

Abstract
A human TcR derived from an autoreactive T cell specific for GAD65, from a subject at high risk for autoimmune diabetes, was introduced into HLA-DR4 transgenic mice. The source of TCR was a CD4+ TH1+T cell clone which responded to an immunodominant epitope of the human islet protein GAD65, an epitope shared with both GAD65 and GAD67 in the mouse. The resulting HLA-DR4/GAD-TcR transgenic mice on a Rag2o/o/I-Abo/o/B6 background exhibited a CD4+ infiltrate into pancreatic islets that correlated with a loss of insulin in infiltrated islets. These mice also exhibited a subclinical impaired tolerance to exogenously fed glucose as assayed by an intraperitoneal glucose tolerance test. T cells containing the GAD65/67 (555–567) responsive TcR undergo strong negative selection as evidenced by a 10-fold lower thymocyte cellularity compared to non-TcR transgenic mice, and clonotype peripheral T cells represented approximately 1 percent of CD4+ T cells in Rag2 sufficient mice. Upon in vitro stimulation, GAD65/67 555–567 responsive T cells secrete IFN-γ, minimal IL2 and TNFα and no IL4, IL5, IL10, or IL17, consistent with a TH1 profile. These data demonstrate that CD4+ T cells specific for a naturally processed epitope within GAD can specifically home to pancreatic islets and lead to impaired islet beta cell function in diabetes-associated HLA-DR4 transgenic mice on the relatively non-autoimmune C57BL/6 background. The relatively slow progression and patchy insulitis are reminiscent of the chronic pre-clinical phase similar to a majority of human at-risk subjects, and models these indolent features of human T1D.
The human HLA- DQB1*0302 and DRB1*04 gene products are strongly associated with autoimmune diabetes, and are also powerful susceptibility genes predisposing to diabetes when expressed as transgenes in the absence of endogenous class II (I-Abo/o) in the relatively non-autoimmune prone C57BL/6 mouse[38].[13;37] We recently described an age-dependent spontaneous loss of tolerance to an epitope within a naturally processed region of the diabetes autoantigen GAD65 (GAD65 555–567) in the presence of the autoimmune accelerator RIP-B7 in these diabetes prone DR4 transgenic mice. The loss of tolerance to GAD65 555–567 precedes overt hyperglycemia and is associated with a loss in glucose tolerance evidenced by an intraperitoneal glucose tolerance test [12;13].
Previous studies using immunization with putative human autoantigens (primarily GAD and Insulin) in HLA transgenic mice have been used to identify, correlate, and confirm human T cell reactive antigenic epitopes that may be targets for autoreactive T cells[1;8;27;39]. The spontaneous islet autoimmunity in the B7/DR4 C57BL/6 mouse, however, offers the opportunity to explore mechanisms of T cell selection and autoreactivity in an unimmunized context. Glutamic acid decarboxylase exists in GAD65 and GAD67 isoforms, with GAD65 the predominant expressed form in human islets and GAD67 in murine islets[21]. It is important to note that GAD65 (555–567) is an ideal epitope for translational studies, as this epitope sequence is identical in all forms of mouse and human GAD (65 and 67). While cellular and humoral reactivity to glutamic acid decarboxylase 65 (GAD65) is readily detected in human T1D and diabetes at-risk subjects[9;17;30], its direct role in the pathogenesis leading to islet insulin-producing β-cell destruction in the human disease is still uncertain. Antibodies to GAD65 are one of three serum antibody markers used in determining susceptibility to T1D in genetically predisposed individuals and imply a temporal relationship between immune reactivity to GAD65 and progression to human diabetes[5;34].
The common murine model for T1D, the NOD mouse, only partially recapitulates this pattern. GAD-mediated tolerogenic protection of diabetes in NOD mice can be afforded by intrathymic injection of GAD protein [7;15;33], inoculation with GAD65 encoding vaccinia virus, [10] rat insulin promoter driven GAD65, [6] and antisense expression of GAD[41]. On the other hand MHC class I promoter-driven expression of GAD65 was shown to exacerbate disease[14]. In T cell directed studies, a GAD65 responsive cell line has been shown to induce diabetes in NOD.scid mice[43] and recent evidence indicates that GAD epitopes are capable of stimulating diabetes-inducing BDC2.5 T cells and cause diabetes in transfer studies[20;40]. However, a protective role of cellular reactivity to GAD65 was shown to delay diabetes when IFN-γ and IL-10 producing GAD65 responsive T cells were transferred in NOD mice from either T cell transgenic mice[22;32], a T cell clone[42] or a T cell line[24]. Unaltered diabetes progression in NOD mice has also been observed in retrogenic expression of other I-Ag7-restricted GAD T cell receptors [4].
In order to assess the characteristics of anti-islet T cell specificities which are prevalent in human T1D we have transgenically expressed a GAD65 (555–567) responsive human T cell receptor (TcR), derived from a diabetes at-risk individual, in DR4 transgenic mice on the C57BL/6 background. We report here that T cells in DR4 mice expressing TcR transgenes specific for GAD65 are strongly negatively selected in the thymus and are limited in numbers in the periphery organs. The percentage of FoxP3+ cells among CD4+ T cells in peripheral organs is about 3-fold greater than non-TcR transgenic mice. TcR transgenic mice were normoglycemic to 40 weeks of age, possibly related to the increase in selected regulatory components. Notably, mice transgenic for this TcR on a Rag2 deficient background are also normal for blood glucose but do exhibit insulitis at around 25 weeks of age that is correlated with a loss in glucose tolerance in an intra-peritoneal glucose tolerance test and a loss in immunoreactive insulin in infiltrated islets. Thus, an anti-GAD T cell specificity associated with human T1D is sufficient to elicit insulitis and impair glucose tolerance in a HLA-transgenic murine model, even in the context of the relatively autoimmune disease-resistant C57BL/6 background. In contrast to the autoimmune-prone NOD model, disease is indolent, insulitis is patchy, and lack of progression to overt diabetes is associated with evidence of T cell regulation, features which may correspond to a large segment of the human T1D and at-risk population.
Research Design and Methods
Mice
DR0404-IE mice (DR4) were generated as previously described[13]. These C57BL/6 I-Abo/o mice express a human-mouse chimeric class II molecule in which the TCR interacting and peptide binding domains of mouse I-E (domains α1 and β1, exon 2 in both genes) have been replaced with the α1 and β1 domains from DRA1*0101 and DRB1*0404 respectively. Retention of the murine α2 and β2 domains allows for the cognate murine CD4-murine MHC interaction. [18].
The GAD65 (555–567) responsive human CD4+ Vα12.1/Vβ5.1 T cell clone 164 was cloned from an HLA DR4 diabetes at-risk individual as previously described[30]. Human-mouse chimeric TcR transgenes were constructed by subcloning PCR amplified regions encoding rearranged VαJα and VβDβJβ domains from the human clone derived TcR sequences into pTαcass and pTβcass TcR transgenic vectors respectively [23]. TcR transgenic vectors pTαcass and pTβcass contain the natural mouse TcR α and β promoter/enhancer elements and mouse Cα and Cβ constant region respectively. DNA injection into C57BL/6/I-Abo/o mouse embryos was performed at the University of Washington (Seattle, WA) in the Comparative Medicine animal facility. Founder mice containing the GAD65 TcR transgene were then crossed onto DR0404-IE mice to generate DR4/164 mice. Additional crosses were made onto Rag2 KO mice.
Blood glucose was performed via saphenous veins bleeds using a One-Touch FastTake glucometer (LifeScan, Milpitas, CA. All animal work was approved by the Benaroya Research Institute (BRI) Animal Care and Use Committee (ACUC) and animals were housed in the BRI AAALAC-accredited animal facility. For intraperitoneal glucose tolerance tests (IPGTT) mice were fasted (given water only) for 6 hours. At the end of 6 hours mice were injected intraperitoneal with 1.0 mg/ml D-glucose (stock solution in PBS) at a dose of 1g/kg body weight). Saphenous blood glucose readings were taken at 0, 15, 30, and 60 minute time points post injection.
Tissue processing and Flow cytometry
Thymus, spleen, and lymph node tissues were processed into single cell suspensions by gently pressing through 0.40 µm cell strainers (BD-Falcon REF 352340, Bedford MD) using the rubber end of a 1 ml tuberculin syringe in DMEM-10 media (DMEM cat 11965-092 (Gibco, Rockville MD.) supplemented with 10% FBS (Hyclone, Logan Utah), 100ug/ml Penicillin, 100U/ml Streptomycin, 50uM βme, 2mM glutamine and 1mM sodium pyruvate (Gibco, Rockville MD)). Cell suspensions were centrifuged at 200g for 10 minutes and resuspended in DMEM-10 media. Splenic RBC were lysed using ACK lysis buffer[2] for 5’ at 37oC at which time ~25 ml of media was added and cells spun down (200g). The following chromophore-labeled antibodies were used in flow cytometric analysis: anti-mouse CD4 (clone RM4-5), CD8 (clone 53-6.7), CD25 (clone PC61), CD62L(Mel-14), and CD44(IM7, BD-Pharmingen, San Jose, CA), and anti-human Vβ5.1-PE (clone IMMU 157 Immunotech-Coulter, Miami, FL) and Vα12.1-FITC (clone 6D6 Endogen Woburn, MA). FACS samples were stained in media on ice for 45 minutes, washed once, and resuspended in FACS stain buffer (PBS containing 1% FBS, 0.1% Na-azide) before being run on a FACS Caliber or LSR II flow cytometer (Becton Dickinson). Internal staining of cells for FoxP3 was performed using eBioscience kit (FJK.16a Ab, San Diego, CA) according to manufacturer’s instructions.
Pancreatic tissues were in either: 1.) fixed in phosphate buffered formalin prior to paraffin embedding for H&E staining or insulin staining or 2.) frozen in Tissue-Tek OCT embedding media (Sakura Finetek, Torrance, CA) for immunofluorescence. For immunofluorescence staining of frozen tissues, 6 µM tissue slices were fixed for 10’ in 4°C acetone and either air dried and stored at −20°C or stained directly. Frozen tissues section were blocked, stained, and washed in PBS containing 0.1%NaN3/1%FBS/2% Horse serum. The following antibodies were used at 1:100 dilution CD4-Alexa-fluor 488 (MCD0420), CD8-Alexa-fluor 488 (MCD0820), control Alexa-fluor 488 (R2a20, Caltag, Burlingame, CA). Islet insulin was detected with primary guinea-pig polyclonal anti-insulin (1:100 dilution, Abcam Ab7842-500 Cambridge, MA) and a secondary goat anti-guinea-pig Alexa-fluor 568 (1:100 dilution, Molecular Probes, Eugene, Or) Immunofluorescence was detected on a Leica DM IRB microscope. For islet infiltrate scoring, at least 8 islets were viewed for each mouse and H&E stained islets we scored as follows: 0-no infiltrate, 1-less than 33% infiltrated, 2- less than 66% infiltrated, and 3- greater than 66% infiltrated.
Proliferation assays
Single cell suspensions of lymph node cells (LNC) from inguinal, mesenteric and brachial lymph nodes and spleen cells were prepared by gently pressing through 0.40 um nylon cell strainers (BD-Falcon REF 352340, Bedford MD) in Hanks buffer (Gibco, Rockville MD) and spun down (1000 rpm, 200g). Splenic RBC were lysed using ACK lysis buffer[2] for 5’ at 37oC at which time ~25 ml of media was added and cells spun down (200g). Splenocytes were resuspended in DMEM-10 (DMEM cat 11965-092 (Gibco, Rockville MD.) supplemented with 10% FBS (Hyclone, Logan Utah), 100ug/ml Penicillin, 100U/ml Streptomycin, 50uM βme, 2mM glutamine and 1mM sodium pyruvate (Gibco, Rockville MD)). In lymph node proliferation assays 1e5 lymph node cells were culture with 2e5 3000 Rads Cs-g irradiated splenocytes. Supernatants for cytokine analysis were taken (50 ul) at 48 hrs. and 1 uCi/well of 3H-thymidine was added at 72 hours. Thymidine incorporation was assayed at 96 hours using a liquid scintillation counter analyzed on a scintillation counter (Wallac-Perkin/Elmer Life Sciences, Boston MD.) at 96 hours. Splenocyte responses were measured in the same manner using 5×105 splenocytes per well.
Cytokine analysis
Cytokines IL-2, IL-4, IL-5, TNF-α, and IFN-γ were assayed using a Mouse Th1/Th2 Cytokine CBA kit (BD Bioscience, San Diego, CA 92121, cat # 551287). IL-10 was assayed using a BD OptEIA mouse IL-10 Elisa Set (BD Bioscience, San Diego, CA 92121 cat # 555252) and IL-17A was assayed using an IL-17A Elisa kit (eBioscience, San Diego, CA 92121, cat # 88-7147-22). Supernatants from triplicate proliferation wells (50 µl/well) were combined for cytokine analysis with 50 µl used for CBA analysis and 50 µl each for IL-10 and IL-17A Elisa.
Results
GAD65 (555–567) is a minimal stimulating epitope within a naturally processed immunodominant epitope (GAD65 552–572) within the diabetes autoantigen GAD65 [25;27]. In studying T1D in human diabetes-correlated HLA transgenic mice, the MHC DR4-binding GAD65 (555–567) epitope is an autologous antigen as mouse and human sequences in GAD65 and GAD67 are all identical[11]. A human CD4+ Vα12.1/Vβ5.1 TcR (Arden nomenclature[3]) T cell clone (164) responsive to GAD65 (555–567) was derived from PBMC of an autoantibody-positive diabetes at-risk individual by in vitro stimulation with a GAD65 (555–567) superagonist APL peptide and single cell sorted from a CD4Hi/CD25+ activated population[30]. 164 TcR transgenic mice were generated using murine TcR cassettes pTαcass and pTβcass[23] in which the variable regions of the mouse TcR were substituted with the human sequences from the 164 human clone TcR. Purified DNA was microinjected directly into C57BL/6 embryos. TcR positive founder mice were crossed onto (I-Abo/o) C57BL/6 DR4 HLA transgenic mice.
Thymic and peripheral cellularity in GAD65 555–567 responsive TcR transgenic mice
Thymocyte cellularity in DR4/164 TcR transgenic mice is severely reduced compared with non-TcR mice, indicating a threshold in negative selection has been crossed in selection of the 164 TcR. (Fig. 1A). In wild type DR4 mice the CD4:CD8 single positive ratio in the thymus is approximately 4:1, the CD4:CD8 ratio in the TcR negative selecting DR4/164 mouse is 1:3.4 and the percentage of CD8 single positive cells in the thymus is ~20% or nearly 10-fold above that seen in wild type DR4 mice (Fig. 1D). A similar type of CD8 skewing has been observed on other self antigen specific TcR transgenic mice[28;29;35]. The presence of the GAD65 (555–567) specific TcR transgenes in DR4 mice results in an increase in the thymic CD4−/CD8− population from less than 5% in wild type DR4 mice to nearly 60% in DR4/164 mice (Fig. 1D).
Figure 1.
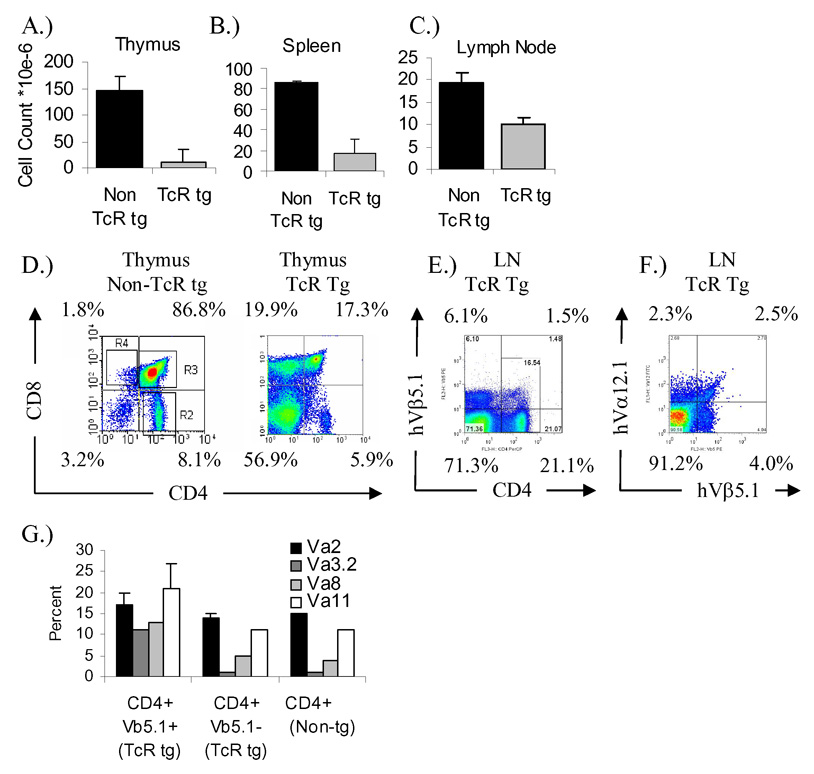
Tissue cellularity and GAD TcR expression in DR4/164 GAD65 555–567 specific TcR transgenic mice. Cellularity for Thymus (A), Spleen (B), and Lymph nodes(C) represent the average counts (n=3 mice each) for Non TcR transgenic (Non TcR tg) and TcR transgenic mice (TcR tg) at 8 weeks of age. CD4 vs. CD8 profiles in thymus of DR4 and DR4/164 GAD TcR mice (D). Expression of human TcR Vβ5.1 transgene on CD4+ lymph node cells from the DR4/164 mouse is shown (E) along with clonotypic expression of the Vα12.1/Vβ5.1 TcR on CD4-gated lymph node cells (F). CD4+ human Vβ5.1+ transgenic T cells express an increase in the use of endogenous mouse TcR Vα compared to Vβ5.1− T cells (G).
A result of the extensive negative selection in the thymus of DR4/164 mice is reflected in the peripheral organs where both splenic and lymph node cellularity are well below non-TcR DR4 mouse levels (Figs. 1B and 1C). In the peripheral lymph nodes the expression of the Vβ5.1 transgene is found on about 6 % of CD4+ T cells (Fig. 1E) and these CD4+/Vβ5.1+ T cells display an increase usage of endogenous Vα TcR compared to CD4+/Vβ5.1− cells (Fig. 1G). The percentage of peripheral CD4+ T cells expressing the clonotypic Vα12.1/Vβ5.1 transgene is around 2% (Fig. 1F).
164 TcR transgenic mice have an increased percentage of FoxP3 positive T regulatory cells
Several other models using TcR transgenic mice have shown an increase in the percentage of FoxP3 expressing cells in the population of T cells responding to antigens which are introduced by transgenesis, and are therefore surrogates for self antigens[19;36]. Consistent with these observations, the percentage of FoxP3 positive cells in the CD4+CD25+ subset of DR4/164 mice is greater in DR4/164 mice compared to non-TcR transgenic DR4 mice (Fig. 2B). However, while an increase in the percentage of CD4+/FoxP3+ cells among the CD4+ subset is observed in inguinal and pancreatic lymph nodes and also in spleen, the increase is only observed in the non-TcR transgenic Vβ5.1− population and not the Vb5.1+/CD4+ cells.
Figure 2.
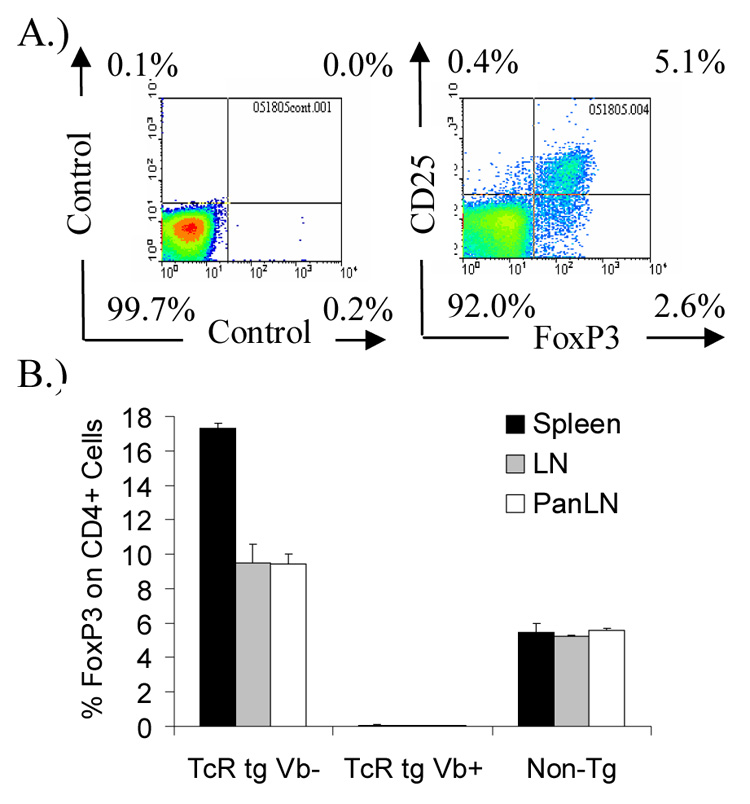
Intracellular FoxP3 expression on CD4+ T cells in GAD65 555–567 TcR transgenic and non-transgenic DR4 mice. FoxP3 expression in lymph node cells from DR4/164 TcR transgenic mice showing a correlation of FoxP3 expression with CD25 expression on CD4+ gated cells (A). Average percentages from 3 mice (8 wks of age) of CD4+ FoxP3+ expressing cells of all CD4+ cells (B). Percentages are shown for CD4+/Vβ5.1+/FoxP3+ (Tg Vβ+) and CD4+/Vβ5.1−/FoxP3+ (TgVβ−) for DR4/164 GAD-TcR mice and for non-TcR transgenic mice (Non-tg).
164 TcR T cells are GAD65 responsive and exhibit a TH1 phenotype
Proliferation and cytokine production of T cells from DR4/164/Rag2o/o mice in response to GAD65 (555–567) and a control peptide derived from diabetes autoantigen islet-specific glucose-6-phosphatase subunit related protein (IGRP 247–258) are shown in Figure. 3. Antigen specific response to GAD65 (555–567) is seen as low as 0.01 µg/ml (6.8 nM, lowest concentration tested) but not to control DR4-binding IGRP (247–258) peptide. A cytokine analysis in response to GAD65 (555–567) stimulation showed that these cells secrete IFN-γ with minimal TNF-α and IL-2 and no IL10, IL-17, IL-4, or IL-5 and is indicative of a TH1 type cell cytokine profile. The same cytokine profile is seen in Rag2 sufficient mice (data not shown) but proliferative values and detectable cytokines are lower, likely due to the large percentage of non-clonotypic T cells and FOXP3+ cells selected in Rag2 sufficient mice (Fig. 1E and Fig. 2B).
Figure 3.
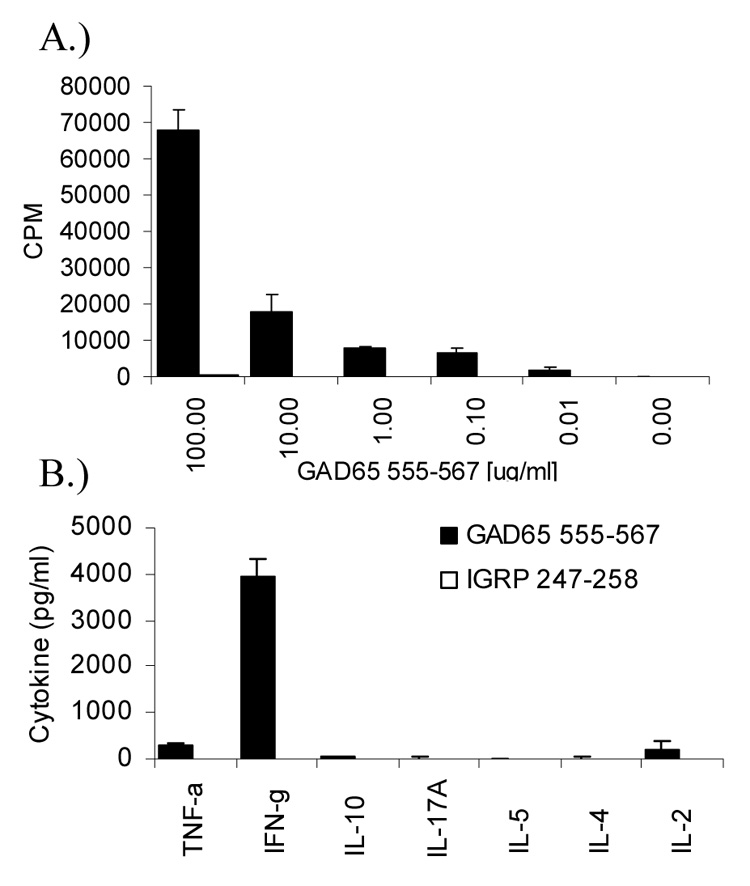
Antigen-specific dose-response and cytokine analysis of in vitro stimulated 164 GAD TcR T cells. Splenocytes from Dw14/164/Rag2o/o mice (2e5) were stimulated with increasing amount of GAD65 555–567 or control IGRP 247–258 peptide for 96 hrs. 3H-thymidine was added at 72 hrs and incorporation was assayed by scintillation counting (A). Cytokines (pg/ml) from supernatants from proliferation assays (at 100 µg/ml antigen) taken at 72 hours (B). Experiment was repeated 3 times with similar results.
GAD65 555–567 TcR transgenic mice on a Rag2o/o background have insulitis
Blood glucose levels in DR4/164 Rag2 sufficient mice (n=20) were monitored up to 40 weeks of age, and no mice showed overt hyperglycemia. Histological examination of pancreata from DR4/164 Rag2 sufficient mice also appeared normal with no indication of an islet infiltrate (data not shown). To increase the expression of the clonotypic transgenic GAD TcR, DR4/164 mice were crossed onto Rag2 knockout mice to generate DR4/164/Rag2o/o mice. As previously seen for Rag2+/+ animals (Fig. 1D), concomitant with the strong negative selection of the 164 TcR in these mice on the Rag2o/o background, a peripheral skewing of the clonotypic TcR towards the CD8+ single positive T cell lineage was observed (Fig. 4). However in contrast to the CD8+ T cells, the CD3+/CD4+ T cells from DR4/164/Rag2o/o mice display an activated CD44Hi/CD62L− phenotype (Fig. 4). In addition, unlike the lack of FoxP3 expression on Vβ5.1+/CD4+ T cells in DR4/164/Rag2+/+ mice (Fig. 2B), a fraction of peripheral CD4+ cells in DR4/164 Rag2o/o do express FoxP3 (2.5%).
Figure 4.
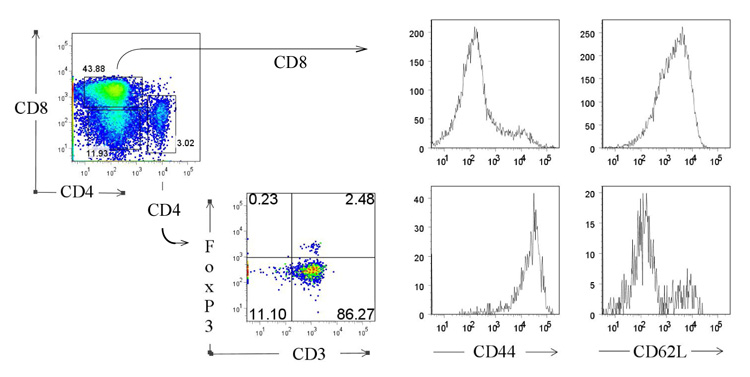
TcR transgenic CD4+ cells in DR4/164/Rag2o/o mice exhibit an activated phenotype. CD4+ lymph node T cells, but not CD8+ cells from DR4/164/Rag2o/o mice (10 weeks of age) are CD44Hi and CD62L−.
Unlike DR4/164/Rag2+/+ mice, DR4/164/Rag2o/o mice exhibit an islet-specific cellular infiltrate into the pancreas beginning at about 25 weeks of age (Fig. 5A). The islet-specific infiltrate is observed primarily in female mice (Figs. 5B and 5C) and is not seen in other organs (supplemental data S1). Immunofluorescence staining of the islets showed CD4+ staining indicative of the 164 T cells infiltrating into these islets (Fig. 6A). CD8+ cells were not detected in the infiltrated islets (data not shown). Correlating with the cellular infiltrate in DR4/164/Rag2o/o islets is a loss of detectable insulin staining in most, but not all, islets (Fig. 6B). To assay if the loss of insulin staining in T cell infiltrated islets is reflected in pancreatic function we performed an intra-peritoneal glucose tolerance test (IPGTT) on DR4/164/Rag2o/o mice. As shown in Figure 7, DR4/164/Rag2o/o mice are impaired in their response to injected glucose at a time when they display a patchy T cell infiltrate into the islets with loss of immunoreactive insulin in these islets.
Figure 5.
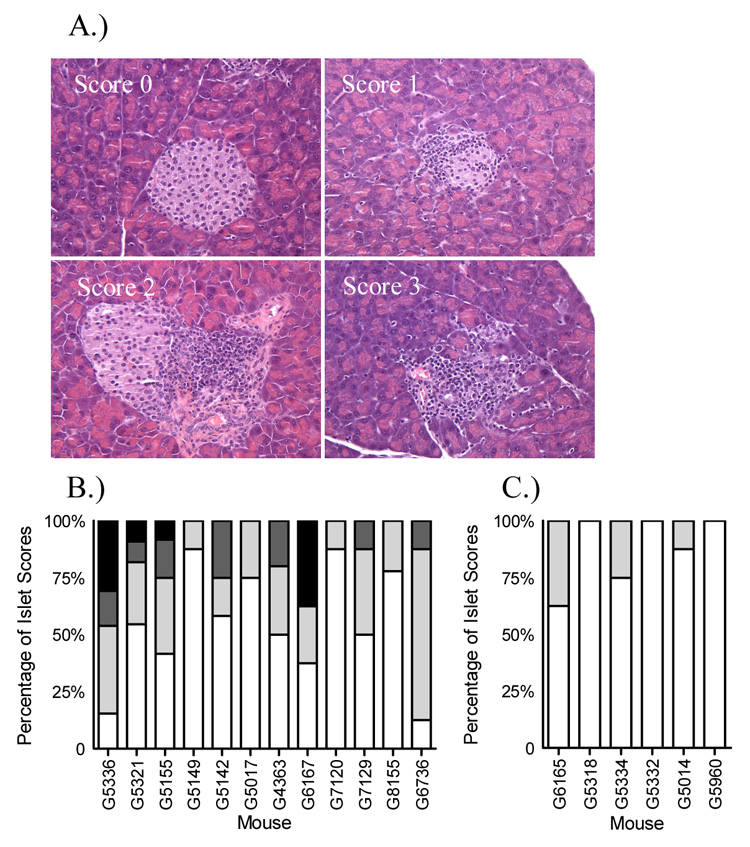
Islet cell infiltrate in DR4/164/Rag2o/o TcR transgenic mice. H & E staining on formalin fixed paraffin embedded pancreatic tissue from a 28 week old female DR4/164/Rag2o/o mouse (A). Islets were scored as 0-no infiltrate, 1-less than 33%, 2-less than 66%, or 3-greater than 66%. A summary of islet infiltrate scores for 12 female and 6 male DR4/164/Rag2o/o mice is shown in (B) and (C) respectively. Islets were scored as 0-no infiltrate (white bars), 1-less than 33% (light grey bars), 2-less than 66% (dark grey bars), or 3-greater than 66% (black bars).
Figure 6.
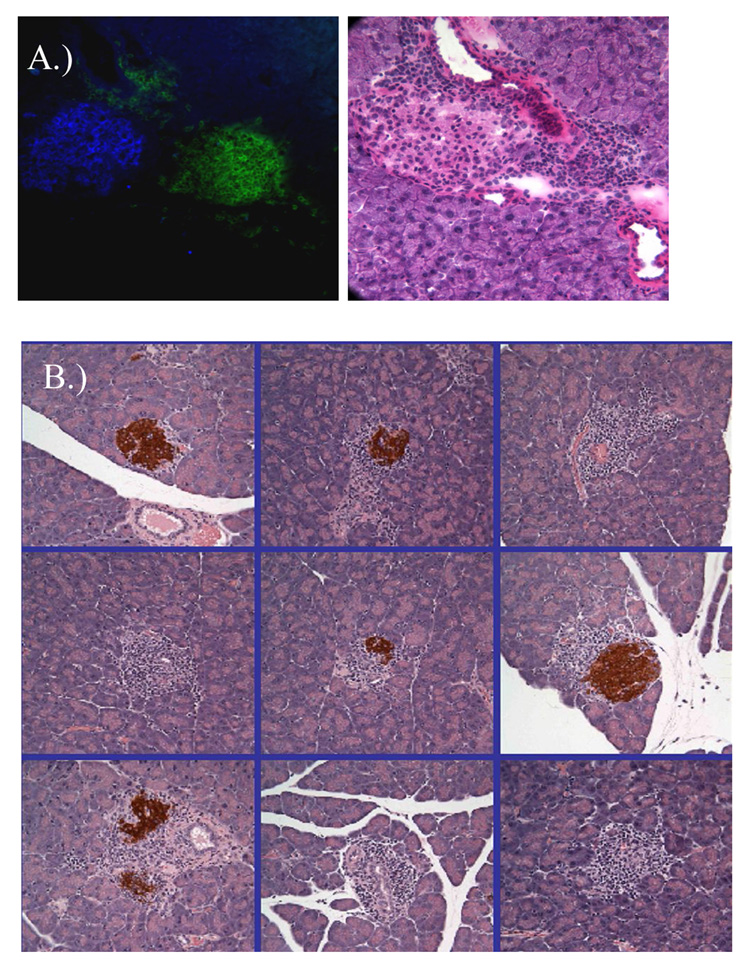
Detection of CD4+ cells and loss of insulin staining in infiltrated islets from DR4/164/Rag2o/o mice. Immunofluorescence staining for insulin (blue) and CD4 (green) in an infiltrated islet from a 28 week old female DR4/164/Rag2o/o mouse (A). Immunohistochemistry staining for insulin (brown) in infiltrated islets from a female DR4/164/Rag2o/o mouse (B).
Figure 7.
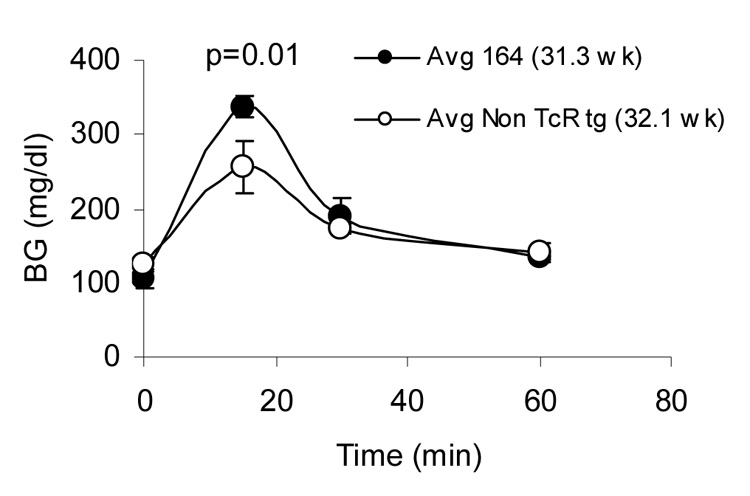
Impaired response to exogenous glucose in DR4/164/Rag2o/o mice. DR4/164/Rag2o/o (164, filled circles) and DR4/Rag2o/o (Non TcR tg, open circles) mice were fasted for 6 hours prior to glucose injection and blood glucose monitoring (see methods). 4 mice 30 weeks of age were used in each group.
Discussion
HLA-DR4 MHC transgenic mice on a relatively non-autoimmune prone C57BL/6 background were evaluated for propensity to autoimmune diabetes by introduction of self-antigen specific GAD65 (555–567) responsive TcR transgenes. The TcR sequence originated from a human T cell clone (164) derived from a diabetes at-risk individual and was chosen because: 1.) the GAD65 (555–567) sequence is identical to both GAD65 and GAD67 forms in both mice and humans with the later form of GAD being the most dominant in the murine pancreas[21]and 2.) the minimal stimulating GAD65 (555–567) sequence is within a naturally processed epitope in both mouse and man [25;27]. These 164 transgenic T cells in DR4 mice were strongly negatively selected against and weakly populate the secondary lymphoid organs. Nevertheless, peripheral transgenic T cells are antigen-specific for GAD65 (555–567) and display a TH1 phenotype by expressing IFN-γ with minimal TNF-α and IL-2 and no IL10, IL-17, IL-4, or IL-5 upon in vitro challenge.
While GAD65 (555–567) TcR transgenic mice on HLA DR4 Rag2 sufficient background did not show evidence for loss in pancreatic function, GAD65 TcR transgenic mice on a Rag2o/o background (DR4/164/Rag2o/o) beginning at about 25 weeks of age showed signs of impaired islet function as demonstrated by a CD4+ T cell islet-specific infiltrate into the pancreas that was correlated with a loss in islet insulin staining and an abnormal response to an intraperitoneal glucose tolerance test. Thus, the GAD65 (555–567) specificity is sufficient for initiation of insulitis, resulting in metabolic compromise characteristic of pre-diabetes. Since this phenotype occurs on the relatively autoimmune resistant C57BL/6 background, it suggests that T cell autoreactivity to GAD65 is sufficient for early immune activation associated with early autoimmunity to pancreatic islets, but that additional autoimmunity predisposition is likely to be necessary for full disease penetrance. This phenotype is distinct from that described by Tarbell, et al ([32]) and Kim et al ([22]) in which GAD65 reactive I-Ag7-restricted TcR transgenic NOD mice appeared to the protected from diabetes and correlated with IL-10 and IFN-γ being secreted by CD4+ T cells. A determination of whether the protection in those GAD TcR models was mediated by T cell produced IL-10 was not addressed. Other islet responsive (including GAD) IL-10 producing T cells have been shown to protect from diabetes in transfer models[16;24;42] with the later study showing abrogating effects by blocking IL-10 signaling. In contrast the insulitic behavior of 164 GAD T cells in DR4 mice shown here do not produce IL-10.
The FOXP3 expression profiles differed in DR4/164 TcR Rag2 sufficient and Rag2−/− mice. In the Rag2-sufficient animals, a substantial population of transgene-negative FOXP3+ T cells was present in the periphery, and in spleen approximately 3 time the frequency found in the non-TcR transgenic DR4 mice. It is possible that these cells arise as a compensatory mechanism suppressing the subpopulation of transgene-positive cells; in any event, the presence of these FOXP3+ cells correlated with a lack of insulitis or hyperglycemia. In the Rag2 deficient mice, we were therefore surprised to find a small percentage of clonotypic CD4+/FoxP3+ cells (Fig. 4). Studies are underway to evaluate the functional suppressive capacity of this small population in animals with insulitis, but lacking overt diabetes.
The insulitis phenotype of GAD TCR and HLA transgenic mice supports the hypothesis that T cell reactivity to GAD restricted by a diabetes-associated human MHC molecule plays a role in diabetes or pre-diabetes pathogenesis. These mice had impaired glucose tolerance but did not become hyperglycemic; potential reasons could be that other T cells specificities (or B cells) are required for disease progression, or alternatively that regulatory mechanisms in the context of the C56Bl/6 genome are sufficient to reduce penetrance. A requirement for B cells in non-TcR transgenic NOD diabetes has been established[26;31]. Mice are presently being crossed onto TcR Cαo/o mice to determine if the presence of B cells in the presence of clonotypic 164 T cells will lead to hyperglycemia. The results from this initial study indicate that TH1 GAD specific T cells can spontaneously migrate specifically to pancreatic islets in DR4 humanized mice on a relatively non-autoimmune background and are capable of mediating a loss in beta cell function.
Supplementary Material
Acknowledgements
We thank Jane Buckner MD for critical review of the manuscript and Helena Reijonen PhD for providing the human T cell clone used in generating the TcR transgenic mice. This research was supported by NIH grant AI050864 and USAMRAA grant PR064261.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Abraham RS, Wilson SB, de SN, Jr, Strominger JL, Munn SR, David CS. NOD background genes influence T cell responses to GAD 65 in HLA-DQ8 transgenic mice. Hum. Immunol. 1999;60:583–590. doi: 10.1016/s0198-8859(99)00057-9. [DOI] [PubMed] [Google Scholar]
- 2.Kruisbeek Ada M. Isolation and Fractionation of Mononuclear Cell Populations. In: Coligan JE, Kruisbeek AM, Margulies DH, Shevach EM, Strober W, editors. Current Protocols in Immunology. John Wiley & Sons, Inc.; 2000. pp. 3.1.1–3.1.5. [Google Scholar]
- 3.Arden B, Clark SP, Kabelitz D, Mak TW. Human T-cell receptor variable gene segment families. Immunogenetics. 1995;42:455–500. doi: 10.1007/BF00172176. [DOI] [PubMed] [Google Scholar]
- 4.Arnold PY, Burton AR, Vignali DA. Diabetes incidence is unaltered in glutamate decarboxylase 65-specific TCR retrogenic nonobese diabetic mice: generation by retroviral-mediated stem cell gene transfer. J. Immunol. 2004;173:3103–3111. doi: 10.4049/jimmunol.173.5.3103. [DOI] [PubMed] [Google Scholar]
- 5.Bingley PJ, Bonifacio E, Williams AJ, Genovese S, Bottazzo GF, Gale EA. Prediction of IDDM in the general population: strategies based on combinations of autoantibody markers. Diabetes. 1997;46:1701–1710. doi: 10.2337/diab.46.11.1701. [DOI] [PubMed] [Google Scholar]
- 6.Bridgett M, Cetkovic-Cvrlje M, O'Rourke R, Shi Y, Narayanswami S, Lambert J, Ramiya V, Baekkeskov S, Leiter EH. Differential protection in two transgenic lines of NOD/Lt mice hyperexpressing the autoantigen GAD65 in pancreatic beta-cells. Diabetes. 1998;47:1848–1856. doi: 10.2337/diabetes.47.12.1848. [DOI] [PubMed] [Google Scholar]
- 7.Cetkovic-Cvrlje M, Gerling IC, Muir A, Atkinson MA, Elliott JF, Leiter EH. Retardation or acceleration of diabetes in NOD/Lt mice mediated by intrathymic administration of candidate beta-cell antigens. Diabetes. 1997;46:1975–1982. doi: 10.2337/diab.46.12.1975. [DOI] [PubMed] [Google Scholar]
- 8.Congia M, Patel S, Cope AP, De Virgiliis S, Sonderstrup G. T cell epitopes of insulin defined in HLA-DR4 transgenic mice are derived from preproinsulin and proinsulin. Proc. Natl. Acad. Sci. U. S. A. 1998;95:3833–3838. doi: 10.1073/pnas.95.7.3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Danke NA, Yang J, Greenbaum C, Kwok WW. Comparative study of GAD65-specific CD4+ T cells in healthy and type 1 diabetic subjects. J. Autoimmun. 2005;25:303–311. doi: 10.1016/j.jaut.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 10.Denes B, Yu J, Fodor N, Takatsy Z, Fodor I, Langridge WH. Suppression of hyperglycemia in NOD mice after inoculation with recombinant vaccinia viruses. Mol. Biotechnol. 2006;34:317–327. doi: 10.1385/MB:34:3:317. [DOI] [PubMed] [Google Scholar]
- 11.Gebe JA, Falk BA, Rock KA, Kochik SA, Heninger AK, Reijonen H, Kwok WW, Nepom GT. Low-avidity recognition by CD4+ T cells directed to self-antigens. Eur. J. Immunol. 2003;33:1409–1417. doi: 10.1002/eji.200323871. [DOI] [PubMed] [Google Scholar]
- 12.Gebe JA, Falk BA, Unrath KA, Nepom GT. Autoreactive T cells in a Partially Humanized Murine Model of T1D. Ann. N. Y. Acad. Sci. 2007 doi: 10.1196/annals.1394.001. [DOI] [PubMed] [Google Scholar]
- 13.Gebe JA, Unrath KA, Falk BA, Ito K, Wen L, Daniels TL, Lernmark A, Nepom GT. Age-dependent loss of tolerance to an immunodominant epitope of glutamic acid decarboxylase in diabetic-prone RIP-B7/DR4 mice. Clin. Immunol. 2006;121:294–304. doi: 10.1016/j.clim.2006.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geng L, Solimena M, Flavell RA, Sherwin RS, Hayday AC. Widespread expression of an autoantigen-GAD65 transgene does not tolerize non-obese diabetic mice and can exacerbate disease. Proc. Natl. Acad. Sci. U. S. A. 1998;95:10055–10060. doi: 10.1073/pnas.95.17.10055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gerling IC, Atkinson MA, Leiter EH. The thymus as a site for evaluating the potency of candidate beta cell autoantigens in NOD mice. J. Autoimmun. 1994;7:851–858. doi: 10.1006/jaut.1994.1068. [DOI] [PubMed] [Google Scholar]
- 16.Han HS, Jun HS, Utsugi T, Yoon JW. A new type of CD4+ suppressor T cell completely prevents spontaneous autoimmune diabetes and recurrent diabetes in syngeneic islet-transplanted NOD mice. J. Autoimmun. 1996;9:331–339. doi: 10.1006/jaut.1996.0045. [DOI] [PubMed] [Google Scholar]
- 17.Reijonen Helena, Mallone Roberto, Heninger Anne-Kristin, Laughlin Else M, Kochik Sharon A, Falk Ben, Kwok William W, Greenbaum Carla, Nepom Gerald T. GAD65-Specific CD4+ T-Cells with High Antigen Avidity Are Prevalent in Peripheral Blood of Patients with Type 1 Diabetes. Diabetes. 2004;53 doi: 10.2337/diabetes.53.8.1987. [DOI] [PubMed] [Google Scholar]
- 18.Ito K, Bian HJ, Molina M, Han J, Magram J, Saar E, Belunis C, Bolin DR, Arceo R, Campbell R, Falcioni F, Vidovic D, Hammer J, Nagy ZA. HLA-DR4-IE chimeric class II transgenic, murine class II-deficient mice are susceptible to experimental allergic encephalomyelitis. J. Exp. Med. 1996;183:2635–2644. doi: 10.1084/jem.183.6.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jordan MS, Boesteanu A, Reed AJ, Petrone AL, Holenbeck AE, Lerman MA, Naji A, Caton AJ. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat. Immunol. 2001;2:301–306. doi: 10.1038/86302. [DOI] [PubMed] [Google Scholar]
- 20.Judkowski V, Pinilla C, Schroder K, Tucker L, Sarvetnick N, Wilson DB. Identification of MHC class II-restricted peptide ligands, including a glutamic acid decarboxylase 65 sequence, that stimulate diabetogenic T cells from transgenic BDC2.5 nonobese diabetic mice. J. Immunol. 2001;166:908–917. doi: 10.4049/jimmunol.166.2.908. [DOI] [PubMed] [Google Scholar]
- 21.Kim J, Richter W, Aanstoot HJ, Shi Y, Fu Q, Rajotte R, Warnock G, Baekkeskov S. Differential expression of GAD65 and GAD67 in human, rat, and mouse pancreatic islets. Diabetes. 1993;42:1799–1808. doi: 10.2337/diab.42.12.1799. [DOI] [PubMed] [Google Scholar]
- 22.Kim SK, Tarbell KV, Sanna M, Vadeboncoeur M, Warganich T, Lee M, Davis M, McDevitt HO. Prevention of type I diabetes transfer by glutamic acid decarboxylase 65 peptide 206-220-specific T cells. Proc. Natl. Acad. Sci. U. S. A. 2004;101:14204–14209. doi: 10.1073/pnas.0405500101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kouskoff V, Signorelli K, Benoist C, Mathis D. Cassette vectors directing expression of T cell receptor genes in transgenic mice. J. Immunol. Methods. 1995;180:273–280. doi: 10.1016/0022-1759(95)00002-r. [DOI] [PubMed] [Google Scholar]
- 24.Maron R, Melican NS, Weiner HL. Regulatory Th2-type T cell lines against insulin and GAD peptides derived from orally- and nasally-treated NOD mice suppress diabetes. J. Autoimmun. 1999;12:251–258. doi: 10.1006/jaut.1999.0278. [DOI] [PubMed] [Google Scholar]
- 25.Nepom GT, Lippolis JD, White FM, Masewicz S, Marto JA, Herman A, Luckey CJ, Falk B, Shabanowitz J, Hunt DF, Engelhard VH, Nepom BS. Identification and modulation of a naturally processed T cell epitope from the diabetes-associated autoantigen human glutamic acid decarboxylase 65 (hGAD65) Proc Natl Acad Sci U. S. A. 2001;98:1763–1768. doi: 10.1073/pnas.98.4.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noorchashm H, Noorchashm N, Kern J, Rostami SY, Barker CF, Naji A. B-cells are required for the initiation of insulitis and sialitis in nonobese diabetic mice. Diabetes. 1997;46:941–946. doi: 10.2337/diab.46.6.941. [DOI] [PubMed] [Google Scholar]
- 27.Patel SD, Cope AP, Congia M, Chen TT, Kim E, Fugger L, Wherrett D, Sonderstrup-McDevitt G. Identification of immunodominant T cell epitopes of human glutamic acid decarboxylase 65 by using HLA-DR(alpha1*0101,beta1*0401) transgenic mice. Proc Natl Acad Sci U. S. A. 1997;94:8082–8087. doi: 10.1073/pnas.94.15.8082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quaratino S, Badami E, Pang YY, Bartok I, Dyson J, Kioussis D, Londei M, Maiuri L. Degenerate self-reactive human T-cell receptor causes spontaneous autoimmune disease in mice. Nat. Med. 2004;10:920–926. doi: 10.1038/nm1092. [DOI] [PubMed] [Google Scholar]
- 29.Ranheim EA, Tarbell KV, Krogsgaard M, Mallet-Designe V, Teyton L, McDevitt HO, Weissman IL. Selection of aberrant class II restricted CD8+ T cells in NOD mice expressing a glutamic acid decarboxylase (GAD)65-specific T cell receptor transgene. Autoimmunity. 2004;37:555–567. doi: 10.1080/08916930400020545. [DOI] [PubMed] [Google Scholar]
- 30.Reijonen H, Novak EJ, Kochik S, Heninger A, Liu A, Kwok W, Nepom G. Detection of GAD65 specific T-cells by MHC class II multimers in type 1 diabetes patients and at-risk subjects. Diabetes. 2002;51:1375–1382. doi: 10.2337/diabetes.51.5.1375. [DOI] [PubMed] [Google Scholar]
- 31.Serreze DV, Chapman HD, Varnum DS, Hanson MS, Reifsnyder PC, Richard SD, Fleming SA, Leiter EH, Shultz LD. B lymphocytes are essential for the initiation of T cell-mediated autoimmune diabetes: analysis of a new "speed congenic" stock of NOD.Ig mu null mice. J. Exp. Med. 1996;184:2049–2053. doi: 10.1084/jem.184.5.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tarbell KV, Lee M, Ranheim E, Chao CC, Sanna M, Kim SK, Dickie P, Teyton L, Davis M, McDevitt H. CD4(+) T cells from glutamic acid decarboxylase (GAD)65-specific T cell receptor transgenic mice are not diabetogenic and can delay diabetes transfer. J. Exp. Med. 2002;196:481–492. doi: 10.1084/jem.20011845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tisch R, Yang XD, Liblau RS, McDevitt HO. Administering glutamic acid decarboxylase to NOD mice prevents diabetes. J. Autoimmun. 1994;7:845–850. doi: 10.1006/jaut.1994.1067. [DOI] [PubMed] [Google Scholar]
- 34.Verge CF, Gianani R, Kawasaki E, Yu L, Pietropaolo M, Jackson RA, Chase HP, Eisenbarth GS. Prediction of type I diabetes in first-degree relatives using a combination of insulin, GAD, and ICA512bdc/IA-2 autoantibodies. Diabetes. 1996;45:926–933. doi: 10.2337/diab.45.7.926. [DOI] [PubMed] [Google Scholar]
- 35.Viret C, Janeway CA., Jr Self-specific MHC class II-restricted CD4- CD8- T cells that escape deletion and lack regulatory activity. J. Immunol. 2003;170:201–209. doi: 10.4049/jimmunol.170.1.201. [DOI] [PubMed] [Google Scholar]
- 36.Walker LS, Chodos A, Eggena M, Dooms H, Abbas AK. Antigen-dependent proliferation of CD4+ CD25+ regulatory T cells in vivo. J. Exp. Med. 2003;198:249–258. doi: 10.1084/jem.20030315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wen L, Chen NY, Tang J, Sherwin R, Wong FS. The regulatory role of DR4 in a spontaneous diabetes DQ8 transgenic model. J. Clin. Invest. 2001;107:871–880. doi: 10.1172/JCI11708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wen L, Wong FS, Tang J, Chen NY, Altieri M, David C, Flavell R, Sherwin R. In vivo evidence for the contribution of human histocompatibility leukocyte antigen (HLA)-DQ molecules to the development of diabetes. J Exp. Med. 2000;191:97–104. doi: 10.1084/jem.191.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wicker LS, Chen SL, Nepom GT, Elliott JF, Freed DC, Bansal A, Zheng S, Herman A, Lernmark A, Zaller DM, Peterson LB, Rothbard JB, Cummings R, Whiteley PJ. Naturally processed T cell epitopes from human glutamic acid decarboxylase identified using mice transgenic for the type 1 diabetes- associated human MHC class II allele, DRB1*0401. J Clin Invest. 1996;98:2597–2603. doi: 10.1172/JCI119079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilson DB. GAD-about BDC2.5: peptides that stimulate BDC2.5 T cells and inhibit IDDM. J. Autoimmun. 2003;20:199–201. doi: 10.1016/s0896-8411(03)00030-1. [DOI] [PubMed] [Google Scholar]
- 41.Yoon JW, Yoon CS, Lim HW, Huang QQ, Kang Y, Pyun KH, Hirasawa K, Sherwin RS, Jun HS. Control of autoimmune diabetes in NOD mice by GAD expression or suppression in beta cells. Science. 1999;284:1183–1187. doi: 10.1126/science.284.5417.1183. [DOI] [PubMed] [Google Scholar]
- 42.You S, Chen C, Lee WH, Brusko T, Atkinson M, Liu CP. Presence of diabetes-inhibiting, glutamic acid decarboxylase-specific, IL-10-dependent, regulatory T cells in naive nonobese diabetic mice. J. Immunol. 2004;173:6777–6785. doi: 10.4049/jimmunol.173.11.6777. [DOI] [PubMed] [Google Scholar]
- 43.Zekzer D, Wong FS, Ayalon O, Millet I, Altieri M, Shintani S, Solimena M, Sherwin RS. GAD-reactive CD4+ Th1 cells induce diabetes in NOD/SCID mice. J Clin Invest. 1998;101:68–73. doi: 10.1172/JCI119878. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.