

Abstract
Targeting viral antigens for proteosomal degradation has previously been proposed as a means for immunogenicity augmentation. However, utilization of modified unstable antigens may be insufficient for potent T-cell cross-presentation by APCs, a mechanism that requires high levels of the antigenic protein. Therefore, we hypothesized that a recombinant vaccine utilizing a combination of proteosome-sensitive and proteosome-resistant versions of an antigen in a prime/boost regimen may provide the most efficient CTL response.
To address this hypothesis, we utilized conserved proteosome-resistant influenza A virus proteins M1 and NS1. Unstable versions of these polypeptides were constructed by destroying their 3-D structure via truncations or short insertions into predicted alpha-helical structures. These modified polypeptides were stabilized in the presence of the proteosome inhibitor MG132, strongly suggesting that they are degraded via a ubiquitin-proteosome pathway. Importantly, with both M1 and NS1antigens, homologous DNA vaccination with a mixture of unstable and proteosome-resistant wt forms of these proteins resulted in significantly higher CTL activity than vaccination with either wt or degradable forms. The most dramatic effect was seen with NS1, where homologous immunization with a mixture of these two forms was the only regimen that produced a notable elevation of CTL response, compared to vaccination with the wt NS1. Additionally, for M1 protein, heterologous vaccination utilizing the unstable form as prime and wild type form as boost, demonstrated significant augmentation of the CTL response. These data indicate that combining proteosome-sensitive and proteosome-resistant forms of an antigen during vaccination is advantageous.
INTRODUCTION
Viral proteins produced by an infected cell are degraded by proteosomes resulting in generation of peptides with the structural features of the major histocompatibility complex (MHC) class I ligands [1, 2]. The ubiquitin (Ub)-proteosome system is directly involved in the production of peptides for antigen presentation by MHC-I [3], although its importance has now been questioned [1, 4]. Details of this important immune defense process have not been elucidated yet and are the subject of intensive discussion [1, 3–6].
There has been considerable interest in vaccine strategies that enhance the induction of antigen (Ag)-specific CTLs via increased proteosome-dependent degradation. To augment the Th1 immunogenicity of a target gene, specifically its ability to induce CTL response, many groups have attempted targeting the gene of interest for proteosome degradation [7–12]. Much attention was focused on directing the immunizing gene into the Ub-proteosome degradation pathway, which as it appears now was based on a somewhat distorted picture of Ub-dependent Ag presentation [1]. The underlying rationale was that the targeting of Ags directly into the MHC-I pathway would provide a higher density of peptide/MHC-I complexes on the surfaces of APCs involved in the induction of the CTL response [10].
Targeting of Ags for rapid proteosomal degradation augmented their processing and MHC-I presentation and enhanced CTL responses [10, 13–15]. The enhancement of CTL responses supported the notion that targeting Ags for rapid cytoplasmic degradation might be a useful vaccine strategy when the induction of CD8+ CTL is desired [14, 15], although several viral proteins were not successfully degraded by their fusion with proteosome-directing signals [10, 12]. Moreover, in many other settings, especially those using viral models, even noticeably higher proteosomal degradation did not lead to augmentation of protective immunity [10].
One of the possible reasons for this result was an underestimation of the complexity of the antigen presentation mechanisms [1, 6, 16, 17]. Recently, it has been demonstrated that generation of peptides for MHC-I presentation may occur in a proteosome-independent manner [18] and, conversely, that proteosome-dependent antigen degradation is important for MHC-II presentation [17, 19]. MHC-II presentation drives CD4+-mediated immunity that, in turn, regulates the CTL response [20] in several experimental systems, including DNA immunization against influenza [21]. Therefore, we hypothesized that the simultaneous utilization of wild-type proteosome-resistant viral protein and its proteosome-degradable form in a single vaccination regimen may be advantageous. In particular, the peptides resulting from proteosomal degradation could serve as CD8-ligands while the full-size protein should be much more efficient in antigen cross-presentation to T cells.
Cross-presentation plays a major role in surveying tissues for foreign antigens and their presentation to T-cells [22, 23]. Immature dendritic cells (DC) most likely acquire viral Ags by internalization of infected cell debris [24]. Ags are then cross-presented either by protease-mediated proteolysis in the phagosome or through the proteosome pathway [25]. In order for this process to be efficient, the presence of full-size Ag is necessary. A highly degradable, unstable protein will be under cross-presented and therefore is unlikely to be sufficiently immunogenic on its own [1, 23]. It is known that even when DC are directly transfected by plasmids [26], rapid antigen degradation is not advantageous [27]. Still, degradable proteins that efficiently generate MHC-I peptides in non-APC cells may contribute to the other stages of immune response development.
Utilization of different vectors for prime and boost stages of vaccination (heterologous prime-boost) results in manifest augmentation of immunogenicity [28–33]. This effect may be connected to the generation of memory T cells with functionally distinct phenotype [34]. In particular, DNA-prime/viral vector-boost immunization was shown to be more potent in many non-viral and viral models, including influenza, than vaccination with either of them alone [35–47]. Additionally, recent data has shown that naïve and memory T cells are stimulated by different subsets of DC, especially in the influenza infection model [48, 49].
Collectively, new insights on the development of immune response and known data on efficiency of heterologous prime-boost vaccination suggest that different forms of antigen: e.g., proteosome degradable in addition to proteosome resistant, may provide for elevated immunity if used simultaneously either in combination or sequentially. Moreover, we propose that for some proteins, resistance to proteosome degradation is at least partially due to the inaccessibility of their three-dimensional structure to proteosome machinery. If so, the targeted destruction of a protein antigens’ native conformation by deletion of a region crucial for protein folding or an insertion of a disruptive element into such a region (i.e., alpha-helical structure), may be enough to generate a proteosome degradable viral antigen. We have chosen M1 and NS1 proteins of influenza A virus as model antigens to test this hypothesis. We constructed degradable forms of these proteins either by truncation or DD insertions into predicted alpha-helical stretches and then assayed their immunogenicity in prime-boost DNA vaccination regimens using proteosome degradable mutants sequentially and/or simultaneously with their wild-type forms.
MATERIALS AND METHODS
Cells and plasmids
All experiments were performed in 293 human embryo kidney (HEK) cells. The full-length genes encoding influenza A/WSN/33 (H1N1) M1 and NS1 proteins were synthesized (GenBank sequence accession numbers L25818 and J02150), inserted into the pcDNA vector (Invitrogen, Carlsbad, CA, USA) and their sequence verified. A SalI site was designed at the 5’-end and an EcoRI site at the 3’-end of both genes. They were then digested with SalI and EcoRI and cloned into the similarly treated pCAGGS expression vector [50], which has been modified to contain a SalI site instead of a 5’-terminal EcoRI site, resulting in pM1wt and pNS1wt. All deletion and point-mutants were constructed by PCR-directed site-specific mutagenesis of the wild-type genes. PCR products were ligated into the pcDNA vector. M1 point mutants (M1pm) were designed to incorporate duplicate DD insertions into alpha-helical stretches of M1 (see Fig. 1A for exact positions). This design was based on the M1 crystal structure which has been resolved and is well-defined [51, 52]. Eight DD di-aminoacid insertions in M1pm1 were encoded by all possible four nucleotide combinations, GACGAC, GACGAT, GATGAC and GATGAT to avoid undesirable recombination, each sexta-nucleotide was utilized twice. M1pm2 and M1pm3 contained five and four non-overlapping (with one exception) DD insertions, all of which were present in M1pm1. M1 deletion mutants were designed to truncate either its four C-terminal alpha-helical stretches (M1del1) or three N-terminal alpha-helical stretches (M1del2). NS1 deletion mutants were designed to truncate in different fashion (two mutants each) either N-terminal RNA-binding domain (amino acids 1–73) or C-terminal effector domain (amino acids 134–230), both of which are well-defined, structurally and biologically [53–58]. An HA-tag (YPYDVPDYA)-encoding sequence was added at the 3’-terminus of all genes to enable their efficient immunological detection. Upon sequence confirmation, all mutant genes were then cloned into the pCAGGS vector similarly to wild-type genes (resulting in pM1del1–2, pM1pm1–3 and pNS1del1–4, nine plasmids total, see Fig. 1A and Fig. 2A).
Fig. 1.
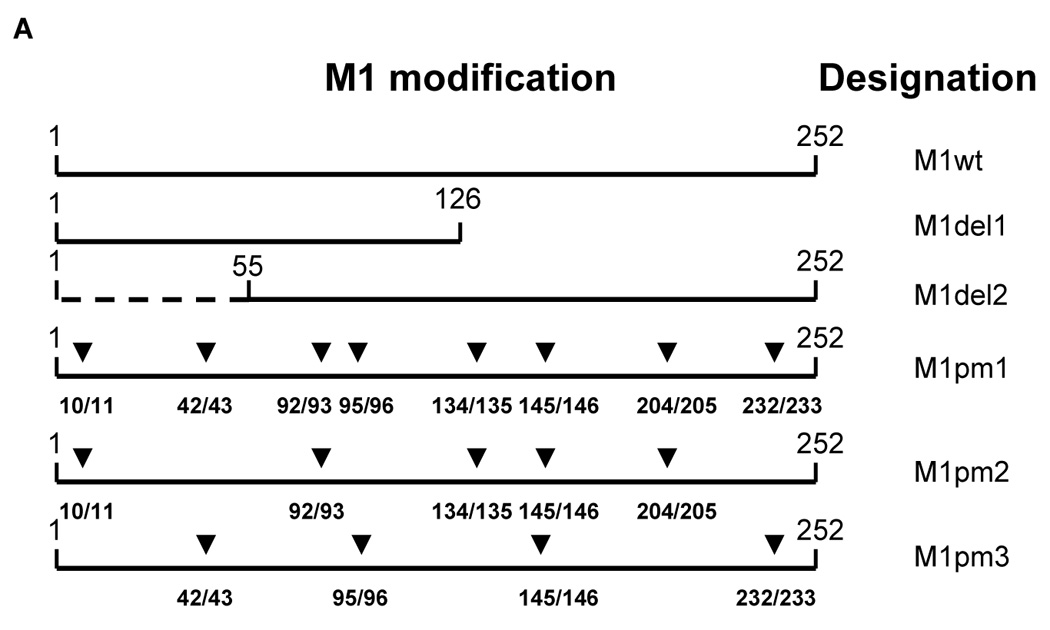
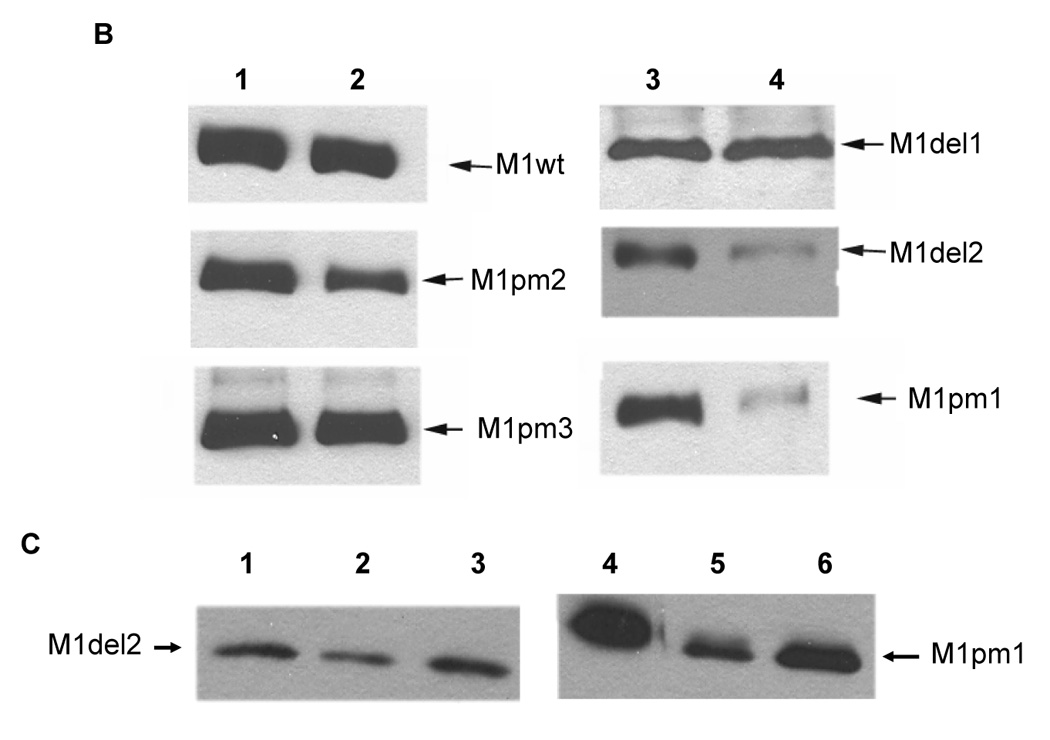
A – Structure of influenza M1 protein mutants. Location of insertions in M1pm1–3 and truncation boundaries for M1del1–2 are shown. ▼ equals the insertion of tandem DD amino acids. M1pm1 has 8 DD insertions, M1pm2 has five and M1pm3 has four. B–C – Proteosomal degradation of wild-type and mutant M1 forms. B – Metabolic stability of different M1 alleles (indicated with arrows). Lanes 1 and 3 – start of chase (0 hours), lane 2 – chase 16 hours, lane 4 – chase 8 hours. C – Proteosomal degradation of the unstable mutant M1 forms in the presence of MG132. Lanes 1–3 – cells transfected with pM1del2, lanes 4–6 – with pM1pm1. Times of chase – 0 hours (lanes 1, 4), 4 hours (lanes 2, 3, 5, 6). Lanes 3 and 6 – MG132 added.
Fig. 2.
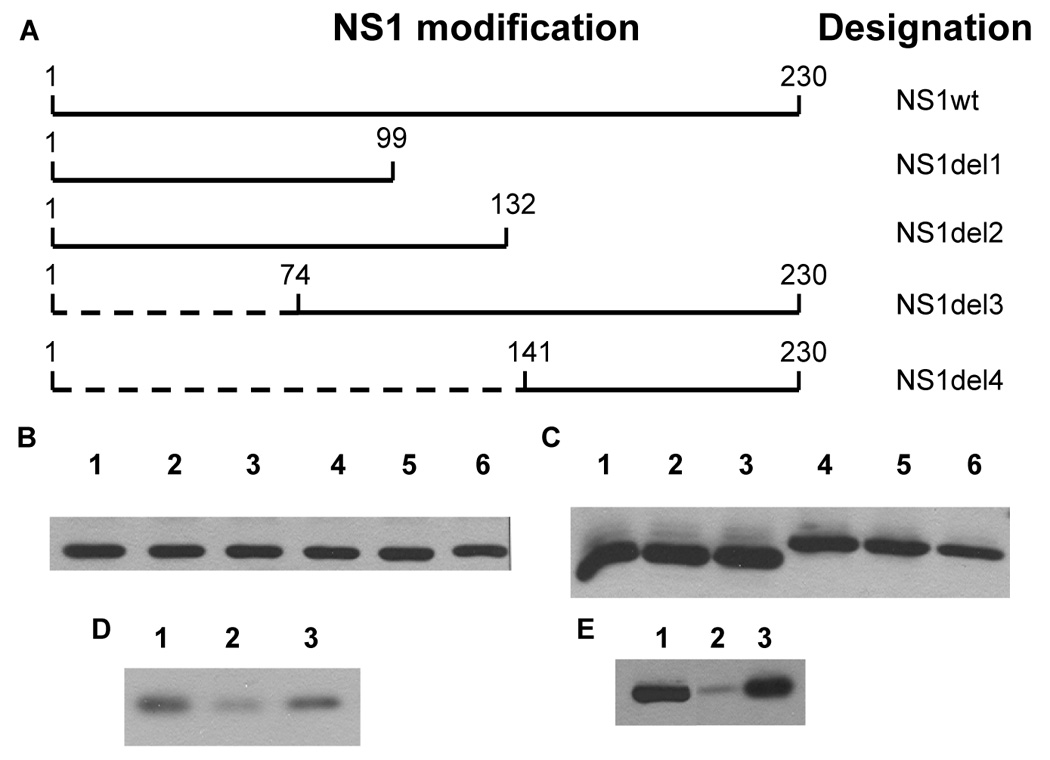
Structure of influenza NS1 protein mutants and their metabolic stability. A – design of NS1 mutants with boundaries of truncations shown, B–E – their metabolic stability and proteosome-dependent degradation. B – NS1 wt. Chase times: 0, 1, 2, 4, 6 and 8 hours (lanes 1–6, correspondingly). C – NS1del1 (lanes 1–3) and NS1del 2 (lanes 4–6). Chase times: 0 hours (lanes 1, 4), 2 hours (lanes 2, 5), 8 hours (lanes 3, 6). D – degradation of NS1del2 in presence of MG132. Chase times: 0 hours (lane 1) and 8 hours (lanes 2, 3). Lane 3 – MG132 added. E – degradation of NS1del4 in presence of MG132. Chase times: 0 hours (lane 1) and 4.5 hours (lanes 2, 3). Lane 3 – MG132 added.
Transfection, protein expression, stability and assessment of proteosome-dependent degradation
M1- and NS1-expressing plasmids were transfected into 293 HEK cells at 60–80% confluence in 35 mm plates using Lipofectamine 2000 (Invitrogen, Carsbad, CA, USA) either for 4 hours or overnight (1.5 µg of total plasmid DNA per 3 µl LF2000). Control cells were transfected with the same amount of empty vector pCAGGS. 40 hours after transfection 12uM emetine was added and samples were taken (2x wash in PBS, −80C) at the indicated time points. All constructs were chased for various time periods falling within 0–24 hours after emetine addition with or without proteosome inhibitor MG132 (10µM). Cells were then homogenized on ice in lysis buffer (about 150µl/ 35mm dish) 150mM NaCl, 50 mM TrisHCl (pH 7.4), 1mM EDTA, 1% Triton X-100 with protease inhibitors. Samples were adjusted for equal total protein concentration and run on SDS-PAGE followed by immunoblotting with either monoclonal anti-HA (6E2) (Cell Signaling Beverly, MA, USA) or anti-HA (Covance, Princeton, NJ, USA) antibodies.
Immunization with pM1 and pNS1 combinations and CTL response in vivo
5 µg of selected pM1 and pNS1 wild-type and mutant forms (2.5 µg when two and 1.7 µg when three plasmids were used concurrently) were injected intramuscularly into each experimental mouse (BALB/c) three times with 14-day interval (in 100 µl of PBS, 5 animals per group). When different plasmid combinations were used for prime and boost, the animals received first combination once (prime) and the other twice (two boosts). The body weight of immunized mice was followed to assess possible toxicity of immunizing DNA (compared to PBS-injected animals). Six days after the third DNA vaccination, mice were sacrificed, their splenocytes purified and stimulated (~108 total, plated at 5×106/ml) in vitro by co-cultivation at 10:1 ratio with the syngeneic feeder splenocytes infected with influenza A/PR/8/34 (H1N1) virus (taken from healthy mice, infected at MOI 20 PFU/cell for 24 hours and UV-inactivated). High levels of M1 and NS1 expression in target spleen cells was demonstrated by immunoblotting with virus protein specific antibodies as previously described [59].
Splenocytes isolated from mice infected intranasally twice at three-week intervals with a sublethal dose of influenza A/Aichi/2/68 (H3N2) virus were used as a positive CTL control. Stimulated splenocytes from each tested mouse were incubated separately in DMEM containing FCS (10%) and 2-mercaptoethanol (2 µM) for 16 days, as previously described [59]. Splenocytes were stimulated and CTL activity was measured on days 8 and 16. In this as well as our previous study [59] we detected only minimal CTL activity against conserved proteins of influenza on day 8 after DNA immunization (10–15% cytotoxicity), even for the highest effector:target ratio (100:1). Therefore, day 16 time-point was selected based on our observation that longer splenocyte stimulation times are necessary to detect CTL activity upon DNA vaccination against conserved proteins of influenza, including M1 and NS1.
Mouse mastocytoma p815 cells infected with influenza A/PR/8/34 virus (MOI 20 PFU/cell) for 24 hrs were used as a target and cytotoxic activity was measured by lactate dehydrogenase (LDH) release (CytoTox 96 Kit; Promega). Target p815 infected cells (0.3 × 105/well) were mixed with 2-fold dilutions of stimulated effector cells starting with 3.0 × 106 cells/well and incubated in 100 µl volume for 6 hrs at 37°C. CTL activity as % of cell lysis was calculated by the following formula: (experimental release–spontaneous release)/(maximum release–spontaneous release) × 100. Target cells incubated in medium alone and with medium containing 1% detergent NP-40 were used to determine spontaneous and maximum LDH release. Uninfected p815 cells were used in a similar layout to control the specificity of CTL-mediated cell killing. Results shown were means (± SD) from each group of four animals. The significance between two percentage values (with probability 0.95) was: t = p1 − p2/√SE12±SE22 ≥ 2.0. The differences at P-value below 0.05 were considered significant.
RESULTS
Protein stability of M1wt and generation of proteosome-degradable influenza M1 forms
A collection of influenza virus M1-expressing mutants was constructed and tested for their metabolic stability as described in Materials and Methods. We constructed a series of M1 mutants that we hypothesized to be less proteosome-resistant than wild-type M1. These mutants bear either truncation at N- or C-termini of the protein or multiple DD insertions in the hydrophobic amino acid stretches designed to destroy the predicted alpha-helical structures (Fig. 1A). Similarly to many other viral proteins, wild-type influenza M1 protein is remarkably resistant to degradation and shows a high degree of stability even after 16 hours of observation in vitro (Fig. 1B). While M1del1 and M1pm3 mutants were of similar stability to M1wt, other tested forms of M1 manifested notably higher degradation kinetics (Fig. 1B), with two of them (M1del2 and M1pm1), being most degradation-prone (with a half-life of 4 to 8 hours). In spite of being unstable, both of these polypeptides were expressed at levels similar to M1wt. These polypeptides were efficiently stabilized by short-term treatment with the proteosome inhibitor MG132 (Fig. 1C). Furthermore, with both polypeptides, no low molecular weight bands appeared in the gel (not shown), strongly suggesting that degradation of M1del2 and M1pm1 is proteosome-dependent. Stability of M1 point-mutants was inversely correlated with the number of DD insertions that were introduced. Since M1pm1 may potentially generate an immune response against all determinants that are present in full-length M1 (compared to M1del2), it was selected for immunogenicity studies in vivo.
Protein stability of NS1wt and generation of proteosome-degradable influenza NS1 forms
Influenza NS1 protein is known to be able to suppress the host immune response via a variety of pathways. Thus, utilization of even partially functional NS1 in future vaccine regimens is unlikely. Therefore, we focused on the construction of NS1 deletion mutants and their subsequent stability testing (mutant structures are shown in Fig. 2A). NS1wt was demonstrated to be extremely resistant to proteosome degradation similar to influenza M1. There was no detectable decrease in protein level even after 8 hours of inhibition of protein synthesis (Fig. 2B). While two of the designed NS1 mutants, NS1del1 (Fig. 2C, lanes 1–3) and NS1del3 (not shown) exhibited no change in protein stability, two other truncation mutants, NS1del2 (Fig. 2C, lanes 4–6) and NS1del4 (Fig. 2E) were markedly less stable. Following 8 hours of protein synthesis inhibition, the levels of NS1del2 dropped significantly, this decrease was even more dramatic for NS1del4 (Figs. 2C, 2E). In both cases, no low molecular weight bands were observed (not shown), and degradation of these mutants was blocked by 10 µM MG132 (Figs. 2D–E), indicating that these polypeptides are degraded in a proteosome-dependent manner. Since collectively the sequences of NS1del2 and NS1del4 cover more than 96% of the NS1 gene, we decided to follow through with both of these plasmids into the next round of immunogenicity studies.
CTL response to the immunization with wild-type and modified forms of M1 and NS1
Several groups of mice were immunized in parallel (prime and two boosts) with plasmids encoding wild-type forms of M1 and NS1 and their selected proteosome-degradable forms (labeled in Fig. 3 as M1modif and NS1modif, the former represents M1pm1, the latter comprises equimolar mixture of NS1del2 and NS1del). The CTL activity in immunized animals was measured as described in Materials and Methods. No significant variation between the test groups was seen when uninfected p815 cells were used as non-specific targets (Fig. 3B), while a different pattern was observed when influenza-infected targets were utilized (Fig. 3A).
Fig. 3.
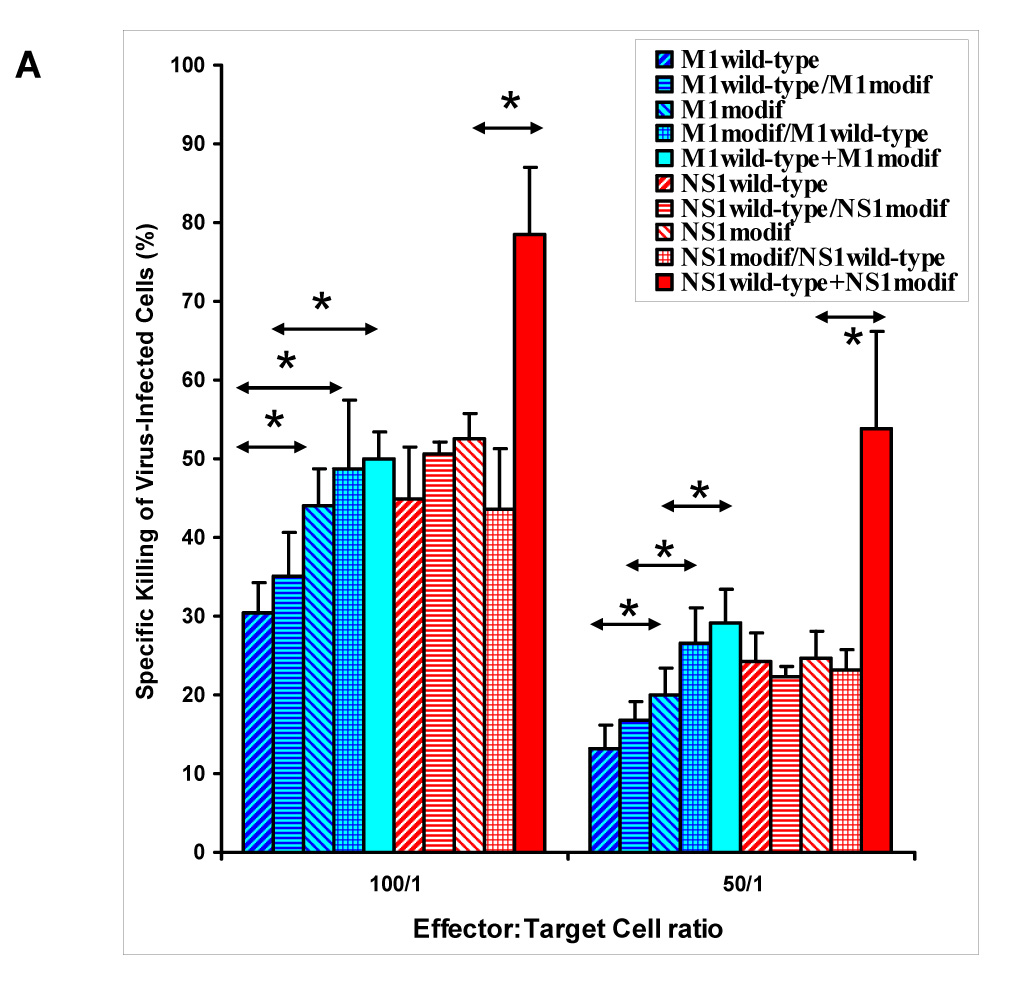
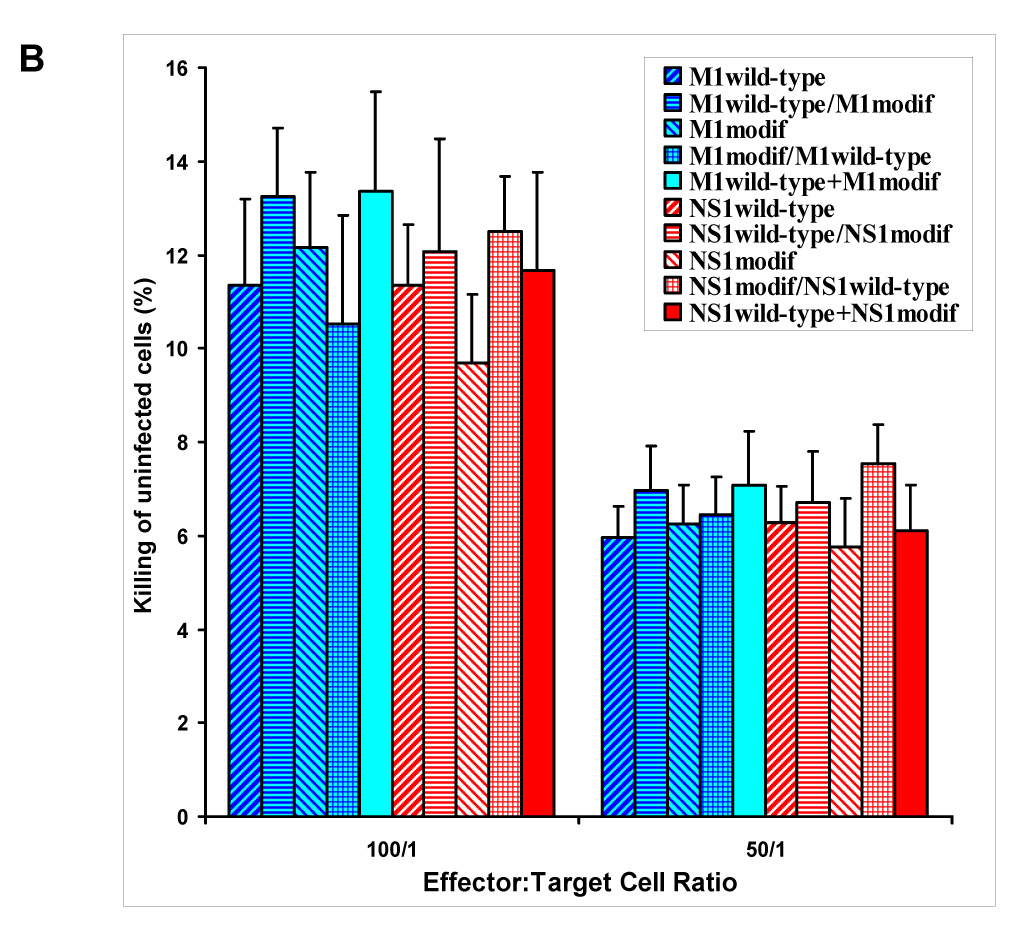
CTL response in animals immunized with various combinations of plasmids encoding wild-type and modified forms of M1 and NS1 as described in text. Wild-type/modif label = subsequent use of two different plasmids; wild-type+modif label = their simultaneous use. BALB/c mice were injected 3 times with 14-day intervals. When different plasmid combinations were used subsequently, then the first combination was utilized as prime and another was injected twice, at both boost immunizations. Virus-infected (A) and uninfected (B) p815 cells were used as targets. Background CTL activity (induced by immunization with empty vector) is subtracted (A). Groups with statistically significant differences are marked (*, P < 0.05).
In both cases homo-allelic triple immunization with wild-type-encoding plasmids resulted in similar CTL activity similar to priming with wild-type plasmids followed by boosts with plasmids encoding modified forms of M1 or NS1 (labeled in Fig. 3 as wild-type/modified immunization groups). In the case of M1, introduction of its modified form at the prime stage led to a noticeable increase of the CTL response, which was evident in M1modified prime/M1wildtype boost group (statistically significant increase over M1wt at 50:1 and 100:1 effector-to-target ratios, and over M1wt/M1modif at 50:1 ratio) and in the group, where the mixture of wild-type and modified forms was used at all stages (M1wild-type+M1modified). The latter regimen resulted in statistically significant augmentation of CTL activity compared to M1wt and M1wt/M1modif regimens at 100:1 and 50:1 ratios and compared to homologous M1modifed immunization at 50:1 ratio (Fig. 3A). Utilization of homologous vaccination with M1modified led to statistically significant elevation of CTL activity over M1wt at 50:1 and 100:1 ratios, but did not provide for augmented CTL immunogenicity over the M1wt/M1modified regimen.
In the animals immunized with combinations of NS1-expressing plasmids, the only regimen that resulted in statistically significant augmentation of CTL activity was the one employing the mixture of wild-type and proteosome degradable NS1 forms at all stages (NS1wild-type+NS1modif group). CTL activity in animals immunized by this plasmid mixture was markedly higher than in all other experimental groups (Fig. 3A). Thus, for both NS1 and M1, immunization with equimolar quantities of plasmids expressing their wild-type as well as modified forms resulted in statistically significant augmentation of the CTL response.
DISCUSSION
The choice of conserved influenza proteins as model antigens for this studies stemmed from necessity to advance the currently existing cross-protective immunization regimens against influenza [12, 21, 38, 46, 59–67]. While both NS1 and M1 proteins alone are not capable of induction of strong anti-influenza immunity compared to nucleoprotein (NP) and minor matrix protein 2 (M2), they present a better model to assess our current hypothesis. In particular, both of them are cytoplasmic, while NP is strongly nuclear [68–70] and M2 predominantly resides on the membrane of infected cells, mostly inducing an antibody response [71–73].
Conventional means of augmentation of viral protein degradation are not particularly effective and novel avenues to increase proteosome-dependent degradation of influenza proteins need to be explored [10, 12]. We hypothesized that high proteosomal stability of viral proteins results from their conformational structure. If proteosome is sterically incapable of processing potentially immunogenic viral proteins, this will dramatically hinder or distort their presentation by MHC class I molecules (the latter seen in [74]). Therefore, in order to increase proteosome-directed degradation of these proteins, one may suggest deletion of a structurally-important region or an insertion of a disruptive element. This approach was shown to be useful since we were able to produce rapidly degradable forms of influenza M1 and NS1 either by introducing DD insertions into hydrophobic amino acid stretches of M1 or by designing deletion mutants that collectively overlap ~ 96% of NS1. Most likely the rapid degradation of these mutant forms is due to their increased susceptibility to the proteosome. Importantly, both M1 and NS1 were initially demonstrated to be extremely proteosome-resistant (Fig. 1 and Fig. 2).
This allowed us to test if the combination of wild-type proteosome-resistant and modified proteosome-degradable forms of a protein antigen may result in augmentation of the CTL response to DNA vaccination. A previously reported approach of substituting a wild-type viral protein with its proteosome-degradable form has mostly resulted in disappointment [10, 66]. We hypothesized that due to the complexity of the antigen presentation network, a combination of proteosome-degradable and resistant forms of an antigenic protein may be beneficial. Both proteosome-dependent and independent pathways for immunogenic peptide generation have been elucidated [16, 18] and their predominance differs between APC and non-APC [16]. Moreover, they are likely to be differentially important at prime and boost stages of vaccination [27, 49]. Therefore, it seemed probable that concurrent utilization of non-degradable wild-type form of viral proteins (capable of APC cross-presentation) and their degradable counterparts (capable of efficient MHC-I peptide generation) should result in higher CTL activity.
Such an augmentation was seen for both M1 and NS1 in a DNA immunization model. Utilization of a mixture of wild-type and degradable forms of said proteins induced higher CTL activities than utilization of either of these forms alone. This phenomenon was more pronounced for NS1 than for M1 (Fig. 3). Heterologous prime-boost vaccination with various forms of NS1 used separately at the different vaccination stages did not manifest elevated immunogenicity. Conversely, a proteosome-degradable form of M1 (M1modif) applied at the prime stage resulted in a significant enhancement of CTL activity and utilization of M1wt form at the boost stage was advantageous (Fig. 3A). It is possible that the exact composition of the most potent wild-type/degradable form-utilizing vaccination regimen should be determined individually for each medicinally important antigen and that the ratio of its wild-type/degradable forms bears additional importance.
While M1 and NS1 proteins of influenza virus were utilized in this study predominantly as model antigens, we are planning to test the protective benefits of immunization with various combinations of these two antigens together with proteosome-degradable and resistant forms of influenza NP protein. NP is known to be capable of inducing a protective T-cell response against influenza [60–62, 75] and we previously reported the protective benefits of a vaccination regimen combining NP, M1 and NS1 [59]. If successful, this undertaking may result in generation of novel NP-based immunogens providing sufficiently broad and effective epitope generation, which is not always a feature of reverse-engineered vaccines [76]. The utilization of other viral proteins of medicinal importance in recombinant vaccines may be advanced as well.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Yewdell JW. Plumbing the sources of endogenous MHC class I peptide ligands. Curr Opin Immunol. 2007;19(1):79–86. doi: 10.1016/j.coi.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 2.Yewdell JW, Haeryfar SM. Understanding presentation of viral antigens to CD8+ T cells in vivo: the key to rational vaccine design. Annu Rev Immunol. 2005;23:651–682. doi: 10.1146/annurev.immunol.23.021704.115702. [DOI] [PubMed] [Google Scholar]
- 3.Loureiro J, Ploegh HL. Antigen presentation and the ubiquitin-proteasome system in host-pathogen interactions. Adv Immunol. 2006;92:225–305. doi: 10.1016/S0065-2776(06)92006-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yewdell JW. The seven dirty little secrets of major histocompatibilty complex class I antigen processing. Immunological reviews. 2005 Oct;207:8–18. doi: 10.1111/j.0105-2896.2005.00309.x. [DOI] [PubMed] [Google Scholar]
- 5.Cresswell P, Ackerman AL, Giodini A, Peaper DR, Wearsch PA. Mechanisms of MHC class I-restricted antigen processing and cross-presentation. Immunol Rev. 2005 Oct;207:145–157. doi: 10.1111/j.0105-2896.2005.00316.x. [DOI] [PubMed] [Google Scholar]
- 6.Monu N, Trombetta ES. Cross-talk between the endocytic pathway and the endoplasmic reticulum in cross-presentation by MHC class I molecules. Curr Opin Immunol. 2007;19(1):66–72. doi: 10.1016/j.coi.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 7.Dobaño C, Rogers WO, Gowda K, Doolan DL. Targeting antigen to MHC Class I and Class II antigen presentation pathways for malaria DNA vaccines. Immunol Lett. 2007;111(2):92–102. doi: 10.1016/j.imlet.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 8.Starodubova E, Boberg A, Kashuba EV, Wahren B, Karpov V, Isaguliants M. HIV-1 reverse transcriptase targeted for proteasomal degradation as a prototype vaccine against drug-resistant HIV-1. Vaccine. 2006;24(21):4541–4547. doi: 10.1016/j.vaccine.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 9.Rodriguez F, Zhang J, Whitton JL. DNA immunization: ubiquitination of a viral protein enhances cytotoxic T-lymphocyte induction and antiviral protection but abrogates antibody induction. J Virology. 1997;71(11):8497–8503. doi: 10.1128/jvi.71.11.8497-8503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wong SBJ, Buck CB, Shen X, Siliciano RF. An evaluation of enforced rapid proteasomal degradation as a means of enhancing vaccine-induced CTL responses. J Immunology. 2004;173(5):3073–3083. doi: 10.4049/jimmunol.173.5.3073. [DOI] [PubMed] [Google Scholar]
- 11.Zhang M, Ishii K, Hisaeda H, Murata S, Chiba T, Tanaka K, et al. Ubiquitin-fusion degradation pathway plays an indispensable role in naked DNA vaccination with a chimeric gene encoding a syngeneic cytotoxic T lymphocyte epitope of melanocyte and green fluorescent protein. Immunology. 2004;112(4):567–574. doi: 10.1111/j.1365-2567.2004.01916.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Altstein AD, Gitelman AK, Smirnov YA, Piskareva LM, Zakharova LG, Pashvykina GV, et al. Immunization with influenza A NP-expressing vaccinia virus recombinant protects mice against experimental infection with human and avian influenza viruses. Arch Virol. 2006;151(5):921–931. doi: 10.1007/s00705-005-0676-9. [DOI] [PubMed] [Google Scholar]
- 13.Restifo NP, Bacik I, Irvine KR, Yewdell JW, McCabe BJ, Anderson RW, et al. Antigen processing in vivo and the elicitation of primary CTL responses. J Immunol. 1995;154(9):4412–4422. [PMC free article] [PubMed] [Google Scholar]
- 14.Townsend A, Bastin J, Gould K, Brownlee G, Andrew M, Coupar B, et al. Defective presentation to class I-restricted cytotoxic T lymphocytes in vaccinia-infected cells is overcome by enhanced degradation of antigen. J Exp Med. 1988;168(4):1211–1224. doi: 10.1084/jem.168.4.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu Y, Kipps TJ. Deoxyribonucleic acid vaccines encoding antigens with rapid proteasome-dependent degradation are highly efficient inducers of cytolytic T lymphocytes. J Immunol. 1997;159(12):6037–6043. [PubMed] [Google Scholar]
- 16.Jensen PE. Recent advances in antigen processing and presentation. Nature Immunol. 2007;8(10):1041–1048. doi: 10.1038/ni1516. [DOI] [PubMed] [Google Scholar]
- 17.Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol. 2007;7(10):767–777. doi: 10.1038/nri2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guil S, Rodriguez-Castro M, Aguilar F, Villasevil EM, Anton LC, Del Val M. Need for tripeptidyl-peptidase II in major histocompatibility complex class I viral antigen processing when proteasomes are detrimental. J Biol Chem. 2006;281(52):39925–39934. doi: 10.1074/jbc.M608522200. [DOI] [PubMed] [Google Scholar]
- 19.Tewari MK, Sinnathamby G, Rajagopal D, Eisenlohr LC. A cytosolic pathway for MHC class II-restricted antigen processing that is proteasome and TAP dependent. Nature Immunol. 2005;6(3):287–294. doi: 10.1038/ni1171. [DOI] [PubMed] [Google Scholar]
- 20.Prlic M, Williams MA, Bevan MJ. Requirements for CD8 T-cell priming, memory generation and maintenance. Curr Opin Immunol. 2007;19(3):315–319. doi: 10.1016/j.coi.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 21.Epstein S, Stack A, Misplon JA, Lo CY, Mostowski H, Bennink J, et al. Vaccination with DNA encoding internal proteins of influenza virus does not require CD8(+) cytotoxic T lymphocytes: either CD4(+) or CD8(+) T cells can promote survival and recovery after challenge. Int Immunol. 2000;12(1):91–101. doi: 10.1093/intimm/12.1.91. [DOI] [PubMed] [Google Scholar]
- 22.Rock KL, Shen L. Cross-presentation: underlying mechanisms and role in immune surveillance. Immunol Rev. 2005 Oct;207:166–183. doi: 10.1111/j.0105-2896.2005.00301.x. [DOI] [PubMed] [Google Scholar]
- 23.Rock KL. Exiting the outside world for cross-presentation. Immunity. 2006;25(4):523–525. doi: 10.1016/j.immuni.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 24.Ackerman AL, Giodini A, Cresswell P. A role for the endoplasmic reticulum protein retrotranslocation machinery during crosspresentation by dendritic cells. Immunity. 2006;25(4):605–617. doi: 10.1016/j.immuni.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 25.Kovacsovics-Bankowski M, Rock KL. A phagosome-to-cytosol pathway for exogenous antigens presented on MHC class I molecules. Science. 1995;267(5195):243–246. doi: 10.1126/science.7809629. [DOI] [PubMed] [Google Scholar]
- 26.Steinman RM, Pope M. Exploiting dendritic cells to improve vaccine efficacy. J Clin Invest. 2002;109(12):1519–1526. doi: 10.1172/JCI15962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tacken PJ, de Vries IJ, Torensma R, Figdor CG. Dendritic-cell immunotherapy: from ex vivo loading to in vivo targeting. Nat Rev Immunol. 2007;7(10):790–802. doi: 10.1038/nri2173. [DOI] [PubMed] [Google Scholar]
- 28.Hsieh MK, Wu CC, Lin TL. Priming with DNA vaccine and boosting with killed vaccine conferring protection of chickens against infectious bursal disease. Vaccine. 2007;25(29):5417–5427. doi: 10.1016/j.vaccine.2007.04.087. [DOI] [PubMed] [Google Scholar]
- 29.Larke N, Murphy A, Wirblich C, Teoh D, Estcourt MJ, McMichael AJ, et al. Induction of human immunodeficiency virus type 1-specific T cells by a bluetongue virus tubule-vectored vaccine prime-recombinant modified virus Ankara boost regimen. J Virol. 2005;79(23):14822–14833. doi: 10.1128/JVI.79.23.14822-14833.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liang R, van den Hurk JV, Babiuk LA, van Drunen Littel-van den Hurk S. Priming with DNA encoding E2 and boosting with E2 protein formulated with CpG oligodeoxynucleotides induces strong immune responses and protection from Bovine viral diarrhea virus in cattle. J Gen Virol. 2006;87(Pt 10):2971–2982. doi: 10.1099/vir.0.81737-0. [DOI] [PubMed] [Google Scholar]
- 31.Otten GR, Schaefer M, Doe B, Liu H, Srivastava I, Megede J, et al. Enhanced potency of plasmid DNA microparticle human immunodeficiency virus vaccines in rhesus macaques by using a priming-boosting regimen with recombinant proteins. J Virol. 2005;79(13):8189–8200. doi: 10.1128/JVI.79.13.8189-8200.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pal R, Venzon D, Santra S, Kalyanaraman VS, Montefiori DC, Hocker L, et al. Systemic immunization with an ALVAC-HIV-1/protein boost vaccine strategy protects rhesus macaques from CD4+ T-cell loss and reduces both systemic and mucosal simian-human immunodeficiency virus SHIVKU2 RNA levels. J Virol. 2006;80(8):3732–3742. doi: 10.1128/JVI.80.8.3732-3742.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Toussaint JF, Letellier C, Paquet D, Dispas M, Kerkhofs P. Prime-boost strategies combining DNA and inactivated vaccines confer high immunity and protection in cattle against bovine herpesvirus-1. Vaccine. 2005;23(43):5073–5081. doi: 10.1016/j.vaccine.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 34.Masopust D, Ha SJ, Vezys V, Ahmed R. Stimulation history dictates memory CD8 T cell phenotype: implications for prime-boost vaccination. J Immunol. 2006;177(2):831–839. doi: 10.4049/jimmunol.177.2.831. [DOI] [PubMed] [Google Scholar]
- 35.Chunling MKY, Jian X, Jian Q, Hua S, Minsheng Z. Enhanced induction of SARS-CoV nucleocapsid protein-specific immune response using DNA vaccination followed by adenovirus boosting in BALB/c mice. Intervirology. 2006;49(5):307–318. doi: 10.1159/000094247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dory D, Fischer T, Béven V, Cariolet R, Rziha HJ, Jestin A. Prime-boost immunization using DNA vaccine and recombinant Orf virus protects pigs against Pseudorabies virus (Herpes suid 1) Vaccine. 2006;24(37–39):6256–6263. doi: 10.1016/j.vaccine.2006.05.078. [DOI] [PubMed] [Google Scholar]
- 37.Dunachie SJ, Walther M, Vuola JM, Webster DP, Keating SM, Berthoud T, et al. A clinical trial of prime-boost immunisation with the candidate malaria vaccines RTS,S/AS02A and MVA-CS. Vaccine. 2006;24(15):2850–2859. doi: 10.1016/j.vaccine.2005.12.041. [DOI] [PubMed] [Google Scholar]
- 38.Epstein S, Kong WP, Misplon JA, Lo CY, Tumpey TM, Xu L, et al. Protection against multiple influenza A subtypes by vaccination with highly conserved nucleoprotein. Vaccine. 2005;23(46–47):5404–5410. doi: 10.1016/j.vaccine.2005.04.047. [DOI] [PubMed] [Google Scholar]
- 39.Gilbert SC, Moorthy VS, Andrews L, Pathan AA, McConkey SJ, Vuola JM, et al. Synergistic DNA-MVA prime-boost vaccination regimes for malaria and tuberculosis. Vaccine. 2006;24(21):4554–4561. doi: 10.1016/j.vaccine.2005.08.048. [DOI] [PubMed] [Google Scholar]
- 40.Goonetilleke N, Moore S, Dally L, Winstone N, Cebere I, Mahmoud A, et al. Induction of multifunctional human immunodeficiency virus type 1 (HIV-1)-specific T cells capable of proliferation in healthy subjects by using a prime-boost regimen of DNA- and modified vaccinia virus Ankara-vectored vaccines expressing HIV-1 Gag coupled to CD8+ T-cell epitopes. J Virol. 2006;80(10):4717–4728. doi: 10.1128/JVI.80.10.4717-4728.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haygreen EA, Kaiser P, Burgess SC, Davison TF. In ovo DNA immunisation followed by a recombinant fowlpox boost is fully protective to challenge with virulent IBDV. Vaccine. 2006;24(23):4951–4961. doi: 10.1016/j.vaccine.2006.03.060. [DOI] [PubMed] [Google Scholar]
- 42.Ma C, Yao K, Zhou F, Zhu M. Comparative immunization in BALB/c mice with recombinant replication-defective adenovirus vector and DNA plasmid expressing a SARS-CoV nucleocapsid protein gen. Cell Mol Immunol. 2006;3(6):459–465. [PubMed] [Google Scholar]
- 43.Perkins SD, O'Brien LM, Phillpotts RJ. Boosting with an adenovirus-based vaccine improves protective efficacy against Venezuelan equine encephalitis virus following DNA vaccination. Vaccine. 2006;24(17):440–3445. doi: 10.1016/j.vaccine.2006.02.020. [DOI] [PubMed] [Google Scholar]
- 44.Rollier C, Verschoor EJ, Paranhos-Baccala G, Drexhage JA, Verstrepen BE, Berland JL, et al. Modulation of vaccine-induced immune responses to hepatitis C virus in rhesus macaques by altering priming before adenovirus boosting. J Infect Dis. 2005;192(5):920–929. doi: 10.1086/432517. [DOI] [PubMed] [Google Scholar]
- 45.Someya K, Ami Y, Nakasone T, Izumi Y, Matsuo K, Horibata S, et al. Induction of positive cellular and humoral immune responses by a prime-boost vaccine encoded with simian immunodeficiency virus gag/pol. J Immunol. 2006;176(3):1784–1795. doi: 10.4049/jimmunol.176.3.1784. [DOI] [PubMed] [Google Scholar]
- 46.Tompkins S, Zhao ZS, Lo CY, Misplon JA, Liu T, Ye Z, et al. Matrix protein 2 vaccination and protection against influenza viruses, including subtype H5N1. Emerg Infect Dis. 2007;13(3):426–435. doi: 10.3201/eid1303.061125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu L, Kong W-P, Nabel GJ. Enhanced breadth of CD4 T-cell immunity by DNA prime and adenovirus boost immunization to human immunodeficiency virus env and gag immunoges. J Virol. 2005;79(13):8024–8231. doi: 10.1128/JVI.79.13.8024-8031.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Belz GT, Smith CM, Kleinert L, Reading P, Brooks A, Shortman K, et al. Distinct migrating and nonmigrating dendritic cell populations are involved in MHC class I-restricted antigen presentation after lung infection with virus. Proc Natl Acad Sci USA. 2004;101(23):8670–8675. doi: 10.1073/pnas.0402644101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Belz GT, Bedoui S, Kupresanin F, Carbone FR, Heath WR. Minimal activation of memory CD8(+) T cell by tissue-derived dendritic cells favors the stimulation of naive CD8(+) T cells. Nat Immunol. 2007;8(10):1060–1066. doi: 10.1038/ni1505. [DOI] [PubMed] [Google Scholar]
- 50.Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108(2):193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- 51.Sha B, Luo M. Structure of a bifunctional membrane-RNA binding protein, influenza virus matrix protein M1. Nat Struct Biol. 1997;4(3):239–244. doi: 10.1038/nsb0397-239. [DOI] [PubMed] [Google Scholar]
- 52.Harris A, Forouhar F, Qiu S, Sha B, Luo M. The crystal structure of the influenza matrix protein M1 at neutral pH: M1-M1 protein interfaces can rotate in the oligomeric structures of M1. Virology. 2001;289(1):34–44. doi: 10.1006/viro.2001.1119. [DOI] [PubMed] [Google Scholar]
- 53.Liu J, Lynch PA, Chien CY, Montelione GT, Krug RM, Berman HM. Crystal structure of the unique RNA-binding domain of the influenza virus NS1 protein. Nat Struct Biol. 1997;4(11):896–899. doi: 10.1038/nsb1197-896. [DOI] [PubMed] [Google Scholar]
- 54.Wang W, Riedel K, Lynch P, Chien CY, Montelione GT, Krug RM. RNA binding by the novel helical domain of the influenza virus NS1 protein requires its dimer structure and a small number of specific basic amino acids. RNA. 1999;5(2):195–205. doi: 10.1017/s1355838299981621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yin C, Khan JA, Swapna GV, Ertekin A, Krug RM, Tong L, et al. Conserved surface features form the double-stranded RNA binding site of non-structural protein 1 (NS1) from influenza A and B viruses. J Biol Chem. 2007;282(28):20584–20592. doi: 10.1074/jbc.M611619200. [DOI] [PubMed] [Google Scholar]
- 56.Bornholdt ZA, Prasad BV. X-ray structure of influenza virus NS1 effector domain. Nat Struct Mol Biol. 2006;13(6):559–560. doi: 10.1038/nsmb1099. [DOI] [PubMed] [Google Scholar]
- 57.Qian XY, Alonso-Caplen F, Krug RM. Two functional domains of the influenza virus NS1 protein are required for regulation of nuclear export of mRNA. J Virol. 1994;68(4):2433–2441. doi: 10.1128/jvi.68.4.2433-2441.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Quinlivan M, Zamarin D, García-Sastre A, Cullinane A, Chambers T, Palese P. Attenuation of equine influenza viruses through truncations of the NS1 protein. J Virol. 2005;79(13):8431–8439. doi: 10.1128/JVI.79.13.8431-8439.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhirnov O, Isaeva EI, Konakova TE, Thoidis G, Piskareva LM, Akopova II, et al. Protection against mouse and avian influenza A strains via vaccination with a combination of conserved proteins NP, M1 and NS1. Influenza and Other Respiratory Viruses. 2007;1(2):71–79. doi: 10.1111/j.1750-2659.2007.00010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen Z, Kadowaki S, Hagiwara Y, Yoshikawa T, Sata T, Kurata T, et al. Protection against influenza B virus infection by immunization with DNA vaccines. Vaccine. 2001;19(11–12):1446–1455. doi: 10.1016/s0264-410x(00)00351-0. [DOI] [PubMed] [Google Scholar]
- 61.Chen Z, Sahashi Y, Matsuo K, Asanuma H, Takahashi H, Iwasaki T, et al. Comparison of the ability of viral protein-expressing plasmid DNAs to protect against influenza. Vaccine. 1998;16(16):1544–1549. doi: 10.1016/s0264-410x(98)00043-7. [DOI] [PubMed] [Google Scholar]
- 62.Chen Z, Yoshikawa T, Kadowaki S, Hagiwara Y, Matsuo K, Asanuma H, et al. Protection and antibody responses in different strains of mouse immunized with plasmid DNAs encoding influenza virus haemagglutinin, neuraminidase and nucleoprotein. J Gen Virol. 1999;80(Pt 10):2559–2564. doi: 10.1099/0022-1317-80-10-2559. [DOI] [PubMed] [Google Scholar]
- 63.De Filette M, Min Jou W, Birkett A, Lyons K, Schultz B, Tonkyro A, et al. Universal influenza A vaccine: optimization of M2-based constructs. Virology. 2005;337(1):149–161. doi: 10.1016/j.virol.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 64.De Filette M, Fiers W, Martens W, Birkett A, Ramne A, Lowenadler B, et al. Improved design and intranasal delivery of an M2e-based human influenza A vaccine. Vaccine. 2006;24(44–46):6597–6601. doi: 10.1016/j.vaccine.2006.05.082. [DOI] [PubMed] [Google Scholar]
- 65.Epstein S, Tumpey TM, Misplon JA, Lo CY, Cooper LA, Subbarao K, et al. DNA vaccine expressing conserved influenza virus proteins protective against H5N1 challenge infection in mice. Emerg Infect Dis. 2002;8(8):796–801. doi: 10.3201/eid0808.010476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fu T, Guan L, Friedman A, Ulmer JB, Liu MA, Donnelly JJ. Induction of MHC class I-restricted CTL response by DNA immunization with ubiquitin-influenza virus nucleoprotein fusion antigens. Vaccine. 1998;16(18):1711–1717. doi: 10.1016/s0264-410x(98)00134-0. [DOI] [PubMed] [Google Scholar]
- 67.Ulmer JB. Influenza DNA vaccines. Vaccine. 2002 Suppl. 2:S74–S76. doi: 10.1016/s0264-410x(02)00136-6. [DOI] [PubMed] [Google Scholar]
- 68.Cros JF, García-Sastre A, Palese P. An unconventional NLS is critical for the nuclear import of the influenza A virus nucleoprotein and ribonucleoprotein. Traffic. 2005;6(3):205–213. doi: 10.1111/j.1600-0854.2005.00263.x. [DOI] [PubMed] [Google Scholar]
- 69.Ozawa M, Fujii K, Muramoto Y, Yamada S, Yamayoshi S, Takada A, et al. Contributions of two nuclear localization signals of influenza A virus nucleoprotein to viral replication. J Virol. 2007;81(1):30–41. doi: 10.1128/JVI.01434-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wu WW, Sun YH, Pante N. Nuclear import of influenza A viral ribonucleoprotein complexes is mediated by two nuclear localization sequences on viral nucleoprotein. Virol J. 2007;4:49. doi: 10.1186/1743-422X-4-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jameson J, Cruz J, Ennis FA. Human cytotoxic T-lymphocyte repertoire to influenza A viruses. J Virology. 1998;72(11):8682–8689. doi: 10.1128/jvi.72.11.8682-8689.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jegerlehner A, Schmitz N, Storni T, Bachmann MF. Influenza A vaccine based on the extracellular domain of M2: weak protection mediated via antibody-dependent NK cell activity. J Immunol. 2004;172(9):5598–5605. doi: 10.4049/jimmunol.172.9.5598. [DOI] [PubMed] [Google Scholar]
- 73.Lamb R, Krug RM. Orthomyxovirida: the viruses and their replication. In: Knipe DMHP, editor. Fields Virology. 4th ed. Philadelphia, PA: Lippincott, Williams & Wilkins; 2001. pp. 1487–1532. [Google Scholar]
- 74.Wherry EJ, Golovina TN, Morrison SE, Sinnathamby G, McElhaugh MJ, Shockey DC, et al. Re-evaluating the generation of a "proteasome-independent" MHC class I-restricted CD8 T cell epitope. J Immunol. 2006;176(4):2249–2261. doi: 10.4049/jimmunol.176.4.2249. [DOI] [PubMed] [Google Scholar]
- 75.Chen Z, Matsuo K, Asanuma H, Takahashi H, Iwasaki T, Suzuki Y, et al. Enhanced protection against a lethal influenza virus challenge by immunization with both hemagglutininand neuraminidase-expressing DNAs. Vaccine. 1999;17(7–8):653–659. doi: 10.1016/s0264-410x(98)00247-3. [DOI] [PubMed] [Google Scholar]
- 76.Sette A, Peters B. Immune epitope mapping in the post-genomic era: lessons for vaccine development. Curr Opin Immunol. 2007;19(1):106–110. doi: 10.1016/j.coi.2006.11.002. [DOI] [PubMed] [Google Scholar]