

Abstract
This study demonstrates that benzo[g]chrysene-11,12-dihydrodiol (B[g]C-11,12-dihydrodiol) derived from the fjord-region parent hydrocarbon B[g]C is oxidized by rat AKR1C9 with a kcat/Km 100 times greater than that observed with the commonly studied bay-region benzo[a]pyrene-7,8-dihydrodiol (B[a]P7,8-dihydrodiol). Conversely, despite its strikingly similar structure to B[g]C-11,12-dihydrodiol, benzo[c]phenanthrene-3,4-dihydrodiol (B[c]Ph-3,4-dihydrodiol) is consumed by AKR1C9 at sluggish rates comparable to those observed with B[a]P-7,8-dihydrodiol. CD spectroscopy revealed that only the (+)-B[g]C-11,12-dihydrodiol stereoisomer was oxidized, while AKR1C9 oxidized both stereoisomers of B[a]P7,8-dihydrodiol and B[c]Ph-3,4-dihydrodiol. The (+)-S,S-and (-)-R,R-stereoisomers of B[g]C-11,12dihydrodiol were purified by chiral RP-HPLC. The 11S,12S-stereoisomer was oxidized at the same rate as the racemate. The 11R,12R-stereoisomer did not act as an inhibitor to AKR1C9, indicating that the (-)-R,R-stereoisomer was excluded from the active site. To understand the basis of stereochemical preference, we screened alanine-scanning mutants of active site residues of AKR1C9. These studies revealed that in comparison to the wild type, F129A, W227A, and Y310A enabled the oxidation of both the B[g]C-11S,12S-dihydrodiol and the B[g]C-11R,12R-dihydrodiol. Molecular modeling revealed that unlike B[a]P-7,8-dihydrodiol and B[c]Ph-3,4-dihydrodiol, B[g]C-11,12-dihydrodiol enantiomers are significantly bent out of plane. As a consequence, the (-)-R,R-stereoisomer was prevented from binding to the active site because of unfavorable interactions with F129, W227, or Y310. Additionally, LC/MS validated that the product of the reaction of B[g]C-11,12-dihydrodiol oxidation catalyzed by AKR1C9 was B[g]C-11,12-dione, which was trapped in vitro with the nucleophile 2-mercaptoethanol. The similarity between rates of trans-dihydrodiol oxidation by the rat and human liver specific AKRs (AKR1C9 and AKR1C4) implicate these enzymes in hepatocarcinogenesis in rats observed with the fjord-region PAH.
Introduction
Polycyclic aromatic hydrocarbons (PAH) are ubiquitous environmental pollutants created during incomplete combustion and pyrolysis (1, 2) that require metabolic activation in order to elicit their carcinogenic effects (3-5). Human exposure to these carcinogens occurs from a myriad of sources including cigarette smoke (6), coal soot, charbroiled meats, fossil fuel exhaust, and petroleum refining (7, 8). Annually, 900-1300 tons of PAH are emitted in the U.S. leading to contamination of soil, water (9), and air in the form of fine particulate matter. A representative sample of PAH contained in samples of particulate size 2.5 μm collected from St. Louis, MO showed that the most studied PAH, the bay-region benzo[a]pyrene (B[a]P), a Group I carcinogen, is present at 3.1 μg/g (10). Group 1 carcinogens refer to compounds ranked as known human carcinogens by the World Health Organization, IARC (11). Comparatively, fjord-region PAHs, such as benzo[g]chrysene (B[g]C) and benzo[c]phenanthrene (B[c]Ph), are present at concentrations of 0.07 and 0.9 μg/g respectively (10). Despite the lower occurrence of the fjord-region PAH, these compounds are of interest because bay-region substitution of PAH (methylation) or introduction of a fjord-region is known to result in an increase in mutagenicity and carcinogenicity over the unsubstituted PAH (12). Because less is known about these PAH, they are currently ranked as Group 2B; “insufficient data to identify as human carcinogens” (13). If more data were available on their mode of activation, a reclassification could occur.
Two major pathways of PAH activation are possible, starting from racemic B[g]C-11,12-dihydrodiol. This compound is anticipated to undergo monooxygenation by P450s to yield (+)- and (-)-anti-diol-epoxide products (14, 15), which can form N2-deoxyguanosine adducts with DNA (16, 17) and act as a potential human carcinogens (18) (Scheme 1). Studies in mouse epidermis have identified P4501A1 as playing a major role in the bioactivation of B[a]P, B[g]C, and B[c]Ph (19).
Scheme 1.

Proposed Competing Pathways for the Metabolic Oxidation of 11S,12S- and B[g]C-11R,12R-dihydrodiols
An alternative fate involves trans-dihydrodiol oxidation catalyzed by aldo-keto reductases (AKRs) to yield a ketol that spontaneously rearranges into a catechol. The unstable catechol undergoes two one-electron oxidations, first forming an o-semiquinone (20) anion radical and H2O2 (21) followed by the formation of an o-quinone (22) with consequential superoxide anion production. In turn, H2O2 undergoes Fenton chemistry to produce a hydroxyl radical, another reactive oxygen species (ROS) (21).
At this point, the fate of the o-quinone bifurcates, where one option is that the o-quinone can be reduced back to the catechol and undergo futile redox cycling in the presence of NAD(P)H to amplify ROS (21, 23). In turn, ROS can inflict large amounts of oxidative damage on cells, causing strand scission (20) and DNA lesions, for example, 8-oxo-2′-deoxyguanosine (23, 24). The second option is that o-quinones are highly reactive Michael acceptors and can react with deoxyguanosine to form bulky or depurinating DNA adducts (25-27). Both oxidative and covalent DNA adducts may lead to G to T transversions in DNA, which are commonly observed in the tumor suppressor p53 in lung cancer (28). Previously, rat liver AKR1C9 oxidized racemic B[a]P-7,8 dihydrodiol to form an o-quinone product, B[a]P-7,8-dione, both in vitro and in transfected MCF-7 cells (22, 29).
The reasons for the high mutagenic potential of the fjord-region PAH have not been fully explained. In this study, we demonstrate that B[g]C-11,12-dihydrodiol is oxidized by AKR1C9 with a kcat/Km over 100 times greater than that observed with the well-characterized bay-region B[a]P-7,8-dihydrodiol. When the oxidation of racemic B[g]C-11,12-dihydrodiol is run to completion, only the S,S-stereoisomer, which corresponds to the (+)-stereoisomer, is consumed. Despite the fact that B[g]C-11,12-dihydrodiol has one additional ring compared to B[c]Ph-3,4-dihydrodiol, B[c]Ph-3,4-dihydrodiol is consumed by AKR1C9at a sluggish rate comparable to that observed with B[a]P-7,8-dihydrodiol. Alanine-scanning mutagenesis of contact residues in the AKR1C9 active site revealed that in comparison to the wild type, the F129A, W227A, and Y310A mutants permit oxidation of B[g]C-11R,12R-dihydrodiol, which corresponds to the (-)-stereoisomer; thus, these mutants oxidize both stereoisomers of B[g]C-11,12-dihydrodiol. The structural basis for stereochemical preference is examined. The toxicological consequence of oxidizing B[g]C-11S,12S-dihydrodiol efficiently by AKRs is discussed.
Experimental Procedures
Caution
PAHs are potentially hazardous chemicals and should be handled with care in accordance with the NIH Guidelines for Use of Chemical Carcinogens.
Chemicals and Reagents
B[c]Ph-3,4-dihydrodiol was kindly provided by Dr. Mahesh K. Lakshman, The City College and The City University of New York (138th Street at Convent Avenue, New York, New York 10031). B[a]P-4,5-dihydrodiol, B[g]C-11,12-dihydrodiol, and B[g]C-11,12-dione were synthesized according to published methods (30). All PAHs were analyzed by LC/MS for identity and purity prior to use. NAD+ and NADP+ were obtained from Boehringer Manheim Biochemicals (Indianapolis, IN). AMPSO and Chromasolv plus DMSO were procured from Sigma-Aldrich Chemical Co. (St. Louis, MO). MOPS was purchased from Fisher Chemicals (Fairlawn, NJ). All other chemicals were of the highest grade available, and all solvents for LC/MS were of HPLC grade. The molar extinction coefficient for B[g]C-11,12-dione was determined to be ε262 = 48,553 M-1 cm-1 in ethanol.
Expression and Purification of Recombinant Wild type and Mutant of AKR1C9
Wild type homogeneous recombinant AKR1C9 was purified as described (31). Enzyme solutions were titered by measuring the oxidation of androsterone to androstanedione under standard assay conditions (31). The specific activity of the aliquots was 1.6 μmol/min/mg at 25 °C. Alanine-scanning mutants used were T24A, L54A, F118A, F129A, T226A, W227A, N306A, and Y310A and have been purified and characterized previously (32).
Determination of the Steady State Kinetic Parameters of B[g]C-11,12-dihydrodiol and B[c]Ph-3,4-dihydrodiol Oxidation via Absorbance Spectroscopy
Initial velocities for the oxidation of the PAH trans-dihydrodiols catalyzed by AKR1C9 were measured using a range of trans-dihydrodiol concentrations (5 to 60 μM) and 2.3 mM NADP+ in 50 mM AMPSO buffer (pH 9.0) in the presence of 8% v/v DMSO as described in Palackal et al. (33). The resultant v versus [S] plots were linear and revealed that it was not possible to observe saturation kinetics because of the poor solubility of the trans-dihydrodiol. By running subsequent reactions at 20 μM, we ensured that reactions were run under pseudo-first-order conditions so that v/[S] provides an estimate of Vmax/[Km]. Control reactions were performed in which one of the following were absent: NADP+, trans-dihydrodiol, or enzyme. Initial velocity measurements were corrected for the 33% inhibition of AKR1C9 by DMSO. Therefore, the velocities measured were multiplied by 1.33 to obtain the true initial velocity.
Determination of the Kinetic Parameters for the Oxidation of B[g]C-11,12-dihydrodiol and B[c]Ph-3,4-dihydrodiol Catalyzed by AKR1C9 Using RP-HPLC Methods
A RP-HPLC method was established to detect the consumption of B[g]C-11,12-dihydrodiol in vitro. Reactions containing 20 μM trans-dihydrodiol and 2.3 mM NADP+ in 50 mM AMPSO buffer (pH 9.0) in the presence of 8% v/v DMSO were preincubated at 37 °C for 5 min. The reaction was initiated by the addition of AKR1C9 and quenched upon mixing with ice-cold acetone. Racemic B[a]P-4,5-dihydrodiol was added to the quenched reaction as an internal standard. Remaining unconsumed trans-dihydrodiol was extracted with ethyl acetate. Samples were dried under vacuum and resuspended in methanol. Aliquots were injected onto a RP-HPLC Column (Zorbax-ODS C18, 5μm, 4.6 mm × 250 mm; Agilent, Santa Clara, CA) on a Waters Alliance 2690 HPLC system with an in line photodiode array detector (PDA). Organic soluble metabolites were eluted at 0.5 mL/min with a water/methanol linear gradient from 60 to 90% methanol over 60 min. PAH metabolites were identified by comparison of retention time to those obtained with authentic synthetic standards. Consumption of the trans-dihydrodiol was monitored directly at 270.9 nm (ε = 60,000 M-1 cm-1). The loss of trans-dihydrodiol during sample workup was corrected for by normalization to the internal standard. Initial velocities were obtained from plots of trans-dihydrodiol consumed versus time.
Identification of the Stereoisomer of B[g]C-11,12-dihydrodiol Oxidized by AKR1C9 Using CD Spectroscopy
To identify the stereoisomer of B[g]C-11,12-dihydrodiol oxidized by AKR1C9, a 50 mL reaction containing 50 μM trans-dihydrodiol and NADP+ was run to completion. Aliquots of the reaction were removed over time to monitor the progress of the reaction via RP-HPLC. At the end point of the reaction (corresponding to 50% substrate depletion), the reaction was quenched by the addition of ethyl acetate, and unreacted trans-dihydrodiol was extracted. The sample was dried with anhydrous sodium sulfate, and ethyl acetate was removed under vacuum. Subsequently, samples were purified by thin layer chromatography on 20 × 20 GF (Analtech) plates using a 55:45 ethyl acetate/hexanes (v/v) solvent system. The unreacted stereoisomer was removed from the plate, purified, and subjected to CD spectroscopy on an AVIV CD spectrometer to determine the Cotton effect of the remaining stereoisomer. The (-)-stereoisomer corresponds to the 11S,12S configuration, and the (-)-stereoisomer corresponds to the 11R,12R configuration (34).
Separation of the B[g]C-11S,12S-dihydrodiol and B[g]C-11R,12R-dihydrodiol Enantiomers Using a Chiralcel OD-RH Column
A chiral RP-HPLC method was used to separate B[g]C-11S,12S-dihydrodiol and B[g]C-11R,12R-dihydrodiol from the racemic B[g]C-11,12-dihydrodiol mixture. Aliquots of racemic trans-dihydrodiol were dissolved in methanol and injected onto a Chiralcel OD-RH column (5μm, 2.1 mm × 150 mm; Chiral Technologies, Inc., West Chester, PA) on a Waters Alliance 2690 HPLC system with an in line PDA. The two enantiomers were eluted at 0.2 mL/min with a water/methanol linear gradient from 60 to 90% methanol over 30 min. The peak at 26 min was identified as B[g]C-11R,12R-dihydrodiol by coelution with B[g]C-11R,12R-dihydrodiol purified from an enzymatic reaction run to completion and identified by CD spectroscopy.
Inhibition Studies with B[g]C-11R,12R-dihydrodiol
The enzymatic oxidation of 20 μM B[g]C-11S,12S-dihydrodiol was monitored while the concentration of B[g]C-11R,12R-dihydrodiol was increased from 0 to 35 μM. The initial velocity (v) in each case was determined as a fraction of the initial velocity (vo), observed in the absence of the R,R-stereoisomer.
Preparation of the 2-Mercaptoethanol Conjugates of B[g]C-11,12-dione
Authentic thio-ether conjugate standards were synthesized by incubating 20 μM B[g]C-11,12-dione, 2.3 mM NADP+, and 400 μM 2-mercaptoethanol in 50 mM MOPS buffer at pH 7.0 at 37 °C for 12 h. The o-quinone products formed by the enzymatic oxidation of racemic B[g]C-11,12-dihydrodiol were isolated under similar conditions except that reactions containing trans-dihydrodiol substrates were initiated by the addition of AKR1C9. Conjugates were analyzed via a RP-HPLC system with the water/methanol gradient described above. The o-quinone thio-ether conjugates from the reaction displayed different retention times and UV/vis spectra (compared to those of trans-dihydrodiols).
LC/MS Analysis of Thio-Ether Conjugates
The identity of thio-ether conjugates were validated by LC/MS. Mass spectrometric data were acquired on a Finnigan LCQ ion trap mass spectrometer (Thermo Electron Corp., San Jose, CA) equipped with an electrospray ionization (ESI) source in the positive ion mode. Instrument operating conditions were as follows: capillary voltage 7 V and capillary temperature at 180 °C, with a needle voltage of 4.5 kV, applied to the ESI source. Nitrogen was used as the sheath gas (65 psi) and auxiliary (15 arbitrary units) gas to assist with nebulization. Full scanning analyses were performed in the range of m/z 150-700. Collision-induced dissociation (CID) experiments coupled with multiple tandem mass spectrometry (MS) employed argon as the collision gas. The relative collision energy was set at 20-40% of the maximum. Online chromatography was performed using a Waters Alliance 2690 HPLC system. A reversed-phased column was used at a flow rate of 0.5 mL/min. Solvent A was 5 mM ammonium acetate in water containing 0.01% formic acid, and solvent B was 5 mM ammonium acetate in methanol containing 0.01% formic acid. Organic soluble metabolites were eluted at 0.5 mL/min with the water/methanol gradient described above.
Results
Oxidation of B[g]C-11,12-dihydrodiol and B[c]Ph-3,4-dihydrodiol by AKR1C9
To further elucidate the role of AKRs in PAH activation, racemic fjord-region trans-dihydrodiols of B[g]C-11,12-dihydrodiol and B[c]Ph-3,4-dihydrodiol were tested as substrates for AKR1C9. Initial velocity measurements were indirectly determined using continuous UV spectrophotometric assays to monitor NADPH formation, which is formed stoichometrically during trans-dihydrodiol oxidation by AKR1C9. Initial velocity measurements were also directly determined using a discontinuous assay via RP-HPLC to monitor the disappearance of trans-dihydrodiol over time. Initial rates obtained from spectrophotometric assays agreed with initial velocities obtained from the RP-HPLC assays.
Initial velocities for the oxidation of racemic B[g]C-11,12-dihydrodiol and racemic B[c]Ph-3,4-dihydrodiol were compared with that obtained for the oxidation of racemic B[a]P-7,8-dihydrodiol (Table 1). It was found that racemic B[g]C-11,12-dihydrodiol was oxidized with a kcat/Km of 1520 min-1 mM-1, which is more than 100 times greater than that observed with B[a]P-7,8-dihydrodiol. Surprisingly, racemic B[c]Ph-3,4-dihydrodiol, which differs from racemic B[g]C-11,12-dihydrodiol by having one less ring, gave a kcat/Km of 39 min-1 mM-1.
Table 1.
Oxidation of Structurally Diverse PAH racemic trans-Dihydrodiols by AKR1C9
| PAH | racemic BP-7,8-dihydrodiol | racemic B[g]C-11,12-dihydrodiol | racemic B[c]Ph-3,4-dihydrodiol |
|---|---|---|---|
| Structure | |||
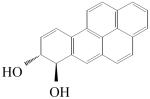 |
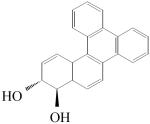 |
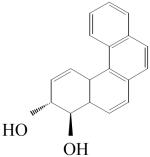 |
|
| Region | Bay | Fjord | Fjord |
| kcat/Kma | 12.3 | 1520 | 39.0 |
kcat/Km = min-1 mM-1, which is derived from v/[S] ) Vmax/Km using the enzyme concentration as a conversion factor. Nine replicates were performed in each case, and the standard error is <10% of the mean value.
Determination of the End Point of Enzymatic Oxidation of Racemic trans-Dihydrodiols by AKR1C9
Using RP-HPLC methods, the AKR1C9 dependent disappearance of racemic trans-dihydrodiol substrates was monitored over time to determine whether AKR1C9 oxidized both stereoisomers of racemic trans-dihydrodiols. Reactions were carried out until no further change in peak area was observed. Time courses of racemic B[a]P-7,8-dihydrodiol and racemic B[c]Ph-3,4-dihydrodiol oxidation validated that AKR1C9 utilized both stereoisomers. By contrast, the reaction of AKR1C9 and racemic B[g]C-11,12-dihydrodiol failed to reach 100% completion (Figure 1). RP-HPLC assays showed that the peak area corresponding to racemic B[g]C-11,12-dihydrodiol only decreased to 50%. One explanation is that the putative o-quinone product covalently modifies and inactivates the free enzyme, thus causing the reaction to halt. For example, naphthalene-1,2-dione leads to the time and concentration dependent irreversible inactivation of AKR1C9 via alkylation (35). However, once the oxidation of racemic B[g]C-11,12-dihydrodiol reached an end point, the addition of more enzyme caused no further oxidation of the trans-dihydrodiol, indicating that the enzyme was not being inactivated (results not shown). An alternative explanation is that AKR1C9 stereospecifically oxidizes either the S,S- or R,R-enantiomer.
Figure 1.
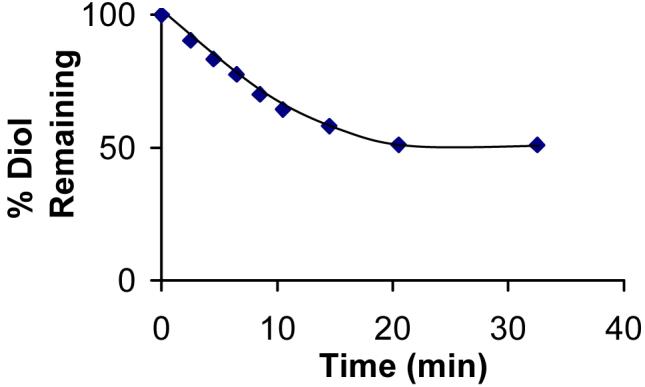
AKR1C9 preferentially oxidizes one stereoisomer of racemic B[g]C-11,12-dihydrodiol. Progress curve for the oxidation of racemic B[g]C-11,12-dihydrodiol with recombinant AKR1C9. Racemic trans-dihydrodiol (20 μM) was incubated with 0.125 μg of AKR1C9 and 2.3 mM NADP+ in 50 mM AMPSO at pH 9.0 at 37 °C. Initial velocities were determined by taking tangents to the progress curve.
Identification of the Stereoisomer of B[g]C-11,12-dihydrodiol Preferentially Oxidized by AKR1C9
CD spectroscopy was employed to determine the stereoisomer of B[g]C-11,12-dihydrodiol preferentially oxidized by AKR1C9. The unreacted stereoisomer was purified after monitoring a large scale enzymatic reaction of racemic B[g]C-11,12-dihydrodiol oxidation to completion. The negative Cotton effect obtained from the unreacted B[g]C-11,12-dihydrodiol substrate showed that the (-)-stereoisomer remained, revealing that only the (+)-stereoisomer is consumed by AKR1C9 (Figure 2). For B[a]P-7,8-dihydrodiol, B[c]Ph-3,4-dihydrodiol, and B[g]C-11,12-dihydrodiol, the (-)-isomer would correspond to the R,R-configuration and the (+)-isomer would correspond to the S,S-configuration (34, 36-38).
Figure 2.
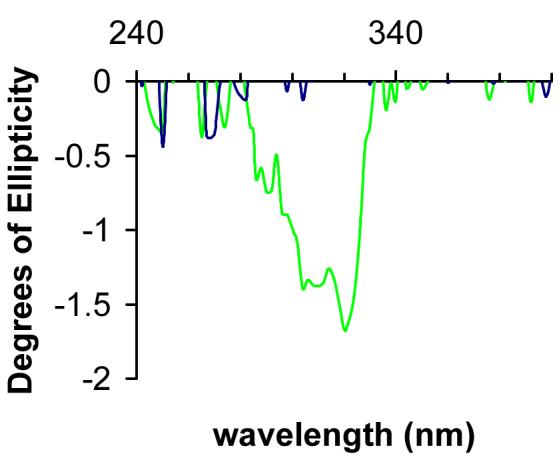
AKR1C9 only metabolizes B[g]C-11S,12S-dihydrodiol. The unreacted stereoisomer was purified via TLC from a 50 mL reaction containing 40 μM racemic B[g]C-11,12-dihydrodiol AKR1C9 and NADP+. Both racemic (in blue) and unreacted trans-dihydrodiol (in green) were dissolved in methanol and recorded in 0.5 nm steps from 240 to 400 nm. The negative Cotton effect observed from the unreacted B[g]C-11,12-dihydrodiol shows that the (-)-R,R-stereoisomer remains, revealing that only the (+)-S,S-stereoisomer is consumed by AKR1C9.
Examination of 11S,12S- and 11R,12R-Enantiomers of Racemic B[g]C-11,12-dihydrodiol as Substrates and Inhibitors of AKR1C9
To validate that B[g]C-11S,12S-dihydrodiol was oxidized by AKR1C9, B[g]C-11S,12S-dihydrodiol and B[g]C-11R,12R-dihydrodiol were purified from racemic B[g]C-11,12-dihydrodiol using a Chiracel OD-RH column. The purification yielded two distinct peaks with retention times of 17 and 26 min (Figure 3A). The peak containing B[g]C-11R,12R-dihydrodiol was identified by coelution with unreacted B[g]C-11R,12R-dihydrodiol identified in the CD experiment. The (-)-stereoisomer corresponded to the peak at 26 min (Figure 3B).
Figure 3.
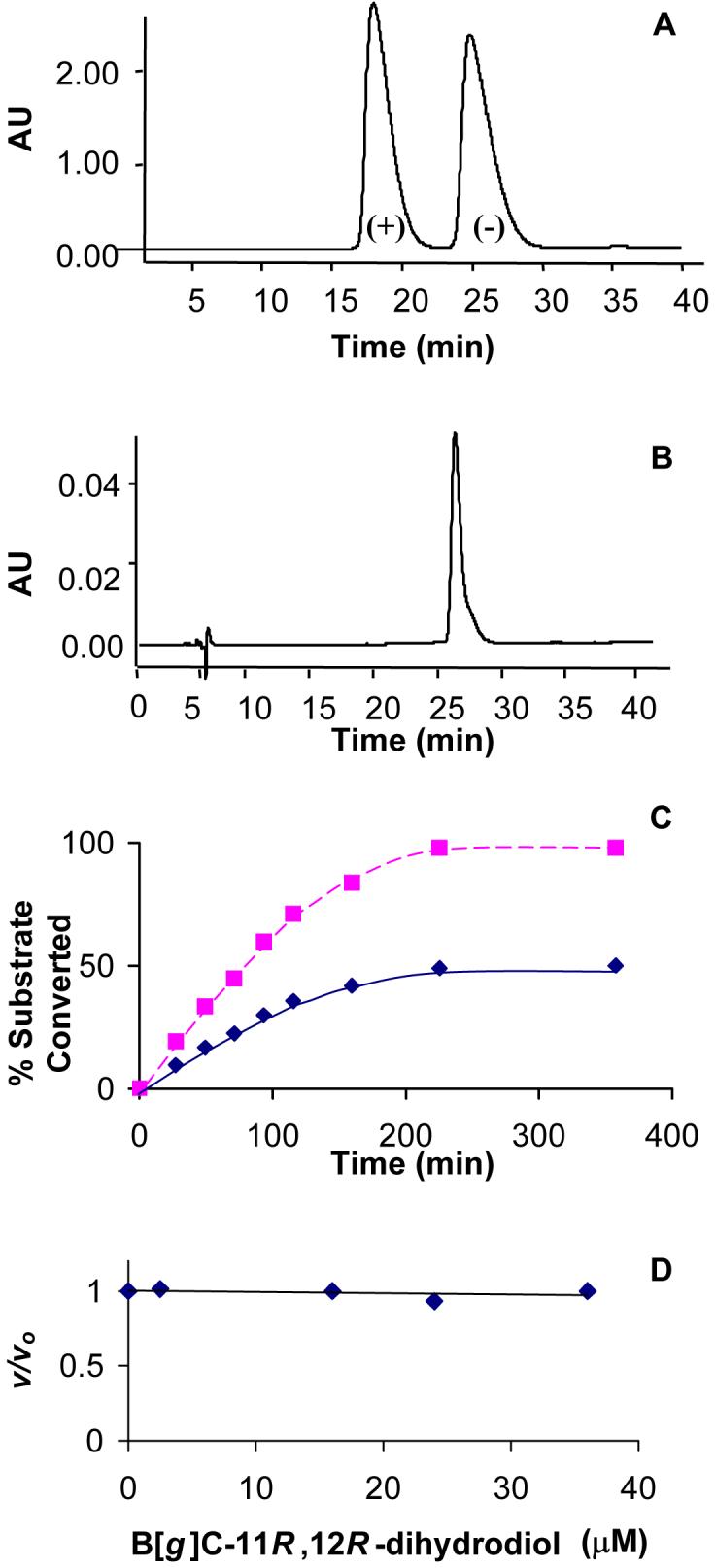
Demonstration that B[g]C-11R,12R-dihydrodiol is neither a substrate nor an inhibitor of AKR1C9. (A) Separation of (+)-S,S- and (-)-R,R isomers of B[g]C-11,12-dihydrodiol from racemic trans-dihydrodiol by chiral RP-HPLC. (B) Coelution of one peak from the racemate with the R,R-isomer identified by CD-spectroscopy. (C) Progress curve for the enzymatic oxidation of 20 μM racemic-B[g]C-11,12-dihydrodiol (red) and 20 μM B[g]C-11S,12S-dihydrodiol (blue) by recombinant AKR1C9. Each reaction was incubated with AKR1C9 in the presence of 2.3 mM NADP+ in 50 mM AMPSO buffer (pH 9.0) at 37 °C. Initial velocities were determined by taking tangents to the progress curves. (D) Effect of increasing concentrations of B[g]C-11R,12R-dihydrodiol on the AKR1C9 mediated oxidation of B[g]C-11S,12S-dihydrodiol. v/vo was measured by dividing v, the initial velocity, in the presence of B[g]C-11R,12R-dihydrodiol, by vo, the initial velocity in the absence of B[g]C-11R,12R-dihydrodiol.
We compared the kcat/Km for the oxidation of 20 μM S,S-stereoisomer and 20 μM racemic B[g]C-11,12-dihydrodiol by AKR1C9. Oxidation of the S,S-stereoisomer went to 100% completion, whereas the oxidation of the racemic trans-dihydrodiol went to 50% completion (Figure 3C). Additionally, 20 μM B[g]C-11S,12S-dihydrodiol was oxidized twice as fast as the 20 μM racemic trans-dihydrodiol since the effective substrate concentration was 2-fold higher. This implies that the unreacted B[g]C-11R,12R-dihydrodiol does not act as an inhibitor of AKR1C9.
To confirm that B[g]C-11R,12R-dihydrodiol is excluded from the active site, we analyzed the ability of the 11R,12R-enantiomer to block the oxidation of B[g]C-11S,12S-dihydrodiol (Figure 3D). Using a concentration range from 0 to 35 μM B[g]C-11R,12R-dihydrodiol, no change in initial velocity for B[g]C-11S,12S-dihydrodiol oxidation was observed. These findings suggest that the R,R-stereoisomer is not an inhibitor for AKR1C9 and that it does not bind to the active site.
Effects of Alanine-Scanning Mutations on B[g]C-11,12-dihydrodiol Oxidation Catalyzed by AKR1C9
Previously, alanine-scanning mutagenesis of active site residues in the ligand-binding cavity of AKR1C9 was used to dissect their role in steroid hormone binding and catalysis (32, 39). In the earlier studies, by far, the largest changes for the oxidation of androsterone were observed in kcat rather than Km. As a result, these changes were reflected in catalytic efficiency, kcat/Km (Table 2). The smallest change in kcat/Km was observed when alanine was used to substitute hydrophilic residues, for example, T24A and T226A. Conversely, W227A, F118A, and F129A revealed the largest changes in kcat/Km, while L54A, N306A, and Y310A showed intermediate changes in kcat/Km versus wild type. The changes in kcat observed were explained by the incorrect positioning of the substrate with respect to the catalytic residues (39). In this study, we used these alanine mutants to determine which residues are important in the exclusion of B[g]C-11R,12R-dihydrodiol from the AKR1C9 active site.
Table 2.
Oxidation of (+/-)-B[g]C-11,12-dihydrodiol by AKR1C9 mutants
| Substrate | B[g]C-11,12-dihydrodiol | Androsterone | |||
|---|---|---|---|---|---|
| Enzyme | kcat/Kma | WT/MUT | Stereoisomer | kcat/Kma,b | WT/MUT |
| WT | 1,520 | 1.00 | S,S | 7,500 | 1.00 |
| T24A | 1,510 | 1.00 | S,S | 4,600 | 1.63 |
| L54A | ND | ND | ND | 290 | 25.9 |
| F118A | ND | ND | ND | 34 | 220.6 |
| F129A | 14.9 | 100 | S,S, R,R | 43 | 174 |
| T226A | 1,508 | 1.00 | S,S | 4,500 | 1.67 |
| W227A | 44.2 | 34 | S,S, R,R | 9.7 | 773 |
| N306A | 209 | 7.3 | S,S | 75 | 100 |
| Y310A | 23.1 | 66 | S,S, R,R | 160 | 46.9 |
kcat/Km = min-1 mM-1, which is derived from v/[S] ) Vmax/Km using the enzyme concentration as a conversion factor. Nine replicates were performed in each case, and the standard error is 10% of the mean value.
Taken from ref 32. Reactions were run in 100 mM potassium phosphate at pH 7.0 and 2.3 mM NADP+ with 6% acetonitrile at 37 °C.
ND = no detectable rate.
Similar to the results observed with androsterone, the smallest change in kcat/Km was with T24A and T226A, Table 2. T226A showed no change in comparison to that of the wild type, implying that the side chain of T226 does not affect the positioning of reactants. The alanine mutants that suffered the largest decline in kcat/Km were L54A, F118A, and F129A. When the kcat/Km for androsterone and racemic B[g]C-11,12-dihydrodiol were compared, the largest change was observed for the F129A mutant.
Identification of the Stereoisomer of B[g]C-11,12-dihydodiol Oxidized by AKR1C9 Alanine Mutants
Oxidation of racemic B[g]C-11,12-dihydrodiol by AKR1C9 alanine mutants was monitored over time to determine whether the mutants retained the stereospecificity of the wild type enzyme. Results showed that reactions with the racemic trans-dihydrodiol went to 50% completion with the T24A, T226A, and N306A mutants (Table 2). Next, using the purified enantiomers of B[g]C-11,12-dihydrodiol, we tested to see if the alanine mutants retained the stereochemical preference for B[g]C-11S,12S-dihydrodiol. The T24A, T226A, and N306A mutants solely utilized B[g]C-11S,12S-dihydrodiol. Conversely, W227A, F129A, and Y310A were also able to oxidize both 11S,12S- and B[g]C-11R,12R-dihydrodiols, eliminating the stereochemical preference of the enzyme for the S,S-stereoisomer (Figure 4).
Figure 4.

Oxidation of 11S,12S- and B[g]C-11R,12R-dihydrodiols by AKR1C9 alanine-scanning mutants. Alanine-scanning mutagenesis was performed for the contact residues in the AKR1C9 active site. With each mutant, the oxidation of (+)-11S,12S- and (-)-11R,12R-isomers of B[g]C-11,12-dihydrodiol (20 μM) was monitored spectrophotometrically over time with 2.3 mM NADP+ in 50 mM AMPSO at pH 9.0 at 37 °C. Initial velocities were determined by taking tangents to the progress curve and converted into kcat/Km using the relationship v/[S] ) Vmax/Km when [S] , Km. Nine replicates were performed in each case, and the standard error was <10% of the mean value.
The oxidation of the purified B[g]C-11S,12S-dihydrodiol and B[g]C-11R,12R-dihydrodiol isomers by the F129A, W227A, and Y310A mutants was monitored over time (Table 3). When the W227A and Y310A mutants were used to oxidize the R,R-stereoisomer, initial velocities accounted for 14.5% and 22.0%, respectively, of the initial velocities observed when the racemic B[g]C-11,12-dihydrodiol was oxidized. Since wild type AKR1C9 oxidized only the S,S-stereoisomer, the data show that both the W227A and Y310A mutants removed the preference for the enantiomeric starting material. When the F129A mutant was used to oxidize the R,R-stereoisomer, the initial velocity accounted for 66.9% of the initial velocity observed when the racemic B[g]C-11,12-dihydrodiol was oxidized. This implies that F129A alters the stereochemical preference from oxidizing predominantly the S,S-stereoisomer to oxidizing predominantly the R,R-stereoisomer.
Table 3.
Contribution of the 11S,12S- and 11R,12R-enantiomers of B[g]C-11,12-dihydrodiol to the oxidation of the racemic compound
| Enzyme | S,S | R,R |
|---|---|---|
| WT | 100 | 0 |
| T24A | 100 | 0 |
| L54A | 0 | 0 |
| F118A | 0 | 0 |
| F129A | 33.1 | 66.9 |
| T226A | 100 | 0 |
| W227A | 85.5 | 14.5 |
| N306A | 100 | 0 |
| Y310A | 78.0 | 22.0 |
Enzymatic reactions were run using 20 μM of either racemic or B[g]C-11R,12R-dihydrodiol in 50 mM AMPSO (pH 9.0) at 37 °C.
Identification of the Product of AKR1C9-Mediated Oxidation of B[g]C-11,12-dihydrodiol
The products of the AKR-mediated oxidation of PAH trans-dihydrodiols are reactive o-quinones and can be trapped in vitro with the nucleophile 2-mercaptoethanol. To trap the products of fjord-region trans-dihydrodiol oxidation, B[g]C-11,12-dione thio-ether conjugates formed with 2-mercaptoethanol were synthesized to aid their identification. LC/MS data from the reaction mixture revealed the formation of two products (Figure 5). The peak corresponding to the unreacted o-quinone was detected at 41 min. The appearance of two product peaks revealed that fjord-region B[g]C-11,12-dione formed mono- and bis-conjugates, as identified by the ion chromatograms and MS data. Similarly, LC/MS data showed that the enzymatic oxidation of B[g]C-11,12-dihydrodiol in the presence of 2-mercaptoethanol yielded two product peaks. These peaks coeluted with the synthetically prepared mono- and bis-thio-ether conjugates and gave MS spectra of m/z = 384 [MH+] and m/z = 461 [MH+], respectively.
Figure 5.

LC/MS analysis of the products of B[g]C-11,12-dihydrodiol oxidation. (A) UV trace of reaction mixtures containing thio-ether conjugates obtained from enzymatic reactions of racemic B[g]C-11,12-dihydrodiol with AKR1C9 in the presence of 2-mercaptoethanol. The peak corresponding to the unreacted trans-dihydrodiol is at 49.5 min. (B) TIC of the of mono-conjugate at 35 min and bis-conjugate at 22 min. (C) Ion chromatogram for mass range 460-461. The peak corresponds to the bis-thio-ether conjugate at 22 min, (D) Ion chromatogram for mass range 384-385. The peak corresponds to the mono-thio-ether conjugate at 35 min. (E) Ion chromatogram for mass range 311-312. The peak corresponding to the unreacted stereoisomer of B[g]C-11,12-dihydrodiol is at 48.8 min. (F) UV trace of reaction mixtures containing thio-ether conjugates formed by reacting B[g]C-11,12-dione with 2-mercaptoethanol. The peak corresponding to unreacted o-quinone is at 41.5 min. (G) TIC of mono- (at 35 min) and bis-conjugates (at 22 min) of B[g]C-11,12-dione. (H) Ion chromatogram for mass range 460-461. The peak corresponds to the bis-thio-ether conjugate at 22 min. (I) Ion chromatogram for mass range 384-385. The peak corresponds to the mono-thio-ether conjugate at 35 min. (J) Ion chromatogram for mass range 309-310. The peak corresponding to the unreacted stereoisomer of B[g]C-11,12-dione is at 41 min. (K) Identification of the mono-thio-ether conjugate (m/z = 385 [MH+]) from 35 min. (L) Identification of the bis-thio-ether conjugate (m/z = 461 [MH+]) from 22 min.
Discussion
Our studies demonstrate that fjord-region B[g]C-11,12-dihydrodiol is a superior substrate for AKR1C9. Although racemic B[g]C-11,12-dihydrodiol has a kcat/Km of 1,520 min-1 mM-1, AKR1C9 only oxidizes the (+)-S,S-stereoisomer. [32P]-Post labeling studies in mouse skin identified that 75% of the adducts were derived from B[g]C-11R,12R-dihydrodiol, suggesting that B[g]C-11S,12S-dihydrodiol is the minor metabolic product (40). In this study, we observed that AKR1C9 catalyzes the NADP+-dependent oxidation of racemic B[g]C-11,12-dihydrodiol 100 times greater than racemic B[a]P-7,8-dihydrodiol and 40 times faster than racemic B[c]Ph-3,4-dihydrodiol.
Fjord-region trans-dihydrodiols are Oxidized by AKRs to Form o-Quinones
Our studies provide evidence for the metabolic activation of B[g]C-11S,12S-dihydrodiol to an o-quinone product. Although B[g]C-11S,12S-dihydrodiol is the minor metabolic isomer formed in vivo (41), the deleterious effects of this minor isomer may be increased by the high kcat/Km values observed for B[g]C-11S,12S-dihydrodiol oxidation with AKRs. Additionally, AKRs would prevent the conversion of B[g]C-11S,12S-dihydrodiol to (+)-syn-diol-epoxides catalyzed by P450 isoforms.
Comparison of Steady State Kinetics of AKR1C9 and AKR1C4 (the Liver Specific Human AKR)
AKR1C4 is the human AKR most similar to AKR1C9. Both enzymes are hepatospecific and display similar kinetic parameters for B[g]C-11,12-dihydrodiol, B[a]P-7,8-dihydrodiol, and B[c]Ph-3,4-dihydrodiol. For example, the kcat/Km of AKR1C4 for B[a]P-7,8-dihydrodiol is 3.7 min-1 mM-1, for B[c]Ph-3,4-dihydrodiol is 8.2 min-1 mM-1, and for B[g]C-11,12-dihydrodiol is 160 min-1 mM-1 (42). It is striking that similar to AKR1C9, the kcat/Km of AKR1C4 for B[g]C-11,12-dihydrodiol is much greater than that for B[a]P-7,8-dihydrodiol, especially because these enzymes are predicted to share only three of the same substrate binding residues within the active site: L54, F118, and W227 (43). The similarities in kcat/Km imply that although the actual active site composition is very different, both of these liver specific AKRs recognize trans-dihydrodiols in a similar matter.
B[g]C-11,12-dihydrodiol and Liver Tumorigenicity
Several studies have identified PAH as causing liver tumors in rodents (44, 45) and causing hepatotoxicity. Importantly, fjord-region B[g]C-diol-epoxide induces more liver tumors than B[c]Ph-diol-epoxide in male mice, both of which were found to be significantly more tumorigenic than B[a]P-diol-epoxide in this end organ (46). When contact-inhibited WB-F344 rat liver epithelial cells were treated with 1 μM B[g]C, P4501A1/1B1 and AKR1C9 mRNA levels were induced, implying that both P450s and AKRs may be involved in liver toxicity (47). The ultimate genotoxic effects caused by P450 and AKR upregulation results in increases in p53 phosphorylation, accumulation of cells in the S phase, and formation of B[g]C-DNA adducts detected by [32P]-postlabeling. These findings may implicate the P450 and AKR pathways in the hepatocarcinogenesis observed with the parent PAH (47).
Role of F129, W227, and Y310 in B[g]C-11,12-dihydrodiol Oxidation by AKR1C9
We previously conducted alanine-scanning mutagenesis of the AKR1C9 steroid binding pocket (32, 39). In this study, we used these mutants to determine their influence on both the turnover of fjord-region trans-dihydrodiols and stereochemical preference (Tables 2 and 3). We found that F129, W227, and Y310 all play roles in the stereochemical preference for B[g]C-11S,12S-dihydrodiol substrate.
By replacing the bulky side chains of the residues with small methyl side chains, the active site could accommodate B[g]C-11R,12R-dihydrodiol. As a result, B[g]C-11R,12R-dihydrodiol occupies the space previously taken up by the side chain of F129, W227, and Y310, thus allowing the mutant to bind this trans-dihydrodiol and permit oxidation. The decrease in kcat/Km for F129A, W227A, and Y310A mutants in comparison to that in the WT suggests that these mutants allow for more wobble of the stereoisomer within the active site.
On the basis of the alanine-scanning mutagenesis study with B[g]C-11,12-dihydrodiol, it is hypothesized that L54 and F118 are important to catalysis by forming critical contacts with B[g]C-11S,12S-dihydrodiol. Without these residues, the mutants display no detectable oxidation of B[g]C-11S,12S-dihydrodiol or B[g]C-11R,12R-dihydrodiol. Previous studies have shown that L54A and F118A mutants retained their ability to oxidize androsterone to androstanedione (39). L54A and F118A play different roles when oxidizing androsterone versus B[g]C-11S,12S-dihydrodiol, which can be attributable to positioning the functional groups. In comparison to androsterone, which has only one reactive hydroxyl group at the C3 position, B[g]C-11S,12S-dihydrodiol has two (at the C11 and C12 positions). This implies that in reference to the general acid, Y55, there is only one mode of binding for androsterone, but B[g]C-11S,12S-dihydrodiol may have several binding modes, and the correct mode may require more guidance from L54 or F118.
Molecular Modeling
We used molecular modeling to understand the structural basis for why wild type AKR1C9 used both stereoisomers of B[c]Ph-3,4-dihydrodiol and B[a]P-7,8-dihydrodiol, while the enzyme was stereoselective for B[g]C-11S,12S-dihydrodiol. Since the X-ray/Cartesian coordinates for B[g]C-11,12-dihydrodiol, B[c]Ph-3,4-dihydrodiol, or B[a]P-7,8-dihydrodiol are unavailable, molecular models of these PAHs were constructed using Chem3D. Previously, Chem3D was used to build a molecule of 7,12-dimethylbenz[a]anthracene, which yielded a structure almost identical to what was found in the Cambridge Crystallographic Database (Jin, Y., personal communication). Molecular modeling of the trans-dihydrodiols revealed that bay-region B[a]P-7,8-dihydrodiol is planar (except for the terminal benzo ring). The presence of a fjord-region in B[c]Ph-3,4-dihydrodiol and B[g]C-11,12-dihydrodiol caused deviation from planarity upon energy minimization. The ring responsible for the fjord-region in B[g]C-11,12-dihydrodiol creates an extra twist in the molecule, which is absent in B[c]Ph-3,4-dihydrodiol (Figure 6). Importantly, models of the S,S- and R,R-stereoisomers of B[g]C-11,12-dihydrodiol show that the structures are twisted in opposite directions.
Figure 6.

Molecular Models of (+)-S,S- and (-)-R,R-stereoisomers of B[a]P-7,8-dihydrodiol, B[c]Ph-3,4-dihydrodiol, and B[g]C-11,12-dihydrodiol. PAH trans-dihydrodiol substrates were built in Chem3D, and energy was minimized using the MM2 program. Structures are shown in ball and stick form minus hydrogen atoms. The starting models for the dihydrodiols were predominately planar, and minimization introduced the twist. The columns show the S,S- and R,R-stereoisomer of each trans-dihydrodiol, respectively. In each panel, the trans-dihydrodiol is shown from the top as well as from the side. The trans-dihydrodiols are (A) B[a]P-7S,8S-dihydrodiol, (B) B[c]Ph-3S,4Sdihydrodiol, (C) B[g]C-11S,12S-dihydrodiol, (D) B[a]P-7R,8R-dihydrodiol, (E) B[c]Ph-3R,4R-dihydrodiol, and (F) B[g]C-11R,12R-dihydrodiol.
Models of B[c]Ph-3S,4S-dihydrodiol and B[c]Ph-3R,4R-dihydrodiol, B[a]P-7S,8S-dihydrodiol and B[a]P-7R,8R-dihydrodiol, and B[g]C-11S,12S-dihydrodiol and B[g]C-11R,12R-dihydrodiol were manually docked into the active site of the AKR1C9-NADP+-testosterone ternary complex crystal structure (48) (Figure 7). For each trans-dihydrodiol, the carbon bonded to the presumptive axial reactive hydroxyl group was aligned with the C3 position of testosterone, and the carbons of the terminal benzo-ring of the trans-dihydrodiol were superimposed over the A ring of testosterone. Studies on steroid substrate specificity with AKR1C9 have previously shown that only the 3α -axial hydroxyl group is oxidized (49). These alignments caused the functional groups of the trans-dihydrodiols and testosterone to assume similar spatial orientations to Y55 and the NADP+ cofactor. These models showed that B[c]Ph-3S,4S-dihydrodiol and B[c]Ph-3R,4R-dihydrodiol, B[a]P-7S,8S-dihydrodiol, B[a]P-7R,8R-dihydrodiol, and B[g]C-11S,12S-dihydrodiol do not clash with active site residues, suggesting that these trans-dihydrodiols bind to the active site and will be oxidized. For example, B[g]C-11S,12S-dihydrodiol was accommodated into the active site so that the aromatic ring that forms the fjord-region is positioned parallel to F129, Y310, and W227.
Figure 7.

Docking of PAH trans-dihydrodiols in the active site of AKR1C9. Molecular modeling was carried out using crystal structure 1AFS, the ternary complex of AKR1C9-NADP+-testosterone (48), in Swiss PDB viewer. The stereoisomers of B[g]C-11,12-dihydrodiol were built as described in Figure 6 and manually docked into the active site. Amino acid residues and trans-dihydrodiols are shown in stick representation. The catalytic tetrad is shown in red and other substrate contact residues are shown in magenta. The trans-dihydrodiols are (A) B[a]P-7S,8S-dihydrodiol (lavender), (B) B[a]P-7R,8R-dihydrodiol (cyan), (C) B[c]Ph-3S,4S-dihydrodiol (orange), (D) B[c]Ph-3R,4R-dihydrodiol (green), (E) B[g]C-11S,12Sdihydrodiol (yellow), and (F) B[g]C-11R,12R-dihydrodiol (white). With the exception of B[g]C-11R,12R-dihydrodiol, none of the PAH trans-dihydrodiols clash with active site residues of AKR1C9. The presence of the ring that forms the fjord-region of B[g]C-11R,12R-dihydrodiol causes clashes with active site residues F129, W227, and Y310A (panel F).
Conversely, when the presumptive reactive 12α-axial alcohol of B[g]C-11R,12R-dihydrodiol is aligned with the carbonyl of C3 on testosterone, the contorted structure of B[g]C-11R,12R-dihydrodiol caused the ring that forms the fjord-region to directly clash with both F129 and Y310. To align the 11α -hydroxyl axial alcohol of B[g]C-11R,12R-dihydrodiol with the carbonyl of C3 of testosterone, the trans-dihydrodiol was flipped 180° in the active site. This caused the R,R-stereoisomer to clash with Y310 and neighboring F311. This suggests that neither the 12α- or 11α-hydroxyl group of B[g]C-11R,12R-dihydrodiol can assume a binding mode that permits oxidation because of direct clashing with active site residues. The modeling is supported by alanine scanning mutagenesis, where F129A and Y310A mutants can accommodate the R,R-stereoisomer because they provide more room for the ring that forms the fjord-region to now bind, resulting in the oxidation of the S,S- and R,R-stereoisomers.
Alanine-scanning mutagenesis data suggests that W227 plays an important role in preventing the oxidation of B[g]C-11R,12R-dihydrodiol. We hypothesize that W227 can rotate in the active site and come in closer contact with B[g]C-11R,12R-dihydrodiol. This is based on the crystal structure of human AKR1C2. In this crystal structure, W227 is rotated 120° around the Cα bond to allow a competitive inhibitor to bind (43). Docking experiments using AKR1C2 also revealed that W227 and L54 form the narrowest part of the active site (50). Therefore, if an analogous movement occurs with W227 in the active site of AKR1C9, this could prevent oxidation of the R,R-stereoisomer. By contrast, the W227A mutant may be able to oxidize both stereoisomers of B[g]C-11,12-dihydrodiol because of active site widening.
Summary
Our studies provide the first evidence of catalysis of fjord-region trans-dihydrodiols B[g]C-11,12-dihydrodiol and B[c]Ph-3,4-dihydrodiol by AKR1C9 to form o-quinone products that may contribute to PAH carcinogenesis. AKR1C9 oxidized B[g]C-11,12-dihydrodiol with a higher catalytic efficiency than other AKRs tested to date. The enzyme was stereoselective for B[g]C-11S,12S-dihydrodiol, but metabolized both stereoisomers of B[c]Ph-3,4-dihydrodiol. Mutation of F129, W227, and Y310 to alanine provides more room to bind for the rings that create the fjord-region, thus permitting oxidation of both S,S- and R,R-stereoisomers. AKR1C4 is the human AKR most similar to AKR1C9. Both enzymes are hepatospecific and display similar kinetic parameters for B[g]C-11,12-dihydrodiol, B[a]P-7,8-dihydrodiol, and B[c]Ph-3,4-dihydrodiol. This suggests that both of these liver specific AKRs recognize trans-dihydrodiols similarly even though their binding pocket residues differ. These findings may implicate the AKRs in the hepatocarcinogenesis observed with the parent PAH.
Acknowledgment
This study was supported by NIH Grants PO1-CA92537, P30-ES 013508, and RO1-CA 039504 (awarded to T.M.P.). We thank Ms. Amy Quinn for her technical assistance, Dr. Yi Jin for her extensive knowledge of AKR crystal structures, Dr. Dalijit K. Vudathala for PAH quality control, Dr. Seon-Hwa Lee for her advice on chiral column chromatography, and Dr. K.S. Reddy for his CD spectroscopy expertise.
References
- (1).Bjorseth A. Handbook of Polycyclic Aromatic Hydrocarbons. CRC Press; OH: 1985. [Google Scholar]
- (2).Grimmer G. Analysis of automobile exhaust condensates. IARC Sci. Pub. 1977;16:29–39. [PubMed] [Google Scholar]
- (3).Conney AH. Induction of microsomal enzymes by foreign chemicals and carcinogenesis by polycyclic aromatic hydrocarbons: G. H. A. Clowes Memorial Lecture. Cancer Res. 1982;42(12):4875–4917. [PubMed] [Google Scholar]
- (4).Gelboin HV. Benzo[α]pyrene metabolism, activation and carcinogenesis: role and regulation of mixed-function oxidases and related enzymes. Physiol. Rev. 1980;60(4):1107–1166. doi: 10.1152/physrev.1980.60.4.1107. [DOI] [PubMed] [Google Scholar]
- (5).Penning TM, Burczynski ME, Hung CF, McCoull KD, Palackal NT, Tsuruda LS. Dihydrodiol dehydrogenases and polycyclic aromatic hydrocarbon activation: generation of reactive and redox active o-quinones. Chem. Res. Toxicol. 1999;12(1):1–18. doi: 10.1021/tx980143n. [DOI] [PubMed] [Google Scholar]
- (6).Hecht SS. Tobacco smoke carcinogens and lung cancer. J. Natl. Cancer Inst. 1999;91(14):1194–1210. doi: 10.1093/jnci/91.14.1194. [DOI] [PubMed] [Google Scholar]
- (7).Fetzer JC. Large (C> ) 24) Polycyclic Aromatic Hydrocarbons: Chemistry and Analysis. Wiley-Interscience; New York: 2000. [Google Scholar]
- (8).Harvey RG. Polycyclic Aromatic Hydrocarbons. Wiley-VCH; New York: 1997. [Google Scholar]
- (9).Hogue C. Dustup Over Pavement Coatings. Chem. Eng. News. 2007;85(7):61–66. [Google Scholar]
- (10).Wise SA, Benner BA, Chesler SN, Hilpert LR, Vogt CR, McAllister JM. Characterization of the polycyclic aromatic hydrocarbons from two standard reference material air particulate samples. Anal. Chem. 1986;58(14):3067–3077. [Google Scholar]
- (11).Straif K, Baan R, Grosse Y, Secretan B, El GF, Cogliano V. Carcinogenicity of polycyclic aromatic hydrocarbons. Lancet Oncol. 2005;6(12):931–932. doi: 10.1016/s1470-2045(05)70458-7. [DOI] [PubMed] [Google Scholar]
- (12).Conney AH. Induction of microsomal enzymes by foreign chemicals and carcinogenesis by polycyclic aromatic hydrocarbons: G. H. A. Clowes Memorial Lecture. Cancer Res. 1982;42(12):4875–4917. [PubMed] [Google Scholar]
- (13).IARC Polycyclic Aromatic Hydrocarbons 5. [accessed Sept 25, 2007];Summary of Data Reported and Evaluation. 2006 IARC website, http://monographs.iarc.fr/ENG/Meetings/92-pahs.pdf.
- (14).Conney AH, Pantuck EJ, Hsiao KC, Kuntzman R, Alvares AP, Kappas A. Regulation of drug metabolism in man by environmental chemicals and diet. Fed. Proc. 1977;36(5):1647–1652. [PubMed] [Google Scholar]
- (15).Jiang H, Vudathala DK, Blair IA, Penning TM. Competing roles of aldo-keto reductase 1A1 and cytochrome P4501B1 in benzo[a]pyrene-7,8-diol activation in human bronchoalveolar H358 cells: role of AKRs in P4501B1 induction. Chem. Res. Toxicol. 2006;19(1):68–78. doi: 10.1021/tx0502488. [DOI] [PubMed] [Google Scholar]
- (16).Koreeda M, Moore PD, Wislocki PG, Levin W, Yagi H, Jerina DM. Binding of benzo[a]pyrene 7,8-diol-9,10-epoxides to DNA, RNA, and protein of mouse skin occurs with high stereoselectivity. Science. 1978;199(4330):778–781. doi: 10.1126/science.622566. [DOI] [PubMed] [Google Scholar]
- (17).Ruan Q, Gelhaus SL, Penning TM, Harvey RG, Blair IA. Aldoketoreductase- and cytochrome P450-dependent formation of benzo[a]pyrene-derived DNA adducts in human bronchoalveolar cells. Chem. Res. Toxicol. 2007;20:424–431. doi: 10.1021/tx060180b. [DOI] [PubMed] [Google Scholar]
- (18).Agarwal R, Coffing SL, Baird WM, Kiselyov AS, Harvey RG, Dipple A. Metabolic activation of benzo[g]chrysene in the human mammary carcinoma cell line MCF-7. Cancer Res. 1997;57(3):415–419. [PubMed] [Google Scholar]
- (19).Kleiner HE, Vulimiri SV, Hatten WB, Reed MJ, Nebert DW, Jefcoate CR, DiGiovanni J. Role of cytochrome P4501 family members in the metabolic activation of polycyclic aromatic hydrocarbons in mouse epidermis. Chem. Res. Toxicol. 2004;17(12):1667–1674. doi: 10.1021/tx049919c. [DOI] [PubMed] [Google Scholar]
- (20).Flowers L, Ohnishi ST, Penning TM. DNA strand scission by polycyclic aromatic hydrocarbon o-quinones: role of reactive oxygen species, Cu(II)/Cu(I) redox cycling, and o-semiquinone anion radicals. Biochemistry. 1997;36(28):8640–8648. doi: 10.1021/bi970367p. [DOI] [PubMed] [Google Scholar]
- (21).Penning TM, Ohnishi ST, Ohnishi T, Harvey RG. Generation of reactive oxygen species during the enzymatic oxidation of polycyclic aromatic hydrocarbon trans-dihydrodiols catalyzed by dihydrodiol dehydrogenase. Chem. Res. Toxicol. 1996;9(1):84–92. doi: 10.1021/tx950055s. [DOI] [PubMed] [Google Scholar]
- (22).Smithgall TE, Harvey RG, Penning TM. Spectroscopic identification of ortho-quinones as the products of polycyclic aromatic trans-dihydrodiol oxidation catalyzed by dihydrodiol dehydrogenase. A potential route of proximate carcinogen metabolism. J. Biol. Chem. 1988;263(4):1814–1820. [PubMed] [Google Scholar]
- (23).Park JH, Gopishetty S, Szewczuk LM, Troxel AB, Harvey RG, Penning TM. Formation of 8-oxo-7,8-dihydro-22-deoxyguanosine (8-oxo-dGuo) by PAH o-quinones: involvement of reactive oxygen species and copper(II)/copper(I) redox cycling. Chem. Res. Toxicol. 2005;18(6):1026–1037. doi: 10.1021/tx050001a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Park JH, Troxel AB, Harvey RG, Penning TM. Polycyclic aromatic hydrocarbon (PAH) o-quinones produced by the aldo-keto-reductases (AKRs) generate abasic sites, oxidized pyrimidines, and 8-oxo-dGuo via reactive oxygen species. Chem. Res. Toxicol. 2006;19(5):719–728. doi: 10.1021/tx0600245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Shou M, Harvey RG, Penning TM. Reactivity of benzo[a]pyrene-7,8-dione with DNA. Evidence for the formation of deoxyguanosine adducts. Carcinogenesis. 1993;14(3):475–482. doi: 10.1093/carcin/14.3.475. [DOI] [PubMed] [Google Scholar]
- (26).Balu N, Padgett WT, Nelson GB, Lambert GR, Ross JA, Nesnow S. Benzo[a]pyrene-7,8-quinone-32-mononucleotide adduct standards for 32P postlabeling analyses: detection of benzo[a]pyrene-7,8-quinone-calf thymus DNA adducts. Anal. Biochem. 2006;355(2):213–223. doi: 10.1016/j.ab.2006.05.023. [DOI] [PubMed] [Google Scholar]
- (27).Balu N, Padgett WT, Lambert GR, Swank AE, Richard AM, Nesnow S. Identification and characterization of novel stable deoxyguanosine and deoxyadenosine adducts of benzo[a]pyrene-7,8-quinone from reactions at physiological pH. Chem. Res. Toxicol. 2004;17(6):827–838. doi: 10.1021/tx034207s. [DOI] [PubMed] [Google Scholar]
- (28).Yu D, Berlin JA, Penning TM, Field J. Reactive oxygen species generated by PAH o-quinones cause change-in-function mutations in p53. Chem. Res. Toxicol. 2002;15(6):832–842. doi: 10.1021/tx010177m. [DOI] [PubMed] [Google Scholar]
- (29).Tsuruda L, Hou Y, Penning TM. Stable expression of rat dihydrodiol dehydrogenase (AKR1C9) in human breast MCF-7 cells results in the formation of PAH-o-quinones and enzyme mediated cell death. Chem. Res. Toxicol. 2001;14(7):856–862. doi: 10.1021/tx0100035. [DOI] [PubMed] [Google Scholar]
- (30).Harvey RG, Dai Q, Ran C, Penning TM. Synthesis of the o-quinones and other oxidized metabolites of polycyclic aromatic hydrocarbons implicated in carcinogenesis. J. Org. Chem. 2004;69(6):2024–2032. doi: 10.1021/jo030348n. [DOI] [PubMed] [Google Scholar]
- (31).Ratnam K, Ma H, Penning TM. The arginine 276 anchor for NADP(H) dictates fluorescence kinetic transients in 3α-hydroxysteroid dehydrogenase, a representative aldo-keto reductase. Biochemistry. 1999;38(24):7856–7864. doi: 10.1021/bi982838t. [DOI] [PubMed] [Google Scholar]
- (32).Heredia VV, Cooper WC, Kruger RG, Jin Y, Penning TM. Alanine scanning mutagenesis of the testosterone binding site of rat 3α-hydroxysteroid dehydrogenase demonstrates contact residues influence the rate-determining step. Biochemistry. 2004;43(19):5832–5841. doi: 10.1021/bi0499563. [DOI] [PubMed] [Google Scholar]
- (33).Palackal NT, Burczynski ME, Harvey RG, Penning TM. The ubiquitous aldehyde reductase (AKR1A1) oxidizes proximate carcinogen trans-dihydrodiols to o-quinones: potential role in polycyclic aromatic hydrocarbon activation. Biochemistry. 2001;40(36):10901–10910. doi: 10.1021/bi010872t. [DOI] [PubMed] [Google Scholar]
- (34).Bushman DR, Grossman SJ, Jerina DM, Lehr RE. Synthesis of optically active fjord-region 11,12-diol 13,14-epoxides and the K-region 9,10-oxide of the carcinogen benzo[g]chrysene. J. Org. Chem. 1989;54:3553–3544. [Google Scholar]
- (35).Penning TM, Sharp RB, Smithgall TE. Non-K-region o-quinones as enzyme-generated inactivators of dihydrodiol dehydrogenase. Biochemistry. 1989;28(10):4505–4511. doi: 10.1021/bi00436a057. [DOI] [PubMed] [Google Scholar]
- (36).Thakker DR, Levin W, Yagi M, Conney AM, Jerina DM. Regio-and stereoselectivity of hepatic cytochrome P-450 toward polycylic aromatic hydrocarbon substrates. AdV. Exp. Med. Biol. 1981;136(Pt A):525–539. doi: 10.1007/978-1-4757-0674-1_39. [DOI] [PubMed] [Google Scholar]
- (37).Thakker DR, Levin W, Yagi H, Yeh HJ, Ryan DE, Thomas PE, Conney AH, Jerina DM. Stereoselective metabolism of the (+)-(S,S)- and (-)-(R,R)-enantiomers of trans-3,4-dihydroxy-3,4-dihydrobenzo[c]phenanthrene by rat and mouse liver microsomes and by a purified and reconstituted cytochrome P-450 system. J. Biol. Chem. 1986;261(12):5404–5413. [PubMed] [Google Scholar]
- (38).Yagi H, Thakker DR, Ittah Y, Croisy-Delcey M, Jerina DM. Synthesis and assignments of absolute configuration to the trans-3,4-dihydrodiols and 3,4-diol-1,2-epoxides of benzo[c]phenanthrene. Tetrahedron Lett. 1983;24(13):1349–1352. [Google Scholar]
- (39).Heredia VV, Kruger RG, Penning TM. Steroid-binding site residues dictate optimal substrate positioning in rat 3α-hydroxysteroid dehydrogenase (3α-HSD or AKR1C9) Chem.-Biol. Interact. 2003;143-144:393–400. doi: 10.1016/s0009-2797(02)00176-x. [DOI] [PubMed] [Google Scholar]
- (40).Giles AS, Seidel A, Phillips DH. Covalent DNA adducts formed in mouse epidermis by benzo[g]chrysene. Carcinogenesis. 1996;17(6):1331–1336. doi: 10.1093/carcin/17.6.1331. [DOI] [PubMed] [Google Scholar]
- (41).Giles AS, Seidel A, Phillips DH. Covalent DNA adducts formed in mouse epidermis by benzo[g]chrysene. Carcinogenesis. 1996;17(6):1331–1336. doi: 10.1093/carcin/17.6.1331. [DOI] [PubMed] [Google Scholar]
- (42).Palackal NT, Lee SH, Harvey RG, Blair IA, Penning TM. Activation of polycyclic aromatic hydrocarbon trans-dihydrodiol proximate carcinogens by human aldo-keto reductase (AKR1C) enzymes and their functional overexpression in human lung carcinoma (A549) cells. J. Biol. Chem. 2002;277(27):24799–24808. doi: 10.1074/jbc.M112424200. [DOI] [PubMed] [Google Scholar]
- (43).Jin Y, Stayrook SE, Albert RH, Palackal NT, Penning TM, Lewis M. Crystal structure of human type III 3α-hydroxysteroid dehydrogenase/bile acid binding protein complexed with NADP(+) and ursodeoxycholate. Biochemistry. 2001;40(34):10161–10168. doi: 10.1021/bi010919a. [DOI] [PubMed] [Google Scholar]
- (44).Della PG. Use of newborn and infant animals in carcinogenicity testing. Food Cosmet. Toxicol. 1968;6(2):243–252. doi: 10.1016/0015-6264(68)90205-8. [DOI] [PubMed] [Google Scholar]
- (45).Toth B, Shubik P. Carcinogenesis in AKR mice injected at birth with benzo(a)pyrene and dimethylnitrosamine. Cancer Res. 1967;27(1):43–51. [PubMed] [Google Scholar]
- (46).Amin S, Desai D, Dai W, Harvey RG, Hecht SS. Tumorigenicity in newborn mice of fjord region and other sterically hindered diol epoxides of benzo[g]chrysene, dibenzo[a,l]pyrene (dibenzo[def,p]chrysene), 4H-cyclopenta[def]chrysene and fluoranthene. Carcinogenesis. 1995;16(11):2813–2817. doi: 10.1093/carcin/16.11.2813. [DOI] [PubMed] [Google Scholar]
- (47).Svihalkova-Sindlerova L, Machala M, Pencikova K, Marvanova S, Neca J, Topinka J, Sevastyanova O, Kozubik A, Vondracek J. Dibenzanthracenes and benzochrysenes elicit both genotoxic and nongenotoxic events in rat liver ‘stem-like’ cells. Toxicology. 2007;232(12):147–159. doi: 10.1016/j.tox.2006.12.024. [DOI] [PubMed] [Google Scholar]
- (48).Bennett MJ, Albert RH, Jez JM, Ma H, Penning TM, Lewis M. Steroid recognition and regulation of hormone action: crystal structure of testosterone and NADP+ bound to 3α-hydroxysteroid/dihydrodiol dehydrogenase. Structure. 1997;5(6):799–812. doi: 10.1016/s0969-2126(97)00234-7. [DOI] [PubMed] [Google Scholar]
- (49).Smithgall TE, Penning TM. Sex differences in indomethacin-sensitive 3α-hydroxysteroid dehydrogenase of rat liver cytosol. Cancer Res. 1985;45(10):4946–4949. [PubMed] [Google Scholar]
- (50).Jin Y, Penning TM. Molecular docking simulations of steroid substrates into human cytosolic hydroxysteroid dehydrogenases (AKR1C1 and AKR1C2): insights into positional and stereochemical preferences. Steroids. 2006;71(5):380–391. doi: 10.1016/j.steroids.2005.12.002. [DOI] [PubMed] [Google Scholar]