

Abstract
Mycobacteria contain an outer membrane composed of mycolic acids and a large variety of other lipids. Its protective function is an essential virulence factor of Mycobacterium tuberculosis. Only OmpA, which has numerous homologs in Gram-negative bacteria, is known to form channels in the outer membrane of M. tuberculosis so far. Rv1698 was predicted to be an outer membrane protein of unknown function. Expression of rv1698 restored the sensitivity to ampicillin and chloramphenicol of a Mycobacterium smegmatis mutant lacking the main porin MspA. Uptake experiments showed that Rv1698 partially complemented the permeability defect of the M. smegmatis porin mutant for glucose. These results indicated that Rv1698 provides an unspecific pore that can partially substitute for MspA. Lipid bilayer experiments demonstrated that purified Rv1698 is an integral membrane protein that indeed produces channels. The main single channel conductance is 4.5 ± 0.3 nanosiemens in 1 m KCl. Zero current potential measurements revealed a weak preference for cations. Whole cell digestion of recombinant M. smegmatis with proteinase K showed that Rv1698 is surface-accessible. Taken together, these experiments demonstrated that Rv1698 is a channel protein that is likely involved in transport processes across the outer membrane of M. tuberculosis. Rv1698 has single homologs of unknown functions in Corynebacterineae and thus represents the first member of a new class of channel proteins specific for mycolic acid-containing outer membranes.
Mycobacteria are classified as Gram-positive bacteria but have evolved a complex cell wall, comprising a peptidoglycan-arabinogalactan polymer with covalently bound mycolic acids of considerable length (up to 90 carbon atoms) and a large variety of extractable lipids (1, 2). Most of these lipids are constituents of the cell envelope that provides an extraordinarily efficient permeability barrier and is an essential part of the intrinsic resistance of mycobacteria to many toxic compounds and antibiotics (3). To account for these observations, Minnikin (4) proposed a model in which the mycolic acids form the inner leaflet of an asymmetrical bilayer. Mutants and treatments affecting mycolic acid biosynthesis and the production of extractable lipids showed an increase in cell wall permeability and a drastic decrease in virulence, underlining the importance of the cell wall integrity for intracellular survival of Mycobacterium tuberculosis (1). Cryoelectron tomography revealed the native organization of the Mycobacterium smegmatis cell envelope. Further, the three-dimensional data and the investigation of ultrathin frozen-hydrated cryosections of M. smegmatis, M. bovis BCG, and Corynebacterium glutamicum identified the outermost layer as a lipid bilayer. Mycolic acids were shown to be essential components of this bilayer, therefore providing the first visualization of mycobacterial outer membranes in their native state (5).
These findings raise the question of how the mycobacterial outer membrane is functionalized for nutrient uptake, signal transduction, and secretion of material to the cell surface and the extracellular medium. To perform these functions, Gram-negative bacteria such as Escherichia coli use more than 60 outer membrane proteins (6). However, only two mycobacterial outer membrane proteins are known so far. The channel-forming protein MspA is located in the outer membrane of M. smegmatis (7) and functions as a classical porin by enabling diffusion of small and hydrophilic solutes (8–10). MspA is an octamer that forms one central channel of 10-nm length (11). By contrast, porins of Gram-negative bacteria are trimeric proteins of 4.5-nm length. Each monomer forms one channel (12). These comparisons provided the proof of principle that mycobacterial outer membrane proteins have structures different from their functional analogs in Gram-negative bacteria.
Both M. tuberculosis and Mycobacterium bovis BCG produce channel-forming proteins that were assumed to be outer membrane proteins (13–15). Recombinant OmpA of M. tuberculosis was purified from E. coli and showed channel activity in lipid bilayer experiments (13). Recently, it was shown that OmpA from M. bovis BCG has channel activity similar to the protein produced in E. coli and that a central domain of ∼150 amino acids is sufficient for channel formation (16). Surprisingly, channel-forming proteins of M. tuberculosis other than OmpA are not known, despite their perceived importance for the physiology and pathogenicity of M. tuberculosis.
Because subcellular fractionation experiments often lead to mixing of inner and outer membrane proteins (17), we have used a bioinformatic approach as an alternative to predict outer membrane proteins of M. tuberculosis (18). In this study, we present conclusive evidence that one of the these proteins, Rv1698, is a protein that forms water-filled channels in lipid membranes. Expression of rv1698 in M. smegmatis increased both uptake rates for nutrients and the susceptibility to small antibiotics. Protease digestion showed that Rv1698 is surface-accessible. All of the mycolic acid-containing bacteria have a single homolog of unknown function. Thus, Rv1698 represents the first member of a new class of proteins specific for the unusual outer membranes of Corynebacterineae.
EXPERIMENTAL PROCEDURES
Bacterial Strains and Growth Conditions—All of the bacterial strains used in this study are listed in supplemental Table S1. Mycobacterial strains were grown at 37 °C in Middlebrook 7H9 liquid medium (Difco Laboratories) supplemented with 0.2% glycerol, 0.05% Tween 80 or on Middlebrook 7H10 agar (Difco Laboratories) supplemented with 0.2% glycerol unless indicated otherwise. E. coli DH5α was used for all cloning experiments and was routinely grown in Luria-Bertani medium at 37 °C. The following antibiotics were used when required at the following concentrations: ampicillin (100 μg ml–1 for E. coli), kanamycin (30 μgml–1 for E. coli; 30 μgml–1 for M. smegmatis), and hygromycin (200 μgml–1 for E. coli, 50 μg ml–1 for M. smegmatis).
Antibiotic Sensitivity Assay—M. smegmatis strains were grown in a 4-ml culture for 2 days at 37 °C to an A600 of 0.6–0.8. The cultures were diluted in Middlebrook 7H9 medium to yield ∼5,000 colony-forming units (cfu)/ml.3 Approximately 500 cfu were streaked out on plates containing the appropriate antibiotic concentrations. As a reference 500 cfu were also plated onto plates without any antibiotic. The number of surviving cells was normalized to the number of cells counted on plates without antibiotic for each strain and expressed as relative colony forming units (% cfu). Colony counts were carried out after 3 days of incubation at 37 °C. The concentrations of the antibiotics were: ampicillin, 32 μg/ml; cephaloridin, 3 μg/ml; and chloramphenicol, 6 μg/ml.
Outer Membrane Permeability for Glucose—The experiments were carried out as previously described (7). To reduce aggregation and clumping, M. smegmatis strains were grown first in 4-ml cultures for 2 days at 37 °C and then filtered through a 5-μm pore size filter (Sartorius). The filtrates were grown for 2 days at 37 °C and then used to inoculate 100-ml cultures that were grown to an A600 of 0.5. The cells were harvested by centrifugation (4 °C, 3000 rpm, 10 min), washed once in 2 mm PIPES, pH 6.5, 0.05 mm MgCl2, and resuspended in the same buffer. The 14C-labeled compounds and their nonlabeled analogs were mixed and added to the cell suspensions to obtain a final concentration of 20 μm with 106 cpm. The mixtures were incubated at 37 °C, and 1 ml-samples were removed at the indicated times. The cells were filtered through a 0.45-μm pore size filter (Sartorius) and washed with 0.1 m LiCl, and their radioactivity was determined using a liquid scintillation counter. The mean dry weight of the cells in these samples was 1.4 ± 0.4 mg. The uptake rate was expressed as nmol/mg cells.
Construction of Overexpression Vectors for rv1698 for Mycobacteria and E. coli—The gene rv1698 of M. tuberculosis and its homologous gene msmeg_3747 of M. smegmatis were amplified by PCR using chromosomal DNA and the oligonucleotide pairs 1698fwd/1698rev and mpoS_SDopt_1/mspTSDrev (supplemental Table S1) introducing the restriction sites PacI and SwaI. The genes were cloned into pMN016 under the control of the psmyc promoter by using those restriction sites (9) to give the expression vectors pMN035 and pMN451, respectively (supplemental Table S1). To purify Rv1698 from mycobacteria, the protein was C-terminally tagged with six histidine residues by PCR using the primer 1698fwd and the 5′-phosphorylated primer mpoTHis (supplemental Table S1). The resulting PCR product was digested with PacI and cloned into the backbone of pMN016 digested with PacI and SwaI to give pML911. To overexpress and analyze the pore forming activity of recombinant Rv1698, the truncated gene lacking the putative signal sequence of 30 amino acids was fused to a C-terminal His tag by cloning it into the vector pET28+ (Novagen). The gene without its putative leader sequence was amplified from pMN035 by using the oligonucleotides pMS-Seq1 and his_rv1698fwd (supplemental Table S1). Both, the PCR fragment and pET28b+ were digested with restriction endonucleases NdeI and HindIII and ligated to give pML122 (supplemental Table S1).
A vector for expression of untagged Rv1698 without the predicted signal peptide in E. coli was constructed using the oligonucleotides mat-rv1698fwd and mat-rvrev introducing the two restriction sites NcoI and NdeI (supplemental Table S1). The digested fragment was cloned into the vector pET-16b, which was treated with the same restriction endonucleases to obtain pML141.
Overexpression, Purification, and Renaturation of Recombinant Rv1698His from E. coli—Cultures of E. coli Rosetta carrying the overexpression vectors pML122 were grown at 37 °C. When the culture reached an A600 of 1, isopropylthio-β-d-galactosidase was added to a final concentration of 0.5 mm to induce gene expression. After a further 4 h of incubation, the bacteria were harvested and resuspended in 20 ml of lysis buffer (50 mm Tris-HCl, 100 mm NaCl, 0.5% Triton X-100). The cell suspension was sonicated four times for 20 s with 12 watts in 30-s intervals. 0.01 mg/ml DNase and 0.1 mg/ml lysozyme were added and incubated at room temperature for 20 min. The broken cells were harvested by centrifugation and resuspended in lysis buffer followed by sonication as described above. This step was repeated twice. Then the broken cells were resuspended in washing buffer (50 mm Tris-HCl, 100 mm NaCl) and sonicated again. Because rRv1698His formed inclusion bodies, the proteins in the pellet were dissolved with 8 m urea, separated from cell debris by centrifugation, and purified using nickel-nitrilotriacetic acid columns (nickel-nitrilotriacetic acid spin kit; Qiagen). The bound proteins were washed and eluted from the column by using a four-step pH gradient with TPU buffer (6 m urea, 0.1 m NaH2PO4, 0.01 m Tris) at different pH levels (pH 6.3/5.9/4.8/4.5) according to the recommendations of the manufacturer. The protein fraction at pH 4.5 contained most of the rRv1698His protein. Purified rRv1698His (180 μg/ml) was diluted 100-fold in 25 mm sodium-phosphate buffer (pH 7.5) containing 0.5% n-octylpolyethylene oxide (OPOE) at room temperature to remove urea and renature the protein. The resulting protein was directly used in black lipid bilayer experiments to determine single channel conductance and ion selectivity of the pore as described below. The BCA kit (Pierce) was used routinely to determine protein concentrations.
Purification of Rv1698His from M. bovis BCG—M. bovis BCG carrying the plasmid pML911 was grown in Middlebrook 7H9 liquid medium supplemented with 10% oleic acid albumin dextrose complex, 0.05% Tween 80, and hygromycin. At an A600 of 1, the bacteria were harvested, incubated in a rotatory shaker (200 rpm) with lysozyme 1 mg/ml in phosphate-buffered saline (PBS; 137 mm NaCl, 10 mm potassium phosphate, 2.7 mm KCl, pH 7.4) for 2 h at 37°C and disrupted at 4 °C using a Sonicator 3000 ultrasonic liquid processor (Misonix) in 2 steps of 30 min (micro tip, pulsar cycle of 1 s, 9 W delivered per cycle). The proteins were solubilized by incubation with 1% SDS in PBS for 18 h (37 °C, 200 rpm). Nonsoluble material was removed by centrifugation (10,000 × g, 4 °C) before purifying Rv1698His on nickel-nitrilotriacetic acid-agarose (Qiagen) using a batch procedure. The bound His-tagged protein was washed and eluted from the resin by using a three-step imidazole gradient (5/20/250 mm) in sodium phosphate buffer (50 mm NaH2PO4, pH 7.6, 0.3 m NaCl) according to the recommendations of the manufacturer. The His-tagged protein was eluted with 250 mm imidazole. Because of the high imidazole concentration, the BCA assay was not used, and the purified Rv1698His protein was quantified by using the Bradford protein assay (Bio-Rad) according to the manufacturer's recommendations. In addition, a calibration curve of band intensities was established with known amounts of bovine serum albumin in Coomassie-stained SDS-polyacrylamide gels using image analysis software (LabWorks 4.6, UVP). Then the amount of Rv1698 was determined relative to the bovine serum albumin reference. Both methods yielded the similar values.
Lipid Bilayer Experiments—The single channel conductance of Rv1698 protein was analyzed on a custom-made lipid bilayer apparatus as described previously (19). Briefly, the Ag/AgCl electrodes were bathed in a solution of 1 m KCl, 10 mm HEPES, pH 7.0. The lipid membranes were painted from a solution of 1% diphytanoylphosphatidylcholine (DPhPC; Avanti Polar Lipids) in n-decane. Before adding the protein, current traces of at least three or more membranes were recorded with the appropriate detergent (0.5% OPOE or 0.1% SDS in 25 mm sodium phosphate, pH 7.5) to exclude any contamination with channel forming activity and to demonstrate that the detergents did not affect the membrane. Then protein was added to both sides of the cuvette. Single channel conductances for more than 100 pores/sample were digitally recorded. The raw data were analyzed using IGOR Pro 5.03 program (WaveMetrics) and a macro provided by Dr. Harald Engelhardt. These data were further analyzed in SigmaPlot 9.0 (Systat Software) to generate the figures shown here. The ion selectivity of Rv1698His was measured exactly as described previously (8).
Preparation of RNA from M. tuberculosis and RT-PCR Experiments—Total RNA of M. tuberculosis H37Rv was isolated by the TRIzol method as recommended by the manufacturer. Briefly, the cultures were grown in 100 ml of 7H9 medium supplemented with 10% oleic acid albumin dextrose complex and 0.05% Tween 80 to log phase. Thirty five ml of the GTC buffer (5 m guanidium thiocyanate, 0.5% sarcosyl, 0.5% Tween 80, 1% β-mercaptoethanol) was added, and the culture was centrifuged at 10,000 × g for 10 min at 4 °C. The pellet was resuspended in 1.5 ml of TRIzol and lysed by agitation with glass beads (FastRNA Tubes-Blue) in a FastPrep© FP120 bead beater apparatus (Bio-101) for 3 × 45 s at level 6.5. The suspensions were cooled on ice for 5 min between agitation steps. 500 μl of chloroform was added, and centrifugation was done for 5 min at 14,000 × g. The upper phase was transferred to a new tube containing an equal volume of isopropanol. The tubes were incubated for 20 min at –80 °C and centrifuged at 14,000 × g for 20 min at 4 °C. The pellet was washed with 70% ethanol, dried, and resuspended in 100 μl of diethylpyrocarbonate-treated water (Ambion). Further purification of samples was performed using Nucleospin→RNAII kit (Macherey-Nagel) following the instructions of the manufacturer. RNA was sonicated to render DNA accessibility to DNase degradation (2 × 20 s at 20% power), 5 min on ice between sonications (20). 5–10 μg of sonicated DNA was used for Turbo DNase treatment, which was done according to the manufacturer protocol (Ambion). cDNA synthesis was performed using SuperScript III first strand synthesis system for RT-PCR (Invitrogen) according to the manufacturer protocol using random primers. AccuPrime Pfx SuperMix (Invitrogen) was employed for the PCR. Primers used were RT-rv1698fwd and RT-rv1698rev (supplemental Table S1). Thirty five cycles (30 s at 95 °C, 30 s at 58 °C, 30 s at 68 °C) were run to amplify the cDNA. The PCR products were analyzed using 1% agarose gels, which were stained with ethidium bromide. Primers specific for 16 S rRNA were used as a positive control for RT-PCR.
Protease Accessibility Assay—Experiments to examine the surface accessibility of Rv1698 were carried out as described previously (21) with minor modifications. M. smegmatis strains carrying the plasmids pMN437, pML911, pML451, and pMV61015.1 were grown in 20 ml of Middlebrook 7H9 medium and harvested as culture reached an A600 of about 3.5. The cells were washed once with Tris-buffered saline buffer (0.5 m Tris-HCl, pH 7.2, 150 mm NaCl, 3 mm KCl) and then resuspended in 1 ml of the same buffer. Two aliquots of 150 μl were taken, and proteinase K (Sigma-Aldrich) was added to one of the aliquots to a final concentration of 100 μg/ml. After 30 min at 4 °C the reaction was stopped by adding Complete© EDTA-free protease inhibitor mixture (Roche Applied Science) from a 7-fold stock solution. The samples were immediately centrifuged, washed once in 250 μl of Tris-buffered saline, and resuspended in 75 μl of Tris-buffered saline plus 25 μl of 4× protein loading buffer (160 mm Tris-HCl pH 7.0, 12% SDS, 32% glycerol, 0.4% bromphenol blue). Finally, the samples were boiled for 20 min to allow cell lysis and centrifuged again to remove insoluble debris, and 50 μl of the protein extracts were separated on a 10% polyacrylamide gel and analyzed in Western blots using standard protocols (22). The program LabWorks 4.6 (UVP) was used for quantitative image analysis to determine the amount of detected Rv1698 protein in treated and untreated samples.
Analysis of Protein Secondary Structures—Secondary structures of Rv1698 and reference proteins were predicted using the JPred 3 server (23, 24) and the Network Protein Sequence Analysis server. SignalP3.0 (25) was accessed to examine the presence of signal peptides for Rv1698. The TMHMM2.0 program (26, 27) was used for prediction of hydrophobic transmembrane α-helices. All of the algorithms were used with standard settings unless otherwise noted. The hydrophobicity profile of Rv1698 was calculated using an algorithm that was developed to detect repetitive oscillations of the hydropathy profile (28) and the Eisenberg hydrophobicity scale of amino acids (29).
Estimation of the Minimal Size of the Rv1698 Pore—The two-dimensional structures of ampicillin (Compound ID 2174), chloramphenicol (Compound ID 5959), and glucose (Compound ID 5793) were downloaded from PubChemCompound data base at NCBI. The Chem3D Pro 8.0 software (Cambridge-Soft) was used to obtain three-dimensional structures of these molecules and to minimize their energy using the MOPAC algorithm. Using the program Chimera (30), the molecules were oriented along their longest axis, and the length of the second longest axis was measured between the nuclei of the most distant atoms along this axis. These values were taken as the widths of the molecules and used for a minimal estimate of the pore size of Rv1698.
RESULTS
The Rv1698 Protein Increases the Susceptibility of an M. smegmatis Porin Mutant to Hydrophilic Antibiotics—A genome-wide secondary structure prediction of all exported proteins identified Rv1698 as a putative outer membrane protein of M. tuberculosis (18). The genomes of all mycobacteria including M. smegmatis (Msmeg_3747) and Mycobacterium leprae (ML1362) and other closely related bacteria such as Nocardia and Corynebacteria encode a single homolog of Rv1698 (supplemental Fig. S1). This indicated that Rv1698 and its homologs might perform a function specific for the mycolic acid-containing outer membrane of these bacteria.
To examine whether this protein might be a porin, we used the strain M. smegmatis MN01, which lacks the major porin gene mspA. The outer membrane permeability of the ΔmspA mutant to hydrophilic β-lactam antibiotics was decreased 9-fold (7). This resulted in a 16-fold increased resistance toward ampicillin compared with wild-type M. smegmatis (31). Constitutive expression of mspA restored the susceptibility of the ΔmspA mutant MN01 to ampicillin and cephaloridine to wild-type levels as determined by using an agar dilution assay (Fig. 1). The susceptibility of the ΔmspA mutant to these antibiotics was also significantly increased by expression of the rv1698 gene. To exclude that this was an antibiotic-specific effect, we examined whether rv1698 had a similar effect on the efficacy of chloramphenicol against M. smegmatis. Indeed, expression of both mspA and rv1698 significantly increased the susceptibility of the ΔmspA mutant MN01 to chloramphenicol. These results indicated that Rv1698 increased the outer membrane permeability of the M. smegmatis ΔmspA mutant.
FIGURE 1.

Expression of rv1698 increases the antibiotic susceptibility of the M. smegmatis porin mutant MN01. The susceptibilities of M. smegmatis wild type with the control vector pMS2 (white bars), the ΔmspA mutant MN01 with the control vector pMS2 (black bars), MN01 with the mspA expression vector pMN014 (light gray bars), and MN01 with the rv1698 expression vector pMN035 (dark gray bars) to 32 μg/ml ampicillin (Amp), 3 μg/ml cephaloridine (Ceph), and 6 μg/ml chloramphenicol (Chl) were determined by agar dilution on 7H10 Middlebrook agar plates containing 2% glycerol. The number of colonies on the antibiotic plates was normalized to the number of colonies on plates without antibiotic for each strain and expressed as relative colony-forming units (% cfu).
Rv1698 Partially Complements the Permeability Defect of Porin Mutants of M. smegmatis—It was shown previously that porin-mediated permeation through the outer membrane is the rate-limiting step for uptake of glucose by M. smegmatis (7, 9). For example, the minimal permeability coefficient of the ΔmspA ΔmspC mutant ML10 for glucose is 50-fold lower than that of wild-type M. smegmatis (9). Therefore, we wanted to examine whether rv1698 could complement the slow uptake of glucose by the porin double mutant ML10. Glucose at a concentration of 20 μm was taken up by the ML10 strain carrying the empty vector pMS2 with an average rate of 42 pmol/min/mg cells. This rate was increased 5-fold to 224 pmol/min/mg cells in the presence of the rv1698 expression vector pMN035 (supplemental Table S1), demonstrating that Rv1698 indeed increased the outer membrane permeability of the porin double mutant ML10 (Fig. 2). However, expression of rv1698 only partially restored the permeability defect of the porin mutant M. smegmatis ML10 in contrast to the endogenous porin gene mspA (Fig. 2).
FIGURE 2.

Rv1698-dependent glucose uptake by a porin mutant of M. smegmatis. Accumulation of [14C]glucose by the ΔmspA ΔmspC double mutant M. smegmatis ML10 containing the empty vector pMS2 (filled circles, control), the mspA expression vector pMN014 (filled squares) and the rv1698 expression vector pMN035 (open circles). Both genes are transcribed from the psmyc promoter. The assay was performed at 37 °C at a final glucose concentration of 20 μm. The uptake experiment was done in triplicate and is shown with standard deviations.
Recombinant Rv1698His Is a Channel-forming Protein—Lipid bilayer experiments provide direct evidence regarding whether a particular protein forms channels within lipid membranes (8). To this end, a truncated rv1698 gene encoding a protein with an N-terminal histidine tag replacing the predicted signal peptide (amino acids 1–30) was cloned under the control of the T7 promoter in the plasmid pML122 (supplemental Table S1). After induction of E. coli Rosetta containing pML122 with isopropylthio-β-d-galactosidase, the bacteria were harvested and disrupted by sonication. Because rRv1698His was insoluble (Fig. 3A, lane 4), the pellet was dissolved with 8 m urea (Fig. 3A, lane 5) and separated from cell debris by centrifugation. The rRv1698His protein was purified by Ni2+ affinity chromatography to apparent homogeneity with a yield of 7.4 mg from a 1-liter culture of E. coli as demonstrated by a silver-stained protein gel (Fig. 3, lane 8). This protein was diluted 1:100 overnight in a 0.5% OPOE-containing buffer to initiate refolding. Nanogram amounts detergent-solubilized rRv1698His protein (3 ng/ml) showed a high channel forming activity after reconstitution in planar lipid membranes (Fig. 3B). No pore was recorded in control experiments when only detergent-containing buffer was added to the lipid bilayer (not shown). The single channel conductance of rRv1698His in 1 m KCl was 4.5 nS as determined from 46 reconstitution events in seven membranes (Fig. 4A). Because this channel conductance is almost identical to that of MspA (8), the activity recorded after addition might have been due to contamination of the bilayer apparatus with minute amounts of highly active and extremely stable MspA (32). Therefore, purification and renaturation of rRv1698His were repeated. Special care was taken to use fresh buffers and equipment that were not in prior contact with MspA. None of the control measurements with detergent alone showed any pore activity. By contrast, 34 reconstitution events were observed in five membranes after addition of rRv1698His to a final concentration of 1 μg/ml (not shown). The single channel conductance of rRv1698His in 1 m KCl was 4.3 nS in excellent agreement with the previous experiments. The reason for the strongly varying channel activity of the recombinant protein is unknown but might be caused by different refolding yields of Rv1698 after dissolving the inclusion bodies.
FIGURE 3.

Single channel recordings of purified recombinant Rv1698His in lipid bilayer experiments. A, expression of rv1698His in E. coli and purification by chromatography. The rv1698 gene was expressed in the E. coli Rosetta strain using the plasmid pML122. The parent plasmid pET-28b+ does not contain the rv1698 gene and was used as a control. The samples were separated on a 10% polyacrylamide gel stained with silver. Lane M, molecular mass marker; lane 1, unsoluble fraction of E. coli Rosetta with pET-28b+; lanes 2 and 3, unsoluble fractions of E. coli Rosetta with pML122 before (lane 2) and after (lane 3) induction with isopropylthio-β-d-galactosidase; lane 4, unsoluble fraction (lane 3) dissolved in 8 m urea; lane 5, flow through from Ni2+ column; lane 6, elution at pH 6.3; lane 7, elution at pH 4.5; lane 8, purified Rv1698 after dilution in Na-P buffer with 0.5% OPOE. B, single channel recordings of purified rRv1698His in lipid bilayer experiments. The current intensity (I) corresponding to the insertion of single channels inside a DPhPC membrane bathing in 1 m KCl was recorded after the addition of 30 ng of purified rRv1698His to both sides of the membrane (final concentration, 3 ng/ml). The data were collected from seven different membranes. The most frequent insertions had a single channel conductance of 4.5 nS. For analysis see Fig. 4A and Table 1.
FIGURE 4.

Analysis of the ion specificity of rRv1698His. The single channel conductance of rRv1698His was determined in different electrolytes. The concentration of each electrolyte was 1 m. The probability P of a conductance step G was calculated from 46 (KCl), 27 (NaCl), 48 (LiCl), 70 (KNO3), and 80 (KAc) insertion events from five to seven membranes. The panels on the left and right sides show the change of the conductance of Rv1698His in dependence on the size of the cation and anions, respectively. The reference electrolyte is KCl and is shown on both sides of this figure for comparison purposes. Thus, A and D are identical. For further information see Table 1.
To exclude that the N-terminal histidine tag modified the channel activity, an rv1698 gene encoding for a rRv1698 protein without the predicted signal peptide and without histidine tag was expressed in E. coli Rosetta using the vector pML141 (supplemental Table S1). This truncated rRv1698 protein was exclusively found in inclusion bodies. The single channel conductance of the purified truncated rRv1698 in 1 m KCl was 4.3 nS as determined from 16 reconstitution events in five membranes (not shown). Taken together, these experiments showed that (i) recombinant Rv1698 is an integral channel-forming membrane protein, (ii) the predicted signal peptide is not necessary for channel formation in vitro, and (iii) the N-terminal histidine tag does not impair the channel activity of Rv1698. Thus, it is concluded that Rv1698 is a channel-forming protein of M. tuberculosis. It should be noted that reconstitution of the Rv1698 pore in lipid bilayers occurred exclusively in a step-like fashion as shown in Fig. 3B, indicating that the recombinant protein formed open channels upon insertion. These results are consistent with the complementation of the permeability defect of porin mutants of M. smegmatis as shown above. The rapid reconstitution within lipid bilayers also demonstrates that recombinant Rv1698 is an integral membrane protein.
Ion Selectivity of the Rv1698 Channel—To further characterize the pore formed by Rv1698 and to identify properties that might distinguish the Rv1698 and MspA channels, the ion selectivity of purified rRv1698His was determined by lipid bilayer experiments. To this end, single channel conductance experiments were done in the presence of 1 m solutions of chloride salts with different cations and potassium salts with different anions. Fig. 4 shows that the single channel conductance of purified rRv1698His was influenced considerably by the salt composition. The channel conductivity of rRv1698His decreased significantly with the increasing radius of the hydrated cation in chloride salts (Fig. 4 and Table 1). For example, the single channel conductance of rRv1698His in 1 m LiCl was less than half of that in 1 m KCl. A similar effect was observed with increasing radii of the hydrated anions in potassium salts (Fig. 4 and Table 1). Thus, it is concluded that cations and anions move with a similar rate through the rRv1698His pore. The conductance of the Rv1698 pore linearly decreased with increasing size of the hydrated cations in the same manner as their specific conductance decreased in water (supplemental Fig. S1). This strongly suggested that rRv1698His forms a wide water-filled channel. It should be noted that the channel activity was extremely low in experiments with RbCl as the electrolyte so that only a few pores were recorded (Table 1). This may indicate that the integration of open channels was inhibited by RbCl either by preventing insertions into the membrane or by blocking the channel.
TABLE 1.
Single channel conductances of purified recombinant Rv1698 in different electrolytes
The lipid bilayer experiments were done in the presence of 1 m of the respective electrolyte. Protein was added on both sides of the DPhPC membrane starting at 6 ng/ml, and its concentration was increased in steps of 6 ng/ml until pores were detected. The salt solutions were buffered in 10 mm MES at pH 6 or as indicated above. The ion radii were taken from Trias and Benz (55) and Tansel et al. (56). No reference was found for the radius of the hydrated acetate anion (NF).
|
Salt
|
Radius of hydrated ion
|
Single channel conductance
|
Number of steps
|
Number of membranes
|
||||
|---|---|---|---|---|---|---|---|---|
| Cation | Anion | Most frequent | Average | |||||
| nm | nS | |||||||
| Tris-HCl, pH 6 | 0.321 | 0.195 | 0.5 | 0.51 | 52 | 6 | ||
| Tris-HCl, pH 8 | 0.321 | 0.195 | 1 | 0.95 | 50 | 6 | ||
| LiCl | 0.216 | 0.195 | 2 | 2.1 | 48 | 5 | ||
| NaCl | 0.163 | 0.195 | 3 | 2.95 | 27 | 5 | ||
| Kac | 0.110 | NF | 3 | 3.04 | 80 | 5 | ||
| KNO3 | 0.110 | 0.340 | 4 | 4.15 | 70 | 6 | ||
| KCl | 0.110 | 0.195 | 4.5 | 4.1 | 46 | 7 | ||
| NH4Cl | 0.110 | 0.195 | 4.5 | 4.94 | 81 | 6 | ||
| RbCl | 0.105 | 0.195 | — | 4.75 | 8 | 6 | ||
| CsCl | 0.106 | 0.195 | 4 | 4.41 | 108 | 5 | ||
Another interesting result was the significant reduction of the single channel conductance in 1 m Tris-HCl at pH 6 compared with pH 8 (Table 1). This may either indicate pH gating of the Rv1698 channel or a proton-induced conformational change of the constriction zone that causes a decreased conductivity for Tris-HCl. Both mechanisms were observed for porins of Gram-negative bacteria (33, 34).
To quantify the ion selectivity of rRv1698His, zero current potentials of membranes containing several hundred rRv1698 pores were measured in the presence of salt gradients. A 3-fold KCl gradient resulted in a potential of 16.0 ± 0.15 mV, which was positive at the more dilute side. Using the Goldman-Hodgkin-Katz equation (35), this is equivalent to a permeability ratio of cations over anions Pc/Pa of 2.5 ± 1.6. This weak preference of cations over anions is consistent with the results of the single channel experiments in different electrolytes. It is concluded that the rRv1698His channel has little charge preference in contrast to the marked cation preference of the MspA pore, which shows a permeability ratio of cations over anions Pc/Pa of 6.6 ± 0.3 (8). These results also demonstrated that the channel activity observed with purified rRv1698His is indeed a genuine activity of the rRv1698His protein and does not result from contamination with the extremely stable MspA pores.
The rv1698 Gene and Its Homologs Are Expressed in Mycobacteria—Reverse transcription PCR was employed to examine whether rv1698 is expressed in M. tuberculosis grown under standard conditions. Total mRNA was purified from wild-type M. tuberculosis H37Rv. As shown in Fig. 5, PCR yielded a 400-bp DNA fragment when the RNA sample was incubated with reverse transcriptase (lane +). This product was identical in length to a PCR product obtained from chromosomal DNA (Fig. 5, lane DNA) and to the theoretical length of the amplified region of the rv1698 gene, indicating that the PCR was specific. By contrast, no PCR fragment was obtained when reverse transcriptase was omitted, demonstrating that the prepared RNA was not contaminated with DNA (Fig. 5, lane –). These results show that the rv1698 gene is transcribed in M. tuberculosis grown under standard growth conditions. Similar results were obtained for M. bovis BCG and M. smegmatis (not shown). Thus, it is concluded that rv1698 is expressed in all three mycobacteria. These findings are supported by the detection of 36-kDa bands in Western blot experiments with SDS extracts of M. tuberculosis, M. bovis BCG, and M. smegmatis using an Rv1698-specific antiserum (not shown).
FIGURE 5.
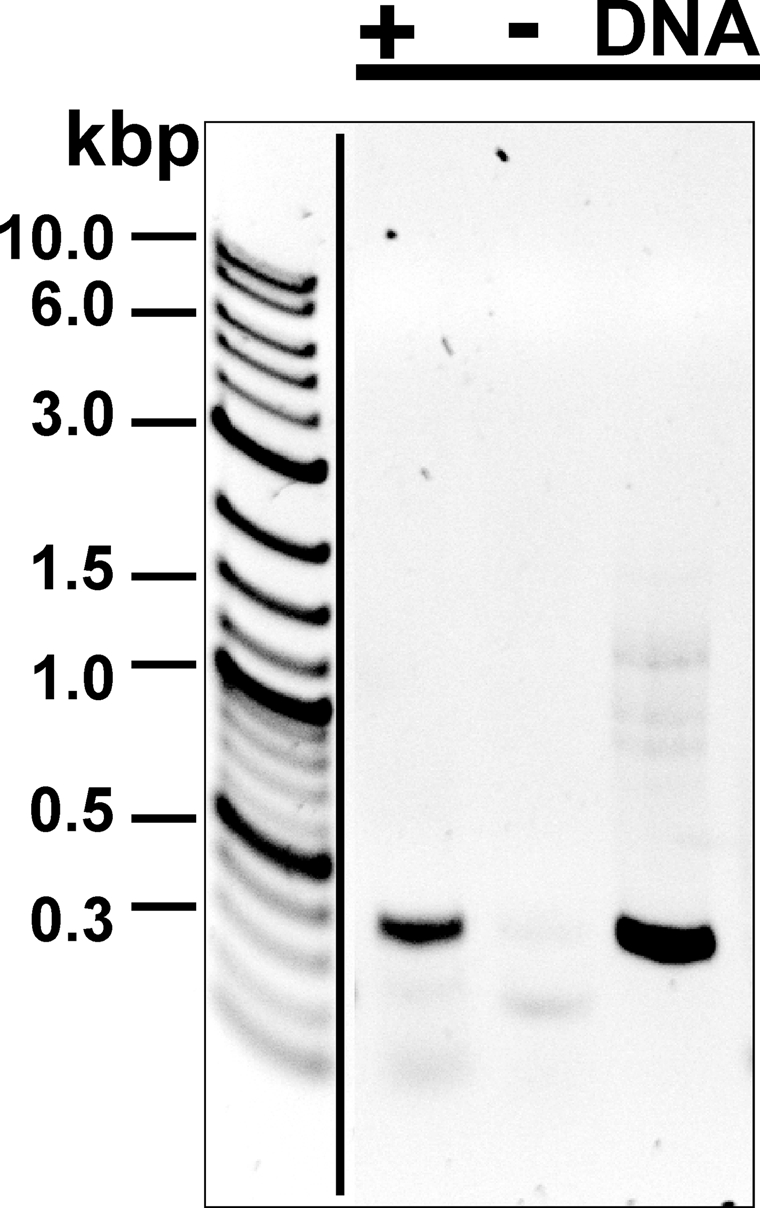
Analysis of expression of rv1698 in M. tuberculosis. The RT-PCR products were separated on a 1% agarose gel. The length of the product is 400 bp. The sample, in which the reverse transcriptase was added for the cDNA synthesis, is marked with +, whereas the–sign denotes the sample, in which reverse transcriptase was omitted to detect contaminations with chromosomal DNA. DNA denotes samples where chromosomal DNA was used as a template for the PCR to analyze the specificity of the primers. The gel was stained with ethidium bromide and is shown as a negative image to enhance the visibility of weak bands.
Channel Forming Activity of Rv1698His Purified from M. bovis BCG—To examine whether the native Rv1698 protein has the same channel activity as the recombinant protein purified from E. coli and to exclude that the channel activity originated from folding artifacts or contaminations from E. coli porins, we set out to purify Rv1698 from M. bovis BCG. To this end, the rv1698 gene encoding a C-terminal histidine tag was expressed from the plasmid pML911 (supplemental Table S1) in M. bovis BCG. The proteins were extracted from cell lysates by 1% SDS, and the SDS extract was purified by nickel affinity chromatography (Fig. 6A). Mass spectroscopy of tryptic fragments revealed that the protein with an apparent molecular mass of 36 kDa is indeed Rv1698 (not shown). Mass spectroscopy also identified the protein with an apparent molecular mass of 57 kDa as GroEL1 (BCG3487c). GroEL1 is a cytoplasmic chaperone that possesses a naturally histidine-rich C-terminal region in mycobacteria (36). Analysis of these samples for reactivity with an Rv1698-specific antiserum in Western blots showed that similar amounts of recombinant Rv1698 were produced from plasmid pML911 in M. bovis BCG and M. smegmatis (Fig. 6B). Further, small amounts of apparently dimeric Rv1698 were detected in the sample purified from M. bovis BCG (Fig. 6B). This indicated that Rv1698 might be an oligomeric protein.
FIGURE 6.

Overexpression and purification of Rv1698His from M. bovis BCG and M. smegmatis. The proteins were extracted from M. bovis BCG/pML911 and M. smegmatis SMR5/pML911 lysates using 1% SDS in PBS. Rv1698His proteins were purified from these extracts by Ni2+ affinity chromatography. All of the samples were analyzed on 10% polyacrylamide gels. A, Coomassie-stained gel. 5μg of protein in raw extracts and 50 ng of the purified Rv1698His proteins were loaded. B, Western blot. 5 μg of protein in raw extracts and 10 ng of the purified Rv1698His proteins were loaded. The proteins were transferred onto a polyvinylidene difluoride membrane and detected using an Rv1698-specific polyclonal antiserum. Lane 1, raw extract from M. bovis BCG/pML911; lane 2, purified Rv1698His from M. bovis BCG/pML911; lane 3, raw extract from M. smegmatis SMR5/pML911; lane 4, purified Rv1698His from M. smegmatis SMR5/pML911; lane M, molecular mass marker. The Rv1698His monomer and its putative dimer are marked with M and D, respectively.
Lipid bilayer experiments were done to examine whether Rv1698 protein isolated from M. bovis BCG also has channel forming activity. No insertion events were detected when buffer was added to the DPhPC membrane. When 300 ng of purified Rv1698His was added to the same membrane, a rapid stepwise current increase was observed (Fig. 7A). More than 100 pores were recorded in seven membranes. The most frequent reconstitution events had conductances of 4.5 to 4.8 nS (Fig. 7B). These results are very similar to the conductance of 4.5 nS determined for the recombinant Rv1698His protein purified from E. coli.
FIGURE 7.

Single channel activity of Rv1698His purified from M. bovis BCG. Rv1698His protein was purified by Ni2+ affinity chromatography from 0.5% OPOE extracts obtained from M. bovis BCG/pML911. Purified Rv1698His protein was added to the cis-side of a DPhPC membrane bathed by 1 m KCl, 10 mm HEPES, pH 7.0, and a –10 mV potential was applied. The channel activity was recorded using a data acquisition card. The boxed traces highlight opening and closing events of different sizes. A, the current trace shows more than 30 opening and closing events recorded 600 s after addition of 300 ng of Rv1698His to the membrane. The total recording time was 1843 s. B, the histogram represents a total of 109 opening and closing events recorded from seven membranes. C, the current trace shows more than eight opening and closing events recorded 400 s after the addition of 1μg of Rv1698His to the membrane. The total recording time was 2862 s.
A significant number of Rv1698His channels with smaller conductances of ∼2–2.5 nS and 1.0–1.5 nS were also recorded (Fig. 7, A and C). Subconductance states are frequently observed for oligomeric porins of E. coli and other Gram-negative bacteria (37), indicating that Rv1698 might indeed form oligomers. It should be noted that lipid bilayer experiments are not really suitable to quantify the activity of channel proteins because different membranes have to be used in each experiment. This can drastically alter the reconstitution frequency of proteins. In conclusion, purified Rv1698 protein expressed in M. bovis BCG and E. coli showed identical single channel activities, demonstrating unequivocally that Rv1698 is a pore protein.
Rv1698 Is a Surface-accessible Protein—Insertion of large, open, water-filled channel proteins such as porins (38) or colicins (39) into the inner membrane of bacteria is a lethal event, most likely because of the immediate breakdown of the proton gradient. Considering its channel characteristics, we therefore assumed that Rv1698 is an outer membrane protein. To determine the subcellular localization of Rv1698 and to examine whether the Rv1698 protein has surface-exposed loops, we employed protease accessibility as previously described for the surface protein PE_PGRS33 encoded by the rv1818c gene of M. tuberculosis (21). Proteinase K cleaves Msmeg_3747 in 160 and Rv1698 in 158 positions evenly distributed along the entire protein molecule. Thus, in principle, even small surface-exposed loops should be cleaved if Rv1698 is accessible to the protease in whole cells. Green fluorescent protein and PE_PGRS33HA were used as controls for a cytoplasmic protein and as a surface-exposed protein (21), respectively. The signal for green fluorescent protein is identical in both samples, indicating that the cell envelope was intact during proteinase K treatment (Fig. 8). By contrast, the PE_PGRS33HA protein disappeared, demonstrating that PE_PGRS33HA is surface-accessible consistent with previous results (21, 40). Importantly, the intensities of the bands of the full-length Msmeg_3747 and Rv1698 was reduced by 60% upon proteinase K treatment, demonstrating that both proteins are surface-exposed. It should be noted that the detection of smaller fragments of Msmeg_3747 and Rv1698 was only possible because of the use of an Rv1698-specific antiserum. This is in contrast to the reference protein PE_PGRS33HA, which disappears completely most likely because of the removal of the hemagglutinin tag from the protein by proteinase K (Fig. 8). Further, the observation of shorter peptides also indicates that some parts of Msmeg_3747 and Rv1698 are protected from proteinase K cleavage, probably because of domains buried in the outer membrane.
FIGURE 8.

Surface accessibility of Rv1698 in M. smegmatis by digestion with proteinase K. Whole cells of M. smegmatis were treated with proteinase K(+) or with PBS as a control (–). After adding protease inhibitors, the cells were washed in PBS buffer, and proteins were extracted with SDS by boiling. The solubilized proteins were analyzed in a 10% polyacrylamide gel and transferred to a polyvinylidene difluoride membrane. The proteins on these blots were specifically detected using the appropriate antibodies. M, molecular mass marker. The samples were extracts from M. smegmatis containing the plasmids pMN437 (green fluorescent protein (Gfp)), pMV61015.1 (PE_PGRS33HA), pML451 (Msmeg_3747His), and pML911 (Rv1698His).
DISCUSSION
Rv1698 Is a Pore Protein of M. tuberculosis—In our search for channel-forming proteins of M. tuberculosis, we focused on Rv1698, a protein of unknown function that was predicted to be an outer membrane protein (18). In this study, we showed that Rv1698 of M. tuberculosis partially complements the permeability defect of a porin mutant of M. smegmatis for glucose (Fig. 2) and alanine (not shown) and restores its sensitivity to ampicillin, cephaloridine, and chloramphenicol (Fig. 1). In a complementary in vitro approach, Rv1698 was purified from E. coli and M. bovis BCG and produced channels of a similar main conductance of 4.5 ± 0.3 nS in 1 m KCl in lipid bilayer experiments. Importantly, the Rv1698 channel showed only a weak preference of cations specificity in contrast to the marked cation preference of MspA (8). These experiments conclusively demonstrate that Rv1698 is a channel protein of M. tuberculosis.
Comparison of Rv1698 with Other Channel Proteins of M. tuberculosis—The single channel conductance under standard conditions is one of the major characteristics that defines a pore protein. Previous experiments revealed pores in gelpurified fractions of a Genapol extract of M. tuberculosis with main conductances (i) of 0.7 nS in fractions containing proteins of 15 and 60 kDa and (ii) of 2.5 to 3 nS containing proteins of 75 kDa and larger (14). However, no channel activity was observed in the fraction containing proteins of the size of Rv1698 (35 kDa). One possible explanation for this apparent discrepancy is that the physiological form of Rv1698 might be an oligomer that may be stable in polyacrylamide gels containing 0.1% SDS. Indeed, Rv1698 remained active after extraction from M. bovis BCG and M. smegmatis with 1% SDS (not shown). Further, weak bands with an apparent molecular mass of 70 kDa, which may represent a dimer, were observed in Western blots of purified Rv1698 (Fig. 6B). An alternative explanation might be that the amount of Rv1698 in detergent extracts from wild-type M. tuberculosis was too low for the detection of channel activity after gel purification. This would be consistent with the very low overall channel activity of detergent extracts of M. tuberculosis and M. bovis BCG as observed in several previous experiments (14, 15). In M. bovis BCG, a pore with a very similar single channel conductance of 4 nS in 1 m KCl was observed (15). However, this pore was found to have a 10-fold preference for cations over anions, whereas recombinant Rv1698 did not exhibit such a pronounced cation preference. This indicated that M. bovis BCG expresses another large conductance channel protein different from Rv1698. The outer membrane channel protein OmpA (35 kDa) of M. tuberculosis runs mainly as a monomer after purification (41) and may have co-purified in the same gel fraction as Rv1698, but it has a very different single channel conductance of 0.7 nS (13, 41).
Rv1698 Can Partially Substitute for the Porin MspA of M. smegmatis—The observation that Rv1698 forms an apparently nonspecific protein channel in a similar manner as MspA (9, 10, 31) raised the question why Rv1698 completely restored the susceptibility of the M. smegmatis mspA porin mutant to antibiotics (Fig. 1), whereas glucose diffusion was slow compared with MspA (Fig. 2). This is in apparent contrast to expectations because differences among outer membrane diffusion pores are more pronounced for larger solutes (42). One explanation might be the very different time scales of both experiments. Whereas the glucose uptake experiments are done within minutes, the plates to determine the susceptibility of the M. smegmatis strains to antibiotics were incubated for 2–3 days. Thus, even a pore with significantly lower diffusion rates would eventually allow enough antibiotic molecules to enter the cell to inactivate the target(s) and to fully restore wild-type susceptibility. The slow glucose uptake in comparison to the mspA expressing porin mutant of M. smegmatis (Fig. 2) could be either due to lower expression levels and/or to intrinsic properties of the Rv1698 pore.
Estimation of the Pore Size of Rv1698—Expression of rv1698 increased the susceptibility of porin mutants of M. smegmatis to ampicillin and chloramphenicol and increased the uptake rate for glucose significantly. These observations enable us to derive a minimal diameter of the Rv1698 pore. To this end, energy-minimized three-dimensional models of these molecules were oriented along their longest axis. The length of the second longest axis was measured for ampicillin, chloramphenicol, and glucose as 0.51, 0.54, and 0.52 nm, respectively. These values were measured between the atomic nuclei of the most distant atoms and provide a lower limit of the channel diameter of the Rv1698 pore. However, the widths of the solutes do not include the van der Waals' spheres and the hydration shell. Furthermore, the diffusion rates for solutes drop drastically when the solute size approaches the pore size (42). Thus, we conclude that the real pore size of the Rv1698 channel is likely significantly larger than 0.54 nm. Indeed, if Rv1698 behaved like a rod of conducting bulk solvent, a pore diameter of 1.0 nm would yield an electric conductance of 5.1 nS in a 1 m KCl solution (11). This is very close to the experimentally observed single channel conductance of 4.5–4.8 nS (Fig. 7).
Rv1698 Represents a Novel Class of Outer Membrane Proteins Specific for Corynebacterineae—The lipid bilayer experiments show that Rv1698 is an integral membrane protein that constitutes a membrane-spanning water-filled channel (Figs. 3, 4, and 7 and supplemental Fig. S1). In principle, this observation does not allow one to distinguish between inner and outer membrane proteins, although insertion of a pore with a large, open channel into the inner membrane is very likely lethal (38). However, only outer membrane and not inner membrane proteins are accessible from the cell surface in Gram-negative bacteria. The situation is similar in mycobacteria because the cell envelope of mycobacteria also contains outer membranes, albeit of a very different chemical composition and architecture (5). Hence, we conclude that Rv1698 is an outer membrane protein. This is strongly supported by the observation that Rv1698 partially complements the permeability defects of an M. smegmatis mutant lacking the porin MspA, which was shown to be located in the outer membrane (7).
A transmembrane β-barrel with a hydrophobic surface facing the membrane lipids and a hydrophilic interior is the hallmark of outer membrane proteins of Gram-negative bacteria (43). The crystal structure of the outer membrane porin MspA shows that similar β-barrel proteins also exist in mycobacteria (11). A comprehensive analysis of the secondary structure using JPred (23, 24) and another consensus method (44) predicted ∼40 and 15% of amino acids of Rv1698 in α-helices and β-strands, respectively (supplemental Fig. S2). The β-strand percentage of Rv1698 is lower than the predicted values for MspA (31%) and OmpA (21%) but higher than those of other outer membrane proteins with large α-helical periplasmic domains such as TolC (8%) of E. coli (45). Significantly, ∼40% of the Rv1698 protein show a regular pattern of alternating hydrophilic and hydrophobic amino acids (supplemental Fig. S3) that is typical for water-filled transmembrane β-barrel pores (46). It is concluded that the predicted secondary structure of Rv1698 is similar to the structural features of other bacterial outer membrane channel proteins. However, Rv1698-like proteins have no known homologs and are only found in the suborder Corynebacterineae of the Actinomycetales, which includes mycolic acid-containing bacteria such as corynebacteria, mycobacteria, and nocardia (47). Each of these species with a known genome sequence, including M. leprae with its decaying genome (48), has a single homolog of the rv1698 gene. Thus, we conclude that Rv1698 represents the first protein specific for the very unusual outer membrane of Corynebacterineae. This is in contrast to OmpA and MspA, the two other outer membrane proteins of mycobacteria known so far. Homologs of OmpA of M. tuberculosis are found in Gram-negative bacteria, in some slow-growing but not in fast-growing mycobacteria,4 whereas MspA-like porins are only present in fast-growing mycobacteria (49).
Conclusions—Considering the importance of matter transport across the outer membrane for the cellular physiology, the lack of knowledge about these processes in M. tuberculosis is surprising. The data presented in this study clearly show that Rv1698 is a novel outer membrane channel protein of M. tuberculosis. As a channel protein, Rv1698 is likely to contribute to matter transport across the outer membrane of M. tuberculosis but may also be involved in host pathogen interactions because it was shown for many outer membrane proteins of Gram-negative bacteria such as the Neisserial porins (50–54). Work to identify the solutes transported by Rv1698 and to identify its role in pathogenicity of M. tuberculosis is in progress.
Acknowledgments
We thank Katrin Stehle and Jennifer Bender for constructing the rv1698 overexpression plasmids pML122 and pML141, respectively; Mikhail Pavlenok for help with computer modelling; Dr. Riccardo Manganelli for the rv1818c expression plasmid pMV61015.1; and Jason Huff for critically reading the manuscript. We are grateful for subcellular fractions of M. tuberculosis H37Rv obtained from Colorado State University and for sequence data for M. smegmatis from the Institute for Genomic Research website.
This work was supported, in whole or in part, by National Institutes of Health Grant AI063432. This work was also supported by European Community 5th Framework Programme Grant QLK2-2000-01761 (to M. N.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement”in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3, Table S1, and references.
Footnotes
The abbreviations used are: cfu, colony-forming unit(s); DPhPC, diphytanoylphosphatidylcholine; OPOE, n-octylpolyethylene oxide; PIPES, piperazine-1,4-bis(2-ethanesulfonic acid); PBS, phosphate-buffered saline; RT, reverse transcription; MES, 2-(N-morpholino)ethanesulfonic acid; nS, nanosiemans.
A. Siroy, unpublished observations.
References
- 1.Barry, C. E., III, Lee, R. E., Mdluli, K., Sampson, A. E., Schroeder, B. G., Slayden, R. A., and Yuan, Y. (1998) Prog. Lipid Res. 37 143–179 [DOI] [PubMed] [Google Scholar]
- 2.Daffé, M., and Draper, P. (1998) Adv. Microb. Physiol. 39 131–203 [DOI] [PubMed] [Google Scholar]
- 3.Brennan, P. J., and Nikaido, H. (1995) Annu. Rev. Biochem. 64 29–63 [DOI] [PubMed] [Google Scholar]
- 4.Minnikin, D. E. (1982) in The Biology of the Mycobacteria: Physiology, Identification and Classification (Ratledge, C., and Stanford, J., eds) pp. 95–184, Academic Press, London
- 5.Hoffmann, C., Leis, A., Niederweis, M., Plitzko, J. M., and Engelhardt, H. (2008) Proc. Natl. Acad. Sci. U. S. A. 105 3963–3967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Molloy, M. P., Herbert, B. R., Slade, M. B., Rabilloud, T., Nouwens, A. S., Williams, K. L., and Gooley, A. A. (2000) Eur. J. Biochem. 267 2871–2881 [DOI] [PubMed] [Google Scholar]
- 7.Stahl, C., Kubetzko, S., Kaps, I., Seeber, S., Engelhardt, H., and Niederweis, M. (2001) Mol. Microbiol. 40 451–464; Correction (2005) Mol. Microbiol. 457, 1509 [DOI] [PubMed] [Google Scholar]
- 8.Niederweis, M., Ehrt, S., Heinz, C., Klöcker, U., Karosi, S., Swiderek, K. M., Riley, L. W., and Benz, R. (1999) Mol. Microbiol. 33 933–945 [DOI] [PubMed] [Google Scholar]
- 9.Stephan, J., Bender, J., Wolschendorf, F., Hoffmann, C., Roth, E., Mailänder, C., Engelhardt, H., and Niederweis, M. (2005) Mol. Microbiol. 58 714–730 [DOI] [PubMed] [Google Scholar]
- 10.Wolschendorf, F., Mahfoud, M., and Niederweis, M. (2007) J. Bacteriol. 189 2435–2442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Faller, M., Niederweis, M., and Schulz, G. E. (2004) Science 303 1189–1192 [DOI] [PubMed] [Google Scholar]
- 12.Cowan, S. W., Schirmer, T., Rummel, G., Steiert, M., Ghosh, R., Pauptit, R. A., Jansonius, J. N., and Rosenbusch, J. P. (1992) Nature 358 727–733 [DOI] [PubMed] [Google Scholar]
- 13.Senaratne, R. H., Mobasheri, H., Papavinasasundaram, K. G., Jenner, P., Lea, E. J., and Draper, P. (1998) J. Bacteriol. 180 3541–3547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kartmann, B., Stenger, S., and Niederweis, M. (1999) J. Bacteriol. 181 6543–6546; Correction (1999) J. Bacteriol. 6181, 7650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lichtinger, T., Heym, B., Maier, E., Eichner, H., Cole, S. T., and Benz, R. (1999) FEBS Lett. 454 349–355 [DOI] [PubMed] [Google Scholar]
- 16.Molle, V., Saint, N., Campagna, S., Kremer, L., Lea, E., Draper, P., and Molle, G. (2006) Mol. Microbiol. 61 826–837 [DOI] [PubMed] [Google Scholar]
- 17.Rezwan, M., Laneelle, M. A., Sander, P., and Daffe, M. (2007) J. Microbiol. Methods 68 32–39 [DOI] [PubMed] [Google Scholar]
- 18.Song, H., Wang, Y., Sandie, R., Andrade-Navarro, M., and Niederweis, M. (2008) Tuberculosis 10.1016/j.tube.2008.02.004 [DOI] [PMC free article] [PubMed]
- 19.Heinz, C., and Niederweis, M. (2000) Anal. Biochem. 285 113–120 [DOI] [PubMed] [Google Scholar]
- 20.Stephan, J., Bail, J. G., Titgemeyer, F., and Niederweis, M. (2004) BMC Microbiol. 4 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Delogu, G., Pusceddu, C., Bua, A., Fadda, G., Brennan, M. J., and Zanetti, S. (2004) Mol. Microbiol. 52 725–733 [DOI] [PubMed] [Google Scholar]
- 22.Ausubel, F. M., Brent, R., Kingston, R. E., Moore, D. D., Seidmann, J. G., Smith, J. A., and Struhl, K. (1987) Current Protocols in Molecular Biology, John Wiley & Sons, New York
- 23.Cuff, J. A., Clamp, M. E., Siddiqui, A. S., Finlay, M., and Barton, G. J. (1998) Bioinformatics 14 892–893 [DOI] [PubMed] [Google Scholar]
- 24.Cuff, J. A., and Barton, G. J. (1999) Proteins 34 508–519 [DOI] [PubMed] [Google Scholar]
- 25.Bendtsen, J. D., Nielsen, H., von Heijne, G., and Brunak, S. (2004) J. Mol. Biol. 340 783–795 [DOI] [PubMed] [Google Scholar]
- 26.Krogh, A., Larsson, B., von Heijne, G., and Sonnhammer, E. L. (2001) J. Mol. Biol. 305 567–580 [DOI] [PubMed] [Google Scholar]
- 27.Melen, K., Krogh, A., and von Heijne, G. (2003) J. Mol. Biol. 327 735–744 [DOI] [PubMed] [Google Scholar]
- 28.Rauch, G., and Moran, O. (1995) Comput. Methods Programs Biomed. 48 193–200 [DOI] [PubMed] [Google Scholar]
- 29.Eisenberg, D., Wilcox, W., and McLachlan, A. D. (1986) J. Cell. Biochem. 31 11–17 [DOI] [PubMed] [Google Scholar]
- 30.Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., and Ferrin, T. E. (2004) J. Comput. Chem. 25 1605–1612 [DOI] [PubMed] [Google Scholar]
- 31.Stephan, J., Mailaender, C., Etienne, G., Daffe, M., and Niederweis, M. (2004) Antimicrob. Agents Chemother. 48 4163–4170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heinz, C., Engelhardt, H., and Niederweis, M. (2003) J. Biol. Chem. 278 8678–8685 [DOI] [PubMed] [Google Scholar]
- 33.Liu, N., and Delcour, A. H. (1998) FEBS Lett. 434 160–164 [DOI] [PubMed] [Google Scholar]
- 34.Varma, S., Chiu, S. W., and Jakobsson, E. (2006) Biophys. J. 90 112–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benz, R., Janko, K., and Läuger, P. (1979) Biochim. Biophys. Acta 551 238–247 [DOI] [PubMed] [Google Scholar]
- 36.Ojha, A., Anand, M., Bhatt, A., Kremer, L., Jacobs, W. R., Jr., and Hatfull, G. F. (2005) Cell 123 861–873 [DOI] [PubMed] [Google Scholar]
- 37.Basle, A., Iyer, R., and Delcour, A. H. (2004) Biochim. Biophys. Acta 1664 100–107 [DOI] [PubMed] [Google Scholar]
- 38.Guilvout, I., Chami, M., Engel, A., Pugsley, A. P., and Bayan, N. (2006) EMBO J. 25 5241–5249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cascales, E., Buchanan, S. K., Duche, D., Kleanthous, C., Lloubes, R., Postle, K., Riley, M., Slatin, S., and Cavard, D. (2007) Microbiol. Mol. Biol. Rev. 71 158–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cascioferro, A., Delogu, G., Colone, M., Sali, M., Stringaro, A., Arancia, G., Fadda, G., Palu, G., and Manganelli, R. (2007) Mol. Microbiol. 66 1536–1547 [DOI] [PubMed] [Google Scholar]
- 41.Alahari, A., Saint, N., Campagna, S., Molle, V., Molle, G., and Kremer, L. (2007) J. Bacteriol. 189 6351–6358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nikaido, H., and Rosenberg, E. Y. (1983) J. Bacteriol. 153 241–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koebnik, R., Locher, K. P., and van Gelder, P. (2000) Mol. Microbiol. 37 239–253 [DOI] [PubMed] [Google Scholar]
- 44.Deleage, G., Blanchet, C., and Geourjon, C. (1997) Biochimie (Paris) 79 681–686 [DOI] [PubMed] [Google Scholar]
- 45.Koronakis, V., Sharff, A., Koronakis, E., Luisi, B., and Hughes, C. (2000) Nature 405 914–919 [DOI] [PubMed] [Google Scholar]
- 46.Schulz, G. E. (2000) Curr. Opin. Struct. Biol. 10 443–447 [DOI] [PubMed] [Google Scholar]
- 47.Stackebrandt, E., Rainey, F. A., and Ward-Rainey, N. L. (1997) Int. J. Syst. Bacteriol. 47 479–491 [DOI] [PubMed] [Google Scholar]
- 48.Cole, S. T., Eiglmeier, K., Parkhill, J., James, K. D., Thomson, N. R., Wheeler, P. R., Honore, N., Garnier, T., Churcher, C., Harris, D., Mungall, K., Basham, D., Brown, D., Chillingworth, T., Connor, R., Davies, R. M., Devlin, K., Duthoy, S., Feltwell, T., Fraser, A., Hamlin, N., Holroyd, S., Hornsby, T., Jagels, K., Lacroix, C., Maclean, J., Moule, S., Murphy, L., Oliver, K., Quail, M. A., Rajandream, M. A., Rutherford, K. M., Rutter, S., Seeger, K., Simon, S., Simmonds, M., Skelton, J., Squares, R., Squares, S., Stevens, K., Taylor, K., Whitehead, S., Woodward, J. R., and Barrell, B. G. (2001) Nature 409 1007–1011 [DOI] [PubMed] [Google Scholar]
- 49.Niederweis, M. (2003) Mol. Microbiol. 49 1167–1177 [DOI] [PubMed] [Google Scholar]
- 50.Massari, P., Ram, S., Macleod, H., and Wetzler, L. M. (2003) Trends Microbiol 11 87–93 [DOI] [PubMed] [Google Scholar]
- 51.Wen, K. K., Giardina, P. C., Blake, M. S., Edwards, J., Apicella, M. A., and Rubenstein, P. A. (2000) Biochemistry 39 8638–8647 [DOI] [PubMed] [Google Scholar]
- 52.Merz, A. J., and So, M. (2000) Annu. Rev. Cell Dev. Biol. 16 423–457 [DOI] [PubMed] [Google Scholar]
- 53.Rudel, T., Schmid, A., Benz, R., Kolb, H. A., Lang, F., and Meyer, T. F. (1996) Cell 85 391–402 [DOI] [PubMed] [Google Scholar]
- 54.Bauer, F. J., Rudel, T., Stein, M., and Meyer, T. F. (1999) Mol. Microbiol. 31 903–913 [DOI] [PubMed] [Google Scholar]
- 55.Trias, J., and Benz, R. (1994) Mol. Microbiol. 14 283–290 [DOI] [PubMed] [Google Scholar]
- 56.Tansel, B., Sager, J., Garland, J., Strayer, R. F., Levine, M., Roberts, M., Hummerick, M., and Bauer, J. (2006) Separ. Purif. Technol. 51 40–47 [Google Scholar]