

Abstract
The cytokine osteopontin (OPN) can be hydrolyzed by thrombin exposing a cryptic α4β1/α9β1 integrin-binding motif (SVVYGLR), thereby acting as a potent cytokine for cells bearing these activated integrins. We show that purified milk OPN is a substrate for thrombin with a kcat/Km value of 1.14 × 105 m–1 s–1. Thrombin cleavage of OPN was inhibited by unsulfated hirugen (IC50 = 1.2 ± 0.2 μm), unfractionated heparin (IC50 = 56.6 ± 8.4 μg/ml) and low molecular weight (5 kDa) heparin (IC50 = 31.0 ± 7.9 μg/ml), indicating the involvement of both anion-binding exosite I (ABE-I) and anion-binding exosite II (ABE-II). Using a thrombin mutant library, we mapped residues important for recognition and cleavage of OPN within ABE-I and ABE-II. A peptide (OPN-(162–197)) was designed spanning the OPN thrombin cleavage site and a hirudin-like C-terminal tail domain. Thrombin cleaved OPN-(162–197) with a specificity constant of kcat/Km = 1.64 × 104 m–1 s–1. Representative ABE-I mutants (K65A, H66A, R68A, Y71A, and R73A) showed greatly impaired cleavage, whereas the ABE-II mutants were unaffected, suggesting that ABE-I interacts principally with the hirudin-like OPN domain C-terminal and contiguous to the thrombin cleavage site. Debye-Hückel slopes for milk OPN (–4.1 ± 1.0) and OPN-(162–197) (–2.4 ± 0.2) suggest that electrostatic interactions play an important role in thrombin recognition and cleavage of OPN. Thus, OPN is a bona fide substrate for thrombin, and generation of thrombin-cleaved OPN with enhanced pro-inflammatory properties provides another molecular link between coagulation and inflammation.
Osteopontin (OPN)2 is a secreted, acidic, phosphorylated glycoprotein that can act as either an extracellular matrix protein important in bone resorption or as a cytokine (1–3). OPN is normally expressed by osteoblasts, osteoclasts, epithelium, and smooth muscle cells. Many inflammatory cells (T cells, macrophages, NK cells, and Kupfer cells) also express OPN, which can be enhanced in response to pro-inflammatory cytokines, tissue injury, and stress (1–3). Studies with OPN-null mice show that OPN has roles in a broad range of homeostatic (e.g. bone remodeling, cellular immunity, wound healing), and pathologic (e.g. tumor metastasis) processes (1, 4–8). OPN is chemotactic for several cell types, in particular monocytes and macrophages, and stimulates cell motility and cell survival. These functions are in part dependent on the RGD sequence in OPN, which interacts with a number of integrins, including αvβ1, αvβ3, αvβ5 (9), α5β1 (10), and α8β1 (11), or binds to either of two variant forms of CD44 (12).
OPN also has a conserved thrombin cleavage site downstream of and contiguous to the RGD domain. Cleavage of OPN by thrombin increases the adhesion, spreading, and migration of a variety of cultured cells in vitro (13, 14). Melanoma cells and HT1080 fibrosarcoma cells will only bind thrombin-cleaved OPN, suggesting that thrombin cleavage is critical in the pathology of certain cancers (13–15). Thrombin cleavage of human OPN (Arg168–Ser169) exposes a cryptic integrin binding motif, 162SVVYGLR168, that specifically binds integrins α4β1, α4β7, and α9β1 (16–19); this sequence is adjacent to the RGD domain. The thrombin-cleaved N-terminal fragment of OPN, where RGD has been mutated to RAA, still binds to α9-transfected cells (19). Cell adhesion can be inhibited by alanine substitution of Tyr165 or by the double deletion of Leu167/Arg168 in the SVVYGLR motif. The integrin α4β1 also recognizes the SVVYGLR motif and is functionally distinct from the RGD domain (16–18). As thrombin and OPN are both present at sites of tissue injury and inflammation, thrombin cleavage of OPN has been postulated to be important in enhancing the pro-inflammatory effect of OPN in vivo. However, although thrombin cleavage of OPN appears to be important in regulating its biological role, its biochemistry has not been studied in detail. This paper establishes OPN as a bona fide substrate of thrombin and defines the domains in that are thrombin important for its recognition and hydrolysis.
EXPERIMENTAL PROCEDURES
Materials—Human wild-type (WT) and alanine-substituted mutant thrombins were expressed, purified, and titrated with PPACK (d-Phe-Pro-Arg-chloromethylketone) as described previously (20). Low molecular weight (LMW) heparin (5 kDa), porcine intestinal mucosal unfractionated heparin, PPACK, and unsulfated hirulog were from Sigma. The peptides SVVYGLR and OPN-(162–197) (SVVYGLR/SKSKKFQRPDIQYPDATDEDITSHMESEE) were synthesized, purified, and quantitated by the peptide synthesis facility at Stanford University School of Medicine.
Purification of Human OPN from Milk—OPN was purified from human milk (Mothers Milk Bank, San Jose, CA) using a previously published procedure with modifications (21). Briefly, 1 liter of human milk pooled from several donors was allowed to separate on ice. The whey was carefully decanted from the curd and clarified by centrifugation at 23,000 × g for 60 min at 4 °C and then clarified further over a 0.2-μm pore size filtration unit. Benzamidine (2 mm) and DTT (2 mm) were added, and the clarified whey fraction was batch-adsorbed to Q-Sepharose (GE Healthcare) for 2 h at 4°C. The resin was washed with 10 mm sodium phosphate/0.2 m NaCl, 2 mm DTT, 2 mm benzamidine, pH 7.4, and then eluted with 10 mm sodium phosphate/1.0 m NaCl, 2 mm DTT, pH 7.4. The eluant was dialyzed overnight in 10 mm sodium phosphate/4 m NaCl, 2 mm DTT, pH 7.4. The dialyzed protein was then loaded onto a 5-ml HiTrap phenyl-Sepharose HP column (GE Healthcare) at 1 ml/min and washed to base line. A linear gradient was developed from 4 to 1 m NaCl at 1 ml/min with 1-ml fractions collected. Pure fractions were pooled and dialyzed extensively against phosphate-buffered saline, pH 7.4. Partially pure fractions were diluted in 10 mm sodium phosphate/4 m NaCl, 2 mm DTT, pH 7.4, and reapplied over the HiTrap phenyl-Sepharose HP column. The concentration of purified protein was estimated at 280 nm using an extinction coefficient (1 cm path length) of 22920.
Hydrolysis of Human Milk OPN by Thrombin and the Quantitation of the Cleaved C-terminal Fragment—OPN (4–31 μm) was reacted with thrombin (≈5–100 nm) in assay buffer containing 20 mm HEPES, 5 mm CaCl2, 0.1% polyethylene glycol 8000, pH 7.5, and at various salt concentrations (≈20–200 mm NaCl) at 37 °C. Reactions were terminated with the addition of PPACK (5 μm) followed by DTT (5 mm). HPLC-gel filtration chromatography (HPLC-GFC) was employed to quantitate the generation of the 15-kDa C-terminal fragment (CTF) liberated by thrombin cleavage. Various amounts (10–40 μl) of the reaction mixture were injected over a Shodex 8 × 300 mm KW-803 column (Showa Denko America Inc, New York), and proteins were resolved in 20 mm sodium phosphate, 300 mm NaCl, 5 mm DTT, pH 7.6, at 1 ml/min. The peak area was detected at 254 nm and was converted to nmoles of product using a calibration curve.
Inhibition of Thrombin Hydrolysis of Human Milk OPN or OPN-(162–197) Peptide by LMW Heparin, Unfractionated Heparin, and Unsulfated Hirulog—Human milk OPN (5 μm) or OPN-(162–197) (10 μm) was preincubated with 5 kDa of LMW heparin, porcine intestinal unfractionated heparin (0.25–1000 μg/ml), or unsulfated hirugen (10 nm–10 μm) at 37°C for 10 min. Thrombin was added to milk OPN (50 nm) and OPN-(162–197) (100 nm) and incubated for 3 and 5 min, respectively, at 37 °C. Reactions containing human milk OPN were terminated with 5 μm PPACK followed by 5 mm DTT. Thrombin hydrolysis of OPN-(162–197) was terminated by the addition of 10% perchloric acid. The concentration of thrombin used and the incubation time were determined empirically to give no more than 30% substrate hydrolysis. The amount of product formed was determined by HPLC-GFC for human milk OPN and reverse-phase HPLC for OPN-(162–197).
Determination of the Michaelis-Menten Parameters for Thrombin Hydrolysis of Milk OPN—Several concentrations of OPN (4–31 μm) were digested with thrombin (typically 5 nm) at 37 °C for various times, such that there was less than 10% hydrolysis of substrate. The reactions were terminated by the addition of 5 μm PPACK and 5 mm DTT. The amount of product formed (OPN CTF) was determined by HPLC-GFC as described above. The values for Km and kcat were determined by plotting the initial velocity of cleavage against the different substrate concentrations and then fitting to the Michaelis-Menten equation by nonlinear regression analysis. Experiments were performed in duplicate, and the data were pooled for analysis.
Estimation of kcat/Km for Cleavage of OPN under First Order Rate Conditions—Values for kcat/Km were determined by full reaction progress curves at a substrate concentration well below Km for WT and representative mutants of thrombin within anion-binding exosite I (ABE-I; R20A, K21A, Q24A, R62A, K65A, H66A, R68A, T69A, R70A, Y71A, R73A, K77A, K106A, K107A), ABE-II (R89A/R93A/E94A, R98A, D122A/R123A/E124A, E169A/K174A/D175A, R178A/R180A/D183A, R245A, K248A), the 50-insertion loop (W50A), and the Na+ binding site (E229A). 5 μm OPN (0.26 × [Km]) was preincubated in assay buffer for 10 min at 37 °C. Reactions were started with the addition of thrombin (typically 50 nm) and incubated at 37 °C. Aliquots were removed at several time points (0–1800 s) and terminated with 5 μm PPACK followed by 5 mm DTT. The amount of product generated (nmoles) for each time point was determined by HPLC-GFC as detailed above. Values for kcat/Km were determined by fitting data from the time course experiments to the following equation (22),
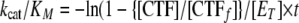 |
(Eq. 1) |
where [CTF] is the concentration of the CTF at a given time, CTFf is the concentration of the CTF at full activation, and [ET] is the total enzyme concentration of thrombin at any given time (t). Curve fitting was performed using Prism 5 (GraphPad Software).
Determination of Km, kcat, and kcat/Km for Hydrolysis of the Peptide OPN-(162–197)—For the estimation of Michaelis-Menten parameters, 10–300 μm OPN-(162–197) peptide was incubated with WT or mutant thrombin in assay buffer containing 20 mm HEPES, 145 mm NaCl, 5 mm CaCl2, 0.1% polyethylene glycol 8000, pH 7.5. Enzyme concentration and incubation time were varied such that there was less than 10% substrate depletion. Reactions were terminated with the addition of 10% perchloric acid. The cleaved and uncleaved peptides were separated by reverse-phase HPLC on a Waters Symmetry C18 (4.6 × 150 mm, 5 μm) column using a 0.2–35% acetonitrile/0.1% trifluoroacetic acid gradient over 30 min at a flow rate of 1.0 ml/min. Cleavage was specific with no secondary cleavage sites. The cleaved peptide peak OPN-(162–168) (SVVYGLR) eluted at 19.8 min and was well resolved from OPN-(162–197) (23.6 min) and the OPN-(169–197) cleavage product (22.3 min). Identity of the cleavage products was confirmed by mass spectrometry. The area under the SVVYGLR peak was converted to nmoles of peptide using a calibration curve constructed with purified and quantitated SVVYGLR peptide. The values for Km and kcat were determined by fitting to the Michaelis-Menten equation by non-linear regression analysis as described above. The deduced value for Km was used to determine the reaction conditions for estimating the specificity constant, kcat/Km. OPN-(162–197) peptide (10 μm, 0.04 × Km) was preincubated in assay buffer (20 mm HEPES, 145 mm NaCl, 5 mm CaCl2, 0.1% polyethylene glycol 8000, pH 7.5) for 10 min. Reactions were started with the addition of WT or mutant thrombin (100–500 nm) with aliquots removed at several time points (0–1800 s) and then terminated with the addition of 10% perchloric acid. The effect of NaCl and choline chloride (ChCl) on kcat/Km was determined in the presence of assay buffer with salt concentrations ranging from 20 to 200 mm. Assays with ChCl were buffered with 5 mm Tris-HCl, pH 7.5, to reduce Na+ ions from pH adjustment. The amount of product (SVVYGLR) formed was quantitated as described above by reverse-phase HPLC analysis, and the values for kcat/Km were determined as described above.
RESULTS
Determination of the Kinetic Parameters for Hydrolysis of Purified Human Breast Milk OPN by Thrombin—The hydrolysis of human milk derived OPN by thrombin gave estimates for Km (19.1 ± 3.9 μm), kcat (0.9 ± 0.1 s–1), and kcat/Km (0.47 × 105 m–1 s–1) under assay conditions looking at initial reaction velocities over several substrate concentrations up to 31 μm (Fig. 1A). However, significant substrate and/or product inhibition was observed at concentrations of 50 μm and higher (data not shown). To account for substrate/product inhibition effects, we estimated a value for the specificity constant kcat/Km of (1.14 ± 0.17) × 105 m–1 s–1 (Fig. 1B) by full reaction progress curves using a substrate concentration estimated to be 0.26 × Km. Under these conditions the estimate for kcat/Km is ∼2.5 times better and most likely reflects the best estimate for the specificity constant.
FIGURE 1.

Kinetic parameters for the hydrolysis of milk OPN by human thrombin. A, human milk OPN was purified to homogeneity and the values for Km and kcat were determined by Michaelis-Menten kinetics using 4–31 μm OPN. B, the derived value for Km was used to approximate kcat/Km using 0.26 x Km for WT thrombin (•) and two representative thrombin ABE-I mutants, H66A (▴) and Y71A (▪).
Unfractionated Heparin, LMW Heparin, and Unsulfated Hirugen Inhibit Thrombin Hydrolysis of OPN—Osteopontin contains two putative heparin-binding domains (23), therefore we hypothesized that heparin could act as a bridge between the heparin-binding domains and the thrombin ABE-II domain, enhancing hydrolysis. OPN can be bound to HiTrap heparin-Sepharose and eluted with 0.37 m NaCl, suggesting the presence of OPN heparin-binding domains (data not shown). However, we found that LMW heparin and unfractionated heparin caused dose-dependent inhibition of thrombin hydrolysis of OPN with IC50 values of 31.0 ± 7.9 and 56.6 ± 8.4 μg/ml, respectively (Fig. 2A). Further, the inhibition curves were not bell-shaped, suggesting that heparin does not function as a macromolecular bridge binding both thrombin and OPN to enhance thrombin cleavage of OPN. Rather, both unfractionated and LMW heparins compete against OPN for binding to the thrombin ABE-II, indicating the importance of ABE-II in thrombin cleavage of OPN.
FIGURE 2.

Heparin and hirugen inhibit thrombin cleavage of milk OPN. A, 5 μm milk OPN was preincubated with 5-kDa LMW heparin (LMWH, ▪) or unfractionated heparin (UFH, •) from 0. 25 to 1000 μg/ml at 37 °C for 10 min. The reaction was started with 50 nm thrombin and incubated for 3 min at 37 °C. Unfractionated heparin (▴; 2.5–1000 μg/ml) was also preincubated with OPN-(162–197) and hydrolyzed by 100 nm thrombin for 5 min. B, milk OPN (▪) and OPN-(162–197) (▴) were also preincubated with unsulfated hirugen (10 nm–10 μm) for 10 min at 37 °C, and then thrombin was added and incubated for 3–5 min at 37 °C. Reactions were terminated and reaction products quantitated as described under “Experimental Procedures.”
Unsulfated hirugen is a tight binding inhibitor specific only for thrombin ABE-I. Hirugen also caused dose-dependent inhibition of thrombin cleavage of OPN with an IC50 of 1.2 ± 0.2 μm (Fig. 2B). The inhibition studies using LMH heparin and hirugen suggest that both exosites play a role in thrombin cleavage of OPN.
Mapping of Residues in ABE-I and ABE-II Important for the Hydrolysis of OPN—A bank of more than 56 mutant thrombins has been used to map domains important in the cleavage of FV (24), FVIII (25), FXI (26), FXIII (22), and fibrinogen (27) for thrombomodulin-dependent activation of protein C and thrombin-activable fibrinolysis inhibitor (27, 28) and for heparin-accelerated inhibition of antithrombin, heparin cofactor II, and protein C inhibitor (29–31). These studies have identified key residues within ABE-I and ABE-II important in defining the interaction interface for all these interactions. The specificity constant was determined for 14 mutants in ABE-I and seven mutants in ABE-II along with two thrombin mutants (W50A and E229A) known to affect the thrombin active site (23–32). Ten thrombin ABE-I mutants had less than 50% WT activity, with mutants K65A (29%), H66A (14%), Y71A (16%), and R73A (20%) severely affected (Table 1), which indicates a very large interaction interface within ABE-I. Three mutants in ABE-II (total of seven alanine-substituted residues) have impaired activity relative to WT thrombin, R98A (41%), R89A/R93A/E94A (31%), and R178A/R180A/D1778A (46%), confirming the role of this ABE from the heparin inhibition studies. The mutant thrombins W50A and E229A were also assessed for their ability to hydrolyze OPN. Trp50 forms part of the 50-insertion loop and is important in defining the insertion loop and the aryl binding site, and Glu229 maintains the integrity of the Na+-binding loop. The crystal structure of E229A shows the collapse of the active site with almost complete occlusion (32). Alanine substitution of both of these residues has a profound effect in the ability of thrombin to hydrolyze OPN, both with 9% WT activity. These mutations clearly show that a functional thrombin active site is needed to cleave at the P1 arginine of SVVYGLR/SKSKKFQ.
TABLE 1.
Impaired hydrolysis of OPN by selected alanine substituted thrombin mutants from ABE-I, ABE-II, and the active site
Values for kcat/Km for the hydrolysis of OPN by WT and mutant thrombins were determined under first order rate conditions in assay buffer fixed at 145 mm NaCl. Values for the mean ± S.E. are based on at least two assays performed in duplicate.
| Thrombina | Chymotrypsinb | Location | kcat/Km × 104 | WT activity |
|---|---|---|---|---|
| m-1s-1 | % | |||
| WT | WT | 11.36 ± 1.72 | 100 | |
| R20A | R35A | ABE-I | 14.22 ± 1.18 | 125 |
| K21A | K36A | 3.56 ± 0.07 | 31 | |
| Q24A | Q38A | 10.42 ± 1.25 | 92 | |
| R62A | R67A | 4.65 ± 0.28 | 41 | |
| K65A | K70A | 3.31 ± 0.20 | 29 | |
| H66A | H71A | 1.58 ± 0.19 | 14 | |
| R68A | R73A | 3.80 ± 0.49 | 34 | |
| T69A | T74A | 7.90 ± 0.95 | 70 | |
| R70A | R75A | 4.65 ± 0.51 | 41 | |
| Y71A | Y76A | 1.84 ± 0.17 | 16 | |
| R73A | R77AA | 2.31 ± 0.15 | 20 | |
| K77A | K81A | 4.73 ± 0.38 | 42 | |
| K106A | K109A | 6.25 ± 0.25 | 55 | |
| K107A | K110A | 5.10 ± 0.36 | 45 | |
| R89A/R93A/E94A | R93A/R97A/E97AA | ABE-II | 3.10 ± 0.19 | 31 |
| R98A | R101A | 4.69 ± 0.28 | 41 | |
| D122A/R123A/E124A | D125A/R126A/E127A | 7.60 ± 0.38 | 67 | |
| E169A/K174A/D175A | E164A/K169A/D170A | 7.02 ± 0.42 | 62 | |
| R178A/R180A/D183A | R173A/R175A/D178A | 5.24 ± 0.47 | 46 | |
| R245A | R233A | 7.28 ± 0.44 | 64 | |
| K248A | K236A | 7.24 ± 0.29 | 64 | |
| W50A | W60DA | Active site | 0.96 ± 0.14 | 8 |
| E229A | E217A | 1.02 ± 0.14 | 9 |
Thrombin-based numbering.
Chymotrypsinogen-based numbering.
Identification of an OPN Domain That Interacts with ABE-I—OPN has a hirudin-like domain, DIQYPDATDEDITSHMESEE (33), adjacent and contiguous to the thrombin cleavage site, that could potentially interact with ABE-I. To address this potential interaction, a peptide was made (OPN-(162–197)) that spanned the cleavage site and hirudin-like ABE-I-binding domain. Values for Km (296.6 ± 10.9 μm), kcat (3.3 ± 0.1 s–1), and kcat/Km (1.11 × 104 m–1 s–1) were initially determined over a range of substrate concentrations; the value derived for Km was used to define experimental conditions to estimate kcat/Km under first order rate conditions (Fig. 3). The specificity constant kcat/Km was determined for the cleavage of this OPN-(162–197) by WT thrombin and was found not to be a good substrate (kcat/Km 1.64 ± 0.06 × 104 m–1 s–1) when compared with milk OPN (kcat/Km 1.14 ± 0.17 × 105 m–1 s–1). Hirugen was found to inhibit thrombin cleavage of OPN-(162–197) (Fig. 2B) with an IC50 = 1.1 ± 0.1 μm showing a requirement for ABE-I. The ability of selected ABE-I and ABE-II mutants to cleave OPN-(162–197) was compared with that of WT thrombin (Table 2). The ABE mutants K65A, H66A, R68A, Y71A, and R73A in general showed similar decreases in activity relative to WT thrombin when compared with the cleavage of milk OPN. Heparin had no effect, excluding the participation of ABE-II (Fig. 2A). Consistent with this finding, ABE-II mutants R98A, E160A/K174A/D175A, and R178A/R180A/D183A in general had no effect on cleavage of the peptide, suggesting that the OPN hirudin-like domain interacts principally with the thrombin ABE-I. The ABE-II R89A/R93A/E94A mutant showed 44% WT activity. The residue Glu94 lies on the rim between ABE-II and the active site, so it is conceivable that mutation of this residue could influence cleavage within the active site.
FIGURE 3.

Determination of Km and kcat for hydrolysis of the OPN-(162–197) peptide by thrombin. The Michaelis-Menten parameters for the hydrolysis of OPN-(162–197) (10–300 μm) by thrombin were determined at 37 °C in assay buffer at either 53 mm (▪) or 145 mm NaCl (•). The deduced value for Km at 145 mm NaCl was used to estimate the values for kcat/Km by progress curves at a substrate concentration well below Km (0.04 × Km).
TABLE 2.
ABE-I alanine-substituted thrombin mutants have impaired catalysis for the OPN-(162–197) peptide
The specificity constant kcat/Km was estimated for the hydrolysis of OPN-(162–197) peptide by WT and selected mutant thrombins under first order rate conditions in assay buffer fixed at 145 mm NaCl. Values for the mean ± S.E. are based on at least two assays performed in duplicate.
| Thrombin | Location | kcat/Km | WT activity |
|---|---|---|---|
| mm-1s-1 | % | ||
| WT | 16.35 ± 0.58 | 100 | |
| K65A | ABE-I | 5.38 ± 1.25 | 33 |
| H66A | 2.87 ± 0.41 | 18 | |
| R68A | 5.80 ± 0.32 | 35 | |
| Y71A | 3.76 ± 0.01 | 23 | |
| R73A | 7.41 ± 0.19 | 45 | |
| R89A/R93A/E94A | ABE-II | 7.13 ± 0.40 | 44 |
| R98A | 15.10 ± 0.83 | 92 | |
| E169A/K174A/D175A | 14.79 ± 1.23 | 90 | |
| R178A/R180A/D183A | 16.33 ± 0.35 | 100 |
Electrostatic Interactions Are Important for Hydrolysis of OPN by Thrombin—Studies between thrombin and hirudin show the importance of complementary electrostatic fields in enhancing the rate of complex formation by “electrostatic steering” (34, 35). To study this phenomenon for thrombin cleavage of OPN, kcat/Km was determined over a range of NaCl concentrations for milk-derived OPN (38–203 mm NaCl). The plot of kcat/Km versus [NaCl] for milk OPN (Fig. 4A) shows that the specificity constant decreased by 1 order of magnitude from 3.18 × 105 m–1 s–1 to 0.28 × 105 m–1 s–1 when the ionic strength was increased from 72 to 203 mm NaCl. The lower value for kcat/Km at 38 mm NaCl could reflect transition from the fast (Na+ bound) to slow (Na+ free) form of thrombin (36). To address potential changes to the ABE-I electrostatic potential made by the fast to slow form transition (37), we examined the cleavage of OPN-(162–197) in the presence of either ChCl (20–200 mm), which favors the slow form of thrombin, or NaCl (53–198 mm). The specificity constant for the cleavage of OPN-(162–197) decreased 3.2-fold from 0.34 × 105 m–1 s–1 at 53 mm NaCl to 0.11 × 105 m–1 s–1 at 198 mm NaCl (Fig. 4B). A 2.7-fold decrease in the specificity constant from 0.19 × 105 m –1 s–1 to 0.71 × 104 m–1 s–1 was also observed from 50 to 200 mm ChCl. As expected, the specificity constant was consistently reduced in the presence of ChCl, consistent with the presence of the slow form of thrombin. At physiological salt concentration (≈150 mm) the kcat/Km for the cleavage of OPN-(162–197) in the presence of NaCl (1.64 ± 0.06 × 104 m–1 s–1) was 1.7-fold greater than the predominantly “slow form” of thrombin (9.75 ± 1.70 × 103 m–1 s–1) in ChCl.
FIGURE 4.

NaCl affects the rate of hydrolysis of OPN by thrombin. A, values for kcat/Km were determined for the cleavage of milk OPN over a range of NaCl concentrations. B, the specificity constant for the cleavage of OPN-(162–197) over a range of NaCl (•) or ChCl (▪) concentrations are shown. C, the Debye-Hückel plot for log kcat/Km versus the square root of the ionic strength was used to estimate Debye-Hückel slopes for milk OPN (▪) and OPN-(162–197) titration with NaCl (•) or ChCl (▴).
A Debye-Hückel slope from 53 to 203 mm1/2 NaCl for the cleavage of milk OPN gave a slope of –4.1 ± 1.0, signifying a high degree of electrostatic interactions in forming the thrombin-OPN encounter complex. A better fit was obtained from 72 to 203 mm1/2 NaCl, with a Debye-Hückel slope of –5.9 ± 0.7, and at an intercept at 0 ionic strength the value for kcat/Km was 1.3 × 107 m–1 s–1. The Debye-Hückel slope for OPN-(162–197) from the NaCl titration was –2.4 ± 0.2 with a kcat/Km at 0 ionic strength estimated to be 1.3 × 105 m–1 s–1. A similar slope (–1.9 ± 0.1) was obtained from the ChCl titration, suggesting that OPN hydrolysis by thrombin is driven primarily by electrostatic steering, with decreased ionic strength in the solution favoring a faster and more productive complex formation between thrombin ABE-I and OPN-(162–197), which is independent of the allosteric state of thrombin.
DISCUSSION
OPN is a multifunctional protein that can act both as an extracellular matrix protein involved in bone resorption and as a cytokine (1–3). As a cytokine, OPN can interact with a number of intergrin-bearing cells through RGD-dependent interactions (1–3). It has been shown in vitro that thrombin can cleave OPN, which exposes the cryptic integrin-binding motif (SVVYGLR) for binding to the integrins α4β1 or α9/β1, independently of RGD or CD44 (13–19). This suggests that thrombin could also act indirectly as a mediator of inflammatory and immune responses by activating OPN. However, the in vivo significance of this is not well understood, and whether OPN is a real substrate for thrombin remains to be established.
In this report we have shown that OPN purified from human milk is a substrate for thrombin with a specificity constant of 1.14 × 105 m–1 s–1. The thrombin active site cleft is topologically similar to that of the archetypal serine protease trypsin, an enzyme that can indiscriminately cleave any protein after an exposed P1 arginine (38, 39). However, the specificity of thrombin is significantly more restricted because of two insertion loops that partially occlude the active site to macromolecular substrates and inhibitors (38, 39). To overcome steric hindrance at the active site, the specific substrates and inhibitors of thrombin have to bind to two distal exosites, which are characterized by a high density of positively charged, solvent-exposed residues (38, 39). ABE-I binds hirudin, PAR1, PAR4, thrombomodulin, actor XIII, and fibrinogen, and ABE-II binds the heparin-bound serpins (antithrombin, heparin cofactor II, and protease nexin 1), the leech inhibitor hemadin, and platelet glycoprotein Ib (40, 41). Both exosites are required for the efficient activation of coagulation factors V, VIII, and XI (40, 41). Hirugen, a specific and tight inhibitor of ABE-I (42), effectively blocked thrombin cleavage of OPN. Unfractionated and LMW heparin also inhibited thrombin cleavage of OPN, presumably through direct competition against OPN binding to ABE-II. The dependence of both ABE-I and ABE-II on optimal cleavage of OPN supports the notion that OPN is a bona fide substrate for thrombin.
To identify the thrombin residues within ABE-I and ABE-II that are important for OPN hydrolysis, we used a library of alanine-substituted thrombin mutants that had been used previously to define specific residues involved with thrombin substrate interactions (23–31). ABE-I showed a large interaction interface with 10 of 14 residues affected by alanine substitution, and ABE-II showed 3 of 5 mutant thrombins (potentially 7 of 15 residues) with impaired activity, confirming the hirugen and heparin studies. Whether this is the result of specific impairment of direct interactions or small conformational changes could not be distinguished, as there is no crystal structure of the thrombin-OPN complex. However it is quite remarkable that the residues identified are similar to those characterized previously as being involved in thrombin cleavage of FV, FVIII, FXI, and FXIII (23–26).
An interesting feature of the OPN polypeptide sequence is a hirudin-like ABE-I-binding domain C-terminal to the SVVYGLR cryptic motif and thrombin cleavage site. The sequence has a large number of acidic amino acids and three serine/threonine phosphorylation sites, along with two prolines and a tyrosine, a common feature of hirudin-like domains seen with thrombomodulin (residues 408–426), factor VIII (residues 716–731), PAR1 (residues 52–69), and heparin cofactor II (residues 56–75), all of which utilize ABE-I. The peptide OPN-(162–197), synthesized to span the cryptic motif, thrombin cleavage site, and hirudin-like ABE-I-binding domain, was used in cleavage studies with WT and selected ABE-I and ABE-II mutant thrombins. The OPN-(162–197) peptide was a poor substrate (kcat/Km 1.64 ± 0.06 × 104 m–1 s–1) for WT thrombin compared with OPN purified from human milk (kcat/Km 1.14 ± 0.17 × 105 m–1 s–1). This result was not wholly unexpected because the peptide lacks the complete functional domains of native OPN and would not be able to present the thrombin cleavage site for hydrolysis in an optimal manner. Further, OPN contains many serine/threonine phosphorylation sites (43), three of which are within the OPN-(162–197) peptide (Thr185, Ser191, and Ser195). The peptide OPN-(162–197) lacks these additional negative charges and thus has a reduced overall electrostatic potential, which may be important in electrostatic steering and ion pair interactions (34, 35, 44). Indeed, native hirudin with a sulfated Tyr63 forms a very tight equimolar complex with thrombin with a Ki of 20 femtomolar. Loss of this charge, as seen with recombinant hirudin, increases the Ki 10-fold to 200 femtomolar (45). Studies are in progress to assess the role of phosphorylated Thr185, Ser191, and Ser195 residues for interaction with the thrombin ABE-I domain. Interestingly, OPN can exist in different forms, based on the extent of phosphorylation, with potentially different biological functions (43, 46–48). Therefore it is possible that in vivo thrombin can have different specificities toward the different OPN species.
An important characteristic of the thrombin ABEs is a high density of basic residues, which produces a large positive electrostatic potential that protrudes into the solvent (35). The electrostatic field generated by thrombin ABE-I has been shown to be important for electrostatic steering with the complementary electrostatic field in its ligands in order to enhance productive complex formation (35, 46). Indeed, studies looking at the binding of thrombin with the thrombomodulin “TM-456” (EGF-456) domain show a very rapid and almost completely electrostatically steered interaction (49). For milk OPN cleavage by thrombin over a range of NaCl concentrations, we deduced a Debye-Hückel slope of –4.1 ± 1.0, which is close to the reported slope of –6.0 ± 1.0 for the interaction of thrombin and TM-456. The slow “Na+-free” form of thrombin could potentially have a long range effect on interactions within ABE-I (37). The Debye-Hückel slopes were very similar between the NaCl (–2.4 ± 0.2) and ChCl (–1.9 ± 0.1) titrations, suggesting that the allosteric form of thrombin does not effect electrostatic steering. It is notable that both of the deduced Debye-Hückel slopes are similar to values reported for the release of fibrinopeptides A and B by thrombin, which have been shown to be dependent on electrostatic steering and independent of the fast or slow form of thrombin (50). Our results demonstrate that OPN hydrolysis by thrombin is highly dependent on electrostatic steering and that the interaction between ABE-I and the hirudin-like motif within the OPN-(162–197) sequence plays an important role.
Although electrostatic steering appears to play an important part in enhancing the formation of productive encounter complexes, the kcat/Km for OPN is considerably lower than that of the other substrates of thrombin. For example, hydrolysis of fibrinogen (1.2 × 107 m–1 s–1 for fibrinopeptide A release and 4.2 × 106 m–1 s–1 for fibrinopeptide B release) (51), the PAR1 receptor peptide (8.0 × 107 m–1.s–1) (52), FV (3.3 × 106 m–1 s–1) (53), and cleavage of FVIII at Arg372 (6.1 × 106 m –1 s–1), Arg740 (1.1 × 108 m –1 s–1), and Arg1689 (7.3 × 106 m–1 s–1) (54) have specificity constants 30–1000-fold higher. The specificity of thrombin for OPN could be restricted by suboptimal binding of the P3 to P3′ residues within the active site. The OPN P2′ (Lys170) and P3′ (Ser171) residues are non-optimal in their interaction with thrombin, which prefers bulky hydrophobic residues at P2′ and positively charged residues at P3′. OPN also has a non-optimal leucine at P2 and glycine at P3, with thrombin preferring a proline at P2 and residues with an aryl side chain at P3 (55). On the other hand, a moderately efficient catalysis would be sufficient in an extravascular compartment, where the high-affinity thrombin inhibitor, antithrombin, is normally not present, and in fact it may have a biological advantage in not generating excessive amounts of a potent cytokine that can exert its influence at very low levels. In contrast, it is imperative for thrombin to function at near maximal catalytic efficiency toward coagulation factors within the vascular compartment, because it needs to form a clot rapidly and to do so despite the presence of a high concentration of antithrombin. Therefore we propose that thrombin utilizes its ABEs to define its specific docking with different substrates but can modulate its ability to turn over substrates by differential binding modes within the active site. In this way, thrombin can link both coagulation and inflammation and generate levels of biologically active mediators appropriate to each process.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 HL057530. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: OPN, osteopontin; WT, wild type; LMW, low molecular weight; PPACK, d-Phe-Pro-Arg-chloromethylketone; DTT, dithiothreitol; ABE, anion-binding exosite; HPLC, high pressure liquid chromatography; GFC, gel filtration chromatography; CTF, C-terminal fragment.
References
- 1.Denhardt, D. T., and Noda, M. (1998) J. Cell. Biochem. Suppl. 30/31 92–102 [PubMed] [Google Scholar]
- 2.Mazzali, M., Kipari, T., Ophascharoensuk, V., Wesson, J. A., Johnson, R., and Hughes, J. (2002) Q. J. Med. 95 3–13 [DOI] [PubMed] [Google Scholar]
- 3.Wai P. Y., and Kuo, P. C. (2004) J. Surg. Res. 121 228–241 [DOI] [PubMed] [Google Scholar]
- 4.Ishijima, M., Rittling, S. R., Yamashita, T., Tsuji, K., Kurosawa, H., Nifuji, A., Denhardt, D. T., and Noda, M. (2001) J. Exp. Med. 193 399–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liaw, L., Birk, D. E., Ballas, C. B., Whitsitt, J. S., Davidson, J. M., and Hogan, B. L. (1998) J. Clin. Investig. 101 1468–2478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ashkar, S., Weber, G. F., Panoutsakopoulou, V., Sanchirico, M. E., Jansson, M., Zawaideh, S., Rittling, S. R., Denhardt, D. T., Glimcher, M. J., and Cantor, H. (2000) Science 287 860–864 [DOI] [PubMed] [Google Scholar]
- 7.Chabas, D., Baranzini, S. E., Mitchell, D., Bernard, C. C. A., Rittling, S. R., Denhardt, D. T., Sobel, R. A., Lock, C., Karpuj, M., Pedotti, R., Heller, R., Oksenberg, J. R., and Steinman, L. (2001) Science 294 1731–1735 [DOI] [PubMed] [Google Scholar]
- 8.Koh, A., da Silva, A. P., Bansal, A. K., Bansal, M., Sun, C., Lee, H., Glogauer, M., Sodek, J., and Zohar, R. (2007) Immunology 122 446–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liaw, L., Skinner, M. P., Raines, E. W., Ross, R., Cherish, D. A., Schwartz, S. M., and Giachelli, C. (1995) J. Clin. Investig. 95 713–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barry, S. T., Ludbrook, S. B., Murrison, E., and Horgan, C. M. T. (2000) Biochem. Biophys. Res. Commun. 267 764–769 [DOI] [PubMed] [Google Scholar]
- 11.Denda, S., Reichardt, L. F., and Müller, U. (1998) Mol. Biol. Cell 9 1425–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weber, G. F., Ashkar, S., Glimcher, M. J., and Cantor, H. (1996) Science 71 509–512 [DOI] [PubMed] [Google Scholar]
- 13.Senger, D. R., Perruzzi, C. A., Papadopoulos-Sergiou, A., and Van De Water, L. (1994) Mol. Biol. Cell 5 565–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Senger, D. R., and Perruzzi, C. A. (1996) Biochim. Biophys. Acta 1314 13–24 [DOI] [PubMed] [Google Scholar]
- 15.Smith, L. L., and Giachelli, C. M. (1998) Exp. Cell Res. 242 351–360 [DOI] [PubMed] [Google Scholar]
- 16.Barry, S. T., Ludbrook, S. B., Murison, E., and Horgan, C. M. T. (2000) Exp. Cell Res. 258 342–351 [DOI] [PubMed] [Google Scholar]
- 17.Bayless, K. J., and Davis, G. E. (2001) J. Biol. Chem. 276 13483–13489 [DOI] [PubMed] [Google Scholar]
- 18.Green, P. M., Ludbrook, S. B., Millar, D. D., Horgan, C. M., and Barry, S. T. (2001) FEBS Lett. 503 75–79 [DOI] [PubMed] [Google Scholar]
- 19.Yokosaki, Y., Matsuura, N., Sasaki, T., Murakami, I., Schneider, H., Higashiyama, S., Saitoh, Y., Yamakido, M., Taooka, Y., and Sheppard, D. (1999) J. Biol. Chem. 274 36328–36334 [DOI] [PubMed] [Google Scholar]
- 20.Tsiang, M., Paborsky, L. R., Li, W. X., Jain, A. K., Mao, C. T., Dunn, K. E., Lee, O. W., Matsumura, S. Y., Matteucci, M. D., Coutré, S. E., Leung, L. L., and Gibbs, C. S. (1996) Biochemistry 35 16449–16457 [DOI] [PubMed] [Google Scholar]
- 21.Bayless, K. J., Davis, G. E., and Meininger, G. A. (1997) Protein Expression Purif. 9 309–314 [DOI] [PubMed] [Google Scholar]
- 22.Philippou, H., Rance, J., Myles, T., Hall, S. W., Ariens, R. A., Grant, P. J., Leung, L. L., and Lane, D. A. (2003) J. Biol. Chem. 278 32020–32026 [DOI] [PubMed] [Google Scholar]
- 23.Kazanecki, C. C., Uzwiak, D. J., and Denhardt, D. T. (2007) J. Cell. Biochem. 102 912–924 [DOI] [PubMed] [Google Scholar]
- 24.Myles, T., Yun, T. H., Hall, S. W., and Leung, L. L. (2001) J. Biol. Chem. 276 25143–25149 [DOI] [PubMed] [Google Scholar]
- 25.Myles, T., Yun, T. H., and Leung, L. L. (2002) Blood 100 2820–2826 [DOI] [PubMed] [Google Scholar]
- 26.Yun, T. H., Baglia, F. A., Myles, T., Navaneetham, D., Lopez, J. A., Walsh, P. N., and Leung, L. L. (2003) J. Biol. Chem. 278 48112–48119 [DOI] [PubMed] [Google Scholar]
- 27.Tsiang, M., Jain, A. K., Dunn, K. E., Rojas, M. E., Leung, L. L. K., and Gibbs, C. S. (1995) J. Biol. Chem. 270 16854–16863 [DOI] [PubMed] [Google Scholar]
- 28.Hall, S. W., Nagashima, M., Zhao, L., Morser, J., and Leung, L. L. (1999) J. Biol. Chem. 274 25510–25516 [DOI] [PubMed] [Google Scholar]
- 29.Tsiang, M., Jain, A. K., and Gibbs, C. S. (1997) J. Biol. Chem. 272 12024–12029 [DOI] [PubMed] [Google Scholar]
- 30.Fortenberry, Y. M., Whinna, H. C., Gentry, H. R., Myles, T., Leung, L. L., and Church, F. C. (2004) J. Biol. Chem. 279 43237–43244 [DOI] [PubMed] [Google Scholar]
- 31.Fortenberry, Y. M., Whinna, H. C., Cooper, S. T., Myles, T., Leung, L. L., and Church, F. C. (2007) J. Thromb. Haemost. 5 1486–1492 [DOI] [PubMed] [Google Scholar]
- 32.Carter, W. J., Myles, T., Gibbs, C. S., Leung, L. L., and Huntington, J. A. (2004) J. Biol. Chem. 279 26387–26394 [DOI] [PubMed] [Google Scholar]
- 33.Rydel, T. J., Tulinsky, A., Bode, W., and Huber, R. (1991) J. Mol. Biol. 221 583–601 [DOI] [PubMed] [Google Scholar]
- 34.Wade, R. C., Gabdoulline, R. R., Lüdemann, S. K., and Lounnas, V. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 5942–5949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karshikov, A., Bode, W., Tulinsky, A., and Stone, S. R. (1992) Protein Sci. 1 727–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wells, C. M., and Di Cera, E. (1992) Biochemistry 31 11721–11730 [DOI] [PubMed] [Google Scholar]
- 37.Ghandi, P. S., Chen, Z., Matthews, F. S., and Di Cera, E. (2008) Proc. Natl. Acad. Sci. U. S. A. 105 1832–1837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stubbs, M. T., and Bode, W. (1993) Thromb. Res. 69 1–58 [DOI] [PubMed] [Google Scholar]
- 39.Bode, W., Turk, D., and Karshikov, A. (1992) Protein Sci. 1 426–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lane, D. A., Philippou, H., and Huntington, J. A. (2005) Blood 106 2605–2612 [DOI] [PubMed] [Google Scholar]
- 41.Adams, T. E., and Huntington, J. A. (2006) Arterioscler. Thromb. Vasc. Biol. 26 1738–1745 [DOI] [PubMed] [Google Scholar]
- 42.Dennis, S., Wallace, A., Hofsteenge, J., and Stone, S. R. (1990) Eur. J. Biochem. 188 61–66 [DOI] [PubMed] [Google Scholar]
- 43.Christensen, B., Nielsen, M. S., Haselmann, K. F., Petersen, T. E., and Sorensen, E. S. (2005) Biochem. J. 390 285–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Myles, T., Le Bonniec, B. F., Betz, A., and Stone, S. R. (2001) Biochemistry 40 4972–4979 [DOI] [PubMed] [Google Scholar]
- 45.Braun, P. J., Dennis, S., Hofsteenge, J., and Stone, S. R. (1988) Biochemistry 27 6517–6522 [DOI] [PubMed] [Google Scholar]
- 46.Katayama, Y., House, C. M., Udagawa, N., Kazama, J. J., McFarland, R. J., Martin, T. J., and Findlay, D. M. (1998) J. Cell. Physiol. 176 179–187 [DOI] [PubMed] [Google Scholar]
- 47.Jono, S., Peinado, C., and Giachelli, C. M. (2000) J. Biol. Chem. 275 20197–20203 [DOI] [PubMed] [Google Scholar]
- 48.Weber, G. F., Zawaideh, S., Hikita, S., Kumar, V. A., Cantor, H., and Ashkar, S. (2002) J. Leukocyte Biol. 72 752–761 [PubMed] [Google Scholar]
- 49.Baerga-Ortiz, A., Rezie, A. R., and Komives, E. A. (2000) J. Mol. Biol. 296 651–658 [DOI] [PubMed] [Google Scholar]
- 50.Vindigni, A., and Di Cera, E. (1996) Biochemistry 35 4417–4426 [DOI] [PubMed] [Google Scholar]
- 51.Higgins, D. L., Lewis, S. D., and Shafer, J. A. (1983) J. Biol. Chem. 258 9276–9282 [PubMed] [Google Scholar]
- 52.Parry, M. A., Myles, T., Tschopp, J., and Stone, S. R. (1996) Biochem. J. 320 335–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Monkovic, D. D., and Tracy, P. B. (1990) Biochemistry 29 1118–1128 [DOI] [PubMed] [Google Scholar]
- 54.Hill-Eubanks, D. C., and Lollar, P. (1990) J. Biol. Chem. 265 17854–17858 [PubMed] [Google Scholar]
- 55.Le Bonniec, B. F., Myles, T., Johnson, T., Knight, C. G., Tapparelli, C., and Stone, S. R. (1996) Biochemistry 35 7114–7122 [DOI] [PubMed] [Google Scholar]