

Abstract
The evaluation of ruthenium olefin metathesis catalysts 4–6 bearing cyclic (alkyl)(amino)carbenes (CAACs) in the cross-metathesis of cis-1,4-diacetoxy-2-butene (7) with allylbenzene (8) and the ethenolysis of methyl oleate (11) is reported. Relative to most NHC-substituted complexes, CAAC-substituted catalysts exhibit lower E/Z ratios (3:1 at 70% conversion) in the cross-metathesis of 7 and 8. Additionally, complexes 4–6 demonstrate good selectivity for the formation of terminal olefins versus internal olefins in the ethenolysis of 11. Indeed, complex 6 achieved 35 000 TONs, the highest recorded to date. CAAC-substituted complexes exhibit markedly different kinetic selectivity than most NHC-substituted complexes.
Olefin metathesis has become a standard method to construct carbon–carbon double bonds.1,2 Although complexes 1–3 are efficient catalysts for many polymerizations and ring-closing, ring-opening, and cross-metathesis reactions, several olefin metathesis processes remain challenging (Chart 1).3 In particular, the development of catalysts that favor the formation of kinetic rather than thermodynamic products is an area of significant interest. Indeed, highly active and stable NHC-containing (N-heterocyclic carbene) catalysts such as 2 and 3 generally produce mixtures of the most stable products containing more trans olefins versus cis olefins or internal olefins versus terminal olefins (ethenolysis).4,5
Chart 1.

Commonly Utilized Olefin Metathesis Catalysts
An E/Z-diastereoselective olefin metathesis catalyst would enable the efficient synthesis of E- or Z-olefins, an attractive goal of synthetic organic chemistry.1 However, the E/Z diastereoselectivity of an olefin metathesis reaction is often controlled by the thermodynamic stability of the olefin isomers rather than the selectivity of the catalyst. The product E/Z ratio of the homodimerization of a terminal olefin is a result of primary and secondary metathesis processes (Figure 1). Primary metathesis is composed of two reactions: the reaction of a ruthenium methylidene species with a terminal olefin to produce a ruthenium alkylidene species, which subsequently reacts with a terminal olefin to generate E- or Z-olefins. The selectivity of the primary metathesis reactions depends on the geometry of the olefin approach and coordination to the ruthenium alkylidene complex.6,7
Figure 1.

Primary and secondary metathesis processes affecting E/Z diastereoselectivity in olefin metathesis.
Secondary metathesis processes involve reactions of the product E- and Z-olefins with ruthenium alkylidene and methylidene species (Figure 1). In general, secondary metathesis results in the interconversion of the isomers, supplying an increased yield of the more thermodynamically stable isomer.8,9 In addition to the different E/Z diastereoselectivities of these reactions, the relative rates of each reaction may also be different because Z-olefins are generally more reactive than E-olefins.10 As a result of these competing processes, the E/Z product ratio at lower conversions is more reflective of the selectivity of primary metathesis processes, whereas at higher conversions an increase in the E/Z ratio is typically observed due to secondary metathesis of the Z-olefin to the more thermodynamically favorable E-olefin.11
Another targeted kinetic process is ethenolysis, the cross-metathesis of ethylene with an internal olefin to provide terminal olefins without significant production of internal olefins. Typically, the observed product distribution reflects the increased stability of internal olefins relative to terminal olefins.4
The ethenolysis catalytic cycle involves two primary metathesis reactions: the reaction of a ruthenium methylidene species with an internal olefin to produce a terminal olefin and ruthenium alkylidene species, which then reacts with ethylene to regenerate the ruthenium methylidene species and yields a second terminal olefin (Figure 2).
Figure 2.

Primary, secondary, and self-metathesis processes during ethenolysis reactions.
Self metathesis and secondary metathesis processes compete with primary metathesis reactions and produce undesired internal olefins (Figure 2).4 Self metathesis of the substrate occurs when a ruthenium alkylidene species binds an internal olefin (instead of ethylene), resulting in the formation of a new internal olefin. Secondary metathesis occurs when a ruthenium alkylidene species reacts with a terminal olefin (rather than ethylene) to generate an internal olefin. As with E/Z-diastereoselective olefin metathesis, secondary metathesis results in the conversion of kinetic products into more thermodynamically stable products.
Although numerous catalysts have been examined for E/Z-diastereoselective olefin metathesis and ethenolysis, no clear trend for ligand development has emerged. Recently, we reported the synthesis and characterization of a series of ruthenium olefin metathesis catalysts bearing cyclic(alkyl)-(amino) carbenes (CAACs) (Chart 2).12 CAACs are more electron-donating than their traditional NHC counterparts and have unique steric properties. In our studies, it was observed that catalysts 4 and 5 require elevated temperatures and extended reaction times to complete ring-closing metathesis reactions, whereas catalyst 6 exhibited activity comparable to catalysts 2 and 3. Herein we report the kinetic selectivity of these ruthenium catalysts.
Chart 2.

Olefin Metathesis Catalysts Bearing CAACs
Results and Discussion
Recently, our group reported the evaluation of catalyst E/Z selectivity by examining the cross-metathesis of 2 equiv of cis-1,4-diacetoxy-2-butene (7) with allylbenzene (8) in the presence of 2.5 mol% catalyst in CH2Cl2 at 25 °C to produce (E)- or (Z)-4-phenylbut-2-enyl acetate (9 and 10, respectively) (eq 1).11 To compensate for the slow or fast reaction rates observed, catalysts were compared via plots of E/Z ratio versus conversion rather than E/Z ratio versus time. Both bis(phosphine) and NHC-containing ruthenium catalysts show similar E/Z ratios (~3–4) at conversions below 60% (Figure 3). At higher conversions, NHC-containing catalysts provide a mixture of products containing a higher E/Z ratio of ~6–10 due to secondary metathesis of 10 to 9.
Figure 3.

Plot of E/Z ratio of cross-products vs % conversion for catalysts 4–6 in comparison with previously studied catalysts 1–3.
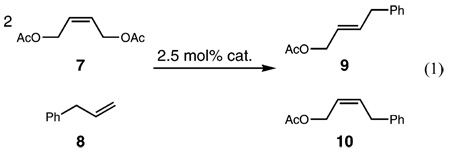
As shown in Figure 3, catalysts 4–6 exhibit enhanced E/Z diastereoselectivity for the formation of Z-olefins over catalysts 1–3. Below 60% conversion to the heterocoupled products 9 and 10, catalysts 4–6 demonstrate E/Z ratios of 1.5–2.5. At 70% conversion, catalysts 4–6 provide E/Z ratios of ~3 compared to catalyst 2, which provides a ratio of ~6. Similar E/Z ratios were observed by Blechert and co-workers utilizing a ruthenium complex bearing an unsymmetrically-substituted NHC.13
Interestingly, catalyst 6 achieves ~60% conversion to product in 1 h at 22 °C, whereas catalysts 4 and 5 require 32 and 48 h at 60 °C, respectively. These results indicate that the higher E selectivity observed is not simply due to a less active catalyst that is slow to isomerize olefins. Rather, these carbenes impart a change in the inherent catalyst selectivity.
Ethenolysis has been investigated for several decades as a method to transform internal olefins derived from seed oils to terminal olefin feedstocks.14 However, an ethenolysis catalyst that is both highly efficient and highly selective has yet to be developed.4
Previous detailed studies of catalysts 14 and 1a15,16 in the ethenolysis of methyl oleate (11) demonstrated their high selectivity for the production of terminal olefins 9-methyl decenoate (12) and 1-decene (13) over self-metathesis products 1,18-dimethyl 9-octadecenoate (14) and 9-octadecene (15) (eq 2). At 100 ppm, catalysts 1 and 1a achieve 58% and 51% conversion to 12 and 13 with 93% and 94% selectivity, resulting in 5400 and 4800 TONs, respectively. Lowering the catalyst loading of 1 from 100 to 35 ppm results in a significant increase in TONs to 12 900 with 94% selectivity for ethenolysis products over self-metathesis products. However, further decreasing the catalyst loading of 1 to 10 ppm did not result in increased TONs. The highest TON reported to date is 14 047 for a bis(9-cyclohexyl-9-phospha-9H-bicyclonane)ruthenium complex.17 The efficiency of first-generation Grubbs-type catalysts is limited by two major factors: catalyst decomposition due to the instability of the propagating methylidene species and catalyst inhibition by the ethenolysis products.4
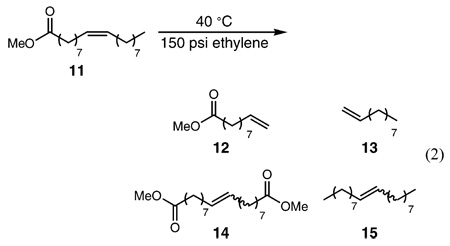
Conversely, NHC-containing systems 2 and 3 demonstrate relatively low selectivity for the synthesis of the desired terminal olefins (Table 1). At 100 ppm, catalysts 2 and 3 produce only 28% and 20% of ethenolysis products 12 and 13 with product selectivities of 44% and 33%, respectively. The remaining products of these reactions are self-metathesis products 14 and 15. Interestingly, in the ethenolysis of 11, bis(phosphine) catalyst 1 outperforms NHC-containing catalysts 2 and 3.
Table 1.
Comparison of Ruthenium Catalysts in the Ethenolysis of 11a
| cat | cat./11 (ppm) | time (min)b | conv.(%)c | selectivity (%)d | yield (%)e | TONf |
|---|---|---|---|---|---|---|
| 1 | 100 | 120 | 58 | 93 | 54 | 5400 |
| 1 | 35 | 240 | 48 | 94 | 45 | 12 900 |
| 1 | 10 | 120 | 13 | >97 | 13 | 12 700 |
| 1a | 100 | 30 | 51 | 94 | 48 | 4800 |
| 2 | 100 | 120 | 64 | 44 | 28 | 2800 |
| 3 | 100 | 30 | 60 | 33 | 20 | 2000 |
| 4 | 100 | 1,320 | 61 | 92 | 56 | 5600 |
| 4 | 50 | 1,200 | 61 | 93 | 57 | 11 400 |
| 5 | 100 | 360 | 46 | 94 | 43 | 4 200 |
| 6 | 100 | <30 | 73 | 73 | 53 | 5300 |
| 6 | 35 | 60 | 75 | 75 | 56 | 16 000 |
| 6 | 10 | <30 | 42 | 83 | 35 | 35 000 |
General conditions: neat 11, 150 psi ethylene, 40 °C.
Time to maximum conversion.
Conversion = 100 − [(final moles of 11) × 100/(initial moles of 11)].
Selectivity = (moles of ethenolysis products 12 + 13) × 100/(moles of total products 12 + 13 + 14 + 15).
Yield = (moles of ethenolysis products 12 + 13) × 100/(initial moles of 11) conversion × selectivity/100.
TON = yield × [(moles of 11)/(moles of cat.)].
Catalysts 4–6 were evaluated for the ethenolysis of methyl oleate (11) under the same conditions (150 psi ethylene, neat 11, 40 °C) (Table 1). At loadings of 100 ppm, catalysts 4–6 exhibited good selectivity (73–94%) for terminal olefins 12 and 13 and achieved TONs ranging from 4200 to 5600. By lowering the catalyst loading of 6 to 10 ppm, TONs of 35 000 were achieved.18 Catalyst 6 exhibits the highest activity for the ethenolysis of methyl oleate to date and represents a new direction of catalyst development in this area.
Summary
Ruthenium complexes 4–6 bearing cyclic (alkyl)(amino)-carbenes exhibit kinetic selectivity in both E,Z-diastereoselective and ethenolysis reactions. Catalysts 4–6 demonstrate increased selectivity for the formation of Z-olefins in the cross-metathesis of Z-1,4-diacetoxy-2-butene (7) and allyl benzene (8) relative to commercially available catalysts 1–3. In the ethenolysis of methyl oleate, catalysts 4 and 5 display high selectivities and TONs for the formation of terminal olefins, which are comparable to those of bis(phosphine) catalyst 1. Complex 6 displays slightly lower selectivity for the formation of terminal olefin relative to internal olefin, but achieves the highest TONs (35 000) observed to date.
Experimental Section
General Cross-Metathesis Procedure
We utilized the procedure outlined in the following for reaction conditions and GC analysis: Ritter, T.; Hejl, A.; Wenzel, A. G.; Funk, T. W.; Grubbs, R. H. Organometallics 2006, 25, 5740. Reactions with catalysts 4–6 were performed in benzene rather than CH2Cl2 to enable higher reaction temperatures. For catalysts 4 and 5, the reactions were performed at 60 °C. For each catalyst, two or three identical reactions utilizing different catalyst batches were performed and the data were averaged together.
General Ethenolysis Procedure
Ethenolysis reactions of research-grade methyl oleate were set up under an inert atmosphere in a glovebox: a Fisher-Porter bottle equipped with a stir bar was charged with methyl oleate (>99%) from Nu-Chek-Prep (Elysian, MN) and further purified by filtration through activated alumina (15.0 g; 50.6 mmol). For ethenolysis reactions run with low catalyst loadings (i.e., catalyst loadings lower than 100 ppm), it is important to use freshly purified methyl oleate. A solution of olefin metathesis catalyst of an appropriate concentration was prepared in anhydrous dichloromethane (from Aldrich), and the desired volume of this solution added to the methyl oleate. The head of the Fisher-Porter bottle equipped with a pressure gauge and a dip-tube was adapted on the bottle. The system was sealed and taken out of the glovebox to an ethylene line. The vessel was then purged three times with ethylene (polymer purity 99.9% from Matheson Tri Gas), pressurized to 150 psi, and placed in an oil bath at 40 °C. The reaction was monitored by collecting samples into vials at different reaction times via the dip-tube. Immediately after collecting a sample, the reaction was stopped by adding 1 mL of a 1.0 M 2-propanol solution of tris-hydroxymethylphosphine (THMP) to the vial. The samples were then heated for at least 1 h at 60 °C, diluted with 1 mL of distilled water, extracted with 1 mL of hexanes, and analyzed by gas chromatography (GC).
GC Analytical Method
The GC analyses were run using a flame ionization detector (FID). Column: Rtx-5 from Restek (30 m × 0.25 mm (i.d.) × 0.25 µm film thickness). GC and column conditions: injector temperature 250 °C; detector temperature 280 °C; oven temperature, starting temperature 100 °C, hold time 1 min, ramp rate 10 °C/min to 250 °C, hold time 12 min; carrier gas helium.
Acknowledgment
D.R.A. acknowledges NSF and NDSEG predoctoral fellowships. G.M., T.U., and Y.S. thank the DOE (DE-FG36-04GO14016) and Cargill, Inc. for financial support. R.H.G. is grateful to the NSF for funding (CHE-0410425). G.B. thanks the NIH for financial support (R01 GM 68825).
References
- 1.Grubbs RH. Handbook of Metathesis. Wiley-VCH: Weinheim; 2003. [Google Scholar]
- 2.Ivin KJ, Mol JC. Olefin Metathesis and Metathesis Polymerization. San Diego, CA: Academic Press; 1997. [Google Scholar]
- 3.Thayer AM. Chem. Eng. News. 2007;85:37. [Google Scholar]
- 4.Burdett KA, Harris LD, Margl P, Maughon BR, Mokhtar-Zadeh T, Saucier PC, Wasserman EP. Organometallics. 2004;23:2027. [Google Scholar]
- 5.Deshmukh PH, Blechert S. Dalton Trans. 2007:2479. doi: 10.1039/b703164p. [DOI] [PubMed] [Google Scholar]
- 6.Romero PE, Piers WE. J. Am. Chem. Soc. 2007;129:1698. doi: 10.1021/ja0675245. [DOI] [PubMed] [Google Scholar]
- 7.Wenzel AG, Grubbs RH. J. Am. Chem. Soc. 2006;128:16048. doi: 10.1021/ja0666598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chatterjee AK, Choi TL, Sanders DP, Grubbs RH. J. Am. Chem. Soc. 2003;125:11360. doi: 10.1021/ja0214882. [DOI] [PubMed] [Google Scholar]
- 9.Connon SJ, Blechert S. Angew. Chem., Int. Ed. 2003;42:1900. doi: 10.1002/anie.200200556. [DOI] [PubMed] [Google Scholar]
- 10.Ulman M, Grubbs RH. Organometallics. 1998;17:2484. [Google Scholar]
- 11.Ritter T, Hejl A, Wenzel AG, Funk TW, Grubbs RH. Organometallics. 2006;25:5740. [Google Scholar]
- 12.Anderson DR, Lavallo V, O'Leary DJ, Bertrand G, Grubbs RH. Angew. Chem., Int. Ed. 2007;46:7262. doi: 10.1002/anie.200702085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vehlow K, Maechling S, Blechert S. Organometallics. 2006;25:25. [Google Scholar]
- 14.Corma A, Iborra S, Velty A. Chem. Rev. 2007;107:2411. doi: 10.1021/cr050989d. [DOI] [PubMed] [Google Scholar]
- 15.Kingsbury JS, Harrity JPA, Bonitatebus PJ, Hoveyda AH. J. Am. Chem. Soc. 1999;121:791. [Google Scholar]
- 16.Newman TH, Rand CL, Burdett KA, Maughon BR, Morrison DL, Wasserman EP. 2006 October 10; 7119216. [Google Scholar]
- 17.Forman GS, McConnell AE, Hanton MJ, Slawin AMZ, Tooze RP, vanRensburg WJ, Meyer WH, Dwyer C, Kirk MM, Serfontein DW. Organometallics. 2004;23:4824. [Google Scholar]
- 18.The purification of 11 is essential at low catalyst loadings, as small amounts of impurities can significantly impact the catalyst efficiency (see Experimental Section).