

Abstract
Plasminogen activator inhibitor type 1 (PAI-1) is a serine protease inhibitor (serpin) in which the reactive center loop (RCL) spontaneously inserts into a central β-sheet, β-sheet A, resulting in inactive inhibitor. Available x-ray crystallographic studies of PAI-1 in an active conformation relied on the use of stabilizing mutations. Recently it has become evident that these structural models do not adequately explain the behavior of wild-type PAI-1 (wtPAI-1) in solution. To probe the structure of native wtPAI-1, we used three conformationally sensitive ligands: the physiologic cofactor, vitronectin; a monoclonal antibody, 33B8, that binds preferentially to RCL-inserted forms of PAI-1; and RCL-mimicking peptides that insert into β-sheet A. From patterns of interaction with wtPAI-1 and the stable mutant, 14-1B, we propose a model of the native conformation of wtPAI-1 in which the bottom of the central sheet is closed, whereas the top of the β-sheet A is open to allow partial insertion of the RCL. Because the incorporation of RCL-mimicking peptides into wtPAI-1 is accelerated by vitronectin, we further propose that vitronectin alters the conformation of the RCL to allow increased accessibility to β-sheet A, yielding a structural hypothesis that is contradictory to the current structural model of PAI-1 in solution and its interaction with vitronectin.
Serpins are a superfamily of proteins with a wide range of functions, such as inhibiting serine or cysteine proteases, serving as carrier molecules for hormones, and regulating the folding of other proteins (1). Despite primary sequence homologies as low as 25%, serpins share a highly conserved tertiary structure (1). The native fold of serpins is metastable and defines the mechanism of inhibition utilized by anti-proteolytic serpins. Serpins contain a solvent-exposed reactive center loop (RCL)3, which holds residues appropriate to the substrate specificity of cognate serine proteases (2–4). Attack by a protease on the P1–P1′ bond of the serpin RCL is initiated via the conventional serine protease mechanism, yielding breakage of the peptide bond and a covalent acyl-enzyme intermediate in which the active site serine residue is ester-linked to the serpin P1 residue (5, 6). Breakage of the P1–P1′ bond leads to a conformational change in the serpin, consisting of insertion of the RCL into β-sheet A in the serpin core, translocation of the covalently attached protease a distance of 70 Å, and concomitant distortion the protease active site, which renders the protease incompetent to resolve the acyl-enzyme intermediate (7–9).
Plasminogen activator inhibitor type-1 (PAI-1) is considered an archetypal serpin and is the most potent inhibitor of tissue-type and urokinase-type plasminogen activators (t-PA and u-PA, respectively) (10). PAI-1 is unlike most serpins in that it spontaneously inactivates under physiologic conditions with an in vitro half-life of 1–2 h (11). During this process, the RCL of PAI-1 inserts into β-sheet A without prior proteolysis of the P1–P1′ bond (12). In the resulting conformation, known as latent PAI-1 (13), the scissile bond of the RCL is no longer accessible to the active sites of target proteases (12). From x-ray crystal structures of active and latent PAI-1, generation of the inactive serpin is thought to require two adjacent β-strands in the central β-sheet of the active conformation to separate enough to allow incorporation of the RCL (14, 15). The inhibitory half-life of PAI-1 is prolonged modestly by binding to its physiologic cofactor, vitronectin (11, 15), which is hypothesized to hinder the formation of latent serpin by impeding the opening of β-sheet A (16–18). A confounder in this model is that the x-ray crystal structures of active PAI-1 and the complex of PAI-1 bound to the relevant domain of vitronectin were resolved using a recombinant PAI-1 variant, known as 14-1B (18). 14-1B contains four mutations that are thought to decrease RCL mobility and hinder β-sheet A from adopting an open conformation, yielding a PAI-1 variant with an inhibitory half-life of about 145 h (19). Therefore, it is uncertain whether models of PAI-1 structure and vitronectin-induced stabilization based on data derived from the stable mutant can yield accurate conclusions regarding the more labile, wild-type inhibitor.
The work presented here demonstrates that the structural model based on the stable mutant, 14-1B, fails to predict how wild-type PAI-1 (wtPAI-1) interacts with certain conformationally sensitive ligands. Data are also provided for an unexpected mechanism by which RCL insertion is regulated upon binding of PAI-1 to vitronectin, and lead to a novel structural hypothesis of the native conformation of PAI-1.
EXPERIMENTAL PROCEDURES
Proteins and Reagents—Recombinant human two-chain u-PA was a generous gift from Dr. Jack Henkin of Abbott Laboratories, Abbott Park, IL. Human t-PA was purchased from EMD Biosciences, Darmstadt, Germany, predominantly in the single-chain form and was converted to the two-chain form using plasmin-Sepharose beads as previously described (20). The active site concentrations of t-PA and u-PA were measured by indirect titration against a calibrated trypsin standard (21). β-Trypsin was a generous gift from Dr. Steve Olson, University of Illinois, Chicago, IL. Wild-type PAI-1, 14-1B, PAI-1AK (which contains R101A and Q123K, substitutions that together disrupt vitronectin binding) (22), 14-1BAK (14-1B carrying R101A and Q123K), PAI-1R (a substrate form of PAI-1 containing T333R and A335R) (23), and PAI-1R-AK (which contains R101A, Q123K, T333R, and A355R, is a substrate for proteases, and does not bind to vitronectin) were expressed in Escherichia coli and purified as previously described (24). The functional concentrations of wtPAI-1, 14-1B, and PAI-1AK were calibrated against an active site-titrated protease as described using u-PA (25). Latent PAI-1 was generated by incubating active wtPAI-1 at 37 °C for 72 h in 0.1 m Tris, pH 7.4, leading to residual anti-u-PA activity of <0.01% in a chromogenic assay. The murine monoclonal antibodies, MA-33B8 (33B8) and MA-31C9 (31C9), raised against recombinant human PAI-1, and the isotype-matched monoclonal antibody, MA-H77A10 (H77A10), raised against mouse u-PA were purchased from Molecular Innovations, Inc., Southfield, MI. Rabbit polyclonal anti-PAI-1 antibodies were also from Molecular Innovations. Another preparation of rabbit polyclonal anti-PAI-1 antibodies was a generous gift of Dr. Peter Andreasen, University of Aarhus, Aarhus, Denmark. Native vitronectin isolated from human plasma under non-denaturing conditions was kindly supplied by Dr. Deane Mosher, University of Wisconsin, Madison, WI. The pentapeptide, Ac-TVASS, corresponding to residues P14–P10 of the PAI-1 RCL, was synthesized by Biopeptide Co., Inc., San Diego, CA, and the octapeptide, Ac-TVASSSTA, corresponding to residues P14–P7, was purchased from Molecular Innovations.
Several chromogenic substrates were used in microplate assays: Spectrozyme UK (Cbo-l-(γ)Glu(α-t-BuO)-Gly-Arg-pNA) and Spectrozyme tPA (CH3SO2-d-CHT-Gly-Arg-pNA) from American Diagnostica, Inc., Stamford, CT; S-2444 (pGlu-Gly-Arg-pNA) from Chromogenix, Inc., Milano, Italy; and Chromozym t-PA (CH3SO2-d-Phe-Gly-Arg-pNA) from Roche Diagnostics Co. Horseradish peroxidase-conjugated goat anti-rabbit IgG secondary antibody was purchased from American Diagnostica, and SuperSignal West Pico chemiluminescent substrate was from Pierce Biotechnology, Inc. Gel electrophoresis components were from Bio-Rad. All supplies and buffers for binding assays with BIAcore 3000 Biosensor were purchased from BIAcore AB (Uppsala, Sweden). All other reagents were of analytical grade or better.
Determination of Functional Half-lives of PAI-1 Variants—100 nm PAI-1 was incubated with or without 150 nm vitronectin for 2 min at 37 °C in 0.1 m Tris, pH 7.4, with 1% bovine serum albumin in a Corning non-binding clear 96-well polystyrene microtiter plate that had been previously blocked with 1% bovine serum albumin. Aliquots were removed after increasing times of incubation, mixed with excess u-PA, and allowed to react for 10 min at 37 °C. The mixture was diluted into 500 μm Spectrozyme UK or S-2444, and cleavage of the substrate was monitored by following changes in OD at 405 nm using a SpectroMax Plus 384 microplate spectrophotometer from Molecular Devices Corp., Sunnyvale, CA. Relative loss of u-PA-inhibiting activity, taken to reflect the generation of latent PAI-1, was plotted against time and was fitted to a single exponential decay equation with a finite span to obtain the rate constant of the latency transition and the functional half-life.
Binding of PAI-1 Variants to the Monoclonal Antibody, 33B8—The interaction between PAI-1 and 33B8 was monitored indirectly by incubating 20 nm wtPAI-1 or 14-1B with 50 nm 33B8 at 37 °C in 0.1 m Tris, pH 7.4, with 1% bovine serum albumin. At various times, the residual PAI-1 activity was measured as described above by adding 1.5-fold molar excess u-PA over PAI-1 and 1 μm latent wtPAI-1. The latent PAI-1 blocked further inactivation of active wtPAI-1 by 33B8 during the incubation with u-PA (data not shown). The data were fit to a single exponential decay equation.
Surface Plasmon Resonance—Direct binding of wtPAI-1 and 14-1B with immobilized ligands was monitored using a BIA-core 3000 optical biosensor (BIAcore AB) as previously described (22). For these studies, CM5 sensor chips (BIAcore AB, Sweden) were prepared by amine-coupling of 33B8, vitronectin, or control monoclonal antibody, 31C9 or H77A10, at the level of ∼1000 response units according to the manufacturer's protocol. A flow cell with immobilized ovalbumin at the level of 500 response units was used as a control for nonspecific protein binding. Remaining binding sites on the chip surfaces were blocked by 1 m ethanolamine, pH 8.5, whereas unbound antibodies were washed out with 0.5% SDS.
All binding reactions were performed in standard HBS-P buffer, pH 7.4 (BIAcore AB, Sweden), at a flow rate of 30 μl/min at 25 °C. After each analyte injection, chip surfaces were regenerated with a 1-min pulse of 10 mm glycine, pH 1.5, followed by washing of the needle, flow cell, and whole flow system with running buffer for ∼2 min for complete removal of regeneration solution and equilibration. All injections were performed using the Wizard “Customized Application” program in an automated mode.
Interaction of PAI-1 with RCL-mimicking Peptides—The effect of RCL-mimicking peptides on PAI-1 was determined by incubating 2 μm wtPAI-1 or 14-1B with or without 1 mm Ac-TVASS or Ac-TVASSSTA for different durations at 37 °C in 0.1 m Tris, pH 7.4. 3 μm u-PA was added, and the reactions were further incubated for 20 min at 37 °C, subjected to reducing SDS-PAGE, the gels stained with Coomassie Brilliant Blue R, and the degree of PAI-1 cleavage determined.
The effect of vitronectin on the incorporation of RCL-mimicking peptides into PAI-1 was monitored via loss of PAI-1 inhibitory activity. PAI-1 variants were incubated with vitronectin as described for the determination of functional half-lives. Then Ac-TVASS or Ac-TVASSSTA were added to the mixture at a final concentration of 1 mm. Residual PAI-1 activity at different times was measured by incubating aliquots with excess u-PA, adding a chromogenic substrate, and monitoring substrate cleavage as described above. For wtPAI-1 and PAI-1AK, 200 nm serpin and 300 nm vitronectin were used. These data were also fit to a single exponential decay equation with a finite span to obtain the pseudo first-order rate constant of inactivation, kobs. This value and the rate constant of the latency transition, klat, as determined above, to account for the loss of PAI-1 activity due to the transition to latency during the incubations, were used to find the second-order rate constant of association between a PAI-1 variant and a peptide, kassoc, according to Equation 1, as previously described (26),
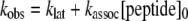 |
(Eq. 1) |
where [peptide]0 is the initial concentration of the peptide.
To determine the effect of vitronectin on the peptide-induced substrate behavior of PAI-1, 1 μm wtPAI-1 in the presence or absence of 1 μm vitronectin was incubated with 1 mm Ac-TVASS or Ac-TVASSSTA at 37 °C in 0.1 m Tris, pH 7.4. At different times, aliquots were withdrawn and mixed with 2 μm u-PA and allowed to further incubate for 10 min. The samples were analyzed via reducing SDS-PAGE and staining with Coomassie Brilliant Blue R. Gels were imaged using a Hewlett-Packard ScanJet 5200C color scanner, and band intensities were quantified using Kodak 1D Image analysis software. The proportion of PAI-1 cleaved was determined by comparing the intensity of the cleaved PAI-1 bands to that of a control lane containing PAI-1 that had not been reacted with protease, and the data were fit to a single exponential association equation.
Cleavage of a Substrate PAI-1 Variant—The effect of vitronectin on the cleavage of a PAI-1 variant that acts as a substrate was determined by preincubating 25 nm PAI-1R with 0 to 30-fold molar excess vitronectin for 5 min at 37 °C in 0.05 m Tris-HCl, pH 7.4, 0.15 m NaCl, 0.01% Tween 20, 100 μg/ml bovine serum albumin, and 1000 units/ml aprotinin, followed by the addition of 0.5 nm u-PA or t-PA for 45 min, or β-trypsin for 10 min using a similar buffer lacking aprotinin. The reactions were quenched with sample loading buffer containing 1% SDS, and analyzed via reducing SDS-PAGE (27). Proteins were transferred after gel electrophoresis onto polyvinylidene difluoride membrane and probed with polyclonal rabbit anti-PAI-1 antibody as described (28). Films were imaged using the Kodak EDAS 290 gel imaging system, band intensities were quantified using Kodak 1D Image software, and the data were fit to a rectangular hyperbolic equation.
Statistical Analysis—Data were evaluated by an unpaired Student's t test.
RESULTS
Binding of Wild-type and Stable PAI-1 to Vitronectin—We incubated wtPAI-1 or 14-1B in the absence and presence of vitronectin for different time periods at 37 °C, and measured the residual inhibitory activity against u-PA. In agreement with previous reports (29–32), vitronectin modestly increased the half-life of wtPAI-1 from 1.28 to 1.73 h (Fig. 1A, Table 1), a factor of 1.35-fold. 14-1B remained active for days, exhibiting a half-life of ∼116 h (Fig. 1A, inset, Table 1). The half-life of 14-1B was enhanced 2.06-fold to 239 h (Fig. 1A, inset, Table 1). No vitronectin-dependent effect on the half-life of PAI-1AK, a variant with nearly undetectable binding to vitronectin, was observed (Table 1), indicating that the binding of vitronectin to PAI-1 and not merely the presence of the cofactor are required for changes in the lifespan of the inhibitor.
FIGURE 1.

Interaction of PAI-1 with vitronectin. In panel A, 100 nm wtPAI-1 (circles) or 14-1B (triangles, inset) was preincubated at 37 °C in the absence (open symbols) or presence (closed symbols) of 150 nm vitronectin as described under “Experimental Procedures.” Then, aliquots were withdrawn from ongoing incubations at the times shown and assayed for residual inhibitory activity toward u-PA. The labels of the axes within the inset are identical as those in the main graph, although the scales are different. Symbols and error bars represent the mean ± S.D. for three experiments. In panel B, real-time vitronectin-PAI-1 binding was monitored using a BIAcore 3000 by injecting 10 nm active wtPAI-1 (tracing 1), active 14-1B (tracing 2), PAI-1AK (wtPAI-1 carrying R101A and Q123K, tracing 3), 14-1BAK (14-1B carrying R101A and Q123K, tracing 4), or latent wtPAI-1 (tracing 5) over a vitronectin-coupled sensor chip, as described under “Experimental Procedures.”
TABLE 1.
Loss of PAI-1 activity in the presence or absence of vitronectin (Vn) 100 nm of each PAI-1 variant was incubated with or without 150 nm vitronectin, as shown, at 37 °C as described under “Experimental Procedures.” At different times, aliquots were removed and the residual PAI-1 activity was measured against u-PA. The loss in PAI-1 activity over time was fit to a single exponential decay equation from which klat and t½, the rate constant and the half-life of the latency transition, respectively, were derived. Shown are the mean ± S.D. for n = 3.
| PAI-1 variant | Vn | klat | t½ | -Fold increase in t½ |
|---|---|---|---|---|
| s–1 | h | |||
| wtPAI-1 | – | 1.51 (±0.09) × 10–4 | 1.28 (±0.07) | 1.35 |
| wtPAI-1 | + | 1.11 (±0.04) × 10–4 | 1.73 (±0.06)a | |
| PAI-1AK | – | 1.35 (±0.08) × 10–4 | 1.43 (±0.09) | 0.93 |
| PAI-1AK | + | 1.45 (±0.02) × 10–4 | 1.33 (±0.02) | |
| 14-1B | – | 1.66 (±0.09) × 10–6 | 116 (±7)b | 2.06 |
| 14-1B | + | 8.33 (±0.69) × 10–7 | 231 (±21)b,c |
Significantly different from corresponding value without vitronectin (p < 0.005)
Significantly different from corresponding value with wtPAI-1 (p < 0.0001)
Significantly different from corresponding value without vitronectin (p < 0.001)
Vitronectin binding was also directly monitored in real-time. Vitronectin was coupled to a sensor chip and binding of PAI-1 variants was studied using a BIAcore 3000 as described under “Experimental Procedures.” Application of wtPAI-1 and 14-1B to the immobilized vitronectin yielded virtually identical real-time affinity binding curves (Fig. 1B, sensorgrams 1 and 2). The variants PAI-1AK and 14–1BAK (Fig. 1B, sensorgrams 3 and 4, respectively) contain R101A and Q123K, substitutions that abolish high affinity binding to the immobilized vitronectin. Because the residues identified in the high affinity vitronectin-binding site of wtPAI-1 are unchanged in the stable mutant (16, 18, 19), these data suggest that the vitronectin-binding sites on active wtPAI-1 and active 14-1B share the same location and a similar conformation. Latent wtPAI-1 also did not bind with high affinity to the immobilized vitronectin (Fig. 1B, sensorgram 5), in agreement with the known structural dissimilarities in the vitronectin-binding sites of active 14-1B and latent wtPAI-1 (12, 33, 34).
Binding of Wild-type and Stable PAI-1 to the Monoclonal Antibody, 33B8—The monoclonal antibody, 33B8, was raised against covalent complexes of human t-PA and PAI-1 (35). The antibody localizes to a neo-epitope at the proximal hinge of the PAI-1 RCL that is formed when that portion of RCL is inserted into β-sheet A, and it extends into the portion of α-helix D that is closest to the RCL (36, 37). Hence, it binds rapidly to cleaved and latent PAI-1 as well as to protease-PAI-1 complexes (38). Transition of active to latent PAI-1 was accelerated by 33B8 (35, 38), which presumably bound to and stabilized a pre-latent intermediate form of the serpin, wherein the RCL is partially inserted into the central β-sheet (36, 37), thus favoring progression to the latent conformation. We used robust binding of 33B8 to the loop-inserted forms of PAI-1, but poor binding to loop-expelled PAI-1, to probe the conformation of the RCL in wtPAI-1 and 14-1B in solution. First, we tested whether a more stably active form of PAI-1 was also more resistant to inactivation by the antibody. Fig. 2A shows that wtPAI-1 rapidly lost anti-u-PA activity in the presence of 33B8 with a half-life of 3.47 min, whereas the stable mutant, 14-1B, retained inhibitory activity for much longer (Fig. 2A, inset), exhibiting a half-life of 142 min, a 41-fold difference.
FIGURE 2.

Interaction of PAI-1 with the murine monoclonal antibody, 33B8. In panel A, 20 nm wtPAI-1 (circles) or 14-1B (triangles, inset) was combined with 50 nm 33B8 at 37 °C. At the times shown, residual anti-u-PA activity was measured as described under “Experimental Procedures,” and the data were fit to a single exponential decay equation. The labels of the axes within the inset are identical as those in the main graph, although the scales are different. Symbols and error bars represent the mean ± S.D. for three experiments. Real-time antibody-PAI-1 binding was directly monitored using a BIAcore 3000 by injection of 10 nm latent wtPAI-1 (tracing 1), active wtPAI-1 (tracing 2), or active 14-1B (tracing 3), a 33B8-coupled sensor chip (panel B), as described under “Experimental Procedures,” or a chip coupled with a control antibody, 31C9, that binds all known conformations of free PAI-1 (panel C).
We then directly monitored the interaction between PAI-1 and the antibody. 33B8 was coupled to a sensor chip and binding of PAI-1 was studied using BIAcore 3000. Fig. 2B shows that, whereas latent PAI-1 binds most rapidly, active wtPAI-1 binds more robustly to immobilized 33B8 than does the stable mutant 14-1B. Fig. 2C shows that latent PAI-1, active wtPAI-1, and active 14-1B all bound robustly to a sensor chip coupled to 31C9, which binds to latent and active PAI-1 (39). The murine monoclonal antibodies, H77A10, 33B8, and 31C9, share the same isotype, IgG1, and no form of PAI-1 bound to the isotype control antibody, H77A10 (data not shown). These data suggest that the relative resistance of 14-1B to inactivation by 33B8 as compared with wtPAI-1 is due to diminished binding between the stable mutant and the antibody, indicating that the two inhibitors are conformationally different at the proximal hinge of the RCL or near α-helix D.
Binding of RCL-mimicking Peptides to Wild-type and Stable PAI-1—Studies employing x-ray crystallography and fluorescently labeled serpins revealed that peptides that carry the amino acid sequence of the RCL bind in β-sheet A between s3A and s5A, becoming s4A (38, 40–43). Peptide binding prevents the RCL from inserting into the central sheet upon engagement of the serpin with a target protease, thereby blocking protease translocation and protease inhibition, allowing the protease to complete cleavage of the serpin. The binding of these peptides to serpins can therefore be accurately followed by monitoring the peptide-dependent loss of inhibitory activity and appearance of substrate behavior in the serpin (30, 44).
Generation of latent PAI-1 requires the insertion of the RCL between strands 3 and 5 of β-sheet A (12). To test the hypothesis that the more rapid transition of wtPAI-1 to latency reflects its central sheet being more accommodating to the insertion of an RCL peptide than is the central sheet of the stable mutant, wtPAI-1 or 14-1B was incubated with either an RCL-mimicking pentapeptide, Ac-TVASS, or octapeptide, Ac-TVASSSTA, which correspond to PAI-1 RCL residues P14–P10 and P14–P7, respectively, and have been shown to bind to the groove between s3A and s5A in PAI-1 (38, 41). Excess u-PA was added, and the substrate behavior of each PAI-1 was monitored by SDS-PAGE as a measure of peptide binding to the groove. As demonstrated in Fig. 3, in the absence of peptide, wtPAI-1 significantly autoinactivated during a 2-h incubation, whereas 14-1B remained almost completely in the active form (lanes 5 and 6), consistent with their known functional half-lives. Either peptide incubated with wtPAI-1 for 2 h(lanes 7 and 9) resulted in less latent PAI-1 (unreacted PAI-1 in the presence of excess u-PA), more cleaved PAI-1, and no detectable acyl-enzyme complexes as compared with the reaction without RCL-mimicking peptides (lane 5). Two-hour incubations of 14-1B with the peptides demonstrated that the pentapeptide was more effective at preventing acyl-enzyme complex formation than was the octapeptide (lanes 8 and 10). However, allowing 14-1B to incubate with the peptides for 20 h showed a time-dependent increase of peptide binding to 14-1B, and facilitated complete binding to 14-1B for the smaller peptide (lane 12), whereas only a small amount of 14-1B that was incubated with the longer peptide retained the ability to complex with u-PA (lane 13). Hence, consistent with the prevailing hypothesis of the nature of the stability within 14-1B, 14-1B incorporates the RCL-mimicking peptides less readily than does wtPAI-1. In addition, the RCL-mimicking peptides appear to inactivate 14-1B in the same manner as has been previously reported for wtPAI-1, by induction of substrate behavior (26).
FIGURE 3.

Interaction of PAI-1 with RCL-mimicking peptides. Wild-type PAI-1 or 14-1B at 2 μm were incubated with or without 1 mm peptide at 37 °C for the times shown. The samples were then reacted with excess u-PA for 20 min at 37 °C and analyzed by reducing SDS-PAGE and Coomassie staining. One representative experiment of three is shown.
The Effect of Vitronectin on the Binding of RCL-mimicking Peptides to wtPAI-1—The current understanding of how vitronectin prolongs the inhibitory activity of PAI-1 predicts that binding of the cofactor to the active serpin should hinder the opening of β-sheet A, thereby hindering autoinsertion of the RCL (16–18). If this model is correct, binding of vitronectin to active PAI-1 should also decrease the likelihood that a peptide with the same sequence as the RCL could insert into the central β-sheet. To test the hypothesis that vitronectin binding results in closure of the s3A–s5A groove, and subsequent slower incorporation of RCL-mimicking peptides into β-sheet A, wtPAI-1 was incubated with either Ac-TVASS or Ac-TVASSSTA in the presence or absence of vitronectin for different amounts of time. After these incubations, excess u-PA was added to each mixture and the reactions were analyzed by following the loss of anti-u-PA activity. As demonstrated in Fig. 4, contrary to the prevailing hypothesis, both pentapeptide and octapeptide insertion (as measured by loss of PAI-1 activity) proceeded more rapidly in the presence of vitronectin (Fig. 4, A and B, respectively). Moreover, direct binding of PAI-1 to vitronectin was required for augmented peptide interaction with PAI-1, as vitronectin had no effect on the peptide-induced loss of inhibitory activity in PAI-1AK, a vitronectin non-binding variant of wtPAI-1 (22) (Fig. 4, A and B, insets).
FIGURE 4.

Effect of vitronectin on the inactivation of PAI-1 by RCL-mimicking peptides. 200 nm wtPAI-1 was briefly incubated in the absence (open circles) or presence (closed circles) of 300 nm vitronectin, and then mixed with 1 mm Ac-TVASS (panel A) or Ac-TVASSSTA (panel B). Aliquots were taken at the times shown and the residual u-PA-inhibiting activity was measured as described under “Experimental Procedures.” Insets, inactivation of 200 nm PAI-1AK by Ac-TVASS (panel A, inset) or Ac-TVASSSTA (panel B, inset) in the absence (open triangles) or presence (closed triangles) of 300 nm vitronectin. The labels of the axes within the inset are identical as those in the main graph, although the scales are different. Symbols and error bars represent the mean ± S.D. for three experiments. Solid lines are the fits of the data to a single-order exponential decay equation.
The loss of inhibitory activity was used to obtain the rate, kobs, of PAI-1 inactivation, and the second-order rate constants, kassoc, for peptide incorporation into the s3A–s5A groove for each form of PAI-1 in the presence and absence of vitronectin (Table 2). Contrary to the currently held structural hypothesis, vitronectin binding was found to accelerate the incorporation of either RCL-mimicking peptide into β-sheet A of PAI-1, suggesting that vitronectin binding resulted in β-sheet A being more open, despite a slower transition to latency. Consistent with this, PAI-1AK, showed essentially identical rates of peptide incorporation in the presence and absence of vitronectin (Table 2).
TABLE 2.
Inactivation of PAI-1 in the presence of an RCL-mimicking peptide PAI-1 variants in the presence and absence of vitronectin, as shown, were combined with either RCL-mimicking peptide as described under “Experimental Procedures.” At increasing times, aliquots were removed, and the residual PAI-1 activity was assayed against uPA. The loss of PAI-1 activity over time was fit to a single exponential decay equation to obtain the observed pseudo first-order rate constant of PAI-1 inactivation, kobs. The second-order rate constant for the association of a PAI-1 variant and a peptide, kassoc, was determined using the first-order rate constant of the latency transition, klat, from Table I, as described under “Experimental Procedures.” Shown are the mean ± S.D. for n = 3.
|
PAI-1 variant
|
Vitronectin
|
Ac-TVASS
|
Ac-TVASSSTA
|
||
|---|---|---|---|---|---|
| kobs | kassoc | kobs, s–1 | kassoc | ||
| s–1 | m–1 s–1 | s–1 | m–1 s–1 | ||
| wtPAI-1 | – | 1.82 (±0.18) × 10–3 | 1.67 (±0.18) | 1.32 (±0.11) × 10–3 | 1.17 (±0.11) |
| wtPAI-1 | + | 2.85 (±0.17) × 10–3 | 2.74 (±0.17)a | 2.09 (±0.10) × 10–3 | 1.98 (±0.10)a |
| PAI-1AK | – | 1.28 (±0.05) × 10–3 | 1.14 (±0.05) | 7.66 (±0.32) × 10–4 | 0.63 (±0.03) |
| PAI-1AK | + | 1.18 (±0.06) × 10–3 | 1.04 (±0.06) | 7.48 (±0.27) × 10–4 | 0.60 (±0.03) |
| 14-1B | – | 7.05 (±0.23) × 10–5 | 6.89 (±0.23) × 10–2b | 1.84 (±0.15) × 10–5 | 1.68 (±0.15) × 10–2b |
| 14-1B | + | 8.93 (±0.80) × 10–5 | 8.85 (±0.80) × 10–2b,c | 5.18 (±0.23) × 10–5 | 5.10 (±0.23) × 10–2b,d |
Significantly different from corresponding value without vitronectin (p < 0.002)
Significantly different from corresponding value with wtPAI-1 (p < 0.0001)
Significantly different from corresponding value without vitronectin (p < 0.02)
Significantly different from corresponding value without vitronectin (p < 0.0001)
To confirm these unexpected findings, we repeated the experiments but monitored the incorporation of peptides by the generation of cleaved PAI-1 via SDS-PAGE. Because the covalent serpin-protease complex co-migrates with vitronectin, progress of the peptide-PAI-1 interaction was followed by analyzing the amount of cleaved serpin. The amount of unreacted serpin did not change, consistent with the peptides preventing the transition to latency. Consistent with the above data but contrary to predictions based on the prevailing structural hypothesis, vitronectin-bound wtPAI-1 demonstrated more rapid cleavage in the presence of RCL-mimicking peptides than did free wtPAI-1 (Fig. 5), implying greater s3A–s5A groove accessibility in the vitronectin·wtPAI-1 complex as compared with wtPAI-1 alone. Importantly, this method of analysis also confirmed that the presence of vitronectin did not change the mechanism by which the RCL-mimicking peptides inactivate PAI-1, namely by inducing serpin cleavage.
FIGURE 5.

Effect of vitronectin on the ability of RCL-mimicking peptides to induce substrate behavior in wtPAI-1. For panel A, 1 μm wtPAI-1 was briefly incubated at 37 °C without (lanes 3–7) or with (lanes 8–12) 1 μm vitronectin, after which 1 mm Ac-TVASS was added. At the times shown, aliquots were removed and mixed with excess u-PA as described under “Experimental Procedures,” and the samples were analyzed by reducing SDS-PAGE and Coomassie staining. Controls include unreacted wtPAI-1 (lane 1) and vitronectin alone (lane 2). Identical experiments were conducted using Ac-TVASSSTA (gel not shown). The amounts of PAI-1 cleavage that were induced by Ac-TVASS (panel B) or Ac-TVASSSTA (panel C) in the absence (open circles) and presence (closed circles) of vitronectin were analyzed as described and are plotted versus time. Symbols and error bars represent the mean ± S.D. for three experiments, and the data were fit to a first-order association equation.
Probing the Structural Basis for Enhanced Stability in 14-1B Using Intercalating Peptides—Both pentapeptide and octapeptide bound to 14-1B about 2 orders of magnitude less efficiently than to wtPAI-1 (Table 2). Also, the pentapeptide inserted into the stable mutant about 4-fold more efficiently than did the octapeptide (Fig. 6, A and B, Table 2). These data indicate that the s3A–s5A groove of the central sheet of 14-1B is less accessible to peptides than that of wtPAI-1 and that there exists some additional hindrance in 14-1B against the binding of a longer peptide.
FIGURE 6.

Effect of vitronectin on the inactivation of 14-1B by Ac-TVASS (panel A) or Ac-TVASSSTA (panel B). 14-1B at 100 nm was incubated briefly at 37 °C without (open circles) or with (closed circles) 150 nm vitronectin, after which 1 mm peptide was added. At the times shown, aliquots were removed and assayed for residual u-PA-inhibiting activity. Symbols and error bars represent the mean ± S.D. for three experiments. Solid lines are the fits of the data to a single-order exponential decay equation.
However, the augmenting effect of vitronectin on the incorporation of 14-1B on the octapeptide was significantly greater than its effect on incorporation of the pentapeptide, or its effect on wtPAI-1 (Fig. 6, A and B, Table 2). Thus vitronectin does not prevent peptides from inserting into the central sheet of 14-1B, but rather relieves some sort of hindrance against peptide binding in β-sheet A. In addition, vitronectin binding seems to allow the s3A–s5A groove in 14-1B to open along a greater length, facilitating binding of a longer peptide, even though the serpin becomes more resistant to loop insertion related to the transition to latency (Fig. 1). These data suggest that the accessibility of β-sheet A in 14-1B to intercalating peptides is regulated in such a way as to confer an enhanced selectivity favoring shorter peptides as compared with wtPAI-1, and that this enhanced selectivity in 14-1B is partially abolished by the binding of vitronectin.
Vitronectin Binding Affects the PAI-1 RCL—Because vitronectin binding modestly hinders latency-associated RCL insertion but does not result in the closing of β-sheet A, we hypothesized that vitronectin caused structural rearrangements in the RCL itself, making it less likely to undergo autoinsertion. To test this hypothesis, we determined the effect of the cofactor on the proteolysis of the PAI-1 P1–P1′ bond, because a change in the RCL conformation would likely result in a change in P1–P1′ bond cleavage. We used a non-inhibitory PAI-1 mutant, PAI-1R, which carries two arginyl residues at positions 333 and 335 in the proximal hinge, corresponding to RCL residues P12 and P14, but is otherwise identical to wtPAI-1 in primary amino acid sequence (23). Bulky charged residues at the proximal hinge have been shown to slow the insertion of the RCL into β-sheet A (45, 46). Thus PAI-1R behaves as a substrate and is completely cleaved by target proteases. Importantly, PAI-1R also binds to vitronectin with an affinity that is comparable with wtPAI-1 (data not shown).
As a test of the accessibility of the RCL to proteases, we determined the effect of vitronectin on the cleavage of PAI-1R. The substrate mutant was incubated briefly with up to 30-fold molar excess of cofactor, and then combined with catalytic amounts of cognate proteases, u-PA or t-PA, or a protease known to exhibit very few exosites, β-trypsin. Fig. 7A shows that as the concentration of vitronectin was increased, more PAI-1R was cleaved by all three target proteases. The effect was saturated between 3- and 10-fold molar excess of vitronectin over PAI-1R, and remained unchanged throughout higher concentrations of cofactor (Fig. 7B), suggesting that vitronectin was not likely acting as a scaffolding molecule onto which PAI-1R and proteases assemble. The augmentation of PAI-1R cleavage was critically dependent on the interaction between PAI-1R and vitronectin, as the variant PAI-1R-AK, which binds poorly to vitronectin, showed no such cofactor-dependent effects (data not shown). Vitronectin does not augment the amidolytic activity of plasminogen activators or β-trypsin (data not shown). Also there are no expected exosite interactions between β-trypsin and PAI-1 that can be enhanced by the presence of vitronectin (47). Thus, the effect of vitronectin on cleavage of the PAI-1 P1–P1′ bond is consistent with vitronectin changing the structural presentation of the RCL to the protease active sites, making that bond more accessible to proteolytic attack, and not with vitronectin altering exosite interactions between PAI-1 and target proteases. These data, combined with the evidence that vitronectin binding does not close β-sheet A, suggests the direct effect of the cofactor on the conformation of the RCL has appreciable stability-enhancing effects.
FIGURE 7.

Effect of vitronectin on the reactivity of the substrate variant, PAI-1R, with target proteases. In panel A, PAI-1R was briefly incubated with vitronectin at the concentrations shown, reacted for 45 min at 37 °C with either u-PA or t-PA or for 10 min with β-trypsin, and then analyzed by reducing SDS-PAGE and immunoblotting with anti-PAI-1 polyclonal antibodies, as described under “Experimental Procedures.” Band intensities were quantified, and the data for cleavage of PAI-1R by t-PA (open circles), u-PA (closed circles), and β-trypsin (open triangles) were fit to a rectangular hyperbolic equation (panel B). Symbols and error bars represent the mean ± ranges for two experiments.
DISCUSSION
We propose a model of PAI-1 that unifies published data and accommodates findings that are inconsistent with the current structural hypothesis for this dynamic serpin. Using three ligands with well defined binding sites, we probed the unknown native solution structure of wtPAI-1 and analyzed our findings in light of the known x-ray crystal structure of the stable PAI-1 mutant, 14-1B. Based on our experiments, native wtPAI-1 binds vitronectin identically to 14-1B, binds the monoclonal antibody, 33B8, more robustly than does 14-1B, and incorporates RCL-mimicking peptides more rapidly than does the stable mutant. We believe that these data reveal three structural features of native wtPAI-1 (Fig. 8): that wtPAI-1 and 14-1B exhibit similar conformations at the vitronectin binding site, suggesting that β-sheet A in the vicinity of α-helix F is in a closed conformation; that the proximal hinge of the RCL of native wtPAI-1 appears more structurally similar to that of latent wtPAI-1 than active 14-1B, suggesting that there is some degree of RCL insertion; and that the region of the central sheet near the RCL (to which we will refer as the “top” of β-sheet A) in native wtPAI-1 is more open than that of 14-1B.
FIGURE 8.

Model of the native conformation of wtPAI-1. In panel A, a model for the native conformation of wtPAI-1 was constructed by mapping the primary sequence of human PAI-1 onto the amino acid backbone of the x-ray crystal structure of native antithrombin (PDB accession number 2BEH) and manually adjusting RCL so that it comports with binding of the monoclonal antibody, 33B8. The x-ray crystal structure of 14-1B (PDB accession number 1B3K, chain A) is shown in panel B. In each panel, the reactive center loop is shown in red, β-sheet A in green, and α-helix F in blue. Structures were generated and modified in DeepView/Swiss-PdbViewer version 3.7 (67), and were rendered in POV-Ray version 3.6 (Persistence of Vision Raytracer Pty. Ltd.).
Review of the known serpin x-ray crystal structures reveals that our model for native wtPAI-1 resembles the structures of myeloid and erythroid nuclear termination stage-specific protein (48), antithrombin (49, 50), protein C inhibitor (51), heparin cofactor II (52), and thyroxine-binding globulin (53). These serpins in their native conformation all exhibit partial insertion of the RCL into the central sheet and a pigeon-toed conformation in β-sheet A, in which the top of the sheet is open, whereas the region of the sheet underneath α-helix F is closed. Of the serpins that can adopt this conformation, antithrombin exhibits a cofactor-induced conformational activation (54, 55) to which the interaction of PAI-1 and vitronectin is thought to be analogous. Upon binding of native antithrombin to heparinpentasaccharide, the RCL is extracted from its partially inserted position as the top of β-sheet A is closed (55). Similar to PAI-1, antithrombin in fully RCL-inserted conformations no longer binds its cofactor with high affinity (44, 50, 56). Importantly, antithrombin has been crystallized in its native partially loop-inserted state while bound to heparin (57, 58). This demonstrates that a pigeon-toed state of the central sheet is indeed compatible with binding to a conformationally sensitive cofactor.
The vitronectin-dependent enhancement of the inhibitory half-life of wtPAI-1 is proposed to arise from a cofactor-induced stabilization of β-sheet A in a closed state, resulting in a diminished likelihood of RCL autoinsertion (16). Comparison of the x-ray crystal structure of latent PAI-1 to that of 14-1B bound to the somatomedin B domain of vitronectin, which contains the high-affinity PAI-1-binding site of the cofactor, seemed to support this straightforward mechanism (18). A similar mechanism for the heparin-induced activation of anti-thrombin has been crystallographically validated (55). Heparin also protects antithrombin from inactivation by RCL-mimicking peptides (44), supporting the hypothesis that the cofactor induces closure of β-sheet A in solution. We tested the hypothesis that vitronectin hinders the opening of β-sheet A by monitoring the effect of vitronectin on the incorporation of RCL peptides by PAI-1. Surprisingly, we found that vitronectin did not protect PAI-1 from such peptides and, in fact, accelerated the peptide-dependent inactivation of PAI-1. It is unlikely that vitronectin enhances partitioning of s3A and s5A and further opening along the length of β-sheet A because serpins predisposed to the central sheet opening due to shutter region mutations exhibit increased polymerization and latency transition in addition to increased incorporation of RCL-mimicking peptides (29, 59). The most parsimonious mechanism by which vitronectin increases the accessibility of the PAI-1 central sheet to peptides while prolonging the half-life of the serpin is by inducing expulsion of the RCL from the native partially inserted position in a manner that is not linked to closure of β-sheet A. Thus vitronectin binding likely reduces the amount of steric hindrance caused by the RCL against peptide insertion.
Other studies have advocated that vitronectin binding induces conformational changes in the PAI-1 RCL. Vitronectin has been shown to restore robust anti-t-PA activity to virtually inactive P1–P1′ mutants of PAI-1 (60), suggesting that the RCL is re-arranged by vitronectin in such a way that improves the fit between an unfavorable peptide and the protease active site. The cofactor has also been shown to increase the orientational freedom of a fluorescent probe bound to the PAI-1 RCL (61). We have found that cleavage of a substrate mutant of PAI-1, PAI-1R, by cognate proteases, t-PA, and u-PA, is accelerated by vitronectin. This effect was also observed when the enzyme used was β-trypsin. For all three proteases, the vitronectin-dependent effect was saturable and did not diminish in 30-fold molar excess vitronectin over PAI-1R. Together the data indicate that vitronectin was not bridging PAI-1R and a target protease nor enhancing exosite interactions between them, but was rather altering the conformation of the RCL such that the P1–P1′ bond is presented more optimally to proteases, in good agreement with previous reports.
We are now in a better position to propose a more complete equilibrium model of the conformational states of wtPAI-1. As shown in Scheme 1, we propose that native active PAI-1 is in equilibrium among three states (states i, ii, and iii). In state i, native PAI-1 is proposed to be in a conformation that is most similar to that of the x-ray crystal structure of the stable mutant, 14-1B, with s3A and s5A closely approximated along their entire lengths, resulting in a closed position in β-sheet A and attendant full expulsion of the RCL. State i is stabilized by low temperature and low pH (11, 62), as these conditions dramatically slow the transition to latency and prevent the binding of RCL-mimicking peptides. In state ii, s3A and s5A are parted near the RCL, with the loop being possibly fully exposed or slightly inserted into the central sheet. In state iii, the RCL is inserted into β-sheet A, perhaps up to residue P11, and the s3A–s5A groove is occupied to a greater degree than that postulated for state ii. State iii correlates with the model that we have depicted in Fig. 8A, a native PAI-1 conformation exhibiting a partially open β-sheet A and a partially inserted RCL, as suggested by rapid binding to 33B8 and RCL-mimicking peptides. Further loop insertion beyond this point highly favors the formation of state iv, latent PAI-1, in which the RCL is completely inserted into β-sheet A. Formation of latent PAI-1 (iv) is a transition that is irreversible under physiologic conditions (12, 63). In the presence of vitronectin, our data suggest that the equilibrium between states ii and iii is shifted toward state ii, resulting in a modest delay in the transition to latency and an increased susceptibility to the incorporation of RCL-mimicking peptides.
SCHEME 1.

Although a pre-latent PAI-1 conformation has been proposed based on the discovery of latency-inducing antibodies, the nature of this state remains elusive. Characterization of the binding sites of these antibodies has yielded insight into the structure of such a PAI-1 intermediate. In human PAI-1, partial insertion of the RCL into β-sheet A at the proximal hinge (36, 38) and detachment of s1C from s2C at the distal hinge (64) have been associated with pre-latency in independent studies. In rat PAI-1, changes in the N terminus in α-helix A (65) have also been associated with pre-latency. However, it is unknown whether these conformational changes at the proximal hinge, distal hinge, and in α-helix A exist within the same state of PAI-1, whether the changes in these regions occur in a specific order in the generation of latent PAI-1, or whether the regions allosterically affect one another. Thus, the term “pre-latent” is unclear and may apply to one or several conformations between states iii and iv in the equilibrium model that we propose in Scheme 1. Further studies are needed to elucidate the specific steps in the transition from the active to the latent forms of PAI-1.
Because both active and latent PAI-1 have been recovered from human plasma and tissues, the transition from active to latent form is thought to be physiologically relevant. Most structural data are derived from the stable mutant of PAI-1, 14-1B, making further delineation of functional and structural differences between wt-PAI-1 and 14-1B important. The differential binding of 33B8 to wtPAI-1 versus 14-1B suggests that the RCL of 14-1B is more extended, with less probability of partial-loop insertion. This is in agreement with a previous study using fluorescent probes to demonstrate that the RCL of wtPAI-1 was folded closely to the serpin core (66), unlike the RCL conformation found in the x-ray crystal structure of 14-1B. As with wtPAI-1, the half-life of 14-1B is prolonged by binding to the cofactor. However, vitronectin binding to 14-1B also relieves a constraint against the incorporation of RCL-mimicking peptides into β-sheet A that is not found in wtPAI-1. This hindrance in 14-1B may be the novel hydrogen bond network between Glu-283 of s6A and the backbone of the loop between s3A and α-helix F (33, 34). The H-bond network is made possible by the unique 310 helix in this loop induced by the N150H,K154T,Q319L substitutions in the stable variant. Substitution of Glu-283 by an alanyl residue decreases the inhibitory half-life of 14-1B by nearly 5-fold (67), suggesting that the novel H-bond network is present in 14-1B in solution and is abrogated by the E283A mutation. Upon the binding of 14-1B to the somatomedin B domain of vitronectin, the H-bond network is similarly broken, and the s3A/hF loop no longer contains the 310 helix (18). We believe this may be the structural correlate to discriminatory behavior of 14-1B toward RCL-mimicking peptides of different lengths, a here-to-fore undescribed functional difference between the stable mutant and the wild-type inhibitor.
In conclusion, we have presented biochemical data suggesting that the current model of the active conformation of PAI-1 does not adequately explain the behavior of serpin in solution. We have therefore presented a more comprehensive structural hypothesis that comports with existing data, that accounts for several states of the molecule, and that provides a clearer understanding of the effect of vitronectin on the active conformation of PAI-1. In doing so, we have identified features of novel conformations of PAI-1 alone and in complex with vitronectin, which may yield new molecular targets for the inactivation of the inhibitor based on the structural properties of the naturally occurring molecule rather than that of a stable variant.
Acknowledgments
We thank Dr. James H. Morrissey of the University of Illinois at Urbana-Champaign (Urbana, IL) for helpful discussions, Evelyn C. Nieves-Li for work on the interaction between PAI-1 and the RCL-mimicking peptides, and Debora McCall for expert secretarial assistance in the preparation of this manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants HL43506 (to B. S. S.), HL055374 and HL055747 (to D. A. L.). This work was also supported by a grant from the Carle Foundation (to B. S. S.). Part of this work was presented in abstract form at the XI International Workshop on Molecular and Cellular Biology of Plasminogen Activation, June 16–20, 2007, Vår Gård Saltsjöbaden, Sweden. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: RCL, reactive center loop; PAI-1, plasminogen activator inhibitor type 1; wtPAI-1, wild-type PAI-1; t-PA, tissue-type plasminogen activator; u-PA, urokinase-type plasminogen activator; pNA, p-nitroanilide.
References
- 1.Gettins, P. G. (2002) Chem. Rev. 102 4751-4804 [DOI] [PubMed] [Google Scholar]
- 2.Bruch, M., and Bieth, J. G. (1989) Biochem. J. 259 929-930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Olson, S. T., and Shore, J. D. (1982) J. Biol. Chem. 257 14891-14895 [PubMed] [Google Scholar]
- 4.Stone, S. R., Nick, H., Hofsteenge, J., and Monard, D. (1987) Arch. Biochem. Biophys. 252 237-244 [DOI] [PubMed] [Google Scholar]
- 5.Egelund, R., Rodenburg, K. W., Andreasen, P. A., Rasmussen, M. S., Guldberg, R. E., and Petersen, T. E. (1998) Biochemistry 37 6375-6379 [DOI] [PubMed] [Google Scholar]
- 6.Lawrence, D. A., Ginsburg, D., Day, D. E., Berkenpas, M. B., Verhamme, I. M., Kvassman, J. O., and Shore, J. D. (1995) J. Biol. Chem. 270 25309-25312 [DOI] [PubMed] [Google Scholar]
- 7.Dementiev, A., Dobo, J., and Gettins, P. G. (2006) J. Biol. Chem. 281 3452-3457 [DOI] [PubMed] [Google Scholar]
- 8.Huntington, J. A., Read, R. J., and Carrell, R. W. (2000) Nature 407 923-926 [DOI] [PubMed] [Google Scholar]
- 9.Stratikos, E., and Gettins, P. G. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 4808-4813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wind, T., Hansen, M., Jensen, J. K., and Andreasen, P. A. (2002) Biol. Chem. 383 21-36 [DOI] [PubMed] [Google Scholar]
- 11.Lindahl, T. L., Sigurdardottir, O., and Wiman, B. (1989) Thromb. Haemostasis 62 748-751 [PubMed] [Google Scholar]
- 12.Mottonen, J., Strand, A., Symersky, J., Sweet, R. M., Danley, D. E., Geoghegan, K. F., Gerard, R. D., and Goldsmith, E. J. (1992) Nature 355 270-273 [DOI] [PubMed] [Google Scholar]
- 13.Hekman, C. M., and Loskutoff, D. J. (1985) J. Biol. Chem. 260 11581-11587 [PubMed] [Google Scholar]
- 14.Lawrence, D. A., Olson, S. T., Palaniappan, S., and Ginsburg, D. (1994) Biochemistry 33 3643-3648 [DOI] [PubMed] [Google Scholar]
- 15.Declerck, P. J., De Mol, M., Alessi, M. C., Baudner, S., Paques, E. P., Preissner, K. T., Muller-Berghaus, G., and Collen, D. (1988) J. Biol. Chem. 263 15454-15461 [PubMed] [Google Scholar]
- 16.Lawrence, D. A., Berkenpas, M. B., Palaniappan, S., and Ginsburg, D. (1994) J. Biol. Chem. 269 15223-15228 [PubMed] [Google Scholar]
- 17.Lawrence, D. A., Palaniappan, S., Stefansson, S., Olson, S. T., Francis-Chmura, A. M., Shore, J. D., and Ginsburg, D. (1997) J. Biol. Chem. 272 7676-7680 [DOI] [PubMed] [Google Scholar]
- 18.Zhou, A., Huntington, J. A., Pannu, N. S., Carrell, R. W., and Read, R. J. (2003) Nat. Struct. Biol. 10 541-544 [DOI] [PubMed] [Google Scholar]
- 19.Berkenpas, M. B., Lawrence, D. A., and Ginsburg, D. (1995) EMBO J. 14 2969-2977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manchanda, N., and Schwartz, B. S. (1991) J. Biol. Chem. 266 14580-14584 [PubMed] [Google Scholar]
- 21.Magnotti, R. A., Jr. (1988) Anal. Biochem. 170 228-237 [DOI] [PubMed] [Google Scholar]
- 22.Xu, Z., Balsara, R. D., Gorlatova, N. V., Lawrence, D. A., Castellino, F. J., and Ploplis, V. A. (2004) J. Biol. Chem. 279 17914-17920 [DOI] [PubMed] [Google Scholar]
- 23.Stefansson, S., Petitclerc, E., Wong, M. K., McMahon, G. A., Brooks, P. C., and Lawrence, D. A. (2001) J. Biol. Chem. 276 8135-8141 [DOI] [PubMed] [Google Scholar]
- 24.Lawrence, D., Strandberg, L., Grundstrom, T., and Ny, T. (1989) Eur. J. Biochem. 186 523-533 [DOI] [PubMed] [Google Scholar]
- 25.Olson, S. T., Swanson, R., Day, D., Verhamme, I., Kvassman, J., and Shore, J. D. (2001) Biochemistry 40 11742-11756 [DOI] [PubMed] [Google Scholar]
- 26.Kvassman, J. O., Lawrence, D. A., and Shore, J. D. (1995) J. Biol. Chem. 270 27942-27947 [DOI] [PubMed] [Google Scholar]
- 27.Laemmli, U. K. (1970) Nature 227 680-685 [DOI] [PubMed] [Google Scholar]
- 28.Barker-Carlson, K., Lawrence, D. A., and Schwartz, B. S. (2002) J. Biol. Chem. 277 46852-46857 [DOI] [PubMed] [Google Scholar]
- 29.Hansen, M., Busse, M. N., and Andreasen, P. A. (2001) Eur. J. Biochem. 268 6274-6283 [DOI] [PubMed] [Google Scholar]
- 30.Jensen, S., Kirkegaard, T., Pedersen, K. E., Busse, M., Preissner, K. T., Rodenburg, K. W., and Andreasen, P. A. (2002) Biochim. Biophys. Acta 1597 301-310 [DOI] [PubMed] [Google Scholar]
- 31.Bodker, J. S., Wind, T., Jensen, J. K., Hansen, M., Pedersen, K. E., and Andreasen, P. A. (2003) Eur. J. Biochem. 270 1672-1679 [DOI] [PubMed] [Google Scholar]
- 32.Gils, A., Pedersen, K. E., Skottrup, P., Christensen, A., Naessens, D., Deinum, J., Enghild, J. J., Declerck, P. J., and Andreasen, P. A. (2003) Thromb. Haemostasis 90 206-217 [DOI] [PubMed] [Google Scholar]
- 33.Nar, H., Bauer, M., Stassen, J. M., Lang, D., Gils, A., and Declerck, P. J. (2000) J. Mol. Biol. 297 683-695 [DOI] [PubMed] [Google Scholar]
- 34.Sharp, A. M., Stein, P. E., Pannu, N. S., Carrell, R. W., Berkenpas, M. B., Ginsburg, D., Lawrence, D. A., and Read, R. J. (1999) Structure 7 111-118 [DOI] [PubMed] [Google Scholar]
- 35.Debrock, S., and Declerck, P. J. (1997) Biochim. Biophys. Acta 1337 257-266 [DOI] [PubMed] [Google Scholar]
- 36.Gorlatova, N. V., Elokdah, H., Fan, K., Crandall, D. L., and Lawrence, D. A. (2003) J. Biol. Chem. 278 16329-16335 [DOI] [PubMed] [Google Scholar]
- 37.Naessens, D., Gils, A., Compernolle, G., and Declerck, P. J. (2003) Thromb. Haemostasis 90 52-58 [PubMed] [Google Scholar]
- 38.Verhamme, I., Kvassman, J. O., Day, D., Debrock, S., Vleugels, N., Declerck, P. J., and Shore, J. D. (1999) J. Biol. Chem. 274 17511-17517 [DOI] [PubMed] [Google Scholar]
- 39.De Taeye, B., Compernolle, G., Dewilde, M., Biesemans, W., and Declerck, P. J. (2003) J. Biol. Chem. 278 23899-23905 [DOI] [PubMed] [Google Scholar]
- 40.Skinner, R., Chang, W. S., Jin, L., Pei, X., Huntington, J. A., Abrahams, J. P., Carrell, R. W., and Lomas, D. A. (1998) J. Mol. Biol. 283 9-14 [DOI] [PubMed] [Google Scholar]
- 41.Xue, Y., Bjorquist, P., Inghardt, T., Linschoten, M., Musil, D., Sjolin, L., and Deinum, J. (1998) Structure 6 627-636 [DOI] [PubMed] [Google Scholar]
- 42.Jankova, L., Harrop, S. J., Saunders, D. N., Andrews, J. L., Bertram, K. C., Gould, A. R., Baker, M. S., and Curmi, P. M. (2001) J. Biol. Chem. 276 43374-43382 [DOI] [PubMed] [Google Scholar]
- 43.Zhou, A., Stein, P. E., Huntington, J. A., Sivasothy, P., Lomas, D. A., and Carrell, R. W. (2004) J. Mol. Biol. 342 931-941 [DOI] [PubMed] [Google Scholar]
- 44.Bjork, I., Ylinenjarvi, K., Olson, S. T., and Bock, P. E. (1992) J. Biol. Chem. 267 1976-1982 [PubMed] [Google Scholar]
- 45.Schulze, A. J., Huber, R., Degryse, E., Speck, D., and Bischoff, R. (1991) Eur. J. Biochem. 202 1147-1155 [DOI] [PubMed] [Google Scholar]
- 46.Lawrence, D. A., Olson, S. T., Muhammad, S., Day, D. E., Kvassman, J. O., Ginsburg, D., and Shore, J. D. (2000) J. Biol. Chem. 275 5839-5844 [DOI] [PubMed] [Google Scholar]
- 47.Blouse, G. E., Perron, M. J., Kvassman, J. O., Yunus, S., Thompson, J. H., Betts, R. L., Lutter, L. C., and Shore, J. D. (2003) Biochemistry 42 12260-12272 [DOI] [PubMed] [Google Scholar]
- 48.McGowan, S., Buckle, A. M., Irving, J. A., Ong, P. C., Bashtannyk-Puhalovich, T. A., Kan, W. T., Henderson, K. N., Bulynko, Y. A., Popova, E. Y., Smith, A. I., Bottomley, S. P., Rossjohn, J., Grigoryev, S. A., Pike, R. N., and Whisstock, J. C. (2006) EMBO J. 25 3144-3155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carrell, R. W., Stein, P. E., Fermi, G., and Wardell, M. R. (1994) Structure 2 257-270 [DOI] [PubMed] [Google Scholar]
- 50.Skinner, R., Abrahams, J. P., Whisstock, J. C., Lesk, A. M., Carrell, R. W., and Wardell, M. R. (1997) J. Mol. Biol. 266 601-609 [DOI] [PubMed] [Google Scholar]
- 51.Li, W., Adams, T. E., Kjellberg, M., Stenflo, J., and Huntington, J. A. (2007) J. Biol. Chem. 282 13759-13768 [DOI] [PubMed] [Google Scholar]
- 52.Baglin, T. P., Carrell, R. W., Church, F. C., Esmon, C. T., and Huntington, J. A. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 11079-11084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou, A., Wei, Z., Read, R. J., and Carrell, R. W. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 13321-13326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huntington, J. A., Olson, S. T., Fan, B., and Gettins, P. G. (1996) Biochemistry 35 8495-8503 [DOI] [PubMed] [Google Scholar]
- 55.Jin, L., Abrahams, J. P., Skinner, R., Petitou, M., Pike, R. N., and Carrell, R. W. (1997) Proc. Natl. Acad. Sci. U. S. A. 94 14683-14688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bjork, I., and Fish, W. W. (1982) J. Biol. Chem. 257 9487-9493 [PubMed] [Google Scholar]
- 57.Johnson, D. J., and Huntington, J. A. (2003) Biochemistry 42 8712-8719 [DOI] [PubMed] [Google Scholar]
- 58.Johnson, D. J., Langdown, J., Li, W., Luis, S. A., Baglin, T. P., and Huntington, J. A. (2006) J. Biol. Chem. 281 35478-35486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou, A., Stein, P. E., Huntington, J. A., and Carrell, R. W. (2003) J. Biol. Chem. 278 15116-15122 [DOI] [PubMed] [Google Scholar]
- 60.Keijer, J., Ehrlich, H. J., Linders, M., Preissner, K. T., and Pannekoek, H. (1991) J. Biol. Chem. 266 10700-10707 [PubMed] [Google Scholar]
- 61.Fa, M., Karolin, J., Aleshkov, S., Strandberg, L., Johansson, L. B., and Ny, T. (1995) Biochemistry 34 13833-13840 [DOI] [PubMed] [Google Scholar]
- 62.Sancho, E., Tonge, D. W., Hockney, R. C., and Booth, N. A. (1994) Eur. J. Biochem. 224 125-134 [DOI] [PubMed] [Google Scholar]
- 63.Wang, Z., Mottonen, J., and Goldsmith, E. J. (1996) Biochemistry 35 16443-16448 [DOI] [PubMed] [Google Scholar]
- 64.Dupont, D. M., Blouse, G. E., Hansen, M., Mathiasen, L., Kjelgaard, S., Jensen, J. K., Christensen, A., Gils, A., Declerck, P. J., Andreasen, P. A., and Wind, T. (2006) J. Biol. Chem. 281 36071-36081 [DOI] [PubMed] [Google Scholar]
- 65.Ngo, T. H., Zhou, Y., Stassen, J. M., and Declerck, P. J. (2002) Thromb. Haemostasis 88 288-293 [PubMed] [Google Scholar]
- 66.Hagglof, P., Bergstrom, F., Wilczynska, M., Johansson, L. B., and Ny, T. (2004) J. Mol. Biol. 335 823-832 [DOI] [PubMed] [Google Scholar]
- 67.Wind, T., Jensen, J. K., Dupont, D. M., Kulig, P., and Andreasen, P. A. (2003) Eur. J. Biochem. 270 1680-1688 [DOI] [PubMed] [Google Scholar]