

Abstract
Obesity is a major risk factor for the development and progression of breast cancer. Increased circulating levels of the obesity-associated hormones leptin and insulin-like growth factor-I (IGF-I), and overexpression of the leptin receptor (Ob-R) and IGF-I receptor (IGF-IR) have been detected in a majority of breast cancer cases and during obesity. Due to correlations between increased leptin, Ob-R, IGF-I, and IGF-IR in breast cancer, we hypothesized that molecular interactions may exist between these two signaling pathways. Co-immunoprecipitation and immunoblotting demonstrated that IGF-IR and Ob-R interact in the breast cancer cell lines MDA-MB-231, MCF7, BT474, and SKBR3. Stimulation of cells with IGF-I promoted Ob-R phosphorylation, which was blocked by IGF-IR kinase inhibition. In addition, IGF-I activated downstream signaling molecules in the leptin receptor and IGF-IR pathways. In contrast to IGFI, leptin did not induce phosphorylation of IGF-IR, indicating that receptor cross signaling is unidirectional, occurring from IGF-IR to Ob-R. Our results demonstrate for the first time a novel interaction and cross talk between the IGF-I and leptin receptors in human breast cancer cells.
Keywords: growth factor receptors, mammary carcinoma, signal transduction
Background
Obesity is an important and manageable risk factor for the development and progression of postmenopausal breast cancer (1). Increased body weight and body mass index are associated with reduced disease-free and overall survival and poorer therapeutic response rates in breast cancer patients, regardless of menopausal status or age (2). Although the exact molecular mechanisms by which obesity influences cancer biology are unknown, there is evidence suggesting that increased production and secretion of adipocyte-derived growth factors and hormones contributes to cellular transformation and tumorigenesis (3,4). The obesity-associated hormones leptin and insulin-like growth factor-I (IGF-I) have been independently implicated in the connection between obesity and breast cancer (5).
Leptin, a product of the obese (ob) gene, is an adipocytokine that regulates appetite, bone formation, reproduction, cellular proliferation, and angiogenesis (6). Because of the strong association between human obesity and elevated levels of circulating leptin, this hormone has been widely studied in the fields of nutrition and weight management (7). More recently, however, leptin has emerged as a potential factor contributing to mammary tumorigenesis. In vitro studies demonstrated that leptin stimulates the growth, survival, and transformation of breast cancer cells (5), primarily by activating the JAK/STAT (Janus kinase/signal transducers and activators of transcription) signaling pathway (8,9) and the phosphoinositol-3-kinase (PI3K)/Akt and mitogen-activated protein kinase (MAPK) pathways (10). Leptin induces cell cycle progression by up-regulating cyclin D1 expression and cyclin-dependent kinase 2 activity, as well as by inactivating the retinoblastoma growth suppressing protein (11). Importantly, leptin and its receptor (Ob-R) were found to be overexpressed in a majority of breast cancer tissues, especially in high grade tumors, but absent or expressed at very low levels in normal mammary epithelium or benign tumors (5,12). In addition, leptin-deficient mice have a decreased incidence of spontaneous and oncogene-induced mammary tumors (13). Thus, leptin signaling appears to play an important role in breast cancer biology.
Similar to leptin, increased levels of IGF-I and its receptor are detected in sera and primary tumors of breast cancer patients (14,15), and transgenic overexpression of IGF-IR has been shown to induce mammary tumor formation (16). IGF-I is an important endocrine, paracrine, and autocrine regulator of breast epithelial cell growth. Increased signaling through the IGF-I receptor (IGF-IR) results in increased cellular proliferation, mitogenesis, and survival, and decreased apoptosis, causing resistance to numerous anti-neoplastic agents (14,17). For these reasons the IGF-I receptor has become an important therapeutic target for drug discovery in breast oncology (17).
Cross talk between different growth factor receptor families is frequently observed in tumors. This mechanism allows cancer cells to enhance downstream signaling resulting in greatly increased proliferation, mitogenesis, and cell survival. The IGF-I receptor has been shown to interact and cross talk with multiple receptors including the epidermal growth factor receptor (18), HER2 (19), platelet-derived growth factor receptor (20), and the estrogen receptor (14). Due to the correlations between elevated levels of leptin, IGF-I, and their associated receptors with obesity and breast cancer, we hypothesized that interactions and/or cross talk may occur between these two signaling pathways.
Results
Insulin-like growth factor-I receptor and leptin receptor interact in human breast cancer cells
The human breast cancer lines MDA-MB-231 (MDA231), MCF7, BT474, and SKBR3 were examined for expression of the IGF-I receptor (IGF-IR) and leptin receptor (Ob-R). Immunoblotting of total protein lysates (Figure 1A) demonstrated that the two major isoforms of Ob-R, called Ob-Rb (longer isoform) and Ob-Rt (shorter isoform) are expressed at similar levels in all cell lines (Figure 1B). IGF-IR is expressed at higher levels in MCF7 and BT474 cells versus SKBR3 and MDA231 cells, with highest levels observed in MCF7 cells (Figure 1B).
Figure 1. Expression of IGF-IR and Ob-R in breast cancer lines.
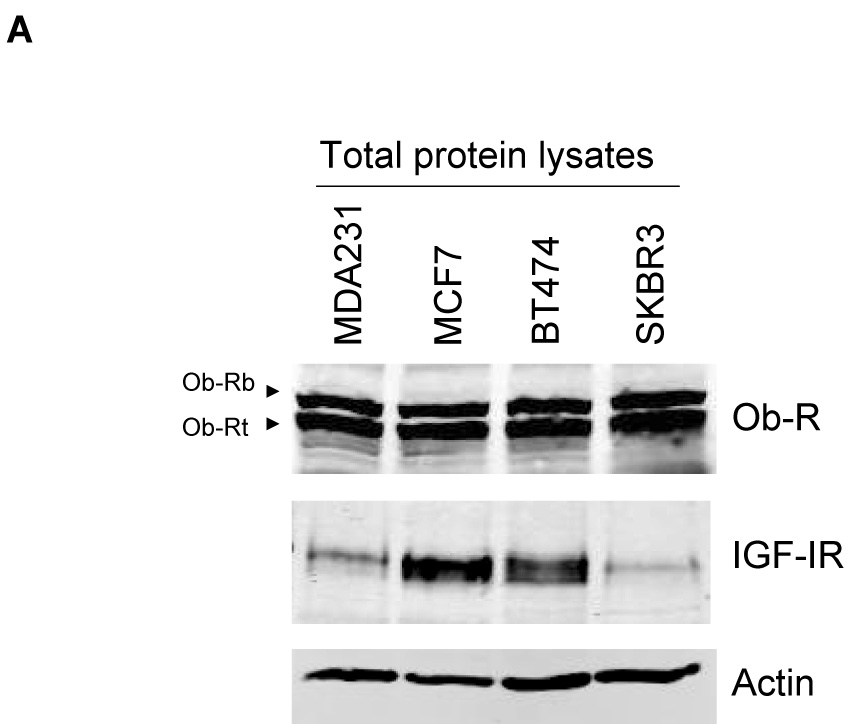
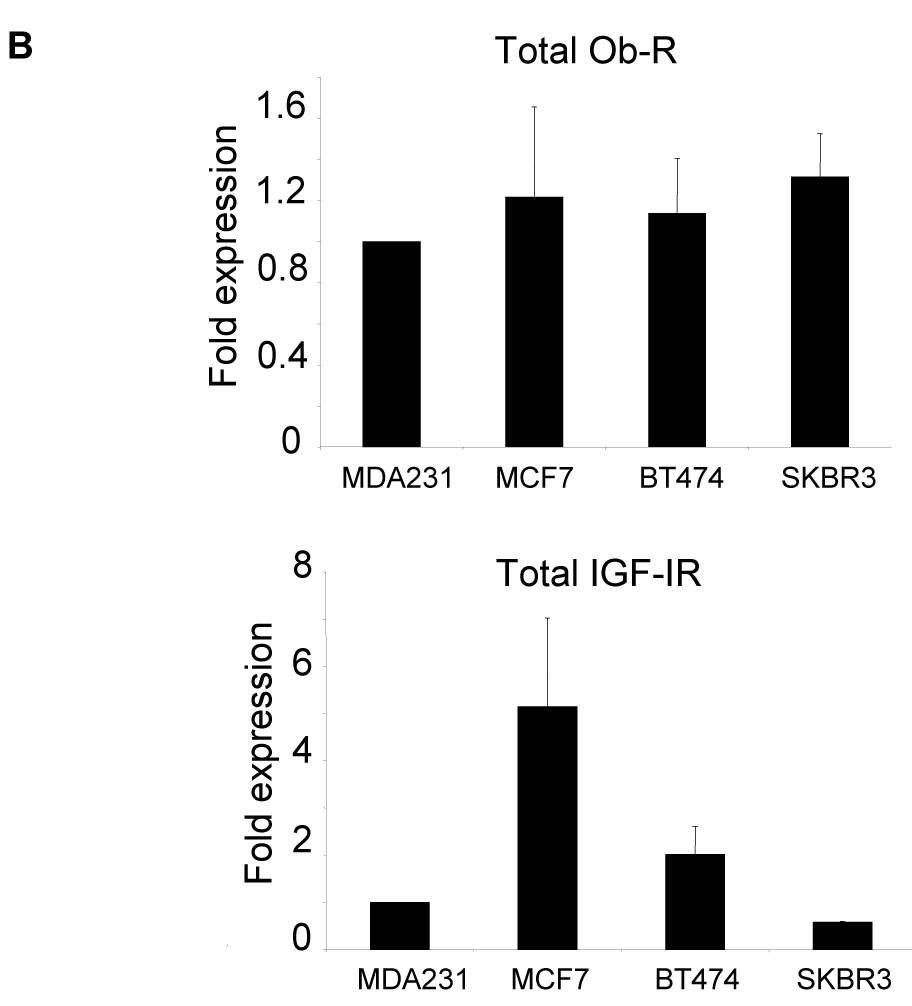
The breast cancer lines MDA-MB-231 (MDA231), MCF7, BT474, and SKBR3 were lysed for total protein. (A) Total protein lysates were immunoblotted for Ob-R using the H-300 polyclonal antibody, which recognizes both the long Ob-Rb isoform and the shorter Ob-Rt isoform of the leptin receptor. Immunoblotting was also performed for total IGF-IR and for actin as a loading control. (B) Bands on immunoblots were quantitated using NIH ImageJ and are expressed relative to expression levels in MDA231 cells (lane 1). Error bars represent standard deviation between three independent experiments. Total Ob-R levels were similar among the four lines; IGF-IR was expressed at the highest level in MCF7 cells, with BT474 cells showing moderate expression compared to the other two lines which expressed the lowest levels of IGF-IR.
Immunoprecipitation of Ob-R with subsequent immunoblotting for IGF-IR showed that Ob-Rb and Ob-Rt are both pulled down with IGF-IR in all four cell lines (Figure 2A). Conversely, IGF-IR immunoprecipitation pulled down Ob-Rb and Ob-Rt in each cell line, with preferential interaction observed with the shorter isoform of Ob-R in MCF7, BT474, and SKBR3 cells (Figure 2B). Quantitation showed that IGF-IR was pulled down with Ob-R to a similar extent in all four lines (Figure 2C). Total Ob-R was pulled down with IGF-IR in all four lines; however, higher levels of Ob-R interacting with IGF-IR was observed in MCF7 cells (Figure 2C), likely due to the higher expression level of total IGF-IR in these cells (Figure 1B). Negative controls in which cell lysates were immunoprecipitated with rabbit IgG confirmed that IGF-IR and Ob-R were not pulled down (Figure 2D). In addition, since IGF-IR has been shown to interact with insulin receptor (Ins-R) (21), we blotted IGF-IR immunoprecipitates for Ins-R as a positive control (Figure 2D). Ins-R was pulled down with IGF-IR in all four lines. Finally, another tyrosine kinase receptor, EGFR, was immunoprecipitated and blotted for Ob-R in all lines (Figure 2D). Collectively, the results of these immunoprecipitation experiments indicate that the insulin-like growth factor-I receptor and leptin receptor interact in human breast cancer cells.
Figure 2. Interaction between IGF-IR and Ob-R in breast cancer.
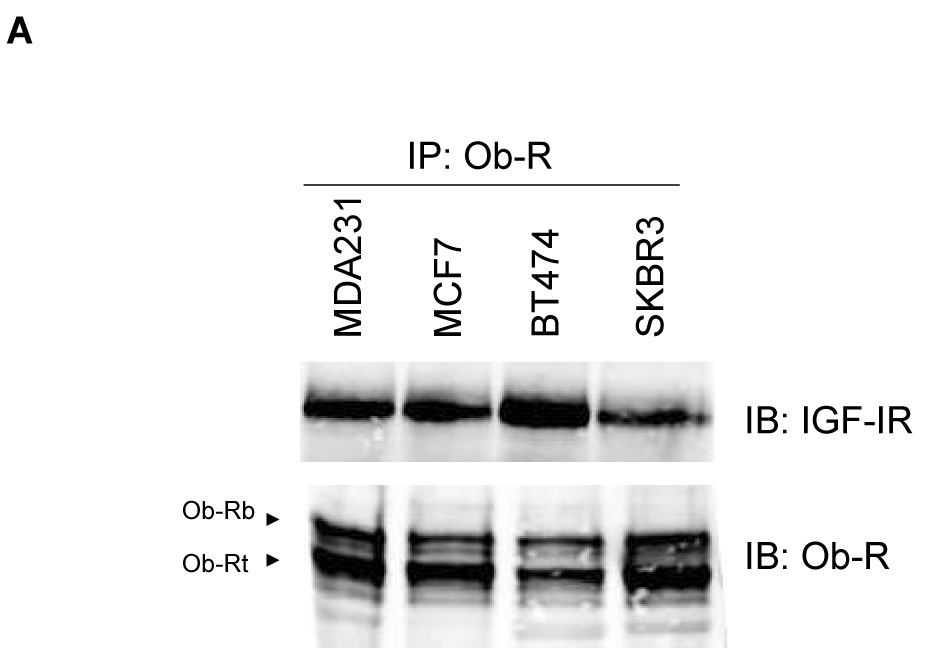
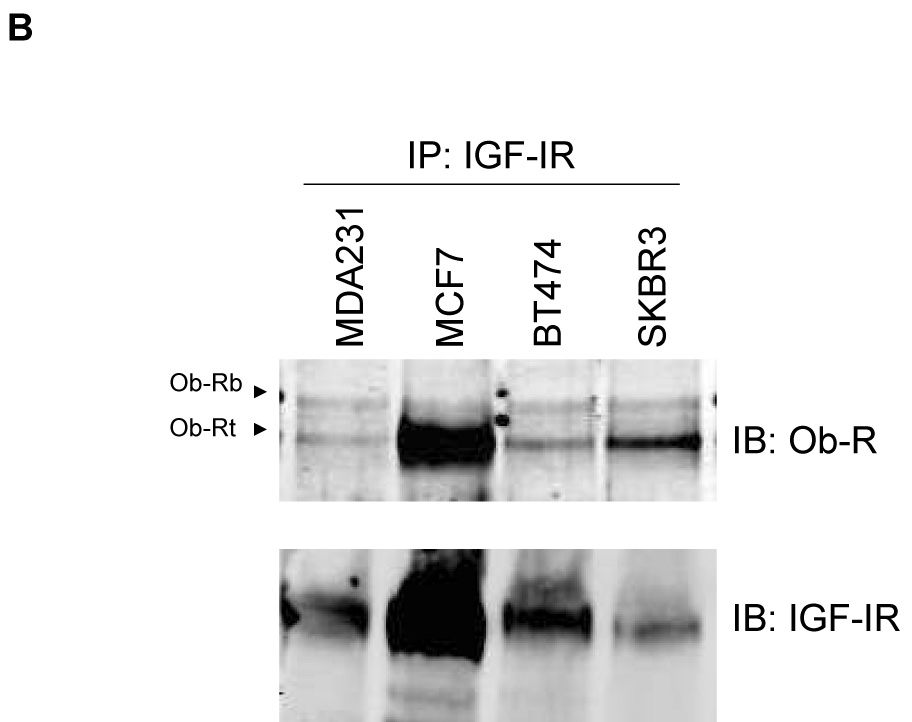
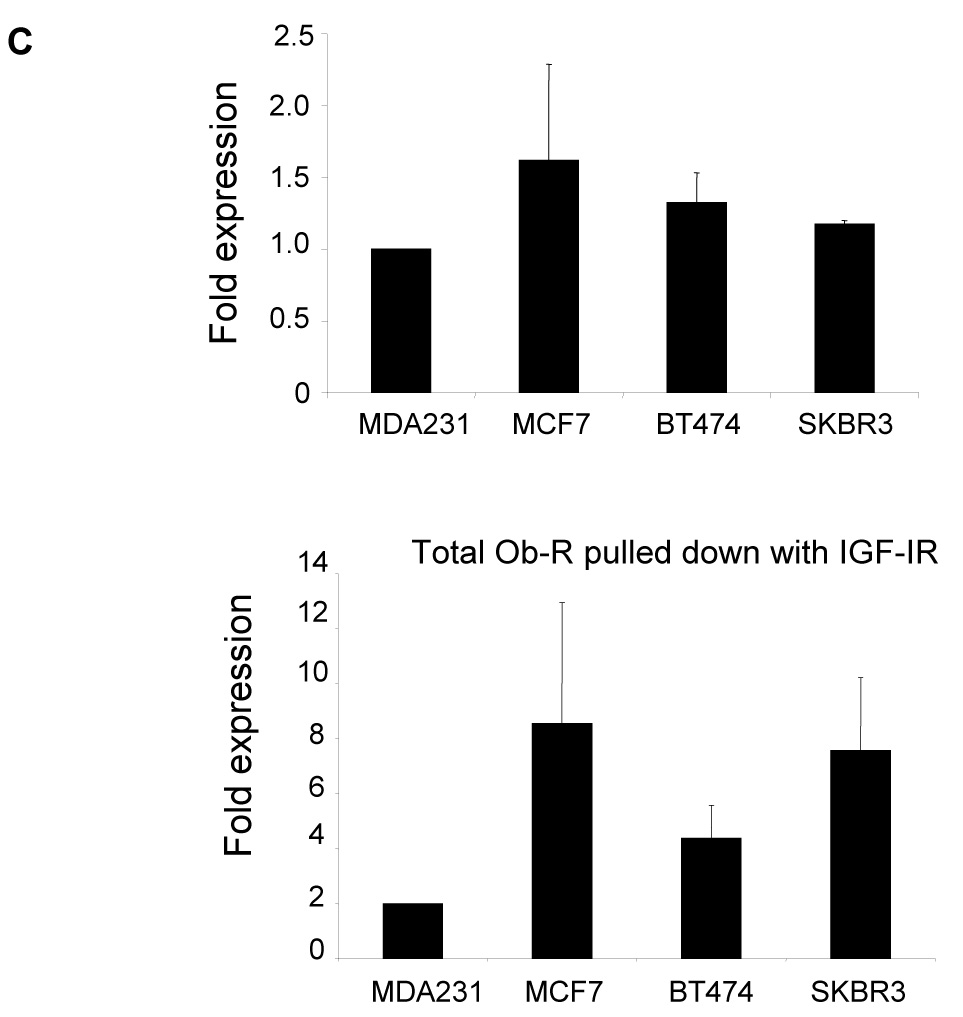
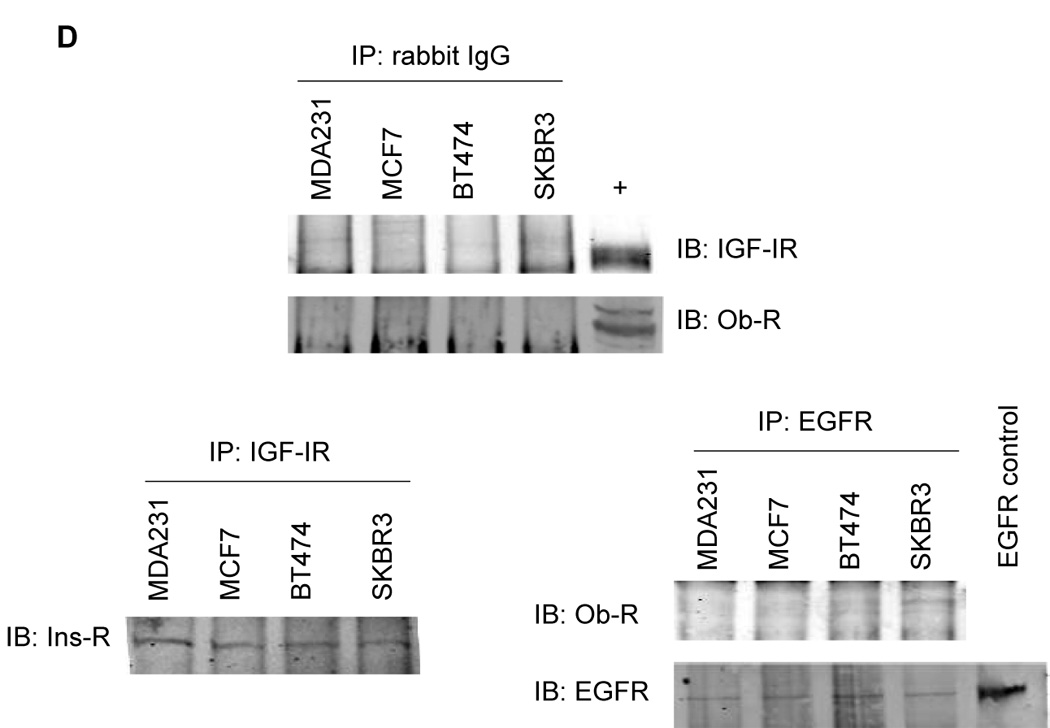
The breast cancer lines MDA-MB-231 (MDA231), MCF7, BT474, and SKBR3 were lysed for total protein. (A) Ob-R and (B) IGF-IR were immunoprecipitated (IP) (1 µg of antibody) from total protein extracts (200 µg) and immunoblotted (IB) to detect IGF-IR and Ob-R. Ob-R immunoprecipitation pulled down IGF-IR; conversely, IGF-IR immunoprecipitation pulled down Ob-R. (C) Quantitation of IP experiments is shown. Error bars represent standard deviation between three independent experiments. Values were normalized to the MDA231 cells in lane 1. (D) Cell lysates were immunoprecipitated using 1 µg rabbit IgG and immunoblotted for IGF-IR and Ob-R as a negative control. On IGF-IR blot, total lysate from MCF7 cells is included as a positive control (+) for the antibody; on Ob-R blot, lysate from COLO320DM cells was purchased as a positive control (+) for the H-300 antibody from Santa Cruz. As a positive IP control, cell lysates were immunoprecipitated with IGF-IR antibody and blotted for insulin receptor (Ins-R), which is known to interact with IGF-IR. EGFR tyrosine kinase receptor was also immunoprecipitated and blotted for Ob-R, with MDA231 total cell lysate added as a positive control for EGFR. Our results demonstrate that the IGF-I receptor and leptin receptor form a protein complex in breast cancer cells.
Insulin-like growth factor-I receptor cross signals to the leptin receptor
To determine the effect of IGF-IR/leptin receptor interaction on receptor signaling, MCF7 cells were serum-starved overnight, and then stimulated with IGF-I (100 ng/mL) for up to one hour. IGF-IR phosphorylation was induced within 5 min (Figure 3A), while total IGF-IR levels were unaltered. Importantly, phosphorylation of Ob-R was also induced within 5 min of IGF-I exposure, suggesting potential cross signaling from IGF-IR to leptin receptor. Similarly, in BT474 cells (Figure 3B) and MDA231 cells (Figure 3C), IGF-I stimulation induced phosphorylation of both IGF-IR and Ob-R within 5 min, without affecting total levels of either receptor. To determine if IGF-I stimulates phosphorylation of the leptin receptor via the IGF-IR kinase, MCF7 cells were treated with the IGF-IR kinase inhibitor I-OMe-AG538 and stimulated with IGF-I (Figure 3D). Immunoblotting demonstrated that inhibition of IGF-IR kinase blocked IGF-I-stimulated phosphorylation of leptin receptor. Thus, IGF-I cross signals to the leptin receptor via the IGF-IR kinase.
Figure 3. Evidence of cross talk from IGF-IR to Ob-R: IGF-I induces phosphorylation of Ob-R which is blocked by IGF-IR kinase inhibition.
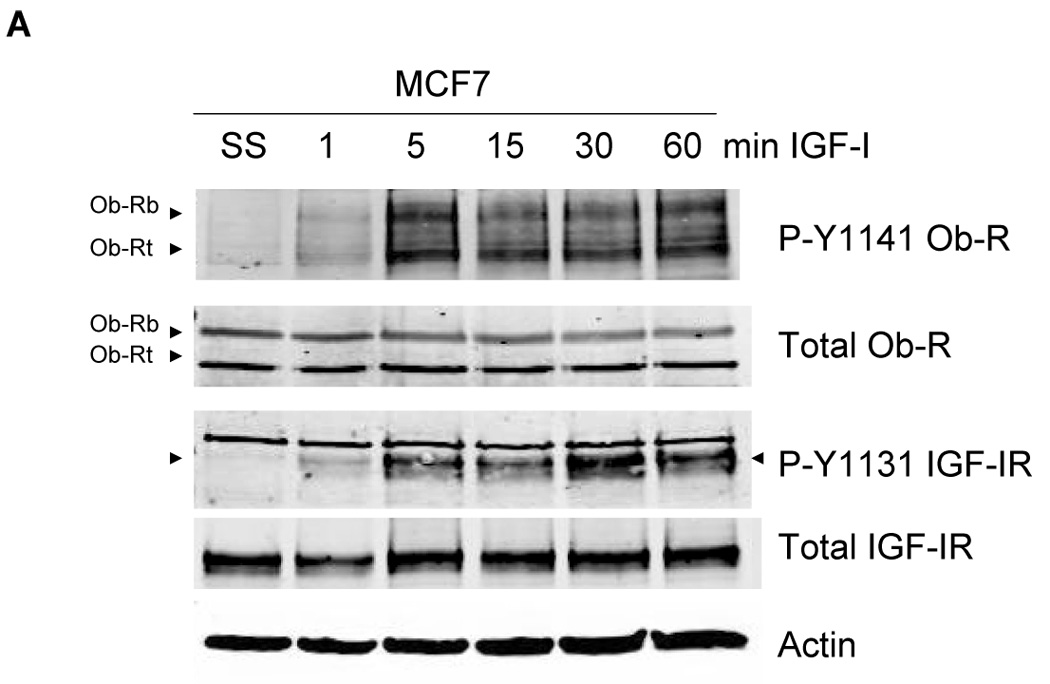
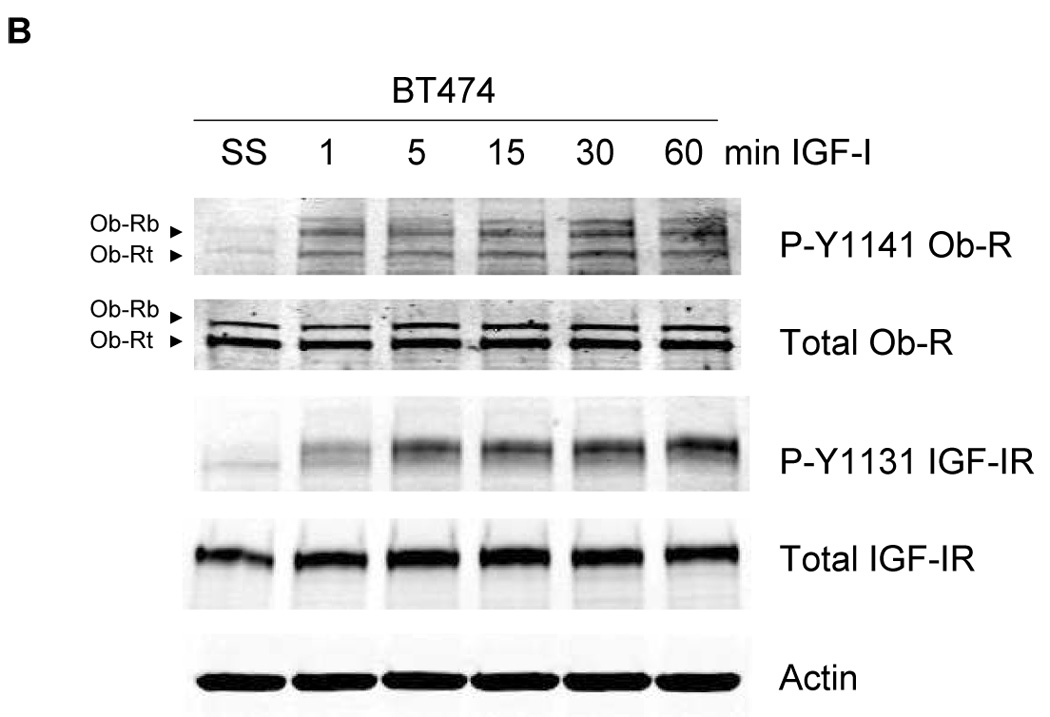
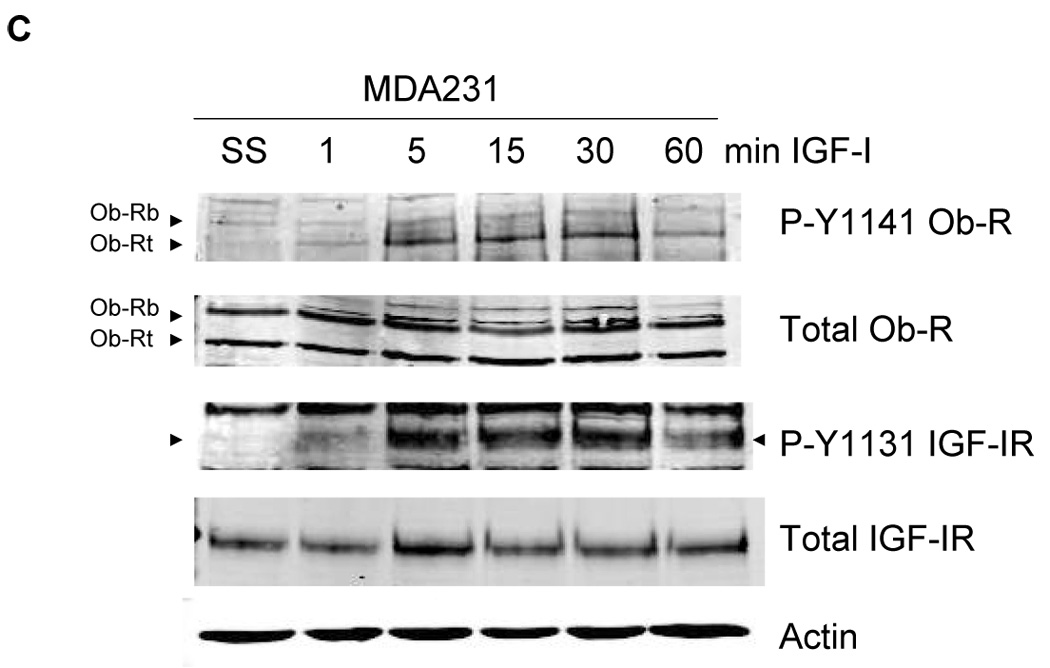
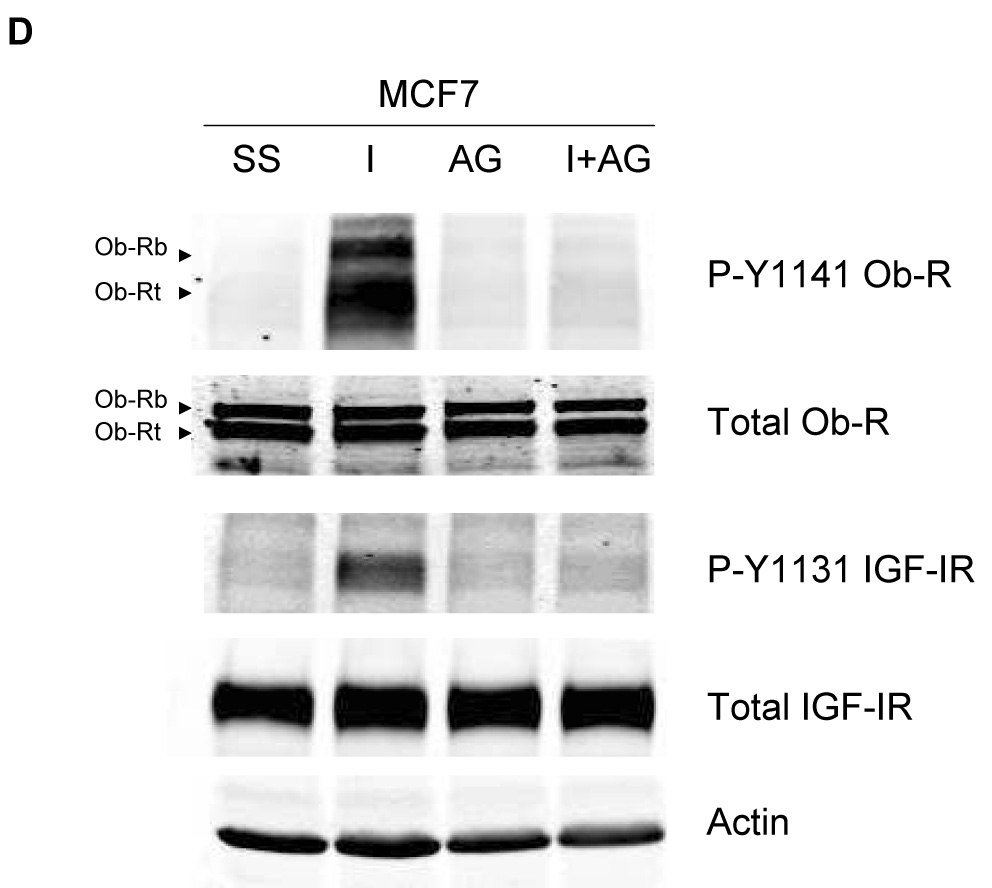
(A) MCF7, (B) BT474, and (C) MDA231 cells were serum-starved overnight, and then stimulated with IGF-I (100 ng/mL) for 1, 5, 15, 30, or 60 min. Cells were lysed for protein and total protein extracts (50 µg) were immunoblotted (SS, serum-starved control) for phosphorylated tyrosine 1141 on leptin receptor (p-Y1141-Ob-R), total Ob-R, phosphorylated tyrosine 1131 on IGF-IR (p-Tyr1131 IGF-IR), IGF-IR beta, and actin as a loading control. IGF-I stimulated phosphorylation of IGF-IR within 5 min in all cell lines. Importantly, phosphorylation of the leptin receptor was also induced within 5 minutes of IGF-I exposure. Total receptor levels did not change. (D) MCF7 cells were serum-starved overnight, then stimulated with IGF-I (100 ng/mL) for 5 min and/or treated with the IGF-IR kinase inhibitor I-OMe-AG538 (10 µM overnight). Total protein was immunoblotted for p-Y1141-Ob-R, total Ob-R, p-Tyr1131 IGF-IR, and total IGF-IR. Experiments were performed at least twice. Inhibition of IGF-IR kinase blocked IGF-I-mediated phosphorylation of leptin receptor, supporting cross talk from the IGF-IR kinase to leptin receptor. (SS, serum-starved control; I, IGF-I; AG, I-OMe-AG538; I+AG, IGF-I + I-OMe-AG538)
Having established that IGF-IR stimulates phosphorylation of the leptin receptor, we examined IGF-I-mediated effects on downstream receptor signaling. MCF7 cells were stimulated with IGF-I and immunoblotted for phosphorylated and total JAK2 and STAT3 (Figure 4A) and for phosphorylated and total Akt, ERK1/2, and p38MAPK (Figure 4B). Significant phosphorylation of JAK2 and STAT3 was observed in response to IGF-I within 5 min. IGF-I also activated the PI3K pathway as shown by phosphorylation of Akt. Phosphorylation of ERK1/2 and p38 MAPK was rapidly activated but transient versus other signaling pathways. Collectively, these results support the concept that IGF-I cross activates the leptin receptor signaling pathway, although the signaling molecules examined are downstream of multiple growth factor receptors, and thus do not strictly confirm activation of leptin receptor signaling. However, as leptin receptor phosphorylation was induced by IGF-I and blocked by IGF-IR kinase inhibitor on tyrosine 1141, which is the phosphorylation site that binds STAT3 and activates downstream signaling, our results strongly suggest that IGF-IR induces activation of the leptin receptor.
Figure 4. IGF-I activates downstream signaling.
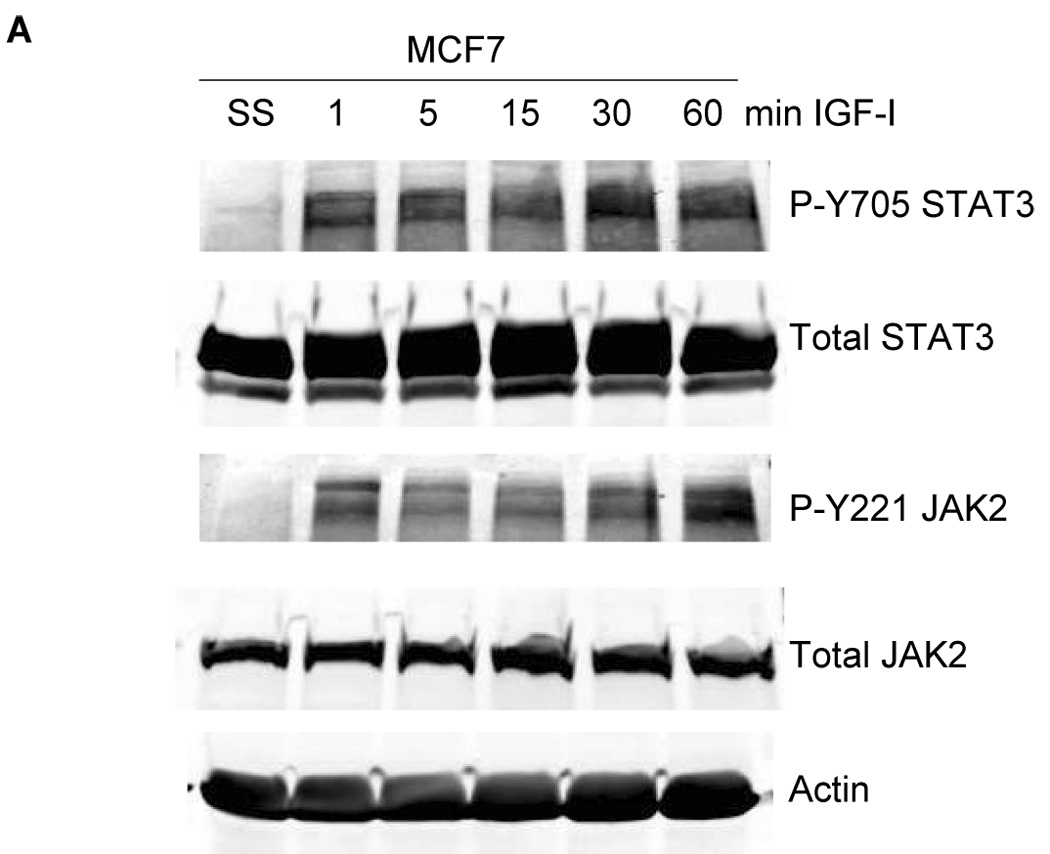
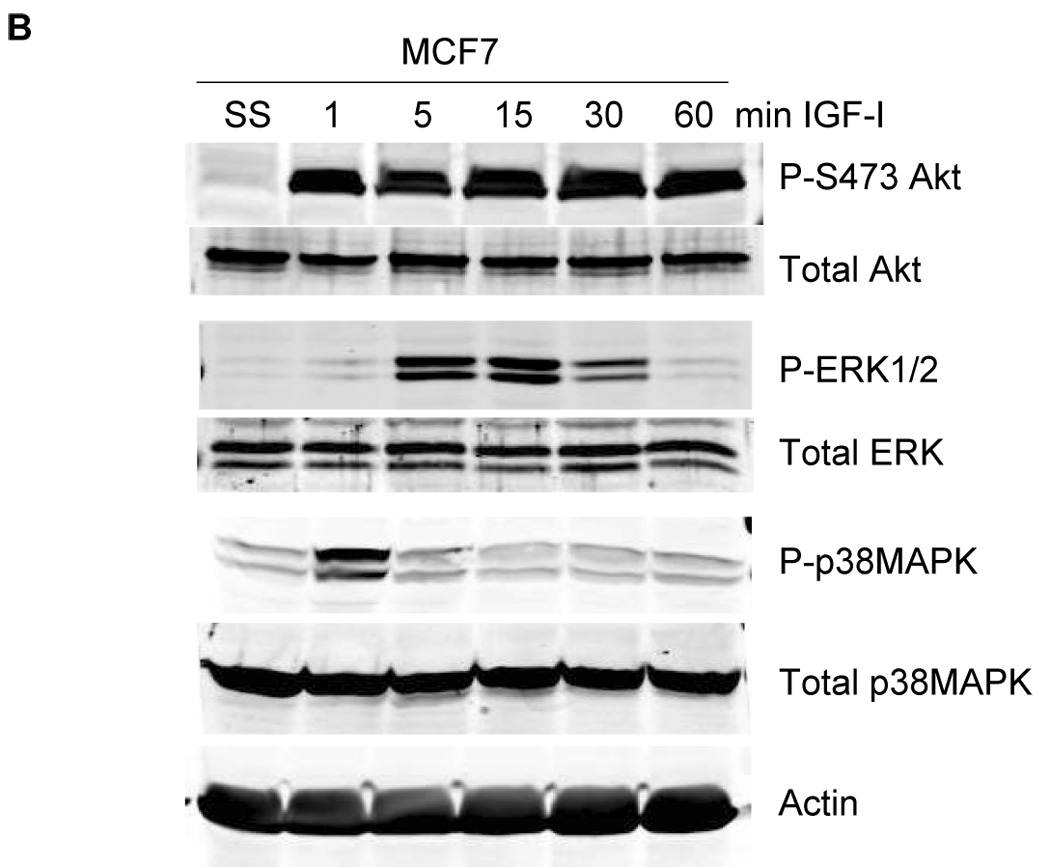
MCF7 cells were serum-starved overnight, and then stimulated with IGF-I (100 ng/mL) for 1, 5, 15, 30, or 60 min. Total protein extracts (50 µg) were immunoblotted for (A) the downstream leptin signaling molecules p-STAT3 (Tyr705), total STAT3, p-JAK2 (Tyr221), and total JAK2 (24B11); and (B) for molecules downstream of both leptin receptor and IGF-IR, p-Akt (Ser 473), total Akt, p-p42/p44 MAPK (Thr202/Tyr204) (ERK1/2), total p42/p44 MAPK (ERK1/2), p-p38MAPK (pThr180/Tyr182), and total p38MAPK. IGF-I induced phosphorylation of STAT3 and JAK2, consistent with IGF-I-mediated activation of leptin signaling, and also activated Akt, ERK1/2, and p38MAPK signaling.
Insulin-like growth factor-I receptor / leptin receptor cross talk is unidirectional
We next examined whether cross talk occurs in the opposite direction, i.e. from the leptin receptor to IGF-IR. MCF7 cells were serum starved and stimulated with leptin (1000 ng/mL) for up to 6 hours. Leptin induced phosphorylation of leptin receptor within 5 min (Figure 5A). However, phosphorylation of IGF-IR at either tyrosine 1131 or tyrosine 1135 and1136 was not stimulated by leptin at these time points up to 6 hours, nor was it stimulated at shorter time point increments or longer time points up to 24 hours or with lower doses of leptin (not shown). As a positive control, IGF-I stimulated phosphorylation of IGF-IR as expected and also induced phosphorylation of leptin receptor as previously observed (Figure 3). Similarly, BT474 cells stimulated with leptin showed phosphorylation of leptin receptor but not of IGF-IR at either of the three sites examined (Tyrosine 1131, 1135, 1136) (Figure 5B). Thus, our results suggest a unidirectional cross talk from the IGF-I receptor to the leptin receptor in breast cancer cells.
Figure 5. Evidence of unidirectional cross talk: leptin does not induce phosphorylation of IGF-IR.
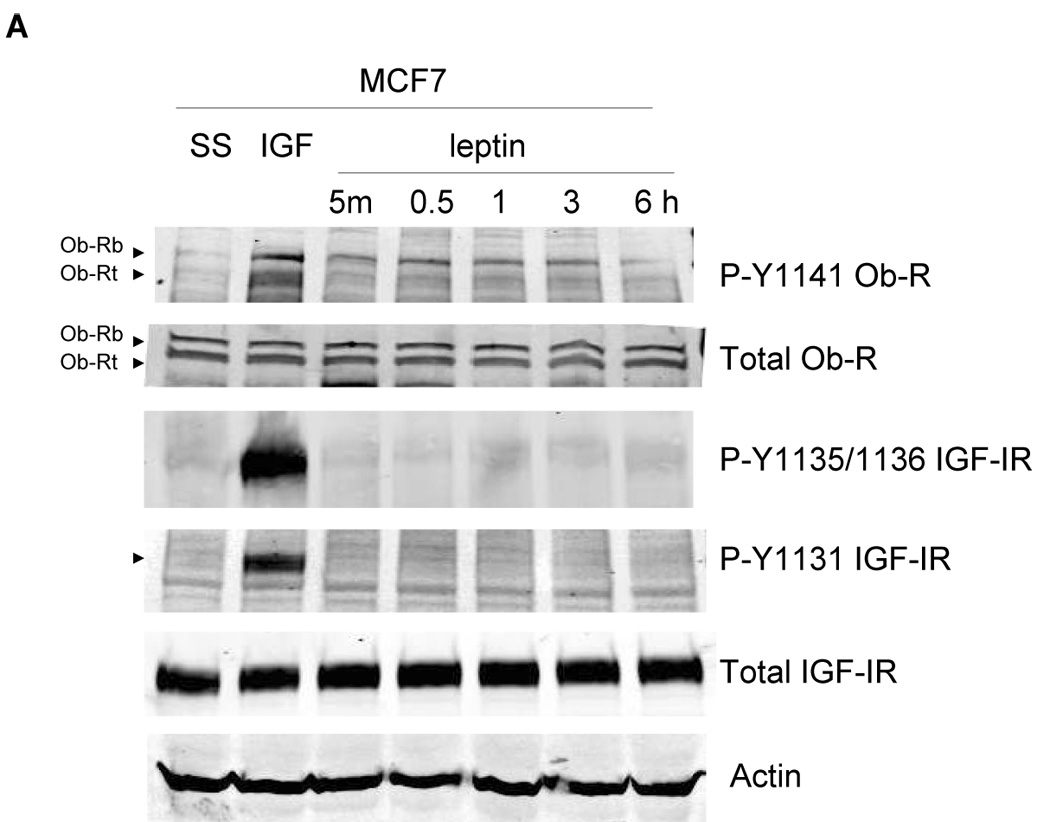
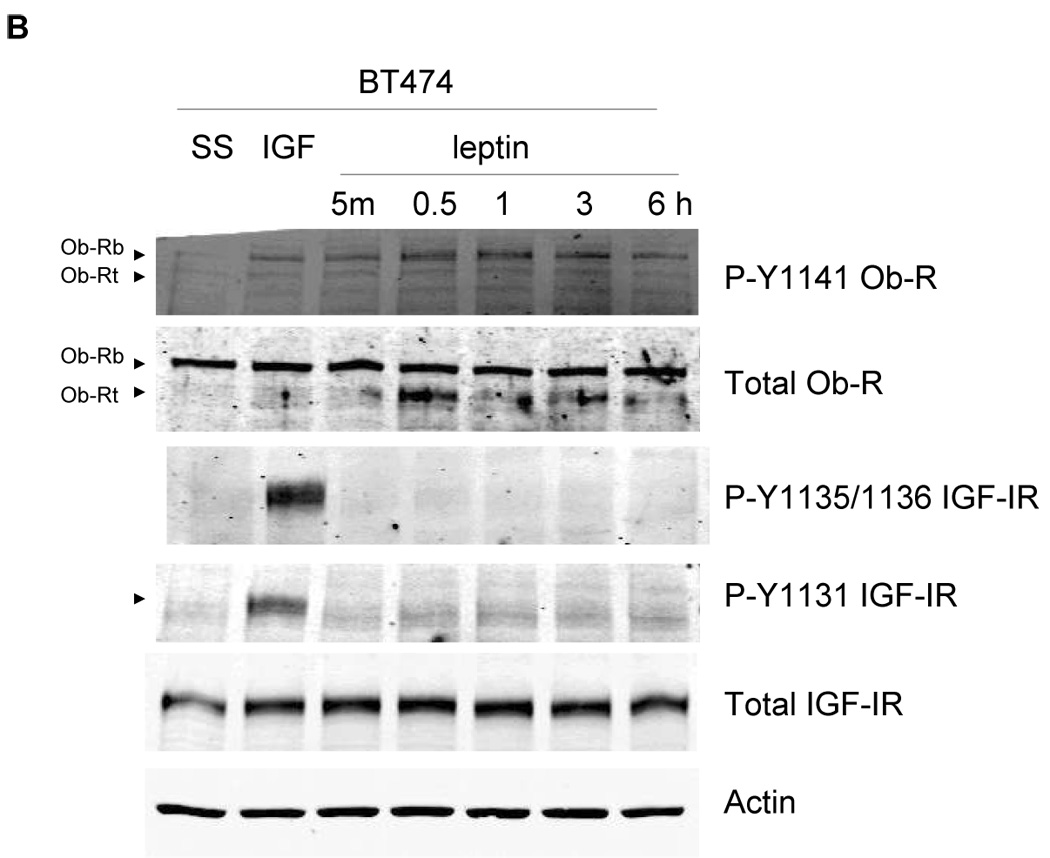
(A) MCF7 and (B) BT474 cells were serum-starved overnight, and then stimulated with IGF-I (100 ng/mL) for 5 min or leptin (1000 ng/mL) for 5 min, or 0.5, 1, 3, or 6 hours. Cells were lysed for protein and total protein extracts (50 µg) were immunoblotted (SS, serum-starved control) for p-Y1141-Ob-R, total Ob-R, p-Tyr1131 IGF-IR, p-Tyr1135/36 IGF-IR, total IGF-IR beta, and actin. Experiments were performed at least twice. IGF-I stimulated phosphorylation of IGF-IR and Ob-R within 5 min in both cell lines as expected, and served as a positive control. Leptin stimulated phosphorylation of Ob-R in both lines, but did not induce phosphorylation of IGF-IR at the phosphorylation sites examined, suggesting that receptor cross talk is unidirectional,occurring from IGF-IR to Ob-R only.
Discussion
Epidemiologic studies estimate that obesity increases the risk of breast cancer by up to 50% (3). The molecular mechanisms guiding obesity-associated breast cancer are not well understood, but are likely to involve an increased production and secretion of obesity-associated hormones (22). IGF-I and leptin are capable of regulating mammary tissue growth at multiple levels (5). Both hormones are secreted by abdominal adipocytes, resulting in endocrine effects on various tissues including the breast. Paracrine growth stimulatory effects occur via IGF-I and leptin released by the adipocyte component of stroma surrounding breast epithelial cells or existing breast tumor cells. In addition, an autocrine signaling component is present as breast cancer cells themselves produce and secrete IGF-I and leptin and express cell surface receptors for both ligands. Thus, IGF-I and leptin represent a molecular link between adipose tissue and mammary tissue.
The IGF-IR and Ob-R signaling pathways have each been independently implicated in the development and progression of breast cancer. High circulating levels of IGF-I have been associated with an increased risk of developing breast cancer, and patients with existing breast cancer expressed high serum levels of IGF-I (17). In addition, transgenic mouse models overexpressing IGF-I, IGF-II, or IGF-IR showed an increased incidence of mammary tumor formation (16,17,23,24). Conversely, liver-specific depletion of IGF-I caused reduced circulating levels of IGF-I in mice, resulting in diminished IGF-I endocrine effects on mammary tissue, and, ultimately, reduced incidence of breast tumors (25). Similar to the IGF-I signaling pathway, leptin signaling has been associated with breast cancer. Leptin and its receptor were shown by immunohistochemistry to be overexpressed in primary and metastatic breast cancers relative to non-cancer tissues (5). Expression of both leptin and Ob-R was most abundant amongst high-grade tumors, supporting a role for this pathway in breast cancer progression. In addition, in vivo models showed that while mice that overexpress transforming growth factor (TGF)-alpha developed mammary tumors, leptin-deficient TGF-alpha mice were resistant to mammary tumor development (13), illustrating the important contribution of the leptin signaling pathway to some forms of breast cancer. Hence, since IGF-I and leptin are frequently detected in the serum of breast cancer patients, and since both receptors are overexpressed in a majority of breast tumors, we sought to determine whether molecular interactions occur between IGF-IR and leptin receptor in breast cancer.
We demonstrated the following novel findings (Figure 6):
Figure 6. A novel unidirectional cross talk from IGF-IR to Ob-R in breast cancer.
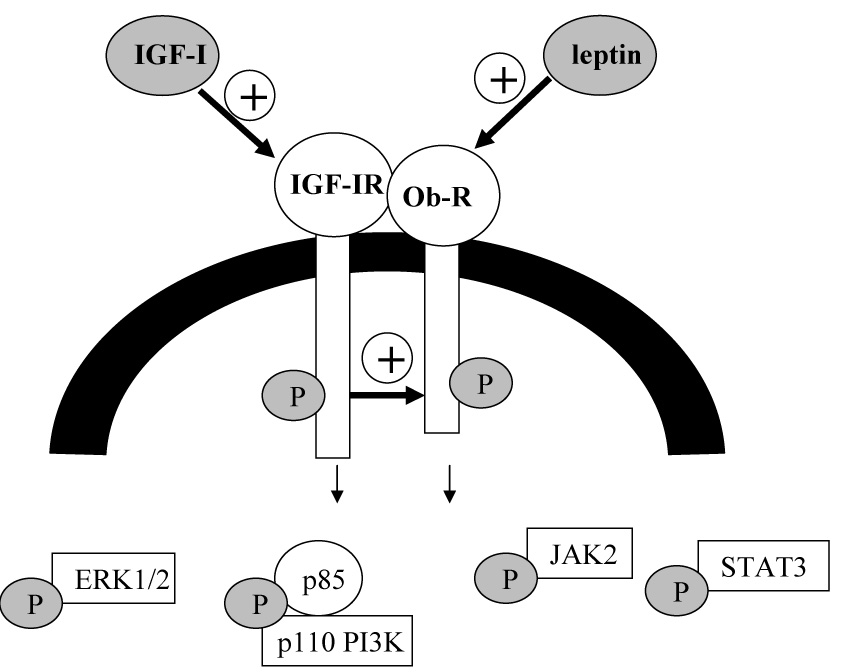
Our results indicate that the IGF-I and leptin receptors interact in human breast cancer cells. Further, cross talk occurs from IGF-IR to Ob-R such that IGF-I stimulation induces phosphorylation and activation of Ob-R. IGF-IR kinase inhibition blocks IGF-I-mediated Ob-R activation. Cross talk is unidirectional, as leptin does not activate IGF-IR.
(1) The IGF-I and leptin receptors interact in human breast cancer cells. Of potential interest, IGF-IR may preferably associate with Ob-Rt versus Ob-Rb in MCF7, BT474, and SKBR3 cells, as more of this isoform was pulled down in the IGF-IR immunoprecipitates (Figure 2B); total levels of both Ob-R isoforms were similar in each line (Figure 1A).
(2) Cross signaling occurs from IGF-IR to Ob-R in breast cancer. IGF-I stimulation induces phosphorylation and activation of Ob-R, while IGF-IR kinase inhibition blocks IGF-Imediated Ob-R activation. Downstream signaling molecules examined included JAK2, STAT3, Akt, and ERK1/2, all of which are functional in the leptin and IGF-IR pathways as well as in multiple other signaling pathways. Thus, the IGF-I signaling experiments do not strictly indicate that IGF-I induces activation of one particular pathway. However, our results clearly indicate that IGF-I induces phosphorylation of Ob-R on tyrosine 1141. Phosphorylation of tyrosine 1141 is required for Ob-R to bind to the STAT3 transcription factor, which is then activated by JAK2 and translocated to the nucleus to stimulate transcription of downstream target genes (26). Thus, our results indicate that IGF-I activates Ob-R via the IGF-IR kinase.
(3) Cross talk is unidirectional, as leptin does not activate IGF-IR. While it is feasible that other phosphorylation sites on IGF-IR may be affected by leptin stimulation, the three sites examined here (Tyrosine 1131 and 1135/1136) were not affected by leptin. These three phosphorylation sites are the critical sites known to be required for IGF-IR mitogenicity and transforming activity (27). Thus, the inability of leptin to induce phosphorylation at these sites suggests that the leptin hormone alone is not likely to affect IGF-IR oncogenic function in breast cancer. However, since IGF-IR cross talks to Ob-R, it is feasible that Ob-R may contribute to IGF-IR molecular or biological effects, and is worthy of further study.
Thus, we have identified a novel receptor interaction and unidirectional cross talk involving the IGF-IR and leptin receptor, which has not been previously described. Interestingly, Garofalo et al. (5) showed that IGF-I can induce leptin transcript levels in MCF7 cells. Our results further support this concept of IGF-I-mediated positive regulation of the leptin pathway.
Cross talk from IGF-IR to other signaling pathways appears to be a potentially common mechanism used by cancer cells to enhance tumor growth, and supports the significance of the IGF-I system to the biology of breast cancer, as well as the relevance of IGF-IR as a therapeutic target. We previously demonstrated that IGF-IR cross talks to the HER2 cell surface receptor in breast cancer cells that have become resistant to the HER2-targeted agent trastuzumab (19). Others have also shown that IGF-IR is capable of cross signaling to the epidermal growth factor receptor (18) and to the estrogen receptor (14). Thus, understanding the mechanisms by which IGF-IR mediates activation of other growth factor signaling pathways is important to breast cancer research. We have examined the role of the Src kinase family in mediating IGF-IR cross talk to leptin receptor, and have found that Src kinase inhibition does not inhibit IGF-IR/Ob-R cross talk (not shown). Future studies will examine the molecular mechanisms mediating this receptor cross talk. In addition, co-targeting leptin receptor and IGF-IR as a strategy to inhibit breast cancer progression, as well as the contribution of leptin receptor to IGF-I-mediated pro-mitogenic and anti-apoptotic effects will be examined in breast cancer cells.
In summary, our results demonstrate for the first time that the IGF-I and leptin receptors physically form a protein complex in breast cancer cell lines, and, further, that there exists a oneway cross talk whereby IGF-IR induces phosphorylation and activation of the leptin receptor in breast cancer.
Materials and Methods
Materials
Human recombinant IGF-I (Sigma, St. Louis, MO) was dissolved at 100 µg/mL in PBS and used at 100 ng/mL in culture. Human recombinant leptin (EMD Biosciences, San Diego, CA) was dissolved at 1 mg/mL in PBS and used at 100 or 1000 ng/mL. I-OMe-AG538 IGF-IR kinase inhibitor (Sigma) was dissolved at 1 mM in PBS and used at 10 µM in culture.
Cell culture
MDA-MB-231 (MDA231), MCF7, BT474, and SKBR3 breast cancer cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA), and maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum.
Ligand stimulation
Cells were serum starved overnight, and then stimulated with IGF-I (100 ng/mL) for 1, 5, 15, 30, or 60 minutes (min), or leptin (1000 ng/mL) for 5 min, 0.5 hour (h), 1 h, 3 h, or 6 h. In addition, a subset of cells were serum starved, treated with the IGF-IR kinase inhibitor I-OMe-AG538 (10 µM overnight), and stimulated with IGF-I (100 ng/mL).
Immunoprecipitation
Total protein lysates (200 µg) were incubated with 1 µg of Ob-R or IGF-IR antibody, or 1 µg rabbit IgG, rotating for 4 hours, followed by addition of protein A/G-agarose (Cell Signaling) and rotating overnight. Beads were then washed three times in PBS containing 0.1% Tween 20 (PBS-T), and immunoblotted to detect Ob-R (H-300, Santa Cruz), IGF-IR (polyclonal, Cell Signaling), EGFR (monoclonal 1F4, Cell Signaling), or insulin receptor beta (polyclonal, Cell Signaling). Blots of immunoprecipitations were quantitated using NIH imaging software ImageJ.
Immunoblotting
Cells were lysed in buffer containing 10 mM Tris (pH 7.5), 100 mM NaCl, 1 mM EDTA, 1% NP40, and protease and phosphatase inhibitor cocktails (Sigma). Total protein extracts (50 µg) were immunoblotted using the following antibodies at the indicated dilutions: IGF-IR beta (polyclonal at 1:1000, Cell Signaling, Beverly, MA); p-Tyr1131-IGFIR/Tyr1146-IR (polyclonal at 1:200, Cell Signaling); p-Tyr1135/1136-IGF-IR/Tyr1150/1151-IR (polyclonal at 1:200, Cell Signaling); leptin receptor (Ob-R) (H-300 polyclonal at 1:200, Santa Cruz Biotechnology, Santa Cruz, CA); p-Y1141-Ob-R (polyclonal at 1:200, Santa Cruz); actin (monoclonal AC-15 at 1:5000, Sigma Chemical Company, St. Louis, MO); from Cell Signaling, polyclonal antibodies against p-STAT3 (Tyr705), total STAT3, p-JAK2 (Tyr221), total JAK2 (24B11), total Akt, p-Thr202/Tyr204 p42/p44 MAPK (ERK1/2), total p42/p44 MAPK (ERK1/2), p-pThr180/Tyr182 p38MAPK, and total p38MAPK, monoclonal 587F11 against p-Ser 473-Akt, each used at 1:1000 dilution, and monoclonal 1F4 anti-EGFR used at 1:200 dilution. Secondary antibodies were chosen according to the species of origin of the primary antibody. Protein bands were detected using the Odyssey Imaging System (Li-Cor Biosciences, Lincoln, NE). Bands were quantitated using NIH imaging software ImageJ.
Acknowledgments
Grant Support: Department of Defense W81XWH0610452 IDEA (R Nahta); National Cancer Institute K01CA118174 (R Nahta); Georgia Cancer Coalition Distinguished Cancer Scholar Award (R Nahta)
Footnotes
Conflicts of interest The authors declare that they have no conflicts of interest.
References
- 1.Reinier KS, Vacek PM, Geller BM. Risk factors for breast carcinoma in situ versus invasive breast cancer in a prospective study of pre- and post-menopausal women. Breast Cancer Res Treat. 2007;103:343–348. doi: 10.1007/s10549-006-9375-9. [DOI] [PubMed] [Google Scholar]
- 2.Chlebowski RT, Aiello E, McTiernan A. Weight loss in breast cancer patient management. J Clin Oncol. 2002;20:1128–1143. doi: 10.1200/JCO.2002.20.4.1128. [DOI] [PubMed] [Google Scholar]
- 3.Calle EE, Thun MJ. Obesity and cancer. Oncogene. 2004;23:6365–6378. doi: 10.1038/sj.onc.1207751. [DOI] [PubMed] [Google Scholar]
- 4.Garofalo C, Surmacz E. Leptin and cancer. J Cell Physiol. 2006;207:12–22. doi: 10.1002/jcp.20472. [DOI] [PubMed] [Google Scholar]
- 5.Garofalo C, Koda M, Cascio S, Sulkowska M, Kanczuga-Koda L, Golaszewska J, et al. Increased expression of leptin and the leptin receptor as a marker of breast cancer progression: possible role of obesity-related stimuli. Clin Cancer Res. 2006;12:1447–1453. doi: 10.1158/1078-0432.CCR-05-1913. [DOI] [PubMed] [Google Scholar]
- 6.Saxena NK, Sharma D, Ding X, Lin S, Marra F, Merlin D, et al. Concomitant activation of the JAK/STAT, PI3K/AKT, and ERK signaling is involved in leptin-mediated promotion of invasion and migration of hepatocellular carcinoma cells. Cancer Res. 2007;67:2497–2507. doi: 10.1158/0008-5472.CAN-06-3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wauters M, Considine RV, Van Gaal LF. Human leptin: from an adipocyte hormone to an endocrine mediator. Eur J Endocrinol. 2000;143:293–311. doi: 10.1530/eje.0.1430293. [DOI] [PubMed] [Google Scholar]
- 8.Bahrenberg G, Behrmann I, Barthel A, Hekerman P, Heinrich PC, Joost HG, et al. Identification of the critical sequence elements in the cytoplasmic domain of leptin receptor isoforms required for Janus kinase/signal transducer and activator of transcription activation by receptor heterodimers. Mol Endocrinol. 2002;16:859–872. doi: 10.1210/mend.16.4.0800. [DOI] [PubMed] [Google Scholar]
- 9.Bjorbaek C, Uotani S, da Silva B, Flier JS. Divergent signaling capacities of the long and short isoforms of the leptin receptor. J Biol Chem. 1997;272:32686–32695. doi: 10.1074/jbc.272.51.32686. [DOI] [PubMed] [Google Scholar]
- 10.Yin N, Wang D, Zhang H, Yi X, Sun X, Shi B, et al. Molecular mechanisms involved in the growth stimulation of breast cancer cells by leptin. Cancer Res. 2004;64:5870–5875. doi: 10.1158/0008-5472.CAN-04-0655. [DOI] [PubMed] [Google Scholar]
- 11.Garofalo C, Sisci D, Surmacz E. Leptin interferes with the effects of the antiestrogen ICI 182,780 in MCF-7 breast cancer cells. Clin Cancer Res. 2004;10:6466–6475. doi: 10.1158/1078-0432.CCR-04-0203. [DOI] [PubMed] [Google Scholar]
- 12.Ishikawa M, Kitayama J, Nagawa H. Enhanced expression of leptin and leptin receptor (OB-R) in human breast cancer. Clin Cancer Res. 2004;10:4325–4331. doi: 10.1158/1078-0432.CCR-03-0749. [DOI] [PubMed] [Google Scholar]
- 13.Cleary MP, Phillips FC, Getzin SC, Jacobson TL, Jacobson MK, Christensen TA, et al. Genetically obese MMTV-TGF-alpha/Lep(ob)Lep(ob) female mice do not develop mammary tumors. Breast Cancer Res Treat. 2003;77:205–215. doi: 10.1023/a:1021891825399. [DOI] [PubMed] [Google Scholar]
- 14.Surmacz E. Function of the IGF-I receptor in breast cancer. J Mamm Gl Biol Neopl. 2000;5:95–105. doi: 10.1023/a:1009523501499. [DOI] [PubMed] [Google Scholar]
- 15.Kahan Z, Gardi J, Nyari T, Foldesi I, Hajnal-Papp R, Ormandi K, et al. Elevated levels of circulating insulin-like growth factor-I, IGF-binding globulin-3 and testosterone predict hormone-dependent breast cancer in postmenopausal women: a case-control study. Int J Oncol. 2006;29:193–200. [PubMed] [Google Scholar]
- 16.Jones RA, Campbell CI, Gunther EJ, Chodosh LA, Petrik JJ, Khokha R, et al. Transgenic overexpression of IGF-IR disrupts mammary ductal morphogenesis and induces tumor formation. Oncogene. 2007;26:1636–1644. doi: 10.1038/sj.onc.1209955. [DOI] [PubMed] [Google Scholar]
- 17.Sachdev D, Yee D. Disrupting insulin-like growth factor signaling as a potential cancer therapy. Mol Cancer Ther. 2007;6:1–12. doi: 10.1158/1535-7163.MCT-06-0080. [DOI] [PubMed] [Google Scholar]
- 18.Knowlden JM, Hutcheson IR, Barrow D, Gee JM, Nicholson RI. Insulin-like growth factor-I receptor signaling in tamoxifen-resistant breast cancer: a supporting role to the epidermal growth factor receptor. Endocrinology. 2005;146:4609–4618. doi: 10.1210/en.2005-0247. [DOI] [PubMed] [Google Scholar]
- 19.Nahta R, Yuan LX, Zhang B, Kobayashi R, Esteva FJ. Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes totrastuzumab resistance of breast cancer cells. Cancer Res. 2005;65:11118–11128. doi: 10.1158/0008-5472.CAN-04-3841. [DOI] [PubMed] [Google Scholar]
- 20.Novosyadlyy R, Dudas J, Pannem R, Ramadori G, Scharf JG. Crosstalk between PDGF and IGF-I receptors in rat liver myofibroblasts: implication for liver fibrogenesis. Lab Invest. 2006;86:710–723. doi: 10.1038/labinvest.3700426. [DOI] [PubMed] [Google Scholar]
- 21.Pandini G, Frasca F, Mineo R, Sciacca L, Vigneri R, Belfiore A. Insulin/insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. J Biol Chem. 2002;277:39684–39695. doi: 10.1074/jbc.M202766200. [DOI] [PubMed] [Google Scholar]
- 22.Schaffler A, Scholmerich J, Buechler C. Mechanisms of disease: adipokines and breast cancer- endocrine and paracrine mechanisms that connect adiposity and breast cancer. Nat Clin Pract Endocrinol Metabol. 2007;3:345–354. doi: 10.1038/ncpendmet0456. [DOI] [PubMed] [Google Scholar]
- 23.Hadsell DL, Murphy KL, Bonnette SG, Reece N, Laucirica R, Rosen JM. Cooperative interaction between mutant p53 and des(1–3)IGF-I accelerates mammary tumorigenesis. Oncogene. 2000;19:889–898. doi: 10.1038/sj.onc.1203386. [DOI] [PubMed] [Google Scholar]
- 24.Bates P, Fisher R, Ward A, Richardson L, Hill DJ, Graham CF. Mammary cancer in transgenic mice expressing insulin-like growth factor II (IGF-II) Br J Cancer. 1995;72:1189–1193. doi: 10.1038/bjc.1995.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu Y, Cui K, Miyoshi K, Henninghausen L, Green JE, Setser J, et al. Reduced circulating insulin-like growth factor I levels delay the onset of chemically and genetically induced mammary tumors. Cancer Res. 2003;63:4384–4388. [PubMed] [Google Scholar]
- 26.Cao Q, Mak KM, Lieber CS. Leptin represses matrix metalloproteinase-1 gene expression in LX2 human hepatic stellate cells. J Hepatol. 2007;46:124–133. doi: 10.1016/j.jhep.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 27.Li S, Ferber A, Miura M, Baserga R. Mitogenicity and transforming activity of the insulin-like growth factor-I receptor with mutations in the tyrosine kinase domain. J Biol Chem. 1994;269:32558–32564. [PubMed] [Google Scholar]