

Abstract
We previously generated genome-wide expression data (microarray) from children with septic shock having the potential to lead the field into novel areas of investigation. Herein we seek to validate our data through a bioinformatic approach centered on a validation patient cohort. Forty-two children with a clinical diagnosis of septic shock and 15 normal controls served as the training data set, while 30 separate children with septic shock and 14 separate normal controls served as the test data set. Class prediction modeling using the training data set and the previously reported genome-wide expression signature of pediatric septic shock correctly identified 95 to 100% of controls and septic shock patients in the test data set, depending on the class prediction algorithm and the gene selection method. Subjecting the test data set to an identical filtering strategy as that used for the training data set, demonstrated 75% concordance between the two gene lists. Subjecting the test data set to a purely statistical filtering strategy, with highly stringent correction for multiple comparisons, demonstrated less than 50% concordance with the previous gene filtering strategy. However, functional analysis of this statistics-based gene list demonstrated similar functional annotations and signaling pathways as that seen in the training data set. In particular, we validated that pediatric septic shock is characterized by large scale repression of genes related to zinc homeostasis and lymphocyte function. These data demonstrate that the previously reported genome-wide expression signature of pediatric septic shock is applicable to a validation cohort of patients.
INTRODUCTION
The application of high throughput technologies, such as microarray, affords unprecedented opportunities to generate novel hypothesis and to gain a more comprehensive understanding of human diseases. We have suggested that complex and heterogeneous phenotypes, such as pediatric septic shock, are particularly amenable to this discovery-oriented approach (15, 18). Septic shock continues to be an important child health problem with significant morbidity and mortality (14, 16). In an ongoing translational research program focused on pediatric septic shock, we have begun to identify the genome-level expression signature of this syndrome through the application of microarray technology (13, 18, 19). The initial data generated through this discovery-oriented approach suggest that altered zinc homeostasis and lymphocyte dysfunction are prevalent problems in children with septic shock, and thus potentially direct the field toward new investigative paradigms given the direct biological links between zinc homeostasis and lymphocyte function (12).
Our current enthusiasm surrounding the genome-wide expression data in pediatric septic shock is tempered by the intricacies and nuances surrounding microarray data analysis, a methodology still in evolution (1, 5, 10, 17). There are currently no definitive, absolute standard statistical approaches for analyzing microarray data. The shear volume of typical microarray data, leading to simultaneous testing of tens of thousands of transcripts, has the potential to produce hundreds of false positive results depending on the statistical approach. Accordingly, the particular statistical and filtering approaches that are applied to a given microarray data set can yield markedly different results (10, 17). Thus, the possibility exists that our previous observations involving gene expression profiling in pediatric septic shock are merely epiphenomena of the bioinformatic approaches used in those initial studies.
Prospective validation of microarray data represents one potential approach for addressing these complex analytical issues. The ultimate goal of validation is to strengthen the initial conclusions derived from microarray data sets, as we have done with selected portions of our previous data (13, 18, 19). Validation of microarray data can be achieved by a variety of strategies (1, 10, 17). For example, real time-polymerase chain reaction can be used for confirmation of differentially expressed gene probes. Proteomics can also be used for data validation and has the advantage of providing gene product information beyond the level of transcripts. An alternative validation strategy involves the use of a “validation” or “test” data set representing an entirely different set of samples from that used for derivation of the original microarray data set.
This paper addresses two equally important and related questions: 1) are the original observations that we reported in children with septic shock also operative in a validation cohort?; and 2) are these observations dependent on the bioinformatic approaches used to analyze the expression data? Herein we have directly addressed these questions by employing a training data set (our original microarray data) and a test data set consisting of an entirely new cohort of patients. We have prospectively applied validation procedures, including the use of statistical approaches that are distinct to that of our previous reports, as a means of assessing the authenticity of our initial observations.
METHODS
Patients
The multi-institutional genomic and clinical database supporting this translational research program has been previously described in detail (13, 19). Briefly, the study protocol was approved by the individual Institutional Review Boards of each participating institution. Children < 10 years of age admitted to the pediatric intensive care unit (PICU) and meeting published, pediatric-specific criteria for septic shock were eligible for the study (7). Normal control patients were recruited from the participating institutions using the following exclusion criteria: a recent febrile illness (within 2 weeks), recent use of anti-inflammatory medications (within 2 weeks), or any history of chronic or acute disease associated with inflammation.
Sample and data collection
After informed consent, blood samples for RNA isolation were obtained within 24 hours of admission to the PICU, heretofore referred to as “day 1” of septic shock. Severity of illness at study entry was calculated using the PRISM III score in the patients with septic shock (11). Clinical and laboratory data were collected daily while in the PICU. Clinical, laboratory, and biological data were entered and stored using a locally developed, web-based database.
RNA extraction and microarray hybridization
The data and protocols described in this manuscript are deposited in the NCBI Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/). The training data set and the test data set can be found under GEO Series accession numbers GSE4607 and GSE9692, respectively.
Total RNA was isolated from whole blood samples using the PaxGene™ Blood RNA System (PreAnalytiX, Qiagen/Becton Dickson, Valencia, CA) according the manufacturer’s specifications. Microarray hybridization was performed by the Affymetrix Gene Chip Core facility at Cincinnati Children’s Hospital Research Foundation as previously described (Human Genome U133 Plus 2.0 GeneChip, Affymetrix, Santa Clara, CA) (13, 19).
Data analysis
Analyses were performed using one patient sample per chip. Image files were captured using an Affymetrix GeneChip Scanner 3000. CEL files were subsequently preprocessed using Robust Multiple-Array Average (RMA) normalization using GeneSpring GX 7.3 software (Agilent Technologies, Palo Alto, CA) (9). All signal intensity-based data were used after RMA normalization, which specifically suppresses all but significant variation among lower intensity probe sets. All chips were then normalized to the respective median values of controls.
Differences in mRNA abundance between patient samples and controls were determined using GeneSpring GX 7.3. All statistical analyses used corrections for multiple comparisons. Cross validation procedures and class prediction modeling were also performed using GeneSpring GX 7.3 using the default parameters of the software. Further details regarding derivation of differentially regulated gene lists, cross validation, class prediction modeling and statistical analyses will be provided in the Results section.
Gene lists of differentially expressed genes were analyzed using D.A.V.I.D. (Database for Annotation, Visualization and Integrated Discovery) and the Ingenuity Pathways Analysis application (IPA, Ingenuity Systems, Redwood City, CA). D.A.V.I.D. allows public access to relational databases of functional gene annotations (6). In the D.A.V.I.D. analytical output, “category” refers to the original database or resource from which the annotations are derived, and “term” refers to the enriched annotation terms associated with the given gene list. The IPA application provides a tool for discovery of signaling pathways within the uploaded gene lists (4, 13, 19). The D.A.V.I.D. and IPA applications are both based on the established biomedical literature and use specific approaches to estimate significance (p values) based on non-redundant representations of the microarray chip and to convert the uploaded gene lists to gene lists containing a single value per gene. The p values for a given functional annotation or signaling pathway provide an estimate of the probability that a given annotation is enriched in a given gene list by chance alone. Because of there is no clear consensus as to the most appropriate bioinformatic approach to use for these types of data sets, we consistently used the default parameters in GeneSpring, D.A.V.I.D., and the IPA application, unless otherwise specified.
RESULTS
General information for the training and test data sets
The training data set (42 children with septic shock and 15 controls) has been previously reported (19). The test data set consisted of 30 separate children with septic shock and 14 separate control subjects that have not been previously reported. The patients for the test data set were selected based on sequential order of enrollment into the database, with an enrollment classification of septic shock or control, but without regard to demographic, clinical, or microbiologic variables.
Summary demographic, clinical, and microbiologic data for the subjects in the test data set are provided in Tables 1 and 2. A total of 44 individual microarray chips, representing 14 individual controls and 30 individual patients with septic shock (test data set), were used for analysis. The microarray data represent the gene expression profiles of “day 1” of septic shock (i.e. within 24 hour of presentation to the PICU). The patients with septic shock in the test data set were similar to the controls, and the training data set (19) with regard to age, race, and gender. In addition, the patients with septic shock in the test data set had a similar severity of illness (PRISM score) as that of the training data set (19). Among the patients in the test data set there were 14 having positive identification of an infecting organism (47%) and 6 deaths (20% mortality). In comparison, the training data set had 67% of the patients with a positively identified infecting organism and 21% mortality (19). Finally, Table 2 demonstrates that the test data set had less heterogeneity of infecting organisms and higher representation of infection with gram-positive bacteria compared to that of the training data set (19).
Table 1.
Clinical and demographic data for all subjects in test data set.
| Controls | Septic Shock | |
|---|---|---|
| No. of individual subjects | 14 | 30 |
| Mean age (years) ± S.D.1 | 2.7 ± 2.4 | 3.2 ± 2.9 |
| Mean PRISM Score ± S.D. 1 | n/a | 18.9 ± 12.3 |
| Gender (Male/Female) 1 | 8/6 | 16/14 |
| Race (no.) 1 | A.A./Black (3) | A.A./Black (2) |
| White (10) | White (26) | |
| Asian (1) | Unreported (2) |
p > 0.05
Table 2.
Microbiology data for test data set patients with septic shock.
| Organism (no.) | Primary source of positive culture (no.) |
|---|---|
| Staphylococcus aureus (5) | Blood (10) |
| Streptococcus pyogenes (2) | Lung (1) |
| Streptococcus agalactiae (2) | Cerebral spinal fluid (1) |
| Neisseria meningitidis (2) | Other (2) |
| Streptococcus pneumoniae (2) | |
| Adenovirus (1) |
Table 3 provides the total white blood cell counts and the absolute leukocyte subpopulation counts for the training and test data sets. The median total white blood cell counts, the median absolute neutrophils counts, and the median absolute monocyte counts were similar between the patients in the training and test data sets. In contrast, patients in the test data set had a significantly lower median absolute lymphocyte count compared to that of the patients in the training data set.
Table 3.
Median absolute white blood cell (WBC) counts for patients with septic shock in the training and test data sets (× 103 per mm3; IQR).
| Training Data Set | Test Data Set | |
|---|---|---|
| Total WBC | 11.6 (4.1 – 22.6) | 6.4 (3.5 – 16.9) |
| Neutrophils (Mature + Immature) | 7.1 (2.6 – 16.5) | 4.5 (1.7 – 11.7) |
| Lymphocytes 1 | 1.8 (1.3 – 3.1) | 0.9 (0.6 – 1.3) |
| Monocytes | 0.4 (0.1 – 1.4) | 0.2 (0.1 – 0.7) |
p < 0.05 by Mann-Whitney Rank Sum Test.
Class prediction modeling based on training and test data sets
The initial approach to validating our microarray data involved class prediction modeling using the previously reported cohort of patients as the training data set, the new cohort of patients described above as the test data set, and the previously published genomic signature of pediatric septic shock (i.e. the list of 2,482 gene probes found to be differentially regulated between patients with septic shock in the training data set and controls (19)). As previously described, the 2,482 gene list was generated by sequential statistical and expression filters as shown in Table 4. For the sake of clarity we will heretofore refer to this 2,482 gene list as “gene list A.”
Table 4.
Gene list designation and derivation.
| Gene list | # of genes | Data set | Statistical filter | Expression filter |
|---|---|---|---|---|
| A | 2,482 | Training | ANOVA, t-test Benjamini-Hochberg False Discovery Rate (5%) | ≥2 fold expression difference between the median of controls and a least 50% of the patients with septic shock |
| B | 3,296 | Test | Same as gene list A | Same as gene list A |
| C | 2,104 | Test | ANOVA, t-test Bonferroni correction for multiple comparisons (p = 0.05) | None |
We first conducted cross validation procedures on the training data set. Two distinct class prediction algorithms were used for this analysis: K-Nearest Neighbors (KNN) and Support Vector Machines (SVM) (3, 20). The class options for cross validation were either “septic shock” or “control.” Table 5 demonstrates that cross validation of the training data set, based on the KNN algorithm, correctly identified 93 to 95% of the subjects in the training data set, depending on the gene selection method. Table 5 also demonstrates that cross validation of the training data set, based on the SVM algorithm, correctly identified 95 to 96% of the subjects in the training data set, depending on the gene selection method.
Table 5.
Results of cross validation of training data set based gene list A, and on K-Nearest Neighbors (KNN) or Support Vector Machines (SVM) algorithms1.
| Gene selection method (algorithm) | # correct predictions | # incorrect predictions | # not predicted |
|---|---|---|---|
| Fisher’s exact test2 | |||
| KNN | 53 | 1 (SS predicted as C) | 3 (2 SS; 1 C) |
| SVM | 54 | 3 (3 C predicted as SS) 100% Sensitivity 80% Specificity | n/a |
| Golub method3 | |||
| KNN | 54 | 1 (1 C predicted as SS) | 2 (2 SS) |
| SVM | 54 | 3 (3 C predicted as SS) 100% Sensitivity 80% Specificity | n/a |
| All genes in list4 | |||
| KNN | 53 | 1 (1 SS predicted as C) | 3 (3 SS) |
| SVM | 55 | 2 (2 C predicted as SS) 100% Sensitivity 87% Specificity | n/a |
The class options for the model were “septic shock” (SS) or “control” (C).
GeneSpring parameters (default): 50 predictor genes, 10 neighbors, 0.2 decision cutoff for p-value ratio.
GeneSpring parameters (default): 50 predictor genes, 10 neighbors, 0.2 decision cutoff for p-value ratio.
GeneSpring parameters (default): 10 neighbors, 0.2 decision cutoff for p-value ratio.
We next applied class prediction modeling to the test data set, again using both the KNN and SVM class prediction algorithms and all possible gene selection methods, as shown in Table 6. All possible combinations of gene selection methods and class prediction algorithms correctly predicted ≥95% of the subjects (controls and septic shock) in the test data set. Using the 50 predictor genes derived from the Golub method of gene selection (derived from the cross validation procedures described above), the KNN algorithm correctly identified 100% of the subjects (controls and septic shock) in the test data set. In a similar manner, using all of the genes in gene list A, the SVM algorithm correctly identified 100% of the subjects in the test data set.
Table 6.
Class prediction modeling of test data set based on cross validation results (Table 5), and K-Nearest Neighbors (KNN) and Support Vector Machines (SVM) algorithms1.
| Gene selection method (algorithm) | # correct predictions | # incorrect predictions | # not predicted |
|---|---|---|---|
| Fisher’s exact test2 | |||
| KNN | 43 | 1 (SS predicted as C) | 0 |
| SVM | 42 | 2 (2 C predicted as SS) 100% Sensitivity 86% Specificity | n/a |
| Golub method3 | |||
| KNN | 44 | 0 | 0 |
| SVM | 42 | 2 (2 C predicted as SS) 100% Sensitivity 86% Specificity | n/a |
| All genes in list4 | |||
| KNN | 42 | 1 (SS predicted as C) | 1 (SS) |
| SVM | 44 | 0 100% Sensitivity 100% Specificity | n/a |
The class options for the model were “septic shock” (SS) or “control” (C).
GeneSpring parameters (default): 50 predictor genes, 10 neighbors, 0.2 decision cutoff for p-value ratio.
GeneSpring parameters (default): 50 predictor genes, 10 neighbors, 0.2 decision cutoff for p-value ratio.
GeneSpring parameters (default): 10 neighbors, 0.2 decision cutoff for p-value ratio.
We further tested the relevance of gene list A, to the test data set, by calculating how many of these genes were differentially regulated among the patients with septic shock and the controls in the test data set. We conducted a two group ANOVA (Benjamini-Hochberg false discovery rate of 5%) using controls and patients with septic shock in the test data set as the comparison groups, and gene list A. This analysis demonstrated that 2,386 of the 2,482 genes (96%) were differentially regulated between the controls and the patients with septic shock in the test data set.
Collectively, these data demonstrate that the genome-wide expression signature originally reported for children with septic shock (i.e. gene list A) can accurately identify a separate test cohort of children with septic shock and controls with a high degree of sensitivity and specificity. The strength of this observation is further supported by the application of multiple combinations of class prediction algorithms and gene selection methods.
Functional analysis of predictor genes
As shown in Tables 5 and 6, the Golub method of gene selection appeared to be the most robust method for class prediction in that it was able to predict 100% of the septic shock patients and the controls in the test data set, based on the KNN class prediction algorithm. Accordingly, we uploaded the 50 predictor genes derived from the Golub-based class prediction (see Supplementary Data for complete 50 predictor gene list) to the IPA application to determine if the gene list corresponded to any specific signaling pathways. Table 7 demonstrates the top 5 signaling pathways represented within the list of 50 predictor genes. All of these signaling pathways are consistent with our current paradigms surrounding the pathobiology of septic shock, thus supporting the biological plausibility of this 50 predictor gene list.
Table 7.
Signaling pathways among 50 predictor genes derived from class prediction modeling and the Golub method of gene selection (see text for details). The analysis is derived from the Ingenuity Pathways Analysis default parameters and the signaling pathways represent the top 5 most significant p values (listed in descending order).
| Signaling Pathway | # of genes | p-value |
|---|---|---|
| NF-κB signaling | 3 | 6.2E-3 |
| Leukocyte extravasation signaling | 3 | 1.3E-2 |
| Liver X receptor/Retinoid X receptor signaling | 2 | 1.3E-2 |
| T cell receptor signaling | 2 | 2.6E-2 |
| p38 MAP kinase signaling | 2 | 2.6E-2 |
Comparison of training and data set gene lists
The next validation step involved the generation of a list of differentially regulated genes between the patients with septic shock and controls in the test data set. In order to make a direct comparison to our previously published data, we applied an identical filtering approach as that used to derive the original gene list A (19). We conducted a two group ANOVA (Benjamini-Hochberg false discovery rate of 5%) using controls and patients with septic shock in the test data set as the comparison groups, and all gene probes within the microarray (54,681 gene probes). This statistical filter yielded a working list of 21,517 gene probes that were differentially regulated between controls and patients with septic shock. To further refine this 21,517 gene list, we next applied an expression filter that selected only the genes, within the above 21,517 gene list, having at least 2-fold expression difference in at least 50% of the patients with septic shock, compared to the median of the controls. This expression filter yielded a final working list of 3,296 gene probes that were differentially regulated between patients with septic shock in the test data set and controls (Table 4, gene list B).
Figure 1 depicts a Venn analysis comparing gene lists A and B. Seventy-five percent of the genes in the training data set (gene list A) were present in the test data set (gene list B). These data demonstrate that the application of an identical filtering strategy, to two separate cohorts of controls and children with septic shock, identifies a relatively similar number (> 50%) of differentially regulated gene transcripts. These data further support the existence of an identifiable, characteristic genome-wide expression signature in children with septic shock.
Figure 1.
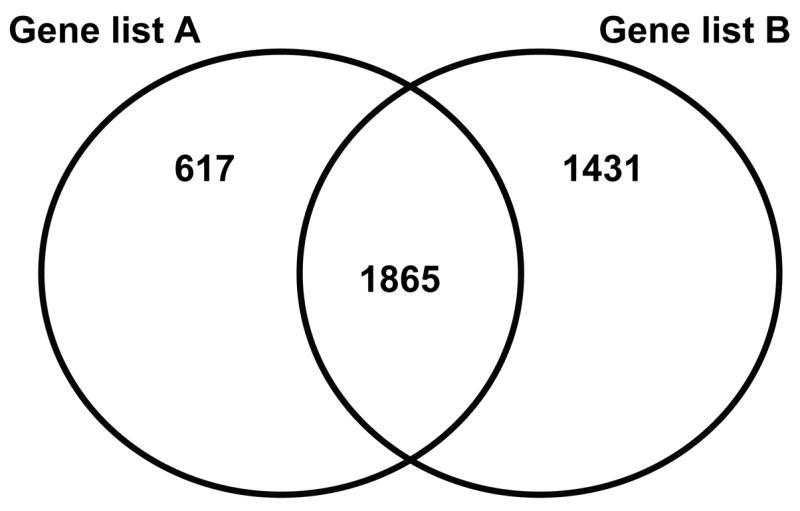
Venn analysis comparing gene lists A and B. See Table 4 and text for gene list derivation.
Alternative gene list derivation strategy in the test data set
As previously stated, the particular statistical and filtering approaches that are applied to a given microarray data set can yield markedly different results (10, 17). Our original report of differentially regulated genes between controls and patients with septic shock was based on sequential statistical and expression filters that used the default parameters in the analysis software (Table 4, gene list A) (19). Furthermore, the statistical filter used in the initial report conducted multiple testing corrections by way of the Benjamini and Hochberg False Discovery Rate.
In this analysis, we generated a list of differentially regulated genes between controls and the patients with septic shock in the test data set by applying only a statistical filter (ANOVA) and conducted multiple testing corrections by way of Bonferroni. In comparison to the Benjamini and Hochberg False Discovery Rate, Bonferroni-based multiple testing correction has a much higher degree of stringency in the context of microarray data, and is generally regarded as being overly stringent for analyzing microarray data (10, 17). Nevertheless, we applied this stringent statistical test to the test data set as another form of validation.
Using this purely statistical approach (i.e. without an additional expression filter), we generated a list of 2,104 gene probes that were differentially regulated between controls and patients with septic shock in the test data set (Table 4, gene list C). Figure 2 depicts a Venn analysis of gene lists B and C. Forty-one percent of the genes in gene list B were present in gene list C. These data illustrate how the particular filtering approach that is applied to a given set of microarray data can profoundly affect the derivation of putative differentially regulated genes.
Figure 2.
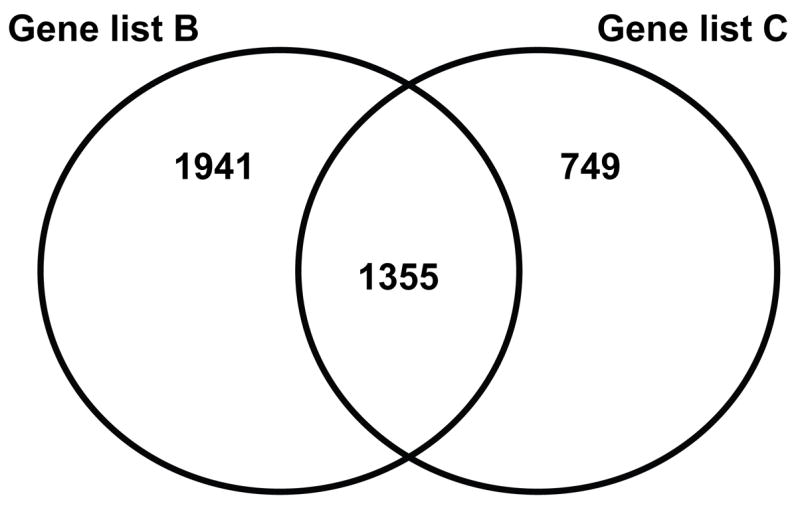
Venn analysis comparing gene lists B and C. See Table 4 and text for gene list derivation.
Functional analysis of the test data set
Having demonstrated the impact of different filtering approaches on gene list derivation in our test data set, we next conducted functional analyses of the test data set to determine if the functional analyses would differ significantly from our original observations (13, 19). All analyses in this section are based on gene list C and were conducted in an analogous manner to that of our previous reports.
To derive biological meaning from gene list C, we uploaded the individual lists of upregulated and downregulated genes, respectively, to both the D.A.V.I.D. database and the IPA application (4, 6, 13, 19). As shown in Tables 8 and 9, the D.A.V.I.D.-dependent analyses yielded several biological relevant functional annotations within both gene lists. Table 8, representing the 846 upregulated genes, is notable for multiple functional annotations related to host defense responses similar to that of our previous data (13, 19). Table 9, representing the 1,258 downregulated genes, is notable for the prevalence of zinc- and metal binding-related ontologies (see Supplementary Data for gene lists corresponding to zinc-related functional annotations), a principal observation of our previous data (13, 19).
Table 8.
Top 20 functional annotations among 846 upregulated genes in gene list C (based on p value and listed in descending order). The analysis is based on the default parameters in D.A.V.I.D. “Category” refers to the original database or resource from which the annotations were derived, and “Term” refers to the enriched annotation terms associated with the gene list.
| Category | Term | # of genes | p-value |
|---|---|---|---|
| SP_PIR_KEYWORDS | membrane | 164 | 2.9E-16 |
| GOTERM_BP_ALL | response to other organism | 52 | 7.7E-12 |
| GOTERM_BP_ALL | response to external stimulus | 47 | 1.9E-11 |
| GOTERM_BP_ALL | response to pest/pathogen/parasite | 49 | 3.3E-11 |
| SP_PIR_KEYWORDS | glycoprotein | 136 | 7.9E-11 |
| GOTERM_BP_ALL | response to wounding | 38 | 5.3E-10 |
| SP_PIR_KEYWORDS | lipoprotein | 40 | 5.6E-10 |
| SP_PIR_KEYWORDS | Direct protein sequencing | 94 | 6.9E-10 |
| SP_PIR_KEYWORDS | phosphorylation | 87 | 2.4E-9 |
| GOTERM_BP_ALL | inflammatory response | 24 | 6.9E-8 |
| SP_PIR_KEYWORDS | signal | 101 | 7.4E-8 |
| GOTERM_BP_ALL | response to stress | 65 | 1.1E-7 |
| SP_PIR_KEYWORDS | transmembrane | 134 | 1.3E-7 |
| GOTERM_BP_ALL | response to biotic stimulus | 73 | 1.5E-7 |
| GOTERM_CC_ALL | membrane | 216 | 3.0E-7 |
| GOTERM_BP_ALL | intracellular signaling cascade | 63 | 9.2E-7 |
| SP_PIR_KEYWORDS | alternative splicing | 130 | 1.4E-6 |
| GOTERM_MF_ALL | catalytic activity | 208 | 1.6E-6 |
| GOTERM_BP_ALL | immune response | 63 | 1.7E-6 |
| SP_PIR_KEYWORDS | transferase | 60 | 1.9E-6 |
Table 9.
Top 20 functional annotations among 1,258 downregulated genes in gene list C (based on p value and listed in descending order). The analysis is based on the default parameters in D.A.V.I.D. “Category” refers to the original database or resource from which the annotations were derived, and “Term” refers to the enriched annotation terms associated with the gene list.
| Category | Term | # of genes | p-value |
|---|---|---|---|
| SP_PIR_KEYWORDS | zinc-finger | 137 | 2.7E-29 |
| SP_PIR_KEYWORDS | nuclear protein | 209 | 2.6E-26 |
| SP_PIR_KEYWORDS | zinc | 144 | 4.0E-24 |
| SP_PIR_KEYWORDS | transcription | 124 | 1.1E-22 |
| SP_PIR_KEYWORDS | transcription regulation | 122 | 5.0E-21 |
| SP_PIR_KEYWORDS | DNA binding | 119 | 2.7E-19 |
| GOTERM_MF_ALL | zind ion binding | 161 | 4.7E-19 |
| SP_PIR_KEYWORDS | metal-binding | 149 | 3.2E-17 |
| GOTERM_MF_ALL | transition metal binding | 174 | 7.9E-16 |
| GOTERM_BP_ALL | transcription | 171 | 9.4E-16 |
| GOTERM_BP_ALL | regulation of transcription | 163 | 7.1E-15 |
| GOTERM_MF_AL | nucleic acid binding | 218 | 1.3E-14 |
| GOTERM_BP_ALL | regulation of biological process | 225 | 3.8E-13 |
| GOTERM_BP_ALL | regulation of cellular process | 214 | 4.4E-13 |
| GOTERM_BP_ALL | regulation of cellular metabolism | 165 | 6.8E-13 |
| GOTERM_BP_ALL | regulation of metabolism | 168 | 9.1E-13 |
| GOTERM_CC_ALL | nucleus | 239 | 9.0E-12 |
| GOTERM_MF_AL | cation binding | 196 | 2.6E-9 |
| GOTERM_MF_ALL | ion binding | 201 | 5.1E-8 |
| GOTERM_MF_AL | metal ion binding | 201 | 5.1E-8 |
As shown in Tables 10 and 11, the IPA-dependent analyses yielded several biologically relevant signaling pathways within both gene lists. Table 10, representing the 846 upregulated genes, is dominated by inflammation-and immunity-related signaling pathways (see Supplementary Data for gene lists corresponding to individual signaling pathways). This observation is consistent with the established literature focused on septic shock, and our previous data (2, 8, 13, 14, 19). Table 11, representing the 1,258 downregulated genes, is most notable for repression of genes corresponding to natural killer cell signaling, T cell receptor signaling, and antigen presentation (see Supplementary Data for gene lists corresponding to individual signaling pathways), which is the other principal observation of our previous data (13, 19).
Table 10.
Signaling pathways among 846 upregulated genes in gene list C. The analysis is derived from the Ingenuity Pathways Analysis default parameters and the signaling pathways represent the top 10 most significant p values (listed in descending order).
| Signaling Pathway | # of genes | p-value |
|---|---|---|
| Toll-like receptor signaling | 10 | 2.6E-6 |
| Interleukin-10 signaling | 10 | 2.8E-5 |
| NF-κB signaling | 17 | 3.6E-4 |
| Acute phase response signaling | 14 | 1.6E-3 |
| p38 MAP kinase signaling | 9 | 5.2E-3 |
| Complement system | 5 | 5.4E-3 |
| Hepatic cholestasis | 11 | 5.5E-3 |
| LXR/RXRα activation | 7 | 6.8E-3 |
| Interleukin-6 signaling | 8 | 9.6E-3 |
| PPARα/RXRα activation | 12 | 1.2E-2 |
Table 11.
Signaling pathways among the 1,258 downregulated genes in gene list C. The analysis is derived from the Ingenuity Pathways Analysis default parameters and the signaling pathways represent the top 5 most significant p values (listed in descending order).
| Signaling Pathway | # of genes | p-value |
|---|---|---|
| Natural killer cell signaling | 18 | 1.0E-7 |
| T-cell receptor signaling | 15 | 3.0E-6 |
| Antigen presentation pathway | 7 | 4.6E-4 |
| Interleukin-4 signaling | 8 | 3.7E-3 |
Collectively, these functional analyses demonstrate that gene list C, derived in a completely different manner to that of our previous data, and representing a test cohort of patients, contains a large number of genes relevant to the pathobiology of septic shock and validate our previous observations.
DISCUSSION
Our initial reports involving the genome-level expression profiles of pediatric septic shock were primarily highlighted by three observations (13, 18, 19): 1) pediatric septic shock is characterized by large scale repression of genes that either depend on normal zinc homeostasis for normal function or directly participate in zinc homeostasis and this observation is associated with abnormally low serum zinc concentrations in nonsurvivors of septic shock; 2) pediatric septic shock is also characterized by large scale repression of genes corresponding to adaptive immunity, T cell function in particular; and 3) longitudinal studies demonstrated that both of these gene repression patterns persist over the first 3 days of illness. While interesting and holding the potential to change our current paradigms regarding the pathobiology of pediatric septic shock, these observations require validation at several levels. In an ongoing approach we are currently seeking functional validation of these observations by conducting laboratory-based experiments focused on altered zinc homeostasis in the context of experimental septic shock and are also conducting functional studies of lymphocyte function in children with septic shock. In the current work, have attempted to complement our efforts at validation through a bioinformatics approach.
The focal point of our bioinformatic approach to validation was a test data set. The test data set consisted of an entirely new set of children with a clinical diagnosis of septic shock and an entirely new set of controls. The test data set patients were similar to the patients in our original report with respect to age and illness severity. In contrast, the test data set appeared to be more homogenous than the original cohort of patients with respect to class of infecting organisms. In addition, the patients with septic shock in the test data set had lower median absolute lymphocyte counts than the patients in the training data set. These potentially confounding factors were the result of our strategy to select patients for the test data set based on chronology of enrollment into the data base. Despite these potentially confounding factors, the key aspects of our original data were validated in the test data set.
One of the major strategies used to validate our previous data involved cross validation procedures on the training data set, then subsequently conducting class prediction procedures on the test data set. The intent of this strategy is not to gain the ability to identify normal versus septic shock in the clinical setting, which can be readily achieved by basic clinical examination and laboratory parameters. Rather, these procedures were conducted as a primary test of the validity of our original data. Using multiple combinations of class prediction algorithms and gene selection methods, we were able to accurately identify ≥95% of the subjects in the test data set, depending on the class prediction algorithm and the gene selection method. In fact, using the KNN algorithm and the 50 predictor genes derived from the Golub method of gene selection, we were able to accurately identify all controls and patients with septic shock in the test data set. One hundred percent prediction accuracy was also achieved using the SVM algorithm and all genes from the training data set (gene list A). In keeping with these observations, the majority of genes in the previously reported gene list were differentially regulated in the test data set as determined by direct statistical analysis of these genes in the context of the test data set. Finally, the 50 predictor genes derived from class prediction modeling correspond to biologically relevant signaling pathways in the context of septic shock. In combination, these data indicate that the previously reported genome-wide expression signature of pediatric septic are applicable to a test data set of children with septic shock and therefore speak to the validity of the original data.
Our current data also well illustrate how different filtering approaches can impact the derivation of differentially regulated gene lists. Using two different filtering strategies (i.e. gene lists B and C) we generated two putative lists of genes in the test data set that were differentially regulated between controls and patients with septic shock in the test data set. Comparison of these two gene lists demonstrated < 50% concordance, thus raising questions of trustworthiness. Accordingly, we submitted the gene list having the least concordance with our previous data (i.e. gene list C) for derivation of functional annotations and signaling pathway associations. These analyses yielded the majority of the key functional annotations and signaling pathways that were previously reported (13, 18, 19). Thus, elucidation and discovery of the key genes coordinately regulated in the context of pediatric septic shock (i.e. the genes that contribute most to enrichment of functional annotations and signaling pathways) may not be highly dependent on the filtering approach. Consequently, these data strongly support the observations made in our previous reports.
The main limitation of our current work is the reliance on whole blood-derived RNA for gene expression profiling, as previously discussed at length (13, 19). Whole blood-derived RNA reflects a mixed population of white blood cells, which has the potential to profoundly confound the resulting microarray data. Our previous data, however, indicate that whole blood-derived RNA can yield consistent and biologically plausible microarray data in children with septic shock. For example, we have previously demonstrated that variation in the absolute number of lymphocytes does not account for the wide spread repression of genes related to adaptive immunity that we have reported in children with septic shock (13).
The potential limitations of using whole blood-derived RNA are now further mitigated by the current data focused on validation in a test data set. For example, the ability to identify controls and patients with septic shock in the current test data set, with 100% accuracy, well supports the concept that whole blood-derived RNA can yield biologically plausible and consistent data in pediatric septic shock. In addition, the ability to derive functional annotations and signaling pathways in the test data set, which are analogous to that of our previous reports, further supports the assertion that using whole blood-derived RNA is a valid approach. Importantly, these functional annotation and signaling pathway data were generated in the context of two potentially strong negative confounders: 1) the data were based on a potentially overly stringent statistical approach (i.e. Bonferroni correction for multiple comparisons), and 2) the patients with septic shock in the test data set had lower median lymphocyte counts than that of the training data set.
In conclusion, we have demonstrated that the previously reported genome-wide expression signature of pediatric septic shock is applicable to a test cohort of children with septic shock at multiple levels. We have also validated, through an entirely different filtering approach, that day 1 of pediatric septic shock is characterized by large scale repression of zinc- and lymphocyte-related genes. Given the importance of zinc homeostasis to normal lymphocyte function, these data have formed a hypothesis surrounding altered zinc homeostasis leading to altered lymphocyte function in children with septic shock as previously proposed (18). This hypothesis is readily testable at the translational and experimental levels.
Supplementary Material
Acknowledgments
Additional Genomics of Pediatric SIRS/Septic Shock Investigators: Allan Doctor (St. Louis Children’s Hospital, Washington University School of Medicine, St. Louis, MO); Douglas Willson (University of Virginia, Charlottesville, VA); Meena Kalyanarman (Newark Beth Israel Medical Center, Newark, NJ); Nancy M. Tofil ((The University of Alabama at Birmingham, Birmingham, AL); Scott Penfil (DuPont Hospital for Children, Wilmington, DE ); Julie Simon (Children’s Hospital and Research Center Oakland, Oakland, CA); Joseph Hess (Penn State Children’s Hospital, Hershey, PA); Margaret Winkler (The University of Alabama at Birmingham, Birmingham, AL); Gwenn McLaughlin, M.D. (Jackson Memorial Hospital, Miami, FL); Cheri Landers (Kentucky Children’s Hospital, Lexington, KY); Gary Kohn, M.D. (Morristown Memorial Hospital, Morristown, NJ);
Supported by a grant from the National Institute of General Medical Sciences (RO1 GM064619)
CONTRIBUTORS
H Wong and T Shanley conceived the original genomic database focused on pediatric septic shock. H Wong directed the overall study, including analysis of the data. H Wong had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. N Cvijanovich, T Shanley, R Lin, G Allen, N Thomas, P Checchia, N Anas, and R Freishtat serve as the local principal investigators at the sites contributing the largest number of patients to the database. M Monaco, and K Odoms manage the clinical database and all clinical samples generated through this program. B Sakthivel provides bioinformatic support to the program. H Wong, N Cvijanovich, and T Shanley drafted the manuscript. All authors reviewed and helped revise the manuscript, and approved the final version of the manuscript.
CONFLICT OF INTEREST STATEMENT
None of the authors have a conflict of interest to declare.
References
- 1.Allison DB, Cui X, Page GP, Sabripour M. Microarray data analysis: from disarray to consolidation and consensus. Nat Rev Genet. 2006;7:55–65. doi: 10.1038/nrg1749. [DOI] [PubMed] [Google Scholar]
- 2.Ayala A, Chaudry IH. Immune dysfunction in murine polymicrobial sepsis: mediators, macrophages, lymphocytes and apoptosis. Shock. 1996;6 (Suppl 1):S27–38. [PubMed] [Google Scholar]
- 3.Byvatov E, Schneider G. Support vector machine applications in bioinformatics. Appl Bioinformatics. 2003;2:67–77. [PubMed] [Google Scholar]
- 4.Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, Cho RJ, Chen RO, Brownstein BH, Cobb JP, Tschoeke SK, Miller-Graziano C, Moldawer LL, Mindrinos MN, Davis RW, Tompkins RG, Lowry SF. A network-based analysis of systemic inflammation in humans. Nature. 2005;437:1032–1037. doi: 10.1038/nature03985. [DOI] [PubMed] [Google Scholar]
- 5.Cobb JP, Mindrinos MN, Miller-Graziano C, Calvano SE, Baker HV, Xiao W, Laudanski K, Brownstein BH, Elson CM, Hayden DL, Herndon DN, Lowry SF, Maier RV, Schoenfeld DA, Moldawer LL, Davis RW, Tompkins RG, Bankey P, Billiar T, Camp D, Chaudry I, Freeman B, Gamelli R, Gibran N, Harbrecht B, Heagy W, Heimbach D, Horton J, Hunt J, Lederer J, Mannick J, McKinley B, Minei J, Moore E, Moore F, Munford R, Nathens A, O'Keefe G, Purdue G, Rahme L, Remick D, Sailors M, Shapiro M, Silver G, Smith R, Stephanopoulos G, Stormo G, Toner M, Warren S, West M, Wolfe S, Young V. Application of genome-wide expression analysis to human health and disease. Proc Natl Acad Sci U S A. 2005;102:4801–4806. doi: 10.1073/pnas.0409768102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 7.Goldstein B, Giroir B, Randolph A. International pediatric sepsis consensus conference: definitions for sepsis and organ dysfunction in pediatrics. Pediatr Crit Care Med. 2005;6:2–8. doi: 10.1097/01.PCC.0000149131.72248.E6. [DOI] [PubMed] [Google Scholar]
- 8.Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138–150. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- 9.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 10.Nadon R, Shoemaker J. Statistical issues with microarrays: processing and analysis. Trends Genet. 2002;18:265–271. doi: 10.1016/s0168-9525(02)02665-3. [DOI] [PubMed] [Google Scholar]
- 11.Pollack MM, Patel KM, Ruttimann UE. The Pediatric Risk of Mortality III--Acute Physiology Score (PRISM III-APS): a method of assessing physiologic instability for pediatric intensive care unit patients. J Pediatr. 1997;131:575–581. doi: 10.1016/s0022-3476(97)70065-9. [DOI] [PubMed] [Google Scholar]
- 12.Rink L, Haase H. Zinc homeostasis and immunity. Trends Immunol. 2007;28:1–4. doi: 10.1016/j.it.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 13.Shanley TP, Cvijanovich N, Lin R, Allen GL, Thomas NJ, Doctor A, Kalyanaraman M, Tofil NM, Penfil S, Monaco M, Odoms K, Barnes M, Sakthivel B, Aronow BJ, Wong HR. Genome-Level Longitudinal Expression of Signaling Pathways and Gene Networks in Pediatric Septic Shock. Mol Med. 2007;13:495–508. doi: 10.2119/2007-00065.Shanley. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shanley TP, Hallstrom C, Wong HR. Sepsis. In: Fuhrman BP, Zimmerman JJ, editors. Pediatric Critical Care Medicine. 3. St. Louis: Mosby; 2006. pp. 1474–1493. [Google Scholar]
- 15.Shanley TP, Wong HR. Molecular genetics in the pediatric intensive care unit. Crit Care Clin. 2003;19:577–594. doi: 10.1016/s0749-0704(03)00004-6. [DOI] [PubMed] [Google Scholar]
- 16.Watson RS, Carcillo JA. Scope and epidemiology of pediatric sepsis. Pediatr Crit Care Med. 2005;6:S3–5. doi: 10.1097/01.PCC.0000161289.22464.C3. [DOI] [PubMed] [Google Scholar]
- 17.Wilkes T, Laux H, Foy CA. Microarray data quality - review of current developments. Omics. 2007;11:1–13. doi: 10.1089/omi.2006.0001. [DOI] [PubMed] [Google Scholar]
- 18.Wong HR. Pediatric septic shock treatment: new clues from genomic profiling. Pharmacogenomics. 2007;8:1287–1290. doi: 10.2217/14622416.8.10.1287. [DOI] [PubMed] [Google Scholar]
- 19.Wong HR, Shanley TP, Sakthivel B, Cvijanovich N, Lin R, Allen GL, Thomas NJ, Doctor A, Kalyanaraman M, Tofil NM, Penfil S, Monaco M, Tagavilla MA, Odoms K, Dunsmore K, Barnes M, Aronow BJ. Genome-level expression profiles in pediatric septic shock indicate a role for altered zinc homeostasis in poor outcome. Physiol Genomics. 2007;30:146–155. doi: 10.1152/physiolgenomics.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yao Z, Ruzzo WL. A regression-based K nearest neighbor algorithm for gene function prediction from heterogeneous data. BMC Bioinformatics. 2006;7 (Suppl 1):S11. doi: 10.1186/1471-2105-7-S1-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.