220. [DOI] [PubMed] [Google Scholar]
- Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, Miyake K, Kimoto M. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J Exp Med. 1999;189:1777–1782. doi: 10.1084/jem.189.11.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibley WA, Bamford CR, Clark K. Clinical viral infections and multiple sclerosis. Lancet. 1985;1:1313–1315. doi: 10.1016/S0140-6736(85)92801-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smiley ST, King JA, Hancock WW. Fibrinogen stimulates macrophage chemokine secretion through Toll-like receptor 4. J Immunol. 2001;167:2887–2894. doi: 10.4049/jimmunol.167.5.2887. [DOI] [PubMed] [Google Scholar]
- Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- Takeshita S, Takeshita F, Haddad DE, Janabi N, Klinman DM. Activation of microglia and astrocytes by CpG oligodeoxynucleotides. Neuroreport. 2001;12:3029–3032. doi: 10.1097/00001756-200110080-00010. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11:443–451. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Hoshino K, Akira S. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J Immunol. 2000a;165:5392–5396. doi: 10.4049/jimmunol.165.10.5392. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Takeda K, Hoshino K, Adachi O, Ogawa T, Akira S. Cellular responses to bacterial cell wall components are mediated through MyD88-dependent signaling cascades. Int Immunol. 2000b;12:113–117. doi: 10.1093/intimm/12.1.113. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Kawaic T, Muhlradt PF, Morr M, Radolf JD, Zychlinsky A, Takeda K, Akira S. Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int Immunol. 2001;13:933–940. doi: 10.1093/intimm/13.7.933. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Sato S, Horiuchi T, Hoshino K, Takeda K, Dong Z, Modlin RL, Akira S. Cutting edge: role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. J Immunol. 2002;169:10–14. doi: 10.4049/jimmunol.169.1.10. [DOI] [PubMed] [Google Scholar]
- Tanga FY, Raghavendra V, DeLeo JA. Quantitative real-time RT-PCR assessment of spinal microglial and astrocytic activation markers in a rat model of neuropathic pain. Neurochem Int. 2004;45:397–407. doi: 10.1016/j.neuint.2003.06.002. [DOI] [PubMed] [Google Scholar]
- Tanga FY, Nutile-McMenemy N, DeLeo JA. The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc Natl Acad Sci U S A. 2005;102:5856–5861. doi: 10.1073/pnas.0501634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapping RI, Akashi S, Miyake K, Godowski PJ, Tobias PS. Toll-like receptor 4, but not Toll-like receptor 2, is a signaling receptor for Escherichia and Salmonella lipopolysaccharides. J Immunol. 2000;165:5780–5787. doi: 10.4049/jimmunol.165.10.5780. [DOI] [PubMed] [Google Scholar]
- Termeer C, Benedix F, Sleeman J, Fieber C, Voith U, Ahrens T, Miyake K, Freudenberg M, Galanos C, Simon JC. Oligosaccharides of hyaluronan activate dendritic cells via Toll-like receptor 4. J Exp Med. 2002;195:99–111. doi: 10.1084/jem.20001858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travassos LH, Girardin SE, Philpott DJ, Blanot D, Nahori MA, Werts C, Boneca IG. Toll-like receptor 2-dependent bacterial sensing does not occur via peptidoglycan recognition. EMBO Rep. 2004;5:1000–1006. doi: 10.1038/sj.embor.7400248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsan MF, Gao B. Endogenous ligands of Toll-like receptors. J Leukoc Biol. 2004;76:514–519. doi: 10.1189/jlb.0304127. [DOI] [PubMed] [Google Scholar]
- Tsunoda I, Tolley ND, Theil DJ, Whitton JL, Kobayashi H, Fujinami RS. Exacerbation of viral and autoimmune animal models for multiple sclerosis by bacterial DNA. Brain Pathol. 1999;9:481–493. doi: 10.1111/j.1750-3639.1999.tb00537.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underhill D, Ozinsky A, Hajjar AM, Stevens A, Wilson CB, Bassetti M, Aderem A. The Toll-like receptor 2 is recruited to macrophage phagosomes and discriminates between pathogens. Nature. 1999a;401:811–815. doi: 10.1038/44605. [DOI] [PubMed] [Google Scholar]
- Underhill DM, Ozinsky A, Smith KD, Aderem A. Toll-like receptor-2 mediates mycobacteria-induced proinflammatory signaling in macrophages. Proc Natl Acad Sci U S A. 1999b;96:14459–14463. doi: 10.1073/pnas.96.25.14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabulas RM, Ahmad-Nejad P, Ghose S, Kirschning CJ, Issels RD, Wagner H. HSP70 as endogenous stimulus of the Toll/interleukin-1 receptor signal pathway. J Biol Chem. 2002a;277:15107–15112. doi: 10.1074/jbc.M111204200. [DOI] [PubMed] [Google Scholar]
- Vabulas RM, Braedel S, Hilf N, Singh-Jasuja H, Herter S, Ahmad-Nejad P, Kirschning CJ, Da Costa C, Rammensee HG, Wagner H, Schild H. The endoplasmic reticulum-resident heat shock protein Gp96 activates dendritic cells via the Toll-like receptor 2/4 pathway. J Biol Chem. 2002b;277:20847–20853. doi: 10.1074/jbc.M200425200. [DOI] [PubMed] [Google Scholar]
- van der Flier M, Geelen SP, Kimpen JL, Hoepelman IM, Tuomanen EI. Reprogramming the host response in bacterial meningitis: how best to improve outcome? Clin Microbiol Rev. 2003;16:415–429. doi: 10.1128/CMR.16.3.415-429.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vikstrom EV. The immunosuppressive activity of chemically modified lipopolysaccharide of Shigella sonnei. Immunol Lett. 2002;80:15–19. doi: 10.1016/s0165-2478(01)00298-x. [DOI] [PubMed] [Google Scholar]
- Viriyakosol S, Kirkland T, Soldau K, Tobias P. MD-2 binds to bacterial lipopolysaccharide. J Endotoxin Res. 2000;6:489–491. [PubMed] [Google Scholar]
- Visintin A, Halmen KA, Latz E, Monks BG, Golenbock DT. Pharmacological inhibition of endotoxin responses is achieved by targeting the TLR4 coreceptor, MD-2. J Immunol. 2005;175:6465–6472. doi: 10.4049/jimmunol.175.10.6465. [DOI] [PubMed] [Google Scholar]
- Visser L, Jan de Heer H, Boven LA, van Riel D, van Meurs M, Melief MJ, Zahringer U, van Strijp J, Lambrecht BN, Nieuwenhuis EE, Laman JD. Proinflammatory bacterial peptidoglycan as a cofactor for the development of central nervous system autoimmune disease. J Immunol. 2005;174:808–816. doi: 10.4049/jimmunol.174.2.808. [DOI] [PubMed] [Google Scholar]
- Waldner H, Collins M, Kuchroo VK. Activation of antigen-presenting cells by microbial products breaks self tolerance and induces autoimmune disease. J Clin Invest. 2004;113:990–997. doi: 10.1172/JCI19388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med. 2004;10:1366–1373. doi: 10.1038/nm1140. [DOI] [PubMed] [Google Scholar]
- Weatherill AR, Lee JY, Zhao L, Lemay DG, Youn HS, Hwang DH. Saturated and polyunsaturated fatty acids reciprocally modulate dendritic cell functions mediated through TLR4. J Immunol. 2005;174:5390–5397. doi: 10.4049/jimmunol.174.9.5390. [DOI] [PubMed] [Google Scholar]
- Weber JR, Freyer D, Alexander C, Schroder NW, Reiss A, Kuster C, Pfeil D, Tuomanen EI, Schumann RR. Recognition of pneumococcal peptidoglycan: an expanded, pivotal role for LPS binding protein. Immunity. 2003a;19:269–279. doi: 10.1016/s1074-7613(03)00205-x. [DOI] [PubMed] [Google Scholar]
- Weber JR, Moreillon P, Tuomanen EI. Innate sensors for gram-positive bacteria. Curr Opin Immunol. 2003b;15:408–415. doi: 10.1016/s0952-7915(03)00078-5. [DOI] [PubMed] [Google Scholar]
- Wieland CW, Knapp S, Florquin S, de Vos AF, Takeda K, Akira S, Golenbock DT, Verbon A, van der Poll T. Non-mannosecapped lipoarabinomannan induces lung inflammation via Toll-like receptor 2. Am J Respir Crit Care Med. 2004;170:1367–1374. doi: 10.1164/rccm.200404-525OC. [DOI] [PubMed] [Google Scholar]
- Williams MJ, Rodriguez A, Kimbrell DA, Eldon ED. The 18-wheeler mutation reveals complex antibacterial gene regulation in Drosophila host defense. EMBO J. 1997;16:6120–6130. doi: 10.1093/emboj/16.20.6120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirenfeldt M, Babcock AA, Ladeby R, Lambertsen KL, Dagnaes-Hansen F, Leslie RG, Owens T, Finsen B. Reactive microgliosis engages distinct responses by microglial subpopulations after minor central nervous system injury. J Neurosci Res. 2005;82:507–514. doi: 10.1002/jnr.20659. [DOI] [PubMed] [Google Scholar]
- Witowski J, Ksiazek K, Jorres A. Interleukin-17: a mediator of inflammatory responses. Cell Mol Life Sci. 2004;61:567–579. doi: 10.1007/s00018-003-3228-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Takeda K, Akira S. TIR domain-containing adaptors define the specificity of TLR signaling. Mol Immunol. 2004;40:861–868. doi: 10.1016/j.molimm.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Yang RB, Mark MR, Gray A, Huang A, Xie MH, Zhang M, Goddard A, Wood WI, Gurney AL, Godowski PJ. Toll-like receptor-2 mediates lipopolysaccharide-induced cellular signalling. Nature. 1998;395:284–288. doi: 10.1038/26239. [DOI] [PubMed] [Google Scholar]
- Yang RB, Mark MR, Gurney AL, Godowski PJ. Signaling events induced by lipopolysaccharide-activated Toll-like receptor 2. J Immunol. 1999;163:639–643. [PubMed] [Google Scholar]
- Yarovinsky F, Zhang D, Andersen JF, Bannenberg GL, Serhan CN, Hayden MS, Hieny S, Sutterwala FS, Flavell RA, Ghosh S, Sher A. TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science. 2005;308:1626–1629. doi: 10.1126/science.1109893. [DOI] [PubMed] [Google Scholar]
- Yoshimura A, Lien E, Ingalls RR, Tuomanen E, Dziarski R, Golenbock D. Cutting edge: recognition of gram-positive bacterial cell wall components by the innate immune system occurs via Toll-like receptor 2. J Immunol. 1999;163:1–5. [PubMed] [Google Scholar]
- Zekki H, Feinstein DL, Rivest S. The clinical course of experimental autoimmune encephalomyelitis is associated with a profound and sustained transcriptional activation of the genes encoding Toll-like receptor 2 and CD14 in the mouse CNS. Brain Pathol. 2002;12:308–319. doi: 10.1111/j.1750-3639.2002.tb00445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Zhang G, Hayden MS, Greenblatt MB, Bussey C, Flavell RA, Ghosh S. A Toll-like receptor that prevents infection by uropathogenic bacteria. Science. 2004;303:1522–1526. doi: 10.1126/science.1094351. [DOI] [PubMed] [Google Scholar]
- Zhang G, Ghosh S. Negative regulation of Toll-like receptor-mediated signaling by Tollip. J Biol Chem. 2002;277:7059–7065. doi: 10.1074/jbc.M109537200. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Guo K, Schluesener HJ. The immunostimulatory activity of CpG oligonucleotides on microglial N9 cells is affected by a polyguanosine motif. J Neuroimmunol. 2005;161:68–77. doi: 10.1016/j.jneuroim.2004.12.009. [DOI] [PubMed] [Google Scholar]
- Zwijnenburg PJ, van der Poll T, Florquin S, Akira S, Takeda K, Roord JJ, van Furth AM. Interleukin-18 gene-deficient mice show enhanced defense and reduced inflammation during pneumococcal meningitis. J Neuroimmunol. 2003a;138:31–37. doi: 10.1016/s0165-5728(03)00088-2. [DOI] [PubMed] [Google Scholar]
- Zwijnenburg PJ, van der Poll T, Florquin S, Roord JJ, Van Furth AM. IL-1 receptor type 1 gene-deficient mice demonstrate an impaired host defense against pneumococcal meningitis. J Immunol. 2003b;170:4724–4730. doi: 10.4049/jimmunol.170.9.4724. [DOI] [PubMed] [Google Scholar]