

Abstract
Objective
Murine genetic models suggest that function of the 12/15 LOX enzyme promotes atherosclerosis. We tested the hypothesis that exonic and/or promoter single nucleotide polymorphisms (SNPs) in the human 12/15-LOX gene (ALOX15) alter the risk of symptomatic coronary artery disease (CAD).
Methods & Results
We resequenced ALOX15 and then genotyped a common promoter and a less common novel coding SNP (T560M) in 1809 subjects with CAD and 1734 controls from Kaiser Permanente including a subset of participants of the Coronary Artery Risk Development in Young Adults study. We found no association between the promoter SNP and the risk of CAD. However, heterozygote carriers of the 560M allele had an increased risk of CAD (adjusted OR, 1.62; P=0.02) compared to non-carriers. In vitro studies demonstrated a 20-fold reduction in the catalytic activity of 560M when compared to 560T. We then genotyped T560M in 12974 participants of the Atherosclerosis Risk in Communities study and similarly found that heterozygote carriers had an increased risk of CAD compared to non-carriers (adjusted HR, 1.31; P=0.06). In both population studies, homozygote carriers were rare and associated with a non-significant decreased risk of CAD compared to non-carriers (adjusted OR, 0.55; P=0.63 and HR, 0.93; P=0.9).
Conclusions
A coding SNP in ALOX15 (T560M) results in a near null variant of human 12/15-LOX. Assuming a co-dominant mode of inheritance, this variant does not protect against CAD. Assuming a recessive mode of inheritance, the effect of this mutation remains unclear, but is unlikely to provide a protective effect to the degree suggested by mouse knockout studies.
Keywords: Arachidonate 15-Lipoxygenase, polymorphism – single nucleotide, coronary disease
The 12/15 lipoxygenases (12/15 LOXs) are members of a diverse family of lipid peroxidizing enzymes, which catalyze the stereospecific oxygenation of polyunsaturated fatty acids even if these substrates are incorporated in biomembranes or lipoproteins1 , 2. The human 12/15-LOX is encoded for by a single gene, ALOX15, localized in the LOX gene cluster on chromosome 17 and oxygenates arachidonic acid to 15-S-hydroperoxyeicosatetraenoic acid (15-HpETE)3. Although 12/15-LOXs have been previously implicated in the pathogenesis of atherosclerosis, their precise role in this context remains controversial as both in vitro and in vivo experiments have yielded conflicting results. In vitro, 12/15-LOXs may facilitate the oxidation of LDL to a more atherogenic form4. In atherosclerotic lesions from rabbits, both the mRNA and the protein have been shown to co-localize with macrophage rich regions and epitopes of oxidized LDL. However, more recent gene expression studies of human atherosclerotic lesions at different stages suggest minimal expression of 12/15-LOX5. In vivo, both 12/15-LOX/apoE and the 12/15-LOX/LDL double knockout mice develop significantly less atherosclerosis than the single apoE and LDL knockouts respectively6. Furthermore, over expression of a human ALOX15 transgene in murine endothelium increased the formation of atherosclerotic lesions in LDL-receptor deficient mice7. In contrast, transgenic rabbits over-expressing the human 12/15-LOX in monocyte/macrophages or systemically are protected from development of atherosclerosis8 , 9, 10.
To better define the role of 12/15-LOX in human atherosclerosis, we sought to test the hypothesis that common polymorphisms in the human 12/15-LOX gene (ALOX15) alter the risk of symptomatic CAD.
Material and Methods
Study Design
The ADVANCE study (Atherosclerotic Disease, VAscular FuNction, and GenetiC Epidemiology) was approved by the Institutional Review Board at both Stanford University and Kaiser Permanente of Northern California (KPNC).
Between October 28, 2001 and December 31, 2003, we recruited a total of 3179 subjects into 5 cohorts: a cohort of subjects with clinically significant CAD at a young age (<45 years for males, < 55 years for females), a cohort of subjects with incident stable angina at an older age, a cohort of subjects with incident acute myocardial infarction (AMI) at an older age, a cohort of young subjects with no history of CAD, and a cohort of subjects aged 60 to 72 with no history of CAD, ischemic stroke, or peripheral arterial disease (PAD). Eligible subjects were identified using the KPNC electronic databases and those who agreed to participate were interviewed and examined at one of several clinics in the San Francisco Bay Area. A sixth cohort of young subjects with no history of CAD included 479 participants in the Coronary Artery Risk Development in Young Adults (CARDIA) Study11 originally recruited through KPNC and attending the study’s year 15 exam in 2000-2001. A detailed description of the source population for all cohorts has been published elsewhere12-14.
The design of the ADVANCE study allowed for several case control comparisons. In this study, we compared subjects with symptomatic early onset CAD (“young cases”) with young subjects without CAD (“young controls”) and older subjects presenting with stable angina or AMI (“older cases”) with older subjects with no history of CAD, CVA, or PAD (“older controls”).
Clinical Measurements
Through a phone interview, a self-administered questionnaire, and the use of the KPNC electronic databases, we documented the presence or absence and age of onset of clinically significant CAD, ischemic stroke, and PAD, as well as traditional risk factors for atherosclerosis. Subjects also provided information on race/ethnicity and were classified into one of nine race/ethnic groups: white/Europeans, black/African Americans, Hispanics, South Asians, East Asians, Pacific Islanders, Native Americans, admixed Hispanics, and admixed non-Hispanics. At the clinic visit, we measured the height and weight of all participants and collected blood for DNA extraction.
Traditional risk factors (smoking, hypertension, high cholesterol, and diabetes) were defined based on self report and were considered to be present only if subjects reported an age of onset of a risk factor that was younger than the age of onset of clinically significant CAD.
Resequencing and Genotyping
Using an automated fluorescent labeling system15, we resequenced the promoter region, the exons, and the intron-exon boundaries of ALOX15 in 24 ethnically diverse males with a history of CAD (SNP discovery set). We then selected a subset of sequenced SNPs to genotype in all participants of the ADVANCE study using the TaqMan® assay. All resequencing and genotyping was performed by the Stanford Human Genome Center.
Statistical Analysis
We excluded from further analysis subjects who did not provide blood for DNA extraction (n=40) and who did not fill out the study questionnaire (n=9). We also excluded South Asians (n=55) because of a lack of controls. Lastly, we excluded Pacific Islanders (n=9) and Native Americans (n=2) because of small numbers.
We compared the distributions and frequencies of all non-genetic covariates of interest in cases and controls using standard parametric and non-parametric methods. For each race/ethnic group, we calculated the minor allele frequency (MAF) and bootstrap derived 95% confidence intervals of all SNPs in both sets of controls combined and tested for Hardy-Weinberg equilibrium (HWE) with the permutation version of the exact test16.
Using Cochran-Mantel-Haenszel (CMH) methods, we derived estimates of the Odds Ratio (OR) and a general association statistic17, 18 for each case/control comparison stratified by race/ethnic group. We compared the heterozygote carriers of the minor allele to non-carriers and homozygote carriers to non-carriers separately. The effect of other covariates of interest was evaluated through a multivariate unconditional logistic regression. To minimize the probability of confounding due to population substructure in our ‘Admixed Hispanic’ and ‘Admixed Non-Hispanic’ race/ethnic groups, we estimated the proportion of our 4 ‘parent’ ancestries (white/European, black/African-America, Hispanic, East Asian) at the individual level19 for all cases and controls in these groups and used these estimates as covariates in our adjusted analyses.
For each SNP genotyped, we estimated the minimal OR we could detect with a power of 80% given the minor allele frequency observed in controls20. For these calculations, we assumed the polymorphism was causal, a population prevalence of symptomatic CAD of 6.2%21, and a Type 1 error of 0.05.
Transcription Factor Binding studies
We examined the possibility that the minor allele of a SNP potentially associated with CAD lead to either the creation or destruction of a transcription binding site using MatInspector22
Mutagenesis studies
For bacterial expression wild-type and mutant 12/15-LOX cDNAs were cloned into the pQE-9 expression plasmid (Qiagen, Hilden, Germany). The enzyme species were expressed as N-terminal his-tag fusion proteins and purified to near homogeneity by Ni-agarose affinity chromatography32. For transient eukaryotic expression the coding region of the 12/15-LOX cDNA was cloned into the pcDNA3.1 expression plasmid (Invitrogen, Hilden, Germany) and HEK293 cells were lipofected with this construct. Site-directed mutagenesis was performed using the QuickChange™ Site-Directed Mutagenesis Kit (Stratagene, Amsterdam, The Netherlands). For each mutant, 5-10 clones were selected, screened for 12/15-LOX expression by immunoblotting and activity assays and were finally sequenced to confirm the T560M exchange and the absence of any other change
Arachidonic acid oxygenase activity of wild-type and mutant 12/15-LOX species was assayed by High Pressure Liquid Chromatography (HPLC) quantification of the oxygenation products after incubating the purified enzyme preparations or the transfected cells for 15 min with exogenous arachidonic acid32. For immunoblotting, transfected cells were lysed in the presence of 5 mM EDTA and aliquots of the lysis supernatants or of purified enzyme preparations were applied to SDS-PAGE. The blots were probed with a polyclonal rabbit antibody raised in guinea pigs against the pure rabbit 12/15-LOX.
Replication of human genetic associations in the ADVANCE study
We planned to replicate suggestive associations between one or more SNPs in ALOX15 and CAD by genotyping the same SNPs in the Atherosclerosis Risk in Communities (ARIC) cohort and assessing their effect on incident CAD. The ARIC Study is an ongoing prospective investigation of atherosclerosis and its clinical sequelae involving 15,792 white and black persons aged 45–64 years at recruitment (1987–1989). Study IRBs approved the ARIC Study and all participants provided written informed consent. A detailed description of the ARIC study design, sampling procedures, methods, definitions of cardiovascular outcomes, and approach to statistical analyses is published elsewhere23 24, 25. Incident CAD in ARIC is defined as documented AMI, coronary artery bypass surgery, unstable angina, or coronary-related death. Follow-up was available until December 31, 2002. Time-to-event was analyzed using Cox proportional hazards modeling. We adopted the methods of Hsieh and Lavori26 to calculate post-hoc the minimal detectable hazard ratio.
For SNPs genotyped in both the ADVANCE and ARIC cohorts, we estimated the OR and 95% confidence interval across both population studies using CMH methods for both the comparison of heterozygotes carriers to non carriers and the comparison of homozygote carriers to non carriers. These OR were adjusted for study and race/ethnic group and were calculated only if there was no significant difference between the minimally and fully adjusted risk ratios derived by regression analyses in each of the two population studies.
Results
Non-Genetic Characteristics of the ADVANCE study sample
The ADVANCE study sample consisted of 3546 subjects, whose clinical and other non-genetic characteristics are summarized in Table 1. The older age of controls in the set of older onset cases and controls was a consequence of our stratified sampling design. Stratified sampling and/or preferential participation also led to differences in the prevalence of certain race/ethnic groups by case/control status and to the low prevalence of young cases that were male.
Table 1.
Non-genetic characteristics of the ADVANCE study according to case/control status (symptomatic coronary artery disease) stratified by two primary predefined comparisons
| Young Cases (n=472) | Young Controls (n=742) | P | Older Cases (n=1337) | Older Controls (n=992) | P | |
|---|---|---|---|---|---|---|
| Mean(SD) | Mean(SD) | T test | Mean(SD) | Mean(SD) | T test | |
| Age(years) at first ever event for cases, study visit date for controls* | 45.3(6.5) | 44.3(5.5) | 0.005 | 62(8.4) | 65.8(2.9) | <0.001 |
|
|
||||||
| Median(Range) | Median(Range) | Wilcoxon | Median(Range) | Median(Range) | Wilcoxon | |
|
|
||||||
| Body Mass Index | 31.1(17.3-61.2) | 26.8(15.8-66.2) | <0.001 | 28.4(16.9-66.1) | 27.5(17.3-52.9) | <0.001 |
| C-Reactive Protein | 2.3(0.1-76.4) | 1.2(0.1-76.8) | <0.001 | 1.7(0.1-77.4) | 1.6(0.1-73.9) | 0.163 |
| Months from first ever event to study visit | 21.3(2.7-222.1)† | -- | -- | 3.5(1.4-26.4)† | -- | -- |
|
|
||||||
| Count(%) | Count(%) | Chi2 | Count(%) | Count(%) | Chi2 | |
|
|
||||||
| Risk Factors(Self Report) | ||||||
| Male | 184(39) | 328(44.2) | 0.072 | 975(72.9) | 618(62.3) | <0.001 |
| Current/Former smoker | 276(58.5) | 266(35.8) | <0.001 | 842(63) | 569(57.4) | 0.006 |
| Hypertension | 154(32.6) | 143(19.3) | <0.001 | 641(47.9) | 407(41) | <0.001 |
| Diabetes Mellitus | 101(21.4) | 51(6.9) | <0.001 | 274(20.5) | 147(14.8) | <0.001 |
| High Cholesterol | 129(27.3) | 148(19.9) | 0.003 | 604(45.2) | 356(35.9) | <0.001 |
| Ancestry | <0.001 | <0.001 | ||||
| White/European | 254(53.8) | 369(49.7) | 947(70.8) | 677(68.2) | ||
| Black/African American | 46(9.7) | 254(34.2) | 50(3.7) | 79(8) | ||
| Hispanic | 25(5.3) | 22(3) | 83(6.2) | 60(6) | ||
| East Asian | 44(9.3) | 35(4.7) | 73(5.5) | 68(6.9) | ||
| Admixed Hispanic | 32(6.8) | 23(3.1) | 52(3.9) | 31(3.1) | ||
| Admixed Non-Hispanics | 71(15) | 39(5.3) | 132(9.9) | 77(7.8) | ||
The age cutoff for a “young case” was not the same for males (<45 years) and females (<55 years).
P < 0.001 for Wilcoxon test. Young cases may have had their qualifying coronary event as early as Jan 1, 1999, while all older cases had their qualifying coronary event after the start of recruitment in October, 2001.
Resequencing and Genotyping
We identified 27 polymorphisms by resequencing ALOX15 in the SNP discovery set. The location and minor allele frequencies of these SNPs have been submitted to dbSNP. Based on their presence in putative functional domains, evolutionary conservation, or being a non-synonymous substitution, we selected a previously identified SNP in the promoter region and a novel SNP in exon 13 to genotype in all participants (Table 2).
Table 2.
Summary of ALOX15 single nucleotide polymorphisms genotyped in the ADVANCE study sample (ordered 5′ to 3′)
| Minor Allele Frequency (95% Confidence intervals) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| SNP Alias |
Public Name |
Details | Major→ Minor Allele* |
White/European | Black/African American |
Hispanic | East Asian | Admixed Hispanic |
Admixed Non-Hispanics |
| ALOX15.7 | rs7220870 | 261bp 5′ transc start | T→G | 22.8(21.2-24.8) | 13.6(11-16.2) | 23.2(16.5-29.3) | 16.3(11.4-21.6) | 22.6(14.2-31.1) | 20.9(15.7-26.1) |
| ALOX15.18 | rs34210653 | Thr→Met in exon 13 | T→C | 1.2(0.7-1.7) | 0.5(0-1.1) | 8(4.3-12.7) | 0(0-0) | 3.7(0.9-7.4) | 1.3(0-3) |
bp = base pairs, transc = transcription
Minor allele is defined as the least prevalent base for a given SNP across both sets of controls in all race/ethnic strata combined.
Association Analyses in ADVANCE
Both SNPs were in Hardy-Weinberg equilibrium in all race/ethnic and case-control strata (details not shown). Across all race/ethnic groups, the minor allele frequency of the coding SNP (ALOX15.18) was considerably lower than the promoter SNP with the highest frequency of ALOX15.18 in Hispanics (Table 2). ALOX15.18 was uncommon in white/Europeans, very rare in black/African Americans, and absent in East Asians. The minor allele of ALOX15.18 was also absent in all East Asian cases (details not shown).
Both the unadjusted and fully adjusted analyses for the promoter SNP revealed no evidence of an association between the minor allele and clinical CAD (details not shown). In contrast, the ORs for ALOX15.18 revealed a significantly increased risk of clinical CAD in heterozygote carriers compared to non-carriers (CMH derived OR, 1.66; P=0.008 and fully adjusted OR, 1.62; P=0.02) (Table 3). This elevated risk in heterozygotes remains significant even after considering a conservative Bonferroni adjusted threshold27 of 0.025 to account for the number of SNPs tested. For homozygote carriers compared to non-carriers, there was a non significant decreased risk of CAD (CMH derived OR, 0.37; P=0.40 and fully adjusted OR, 0.55; P=0.63).
Table 3.
Genotypes counts and Odds Ratios for the ALOX15.18 SNP in the ADVANCE study, stratified by case/control set and race/ethnic group
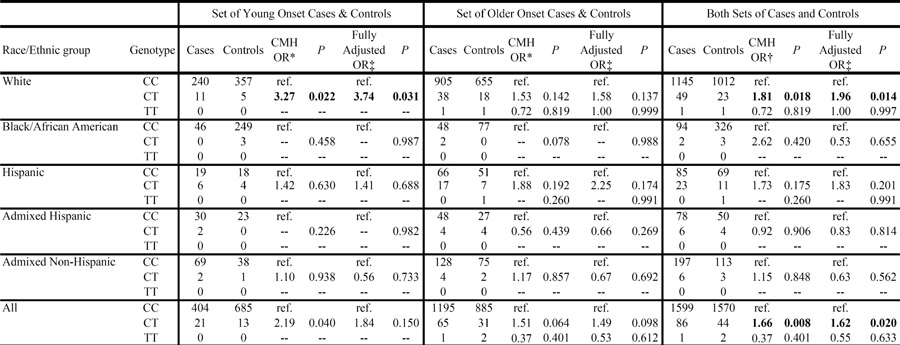 |
CMH = Cochran-Mantel-Haenszel, OR = Odds Ratio, ref. = reference group, -- = unable to compute, OR with p values < 0.05 in bold
Combined analyses adjusted for Race/ethnic group.
Combined analyses adjusted for race/ethnic group, case-control set, or both.
*,† For all combined analyses, Breslow-Day test of homogeneity of ORs across strata was not significant.
Adjusted for age, sex, BMI, smoking status, hypertension, diabetes, high cholesterol. “Admixed” strata further adjusted for individual proportion of white, black, hispanic, and east asian ancestry derived by the program STRUCTURE. Non stratified analyses further adjusted for race/ethnic group, case/control set, or both.
The minimal detectable ORs for the promoter SNP was 1.2 (or 0.83 assuming a low risk minor allele) for the log additive co-dominant mode of inheritance and 1.8 (0.25) for the recessive mode of inheritance. For ALOX15.18 the minimal detectable OR was 1.7 (or 0.53 assuming a low risk minor allele) and 46.0 (0.00), respectively.
Transcription Factor Binding studies
The minor allele of ALOX15.18 results in a non-synonymous amino acid exchange from threonine at position 560 to methionine (T560M). MatInspector analysis of the DNA sequence in the region of the polymorphism did not find a concensus binding site for known transcriptional regulators with either the major or the minor allele base at the ALOX15.18 polymorphic site.
Mutagenesis studies
Expression of the wild type (wt) 12/15-LOX in E. coli resulted in large amounts of enzyme protein. In contrast, expression of the 560M mutant was strongly reduced (Fig. 1, inset A). To directly compare the specific catalytic activities, the two enzyme species were purified by affinity chromatography and equal amounts of 12/15-LOX protein were subjected to activity assays, in which the 560M mutant exhibited a residual catalytic activity of about 5% compared to the wt (Fig 1, Table 4). To exclude possible artefacts that might be related to the prokaryotic expression system we transiently transfected HEK792 (human kidney carcinoma) cells with mammalian expression plasmids containing either the wt or the mutant 12/15-LO cDNA. Transfection efficiency was normalized to a control plasmid (luciferase) and 12/15-LOX protein expression was quantified by immunoblotting. In this system we did not find major differences in the level of protein expression. Again the 560M mutant exhibited a residual catalytic activity of about 7% when compared with the wt enzyme (Table 4).
Fig. 1. Expression of wt-human 12/15-LOX and its T560 M mutant in E. coli.
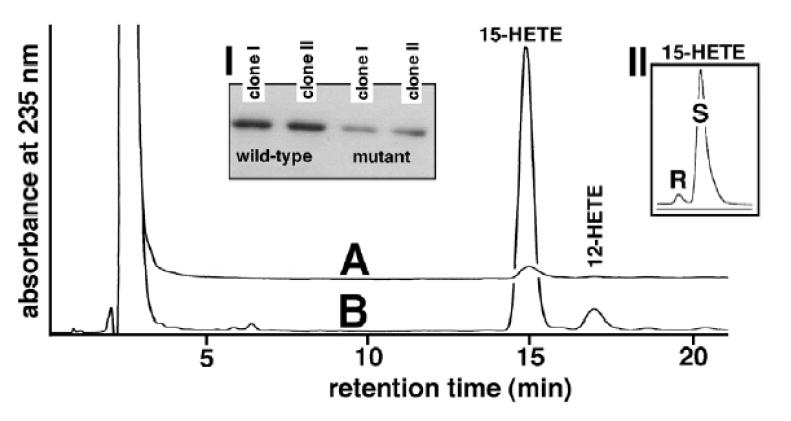
The two enzyme species were expressed in E. coli, purified on a Ni-agarose column and used in activity assays and immunoblotting (see Material and methods). A: T560M mutant, B: wild-type enzyme, Inset I: Comparison of the absolute expression levels of the two enzyme species (immunoblotting of lysis supernatant). Inset II: Enantiomer composition 15-HETE formed by the mutant enzyme.
Table 4.
Relative expression levels and specific activities* of 12/15-LOX species expressed in pro- and eukaryotic expression systems
| Expression system | rel. expression level | rel. specific activity | ||
|---|---|---|---|---|
| wt enzyme | T560M | wt enzyme | T560M | |
| E. coli | 100 | 31 | 100 | 5 |
| HEK 297 cells | 100 | 91 | 100 | 7 |
The expression level and specific activities observed with the wild-type enzyme in the different expression systems was set 100. The expression levels were quantified by immunoblotting and the specific activities by activity assays (see Material and methods)
We repeated the HPLC quantification of oxygenation products using an internal loading control an obtained essentially identical results (details not shown).
Association Analysis in the ARIC study
The frequency of ALOX15.18 (T560M) in the ARIC study controls was similar to that of the ADVANCE controls in both whites (1.8%, 95% CI 1.6%-2.0%) and blacks (0.3%, 95% CI 0.16%-0.44%). The minor allele was in HWE in both race/ethnic strata (details not shown).
Table 5 summarizes the association analyses from the ARIC study. Compared to non-carriers, unadjusted analyses revealed that neither heterozygote nor homozygote carriers were at increased risk of incident CAD. However, both unadjusted estimates of the relative risk were greater than one (RR 1.23, P=0.18 and RR 1.3, P=0.81). Cox proportional hazards ratios adjusting for all traditional risk factors demonstrated a trend towards an increased risk in heterozygote carriers (HR 1.31, P=0.06) and a non significant slight decrease risk in homozygote carriers (HR 0.93, P=0.9). The minimal detectable hazard rate ratio was 1.6 (0.63 assuming a low risk minor allele) for the log additive co-dominant mode of inheritance and 27 (0.04) for the recessive mode of inheritance.
Table 5.
Genotypes counts and Risk Ratios for the ALOX15.18 SNP in 1407 incident CHD cases and 11567 non-cases from the ARIC study, stratified by race
| Race | Genotype | CHD cases | Non-cases | RR* | P | Fully Adjusted HR† | P |
|---|---|---|---|---|---|---|---|
| White | CC | 1057 | 8267 | ref. | -- | ||
| CT | 47 | 304 | 1.21 | 0.23 | 1.29 | 0.09 | |
| TT | 1 | 6 | 1.30 | 0.81 | 0.90 | 0.90 | |
|
| |||||||
| Black | CC | 299 | 2972 | ref. | -- | ||
| CT | 3 | 18 | 1.66 | 0.42 | 1.83 | 0.30 | |
| TT | 0 | 0 | -- | -- | -- | -- | |
|
| |||||||
| All | CC | 1356 | 11239 | ref. | -- | ||
| CT | 50 | 322 | 1.23 | 0.18 | 1.31 | 0.06 | |
| TT | 1 | 6 | 1.30 | 0.81 | 0.93 | 0.90 | |
CHD = Coronary Heart Disease, CMH = Cochran-Mantel-Haenszel, RR = Relative Risk, ref. = reference group, -- = unable to compute due to cells with zero counts
Adjusted for race in non stratified analyses.
Adjusted for age, gender, center, smoking, diabetes and hypertension status (and race in non-stratified analyses)
The CMH derived OR across both population studies for the effect of the heterozygotes carriers to non-carriers was 1.4 with 95% CI of 1.1 to 1.8 (p = 0.004). For the comparison of homozygous carriers to non carriers, the OR was 0.8 with 95% CI of 0.2 to 3.9 (p = 0.7).
Discussion
We investigated the effect of specific targeted genetic variation in ALOX15 on the risk of CAD. We found no association between a previously identified SNP in the promoter region and clinical CAD. We also identified a novel SNP in exon 13, ALOX15.18, which produces a near null variant of 12/15-LOX (T560M). In the race/ethnic groups we studied, the minor allele of ALOX15.18 was most common in Hispanics followed by white/Europeans. The minor allele was exceedingly rare in black/African Americans and absent in East Asians. Two independent cohort studies reveal interesting trends on the risk of CAD in carriers of the ALOX15.18 minor allele that warrant further investigation.
The mutagenesis data indicate an important role of Thr560 for the catalytic activity of the 12/15-LOX. In the 3D-structure of the rabbit enzyme, the corresponding amino acid is located in proximity to residues that are critical for catalysis28, 29 Using the DNA-Strider software package, multiple sequence alignments of mammalian LOX isoforms reveal a high level of conservation of this residue (details not shown). The human and murine 12R-LOXs as well as the human epidermis-type 15-LOX (15-LOX2) have a Ser at this position. However, both amino acids (Thr and Ser) contain a hydroxyl group in their side chains suggesting that such OH-groups may play a role in the catalytic activity. In fact, when we mutated Thr560 to a Ser in the human 12/15-LOX we obtained an active enzyme species (data not shown). Modeling studies suggest that this amino acid may not directly interact with fatty acid substrates but appears to contact the primary determinants for the positional specificity (Phe353, Ile418, Ile593) located at the bottom of the substrate binding pocket29. Mutation of these residues alters the catalytic activity of the enzyme and impacts reaction specificity29. A larger Met residue at this position may lead to conformational changes within this critical region impairing catalytic activity.
Studies in mice show that loss of both functional ALOX15 alleles significantly decreases the formation of aortic atherosclerosis30, 31. While an intermediate protective effect of single functional allele loss has not been definitively established it cannot be ruled out30. In the apo E−/−/L-12LO−/− double-knockout mice fed normal chow, a marked 98.7% reduction in the lesion area of aortic atherosclerosis was witnessed compared with the apo E−/−/L-12LO+/+ mice at 15 weeks30. Reduction of aortic atherosclerosis was also observed in the LDL receptor knockout model, LDL-R−/−/L-12LO−/− mice compared with LDL-R−/−/L-12LO+/+ mice showed approximately 94% reduction in the percent surface coverage of the aorta at 9 weeks and 50% reduction at 12 and 18 weeks31. In our genetic association studies in humans, our estimated lower 95% confidence interval for the OR between heterozygote carriers of 560M and non carriers across both population studies argues strongly against any protective effect of a near null variant of this enzyme when assuming a co-dominant mode of inheritance. In fact, assuming this mode of inheritance, 560M may increase the risk of CAD, a situation more consistent with experimental findings in transgenic rabbits 8 , 9, 10. The lack of an elevated risk in our very small number of homozygote carriers is inconsistent with this possibility, although the low allele frequencies of 560M found in all race/ethnic groups studied limited our ability to examine the risks associated with homozygosity of the 560M allele in a meaningful way. Importantly, our estimated lower 95% confidence interval of 0.2 across both population studies argues against the presence of a protective effect to the degree suggested by the authors of the two mouse knockout studies to date30, 31.
In conclusion, we identified a SNP in ALOX15 that considerably impairs the enzymatic activity of 12/15-LOX, leading to a near null variant of this enzyme. Among the race/ethnic groups we studied, the frequency of the 560M allele was highest in Hispanic subjects. Assuming a co-dominant mode of inheritance, this variant does not protect against clinically significant CAD and may actually increase risk. Assuming a recessive mode of inheritance, the effect of this mutation remains unclear, but a protective effect to the degree noted in mouse knockouts appears unlikely. Additional genetic studies are required before firm conclusions can be made on whether this mutation alters the risk of CAD.
Acknowledgments
The authors thank the staff and participants of the ADVANCE and ARIC studies for their important contributions.
Sources of Funding The ADVANCE Study was supported by a grant from the Donald W. Reynolds Foundation, Las Vegas, NV and the Stanford Cardiovascular Institute, Stanford, CA. The CARDIA study is supported by contracts N01-HC-48047, N01-HC-48048, N01-HC-48049, N01-HC-48050, and N01-HC-95095 from the National Heart, Lung, and Blood Institute. The Atherosclerosis Risk in Communities Study is carried out as a collaborative study supported by the National Heart, Lung, and Blood Institute contracts N01-HC-55015, N01-HC-55016, N01-HC-55018, N01-HC-55019, N01-HC-55020, N01-HC-55021, N01-HC-55022. Financial support to H.K. was provided by Deutsche Forschungsgemeinschaft (Ku 961/8-2) and by the European Commission (FP6, LSHM-CT-2004-0050333).
Role of the Sponsors The funding agencies had no role in the design or conduct of the study; collection, management, analysis or interpretation of the data; or in preparation of the manuscript.
Footnotes
Disclosures See disclosures forms for all co-authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schewe T, Halangk W, Hiebsch C, Rapoport SM. A lipoxygenase in rabbit reticulocytes which attacks phospholipids and intact mitochondria. FEBS Lett. 1975;60(1):149–152. doi: 10.1016/0014-5793(75)80439-x. [DOI] [PubMed] [Google Scholar]
- 2.Belkner J, Wiesner R, Rathman J, Barnett J, Sigal E, Kuhn H. Oxygenation of lipoproteins by mammalian lipoxygenases. Eur J Biochem. 1993;213(1):251–261. doi: 10.1111/j.1432-1033.1993.tb17755.x. [DOI] [PubMed] [Google Scholar]
- 3.Kuhn H, Thiele BJ. The diversity of the lipoxygenase family. Many sequence data but little information on biological significance. FEBS Lett. 1999;449(1):7–11. doi: 10.1016/s0014-5793(99)00396-8. [DOI] [PubMed] [Google Scholar]
- 4.Cathcart MK, Folcik VA. Lipoxygenases and atherosclerosis: protection versus pathogenesis. Free Radic Biol Med. 2000;28(12):1726–1734. doi: 10.1016/s0891-5849(00)00230-6. [DOI] [PubMed] [Google Scholar]
- 5.Spanbroek R, Grabner R, Lotzer K, Hildner M, Urbach A, Ruhling K, Moos MP, Kaiser B, Cohnert TU, Wahlers T, Zieske A, Plenz G, Robenek H, Salbach P, Kuhn H, Radmark O, Samuelsson B, Habenicht AJ. Expanding expression of the 5-lipoxygenase pathway within the arterial wall during human atherogenesis. Proc Natl Acad Sci U S A. 2003;100(3):1238–1243. doi: 10.1073/pnas.242716099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Funk CD, Cyrus T. 12/15-lipoxygenase, oxidative modification of LDL and atherogenesis. Trends Cardiovasc Med. 2001;11(34):116–124. doi: 10.1016/s1050-1738(01)00096-2. [DOI] [PubMed] [Google Scholar]
- 7.Harats D, Shaish A, George J, Mulkins M, Kurihara H, Levkovitz H, Sigal E. Overexpression of 15-lipoxygenase in vascular endothelium accelerates early atherosclerosis in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2000;20(9):2100–2105. doi: 10.1161/01.atv.20.9.2100. [DOI] [PubMed] [Google Scholar]
- 8.Shen J, Kuhn H, Petho-Schramm A, Chan L. Transgenic rabbits with the integrated human 15-lipoxygenase gene driven by a lysozyme promoter: macrophage-specific expression and variable positional specificity of the transgenic enzyme. Faseb J. 1995;9(15):1623–1631. doi: 10.1096/fasebj.9.15.8529842. [DOI] [PubMed] [Google Scholar]
- 9.Shen J, Herderick E, Cornhill JF, Zsigmond E, Kim HS, Kuhn H, Guevara NV, Chan L. Macrophage-mediated 15-lipoxygenase expression protects against atherosclerosis development. J Clin Invest. 1996;98(10):2201–2208. doi: 10.1172/JCI119029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trebus F, Heydeck D, Schimke I, Gerth C, Kuhn H. Transient experimental anemia in cholesterol-fed rabbits induces systemic overexpression of the reticulocyte-type 15-lipoxygenase and protects from aortic lipid deposition. Prostaglandins Leukot Essent Fatty Acids. 2002;67(6):419–428. doi: 10.1054/plef.2002.0452. [DOI] [PubMed] [Google Scholar]
- 11.Hughes GH, Cutter G, Donahue R, Friedman GD, Hulley S, Hunkeler E, Jacobs DR, Jr, Liu K, Orden S, Pirie P, et al. Recruitment in the Coronary Artery Disease Risk Development in Young Adults (Cardia) Study. Control Clin Trials. 1987;8(4 Suppl):68S–73S. doi: 10.1016/0197-2456(87)90008-0. [DOI] [PubMed] [Google Scholar]
- 12.Go AS, Iribarren C, Chandra M, Lathon PV, Fortmann SP, Quertermous T, Hlatky MA. Statin and beta-blocker therapy and the initial presentation of coronary heart disease. Ann Intern Med. 2006;144(4):229–238. doi: 10.7326/0003-4819-144-4-200602210-00004. [DOI] [PubMed] [Google Scholar]
- 13.Iribarren C, Go AS, Husson G, Sidney S, Fair JM, Quertermous T, Hlatky MA, Fortmann SP. Metabolic syndrome and early-onset coronary artery disease: is the whole greater than its parts? J Am Coll Cardiol. 2006;48(9):1800–1807. doi: 10.1016/j.jacc.2006.03.070. [DOI] [PubMed] [Google Scholar]
- 14.Taylor-Piliae RE, Norton LC, Haskell WL, Mahbouda MH, Fair JM, Iribarren C, Hlatky MA, Go AS, Fortmann SP. Validation of a New Brief Physical Activity Survey among Men and Women Aged 60-69 Years. Am J Epidemiol. 2006 doi: 10.1093/aje/kwj248. [DOI] [PubMed] [Google Scholar]
- 15.Strachan T, Read AP. Human molecular genetics 2. 2. New York: Wiley-Liss; 1999. [Google Scholar]
- 16.Guo SW, Thompson EA. Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics. 1992;48(2):361–372. [PubMed] [Google Scholar]
- 17.Landis RJ, Heyman ER, Koch GG. Average Partial Association in Three-way Contingency Tables: A Review and Discussion of Alternative Tests. International Statistical Review. 1978;46:237–254. [Google Scholar]
- 18.Birch MW. The Detection of Partial Association, II: The General Case. Journal of the Royal Statistical Society. 1965;B(27):111–124. [Google Scholar]
- 19.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155(2):945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Purcell S, Cherny SS, Sham PC. Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics. 2003;19(1):149–150. doi: 10.1093/bioinformatics/19.1.149. [DOI] [PubMed] [Google Scholar]
- 21.Holtby S, Zahnd E, Yen W, Lordi N, McCain C, SDiSogra C. Health of California’s Adults, Adolescents and Children: Findings from CHIS 2001. Los Angeles: UCLA Center for Health Policy Research; 2004. [Google Scholar]
- 22.Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M, Werner T. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics (Oxford, England) 2005;21(13):2933–2942. doi: 10.1093/bioinformatics/bti473. [DOI] [PubMed] [Google Scholar]
- 23.The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives. The ARIC investigators. Am J Epidemiol. 1989;129(4):687–702. [PubMed] [Google Scholar]
- 24.White AD, Folsom AR, Chambless LE, Sharret AR, Yang K, Conwill D, Higgins M, Williams OD, Tyroler HA. Community surveillance of coronary heart disease in the Atherosclerosis Risk in Communities (ARIC) Study: methods and initial two years’ experience. J Clin Epidemiol. 1996;49(2):223–233. doi: 10.1016/0895-4356(95)00041-0. [DOI] [PubMed] [Google Scholar]
- 25.Volcik KA, Ballantyne CM, Coresh J, Folsom AR, Wu KK, Boerwinkle E. P-selectin Thr715Pro polymorphism predicts P-selectin levels but not risk of incident coronary heart disease or ischemic stroke in a cohort of 14595 participants: the Atherosclerosis Risk in Communities Study. Atherosclerosis. 2006;186(1):74–79. doi: 10.1016/j.atherosclerosis.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 26.Hsieh FY, Lavori PW. Sample-size calculations for the Cox proportional hazards regression model with nonbinary covariates. Control Clin Trials. 2000;21(6):552–560. doi: 10.1016/s0197-2456(00)00104-5. [DOI] [PubMed] [Google Scholar]
- 27.Sankoh AJ, Huque MF, Dubey SD. Some comments on frequently used multiple endpoint adjustment methods in clinical trials. Statistics in medicine. 1997;16(22):2529–2542. doi: 10.1002/(sici)1097-0258(19971130)16:22<2529::aid-sim692>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 28.Gillmor SA, Villasenor A, Fletterick R, Sigal E, Browner MF. The structure of mammalian 15-lipoxygenase reveals similarity to the lipases and the determinants of substrate specificity. Nat Struct Biol. 1997;4(12):1003–1009. doi: 10.1038/nsb1297-1003. [DOI] [PubMed] [Google Scholar]
- 29.Kuhn H. Structural basis for the positional specificity of lipoxygenases. Prostaglandins Other Lipid Mediat. 2000;62(3):255–270. doi: 10.1016/s0090-6980(00)00084-8. [DOI] [PubMed] [Google Scholar]
- 30.Cyrus T, Witztum JL, Rader DJ, Tangirala R, Fazio S, Linton MF, Funk CD. Disruption of the 12/15-lipoxygenase gene diminishes atherosclerosis in apo E-deficient mice. J Clin Invest. 1999;103(11):1597–1604. doi: 10.1172/JCI5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.George J, Afek A, Shaish A, Levkovitz H, Bloom N, Cyrus T, Zhao L, Funk CD, Sigal E, Harats D. 12/15-Lipoxygenase gene disruption attenuates atherogenesis in LDL receptor-deficient mice. Circulation. 2001;104(14):1646–1650. doi: 10.1161/hc3901.095772. [DOI] [PubMed] [Google Scholar]