

Abstract
G-protein-coupled receptor signaling is terminated by arrestin proteins that preferentially bind to the activated phosphorylated form of the receptor. Arrestins also bind active unphosphorylated and inactive phosphorylated receptors. Binding to the non-preferred forms of the receptor is important for visual arrestin translocation in rod photoreceptors and the regulation of receptor signaling and trafficking by non-visual arrestins. Given the importance of arrestin interactions with the various functional forms of the receptor, we performed an extensive analysis of the receptor-binding surface of arrestin using site-directed mutagenesis. The data indicated that a large number of surface charges are important for arrestin interaction with all forms of the receptor. Arrestin elements involved in receptor binding are differentially engaged by the various functional forms of the receptor, each requiring a unique subset of arrestin residues in a specific spatial configuration. We identified several additional phosphate-binding elements in the N-domain and demonstrated for the first time that the active receptor preferentially engages the arrestin C-domain. We also found that the interdomain contact surface is important for arrestin interaction with the non-preferred forms of the receptor and that residues in this region play a role in arrestin transition into its high affinity receptor binding state.
Arrestins regulate G-protein-coupled receptor signaling by binding to the activated phosphorylated receptor with high affinity to prevent its further interaction with G proteins. Receptor-bound arrestin can serve as an adaptor linking the complex to the internalization machinery of the coated pit through its binding to clathrin and AP2 (1) and as a scaffold through the formation of multiprotein signaling complexes with mitogen-activated protein (MAP) kinases and other partners (2). The fact that several of these proteins preferentially bind to the “active” receptor-bound form of arrestin (3) suggests that arrestin undergoes a significant conformation rearrangement upon receptor binding. This was initially proposed based on the observation that arrestin-receptor binding has an unusually large activation energy (4) and also by an increase of the sensitivity of receptor-bound arrestin to proteolysis (5). The crystal structure of arrestin (6) and numerous mutagenesis studies (7-10) support the idea that arrestin undergoes a significant conformational rearrangement upon binding. The molecular mechanism that governs arrestin-receptor interaction was first elucidated on the visual arrestin-rhodopsin model. Visual arrestin binds light-activated phosphorylated rhodopsin (P-Rh*)3 with remarkable selectivity, whereas its binding to an equal amount of dark (inactive) phosphorhodopsin (P-Rh) or light-activated unphosphorylated rhodopsin (Rh*) is 10–20 times lower (7). A model of sequential multisite binding was proposed to explain the selectivity of arrestin for P-Rh*. This model posits that arrestin has two “sensor” sites, detecting the phosphorylation and activation status of the receptor, respectively, and that simultaneous engagement of both sensors causes a global conformation change in arrestin, i.e. its transition into the active high affinity receptor binding state (reviewed in Ref. 11). Only the “phosphate sensor” in arrestin has been characterized extensively (6, 8, 9, 12-15). It was shown that receptor-attached phosphates must break the salt bridge between Arg-175 and Asp-296 to destabilize the polar core and make high affinity arrestin binding possible (9). Although the “activation sensor” has yet to be identified, it has been proposed to be at the contact point between the N- and C-domains since it is the only stabilizing intramolecular interaction in arrestin that is not affected by phosphates (11).
Arrestin elements implicated in receptor binding all map to the concave sides of both arrestin domains (11). Collectively, these data identify a fairly extensive surface containing the side chains of more than 70 residues (Fig. 1). With the exception of phosphate-binding elements and a few adjacent hydrophobic ones in β-strand X (8, 15, 16), the importance of the individual residues on this surface for arrestin interactions with the different functional forms of the receptor has not been systematically probed. The size of the receptor-binding surface indicates that either a small number of interacting residues are scattered throughout the whole span of the molecule or a large number of arrestin residues directly participate in receptor binding. Here we used site-directed mutagenesis to discriminate between these two possibilities and to determine the importance of individual charged residues on the surface of arrestin in its binding to rhodopsin. Our results showed that >25 charged residues are differentially involved in arrestin interactions with the various functional forms of the receptor and demonstrated for the first time that the stability of the interdomain region is important for high affinity arrestin binding.
FIGURE 1. Identified receptor-binding elements and regulatory intramolecular interactions in arrestin.

Shown is the visual arrestin crystal structure (6), highlighting receptor-binding elements as follows: blue, phosphate-binding residues (6, 8, 9, 12-15); green, β-strands and loops implicated in receptor binding by peptide competition (25, 26) and element swapping (15, 19). Residues participating in regulatory intramolecular interactions are indicated as follows: pink, polar core (9) (except phosphate-binding Arg-175, indicated in dark blue); orange, three-element interaction (8). Top panel, side view. Lower panel, top view (from the receptor-binding side).
EXPERIMENTAL PROCEDURES
Materials
[γ-32P]ATP, [14C]leucine, and [3H]leucine were from PerkinElmer Life Sciences. All restriction enzymes were from New England Biolabs. Sepharose 2B and all other chemicals were from sources previously described (17). Rabbit reticulocyte lysate was from Ambion, and SP6 RNA polymerase was prepared as described (18). 11-cis-retinal was generously supplied by Dr. R. K. Crouch.
Site-directed Mutagenesis
Bovine visual arrestin cDNA was a gift from Dr. T. Shinohara. The plasmid pARR-VSP was constructed and modified as described (15). Mutations were introduced by PCR using an appropriate mutagenizing oligonucleotide as a forward primer and an oligonucleotide downstream from the far restriction site to be used for subcloning as a reverse primer. Resulting fragments of various lengths and an appropriate primer upstream of the near restriction site were then used as reverse and forward primers, respectively, for the second round of PCR. Sites BamHI and SacII were used for mutations in the N-domain, and sites SacII and HindIII were used for mutations in the C-domain. The resulting fragments were purified, digested with the appropriate enzymes, and subcloned into the appropriately digested pARR-VSP plasmid. The sequences of all constructs were confirmed by dideoxy sequencing.
In Vitro Transcription, Translation, and Evaluation of Mutant Stability
pGEM2-based plasmids with the arrestin coding sequence equipped with the “idealized” 5′-untranslated region (18) under the control of the SP6 promoter were linearized using the HindIII site downstream of the coding sequence before in vitro transcription. In vitro transcription and translation were performed as described previously (17). All arrestin proteins were labeled by the incorporation of [3H]leucine and [14C]leucine with the specific activity of the mix between 1.5 and 3 Ci/mmol, resulting in the specific activity of arrestin proteins within the range of 66–85 Ci/mmol (150–230 dpm/fmol). The translation of every mutant used in this study produced a single labeled protein band with the expected mobility on SDS-PAGE. The relative stability of all mutants (assessed as described (14)) exceeded 80%.
Direct Arrestin Binding to Different Functional Forms of Rhodopsin
The assay was performed as described previously (17). Briefly, in vitro translated radiolabeled arrestins (100 fmol) were incubated in 50 mm Tris-HCl, pH 7.5, 0.5 mm MgCl2, 1.5 mm dithiothreitol, 1 mm EGTA, 50 mm potassium acetate for 5 min at 37°C with 0.3 μg of the indicated form of rhodopsin in room light (Rh* and P-Rh*) or in the dark (Rh and P-Rh) and then quickly cooled on ice. Rhodopsin-containing membranes along with bound arrestin were separated by size-exclusion chromatography on 2-ml columns of Sepharose 2B-CL at 4°C. Parallel samples with arrestin alone served as controls. The rhodopsin with bound arrestin eluted between 0.5 and 1.1 ml directly into scintillation vials, and bound arrestin was quantified by liquid scintillation counting.
RESULTS
Exposed charges in arrestin are remarkably concentrated on the putative receptor-binding surface (6). Moreover, arrestin binding to all forms of rhodopsin (P-Rh*, P-Rh, Rh*, and Rh) is very sensitive to high salt inhibition, indicating the involvement of ionic interactions in this process (7). Therefore, to determine the importance of individual charges in the arrestin-receptor interaction, we performed charge reversal mutagenesis of almost all of the exposed positive charges and reversal or neutralization of selected negative charges and then introduced several additional surface charges into the arrestin molecule. All mutants were expressed in radiolabeled form in cell-free translation, and their binding to the four functional forms of rhodopsin was measured in our direct binding assay (7).
Binding of Arrestin Charge Reversal Mutants to Rhodopsin
We found that as many as 22 point mutations (in 18 different positions) of the surface charges in arrestin reduce its binding to P-Rh* by 15–47%, and one substitution (R291E) appreciably increases it (Fig. 2, A and D). These mutations are spread across the molecule clustering in two distinct “patches”, one in each domain (Fig. 3A). The effects of some mutations on arrestin binding to the non-preferred forms of rhodopsin tend to be more dramatic: eight mutations (in five positions) increase binding to dark P-Rh 1.9–3.4-fold (Fig. 2, B and E), and four mutations enhance binding to light-activated unphosphorylated Rh* 1.8–4.5-fold (Fig. 2, C and F). In addition, removal of several other individual positive charges significantly reduces binding to dark P-Rh and/or Rh* (Fig. 2, B, C, E, and F). In contrast to P-Rh* binding, the mutations affecting binding to dark P-Rh are evenly spread over the entire length of the arrestin molecule on the receptor-binding side, clustering at the distal tips of both domains and around the central “crest” where the two domains meet (Fig. 3B). Interestingly, mutations affecting Rh* binding are located exclusively in the C-domain (with the exception of K141E, which is located in a loop projecting toward the C-domain) (Fig. 3C), implicating the positive charges in this region as important for recognition of the activated form of the receptor.
FIGURE 2. The exposed charged residues in arrestin implicated in receptor binding.
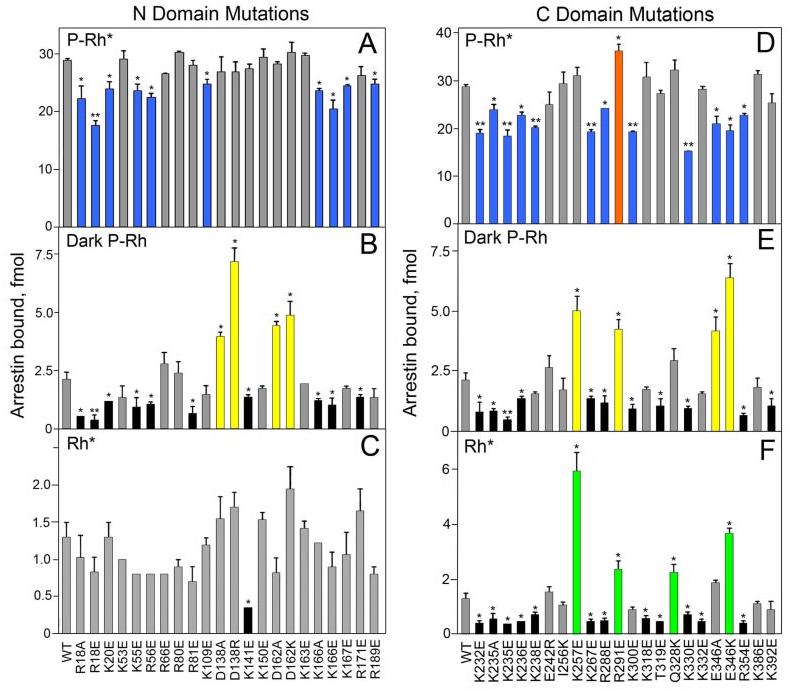
The binding of radiolabeled arrestins produced in cell-free translation to P-Rh* (A and D), dark P-Rh (B and E), and Rh* (C and F) was measured as described under “Experimental Procedures.” Binding to dark Rh was negligible in all cases (data not shown). Mutations in the N-domain (A–C) and C-domain (D–F) are color-coded as follows: blue, those that significantly reduce P-Rh* binding; orange, those that enhance P-Rh* binding (R291E); yellow, those that enhance dark P-Rh binding; green, those that enhance Rh* binding; black, those that reduce dark P-Rh and/or Rh* binding; gray, those that do not significantly affect binding. Means ± S.D. from two experiments performed in duplicate are shown. *, p < 0.05, **, p < 0.01, as compared with wild-type (WT) arrestin.
FIGURE 3. Map of exposed charged residues implicated in receptor binding.
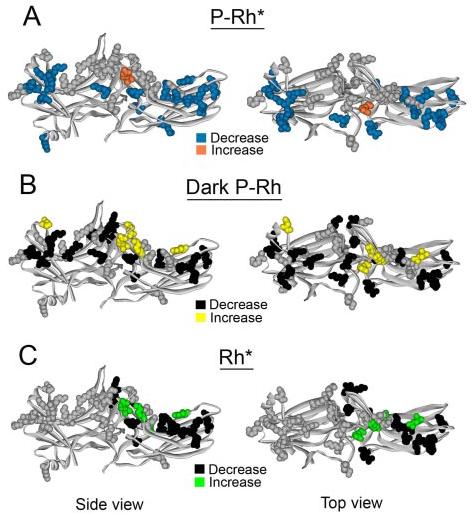
A–C, visual arrestin crystal structure highlighting mutations of exposed charges as follows. A, blue, those that significantly reduce P-Rh* binding; orange, those that enhance P-Rh* binding (R291E); gray, that do not affect P-Rh* binding. B, yellow, those that enhance dark P-Rh binding; black, that reduce dark P-Rh binding; gray, that do not affect dark P-Rh binding. C, green, those that enhance Rh* binding; black, those that reduce Rh* binding; gray, those that do not affect Rh* binding. (This figure was based on data in Fig. 2). Left panel, side view. Right panel, top view.
The mutations of several positive charges in the C-domain cluster were the most detrimental to P-Rh* binding (Fig. 2D). Element swapping between visual arrestin and the non-visual arrestin2 has shown that β-strands XV and XVI (residues 237–268) in this region of the C-domain are partially responsible for the receptor specificity of arrestin proteins (Fig. 1) (19). Within this region, there are three exposed sites where the surface charge differs between the two arrestins (visual residues Glu-242, Ile-256, and Lys-267, corresponding to Arg-236, Lys-250, and Thr-261 in arrestin2). When these residues were mutated in visual arrestin, only charge reversal of Lys-267 had any significant effect, decreasing binding to all forms of rhodopsin (Fig. 2, D-F). Effects of charge reversal mutations adjacent to these sites that are also different between the two arrestins (Thr-319, Gln-328, and Arg-354, corresponding to Glu-313, Lys-322, and Thr-350 in arrestin2) were more dramatic. The R354E mutation significantly reduced binding to all forms of rhodopsin and T319E reduced binding to both dark P-Rh and Rh*, whereas Q328K increased binding to Rh* (Fig. 2, D-F). Collectively, these results suggest that specific charges present in the C-domain of arrestin are important for recognizing the activated form of the receptor (Fig. 3C).
The cluster of mutations affecting P-Rh* in the N-domain is located near previously identified phosphate-binding residues (Fig. 1) (8, 15, 16), such as Arg-18 and Arg-171 (Fig. 2, A and B). Our data indicate that mutations of positively charged residues Lys-20, Lys-55, Arg-56, and Lys-166 in this region, as well as Lys-300 near the polar core, result in reduced binding to P-Rh* and dark P-Rh, whereas removal of a negative charge (D162A) or addition of an extra positive charge (D162K) in this region significantly increases arrestin binding to dark P-Rh (Fig. 2, A and B). These results suggest that several previously unidentified positive charges may participate in phosphate binding and further demonstrate the importance of the N-domain in the recognition of receptor-attached phosphates.
Mutations Affecting the Interdomain Contact
The data also show that several of the mutations that affected arrestin binding to the non-preferred forms of the receptor, especially dark P-Rh, are clustered at the point of contact between the two arrestin domains (Fig. 3B). These residues include Arg-81, Lys-141, Asp-138, Lys-257, Arg-288, Arg-291, Thr-319, and Lys-392. This suggests that the interaction between the two domains is important for low affinity arrestin binding to dark P-Rh and Rh*. Residues Asp-138 and Lys-257 actually form a salt bridge between the two domains. When these charges are neutralized (D138A) or reversed (D138R or K257E), binding to dark P-Rh (and Rh* in the case of K257E) is dramatically enhanced (Fig. 2, B and E). These data support the notion that destabilization of the interdomain contact is important for the global conformational change that takes place in arrestin upon receptor binding.
Aside from this salt bridge, the interdomain contact is almost exclusively mediated by hydrophobic interactions and H-bonds. To further test the hypothesis that the stability of this region may be important for receptor binding, we mutated two hydrophobic residues (Phe-65 and Tyr-250) that interact with a number of partners at the interdomain contact point. We also introduced different mutations reversing the charges of the salt bridge partners, D138K and K257D, so that their combination in a double mutant (D138K,K257D) would recreate this important salt bridge in the opposite configuration (Fig. 4A). The disruption of the salt bridge by D138K and K257D enhanced arrestin binding to dark P-Rh (and to Rh* in the case of K257D) (Fig. 4B), similar to the D138R and K257E mutations (Fig. 2). Interestingly, even the combination mutant (D138K,K257D), in which the salt bridge is restored, demonstrated increased dark P-Rh binding. We also found that although F65A alone had no effect on rhodopsin binding, the Y250A mutation and F65A,Y250A combination significantly reduced P-Rh* binding (by 25–40%; Fig. 4B). These data suggest that either the exact structure of the contact between domains is important for high affinity receptor binding or that Tyr-250 and Phe-65 in the active form of arrestin are exposed and directly participate in P-Rh* interaction.
FIGURE 4. The effects of mutations in the interdomain region.
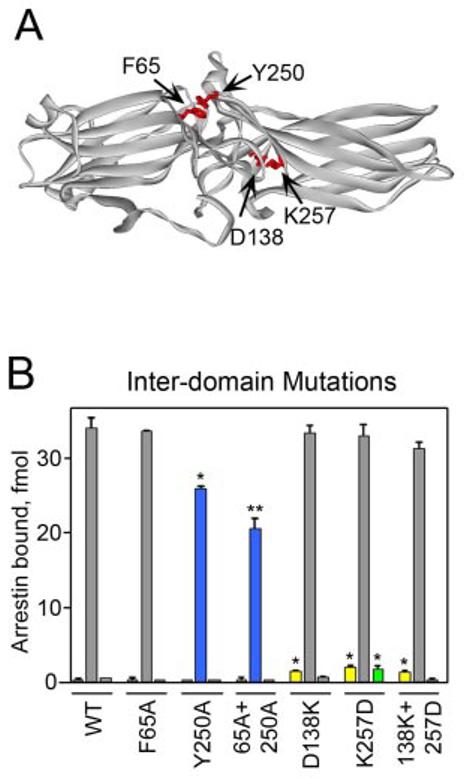
A, top view of the visual arrestin crystal structure highlighting residues between the two domains that were mutated. B, the binding of each radiolabeled arrestin produced in cell-free translation to dark P-Rh (left), P-Rh* (center), and Rh* (right) is shown. Results are color-coded as follows: blue, mutations that significantly reduce P-Rh* binding; yellow, mutations that enhance dark P-Rh binding; green, mutations that enhance Rh* binding; gray, mutations that do not significantly affect binding. Means ± S.D. from two experiments performed in duplicate are shown. *, p < 0.05, **, p < 0.01, as compared with wild-type (WT) arrestin.
DISCUSSION
Several studies using a variety of methods including truncation mutagenesis (7, 20), differential chemical modification and H/D exchange (21), site-directed mutagenesis (8, 9, 22-24), construction of chimeric arrestins (15, 19), peptide competition (25, 26), and epitope insertion (27) have shown that arrestin elements important for receptor binding are localized to the concave surfaces of both arrestin domains (Fig. 1). However, the contribution of individual residues on these surfaces to its binding to the various functional forms of the receptor has never been systematically tested. Ionic interactions play an important role in arrestin binding to the receptor (7). Therefore, we performed targeted mutagenesis of virtually all of the positive charges and selected negative charges on the arrestin surface (Fig. 2). We found that a subset individual charges spanning the entire length of the molecule is important for phosphoreceptor binding (Fig. 3, A and B), whereas binding to the activated unphosphorylated receptor was only significantly affected by mutations in the C-domain (Fig. 3C), implicating this region as a possible point of first contact with the active form of the receptor. These data indicate that the receptor-binding surface of arrestin is indeed extensive, directly involving a large number of residues on the concave surfaces of the entire molecule (rather than a small number of key residues).
Several phosphate-binding residues in visual arrestin have been identified previously including Arg-171, the phosphate sensor Arg-175 (15), Lys-14, Lys-15 (8), and Arg-18 (16), all of which are localized to a small region in the arrestin N-domain (Fig. 1). In this study, we identified several adjacent positive charges including Lys-20, Lys-55, Arg-56, Lys-166, and Lys-300, which are important for both P-Rh* and dark P-Rh binding. These data suggest that these additional N-domain residues may also interact with receptor-attached phosphates (Fig. 2,A,B,D, and E). In contrast, several charges in the C-domain were found to be important for Rh* interaction. Some of these residues are different from the corresponding residues in arrestin2. Replacement of Lys-267, Thr-319, and Arg-354 was detrimental to Rh* (and in some cases P-Rh*) binding, whereas the Q328K mutation actually helps visual arrestin bind Rh* better. These results suggest that several specific residues in the C-domain participate in binding to the active form of the receptor and support the idea that this region is important for determining receptor preference of arrestin (which is independent of phosphate interaction and therefore must be mediated solely by the binding to the active conformation of the receptor) (19).
Several C-domain positive charges important for Rh* binding were also found to affect P-Rh* interaction. These include the cluster of lysines 232, 235, 236, 267, and 330 on three adjacent β-strands. To test the importance of this concentrated positive patch, we removed and reversed a nearby negative charge (mutations E346A and E346K, respectively). Both mutations severely impede P-Rh* binding but dramatically increase dark P-Rh and Rh* binding (Fig. 2, D-F). This effect likely indicates that disruption of this network of charges “loosens up” the C-domain, causing increased binding to the non-preferred forms of the receptor, whereas the reduction in P-Rh* binding suggests that Glu-346 directly participates in interaction with the light-activated phosphorylated form of the receptor. These data further demonstrate the importance of having particular charges in arrestin in a specific spatial configuration for its differential interaction with the various forms of the receptor.
Binding of several mutants was of particular interest. First, charge reversal of Arg-291 was the only mutation to increase binding to all forms of the receptor (P-Rh*, P-Rh, and Rh*) (Fig. 2, D-F). This result was not surprising since this arginine forms a salt bridge with Glu-390 in the acidic arrestin C-tail. Several indirect lines of evidence suggest the C-tail is released upon binding (5, 8, 10, 14). Truncated mutants of arrestin that lack the C-tail, as well as mutants in which the three-element interaction between β-strand I, α-helix I, and the C-tail (Fig. 1) is destabilized, demonstrate enhanced binding to all forms of the receptor (14). Thus, charge reversal of Arg-291 likely destabilizes the interaction of the loop containing this arginine with the C-tail, thereby facilitating its release and increasing binding to the receptor.
Interestingly, two mutations on the “back” of the molecule reduce arrestin binding to P-Rh* without affecting binding to any other functional form (Fig. 3). The first, K109E, is located in α-helix I. The α-helix is part of the three-element interaction that stabilizes the basal conformation of arrestin (Fig. 1) (8, 14). It apparently participates in receptor binding and trafficking (12, 28); therefore, it is not surprising that mutation in this helix affects binding. The second, Arg-189, is at the edge of the interdomain hinge and is next to several other charges including Glu-191 and Lys-211. Charge reversal at 189 could affect the flexibility of the hinge, which is important for high affinity P-Rh* binding (10).
The sequential multisite model of arrestin-receptor interaction explains the remarkable selectivity of arrestin for P-Rh* over other forms of the receptor by the existence of “sensor sites,” which recognize the phosphorylated and activated state of the receptor, respectively. Only when both sensors are engaged will arrestin assume its active high affinity receptor binding conformation (11). The phosphate sensor has been extensively studied. Residues in the polar core, as well as several positive charges on the concave surface of the N-domain, have been shown to be critical for phosphoreceptor recognition (Figs. 1-3) (6, 8, 9, 12-15). In contrast, the activation sensor has remained elusive, although it has been proposed that the point of contact between the N- and C-domains might play a role in this process (11).
Mutations that turn the activation sensor “on” would be expected to enhance arrestin binding to the phosphorylated inactive receptor. The interface between domains is hydrophobic, making this site highly adaptable. Both N- and C-domain elements that determine the receptor specificity of arrestin proteins extend to this interdomain contact point (19). We found that breaking a salt bridge connecting the two domains (Asp-138–Lys-257) results in increased binding to the non-preferred forms of the receptor, especially dark P-Rh (Figs. 2 and 4), supporting the hypothesis that the interdomain contact point may serve as the activation sensor in arrestin. To further test this idea, we mutated two residues, Phe-65 and Tyr-250, each of which participates in multiple interdomain interactions. We found that Y250A and the F65A,Y250A combination significantly reduce P-Rh* binding (Fig. 4B), although neither affects dark P-Rh binding or Rh* binding. It is possible that Phe-65 and Tyr-250 directly participate in receptor interaction. Although these residues are buried in the basal state, they may become exposed when arrestin undergoes its conformational change into its active receptor-bound form. This would explain why their removal reduces P-Rh* interaction but does not affect binding to other forms of the receptor. Our data indicate that disruption of the interdomain contact is important for arrestin transition into its high affinity receptor binding state, but whether it serves specifically as the activation-recognition site in arrestin remains to be elucidated.
Visual arrestin binds to dark P-Rh and Rh* with very low affinity such that the absolute level of binding in our direct assay is ∼10–20 times lower than to P-Rh*. However, the concentration of visual arrestin in rod photoreceptors is ∼150 μm (29), enabling even these low affinity interactions and making them biologically relevant. Visual arrestin moves between compartments of the rod photoreceptor cell in a light-dependent manner (30). We recently demonstrated that the presence of Rh* (even in the absence of its high affinity target P-Rh*) is sufficient to induce arrestin movement to the outer segments of rods. Similarly, the presence of dark P-Rh and phosphoopsin keeps arrestin from moving back to the inner segments in the dark (30). Both non-visual arrestins interact with various unphosphorylated G-protein-coupled receptors (reviewed in Ref. 31). Their interaction with phosphorylated inactive receptors regulates receptor trafficking and the ultimate fate of the internalized receptor (32). Thus, the binding of different arrestins to the non-preferred forms of their cognate receptors is biologically important. Our results demonstrate for the first time that different elements of visual arrestin are engaged by phosphorylated (P-Rh), activated (Rh*), and activated-phosphorylated (P-Rh*) receptors and identify a unique subset of residues that are important for arrestin binding to each form of the receptor.
Acknowledgments
We are grateful to Dr. Jeffrey L. Benovic for purified receptor kinases, Dr. T. Shinohara for bovine visual arrestin cDNA, and rotation students Todd Townsend and Courtney Frederick for the construction of some of the arrestin mutants used in this study.
Footnotes
This work was supported in part by National Institutes of Health Grants EY11500 and GM63097 (to V. V. G.).
The abbreviations used are: P-Rh*, light-activated phosphorylated rhodopsin; Rh, light-activated unphosphorylated rhodopsin; P-Rh, inactive phosphorhodopsin.
REFERENCES
- 1.Ferguson SS, Downey WE, Colapietro AM, Barak LS, Menard L, Caron MG. Science. 1996;271:363–366. doi: 10.1126/science.271.5247.363. [DOI] [PubMed] [Google Scholar]
- 2.Perry SJ, Lefkowitz RJ. Trends Cell Biol. 2002;12:130–138. doi: 10.1016/s0962-8924(01)02239-5. [DOI] [PubMed] [Google Scholar]
- 3.Gurevich VV, Gurevich EV. Structure (Camb.) 2003;11:1037–1042. doi: 10.1016/s0969-2126(03)00184-9. [DOI] [PubMed] [Google Scholar]
- 4.Schleicher A, Kuhn H, Hofmann KP. Biochemistry. 1989;28:1770–1775. doi: 10.1021/bi00430a052. [DOI] [PubMed] [Google Scholar]
- 5.Palczewski K, Pulvermuller A, Buczylko J, Hofmann KP. J. Biol. Chem. 1991;266:18649–18654. [PubMed] [Google Scholar]
- 6.Hirsch JA, Schubert C, Gurevich VV, Sigler PB. Cell. 1999;97:257–269. doi: 10.1016/s0092-8674(00)80735-7. [DOI] [PubMed] [Google Scholar]
- 7.Gurevich VV, Benovic JL. J. Biol. Chem. 1993;268:11628–11638. [PubMed] [Google Scholar]
- 8.Vishnivetskiy SA, Schubert C, Climaco GC, Gurevich YV, Velez MG, Gurevich VV. J. Biol. Chem. 2000;275:41049–41057. doi: 10.1074/jbc.M007159200. [DOI] [PubMed] [Google Scholar]
- 9.Vishnivetskiy SA, Paz CL, Schubert C, Hirsch JA, Sigler PB, Gurevich VV. J. Biol. Chem. 1999;274:11451–11454. doi: 10.1074/jbc.274.17.11451. [DOI] [PubMed] [Google Scholar]
- 10.Vishnivetskiy SA, Hirsch JA, Velez MG, Gurevich YV, Gurevich VV. J. Biol. Chem. 2002;277:43961–43968. doi: 10.1074/jbc.M206951200. [DOI] [PubMed] [Google Scholar]
- 11.Gurevich VV, Gurevich EV. Trends Pharmacol. Sci. 2004;25:59–112. doi: 10.1016/j.tips.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 12.Han M, Gurevich VV, Vishnivetskiy SA, Sigler PB, Schubert C. Structure (Camb.) 2001;9:869–880. doi: 10.1016/s0969-2126(01)00644-x. [DOI] [PubMed] [Google Scholar]
- 13.Celver J, Vishnivetskiy SA, Chavkin C, Gurevich VV. J. Biol. Chem. 2002;277:9043–9048. doi: 10.1074/jbc.M107400200. [DOI] [PubMed] [Google Scholar]
- 14.Gurevich VV. J. Biol. Chem. 1998;273:15501–15506. doi: 10.1074/jbc.273.25.15501. [DOI] [PubMed] [Google Scholar]
- 15.Gurevich VV, Dion SB, Onorato JJ, Ptasienski J, Kim CM, Sterne-Marr R, Hosey MM, Benovic JL. J. Biol. Chem. 1995;270:720–731. doi: 10.1074/jbc.270.2.720. [DOI] [PubMed] [Google Scholar]
- 16.Sutton RB, Vishnivetskiy SA, Robert J, Hanson SM, Raman D, Knox BE, Kono M, Navarro J, Gurevich VV. J. Mol. Biol. 2005;354:1069–1080. doi: 10.1016/j.jmb.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 17.Gurevich VV, Benovic JL. Methods Enzymol. 2000;315:422–437. doi: 10.1016/s0076-6879(00)15859-8. [DOI] [PubMed] [Google Scholar]
- 18.Gurevich VV. Methods Enzymol. 1996;275:382–397. doi: 10.1016/s0076-6879(96)75023-1. [DOI] [PubMed] [Google Scholar]
- 19.Vishnivetskiy SA, Hosey MM, Benovic JL, Gurevich VV. J. Biol. Chem. 2004;279:1262–1268. doi: 10.1074/jbc.M308834200. [DOI] [PubMed] [Google Scholar]
- 20.Gurevich VV, Benovic JL. J. Biol. Chem. 1992;267:21919–21923. [PubMed] [Google Scholar]
- 21.Ohguro H, Palczewski K, Walsh KA, Johnson RS. Protein Sci. 1994;3:2428–2434. doi: 10.1002/pro.5560031226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gurevich VV, Benovic JL. J. Biol. Chem. 1995;270:6010–6016. doi: 10.1074/jbc.270.11.6010. [DOI] [PubMed] [Google Scholar]
- 23.Gurevich VV, Benovic JL. Mol. Pharmacol. 1997;51:161–169. doi: 10.1124/mol.51.1.161. [DOI] [PubMed] [Google Scholar]
- 24.Gray-Keller MP, Detwiler PB, Benovic JL, Gurevich VV. Biochemistry. 1997;36:7058–7063. doi: 10.1021/bi963110k. [DOI] [PubMed] [Google Scholar]
- 25.Pulvermuller A, Schroder K, Fischer T, Hofmann KP. J. Biol. Chem. 2000;275:37679–37685. doi: 10.1074/jbc.M006776200. [DOI] [PubMed] [Google Scholar]
- 26.Kieselbach T, Irrgang KD, Ruppel H. Eur. J. Biochem. 1994;226:87–97. doi: 10.1111/j.1432-1033.1994.tb20029.x. [DOI] [PubMed] [Google Scholar]
- 27.Dinculescu A, McDowell JH, Amici SA, Dugger DR, Richards N, Hargrave PA, Smith WC. J. Biol. Chem. 2002;277:11703–11708. doi: 10.1074/jbc.M111833200. [DOI] [PubMed] [Google Scholar]
- 28.Dinh DT, Qian H, Seeber R, Lim E, Pfleger K, Eidne KA, Thomas WG. Mol. Pharmacol. 2005;67:375–382. doi: 10.1124/mol.104.004721. [DOI] [PubMed] [Google Scholar]
- 29.Hamm HE, Bownds MD. Biochemistry. 1986;25:4512–4523. doi: 10.1021/bi00364a010. [DOI] [PubMed] [Google Scholar]
- 30.Nair KS, Hanson SM, Mendez A, Gurevich EV, Kennedy MJ, Shestopalov VI, Vishnivetskiy SA, Chen J, Hurley JB, Gurevich VV, Slepak VZ. Neuron. 2005;46:555–567. doi: 10.1016/j.neuron.2005.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gurevich VV, Gurevich EV. Pharmacol. Ther. 2005 in press. [Google Scholar]
- 32.Pan L, Gurevich EV, Gurevich VV. J. Biol. Chem. 2003;278:11623–11632. doi: 10.1074/jbc.M209532200. [DOI] [PubMed] [Google Scholar]