

Abstract
Neutrophil serine proteases are granule-associated enzymes known mainly for their function in the intracellular killing of pathogens. Their extracellular release upon neutrophil activation is traditionally regarded as the primary reason for tissue damage at the sites of inflammation. However, studies over the past several years indicate that neutrophil serine proteases may also be key regulators of the inflammatory response. Neutrophil serine proteases specifically process and release chemokines, cytokines, and growth factors, thus modulating their biological activity. In addition, neutrophil serine proteases activate and shed specific cell surface receptors, which can ultimately prolong or terminate cytokine-induced responses. Moreover, it has been proposed that these proteases can impact cell viability through their caspase-like activity and initiate the adaptive immune response by directly activating lymphocytes. In summary, these studies point to neutrophil serine proteases as versatile mediators that fine-tune the local immune response and identify them as potential targets for therapeutic interventions.
Keywords: neutrophil, cathepsin G, neutrophil elastase, proteinase 3, inflammation
Introduction
Neutrophils are the first line of defense against infection and their ability to phagocytose and kill invading microorganism is critical for the host defense. In response to infection, neutrophils engulf pathogens into an intracellular membrane-bound organelle called a phagosome where these microorganisms are destroyed by a combination of reactive oxygen species (ROS), proteases and antimicrobial peptides, aided by the acidic and nutrient deprived environment of the phagosome (Reeves et al., 2002; Stuart and Ezekowitz, 2005). Disorders of neutrophil number and function lead to increased risks of infection, as seen in patients with severe chronic neutropenia, and confirm that neutrophils are vital components of the immune defense (Newburger, 2006). However, once recruited to the site of inflammation, neutrophils must undergo apoptosis and their clearance is essential for the resolution of the infection (Eyles et al., 2006). Therefore, prolonged neutrophil accumulation can damage the host tissue and has been linked to chronic inflammation, which in turn is thought to contribute to the development of autoimmunity (Nathan, 2006). Indeed, neutrophils are a common feature of several inflammatory diseases, including rheumatoid arthritis, inflammatory bowel disease, and chronic lung diseases, to name a few (Weiss, 1989). The importance of neutrophils in initiating and maintaining inflammatory response is demonstrated by several experimental animal models. Depletion of neutrophils abrogates the inflammation in experimental arthritis (Tanaka et al., 2006; Wipke and Allen, 2001), experimental allergic airway disease (Park et al., 2006), experimental colitis (Qualls et al., 2006), and several ischemia/reperfusion injury models (Crinnion et al., 1994; Kyriakides et al., 2000; Sisley et al., 1994). However, neutrophils are more than destructive cells whose main function is to kill and digest pathogens while damaging the host’s tissues. Recent literature indicates that neutrophils have the unique capacity to directly and specifically shape the immune response (Nathan, 2006).
Neutrophils express a family of neutral serine proteases including cathepsin G (CG), neutrophil elastase (NE), and proteinase 3 (PR3) in their azurophil granules (Faurschou and Borregaard, 2003). In addition, human neutrophils also express azurocidin (AZU), an inactive serine protease homologue with chemotactic and antimicrobial activities (Almeida et al., 1996; Chertov et al., 1997). The bactericidal activity of neutrophils depends partly on the presence of these granule-associated proteases and the generation of neutrophil serine protease-deficient mice confirmed the relative importance of CG and NE in the killing of certain Gram-negative and Gram-positive bacteria (Belaaouaj et al., 2000; Belaaouaj et al., 1998; Reeves et al., 2002), as well as the clearance of certain fungi (Reeves et al., 2002; Tkalcevic et al., 2000). In addition, neutrophil serine proteases are implicated in several non-infectious inflammatory diseases, as indicated by animal models of inflammatory arthritis (Adkison et al., 2002), bullous pemphigoid (Liu, 2004; Liu et al., 2000), chronic inflammatory lung diseases (Chua et al., 2007; Shapiro et al., 2003), and tissue damage following ischemia/reperfusion injury (Shimoda et al., 2007). In some instances, the activity of these proteases goes beyond simple degradation of extracellular matrix. It is recently recognized that, although azurophil granules only undergo limited exocytosis, the extracellularly released serine proteases play an important role in the modulation of the inflammatory response by proteolytically modifying chemokines and cytokines, activating latent forms of cytokines and growth factors, and cleaving specific cell surface receptors. In addition, neutrophil serine proteases can induce caspase-independent cell apoptosis and initiate the adaptive immune response by activating lymphocytes. The aim of this review is to present the current knowledge regarding neutrophil serine proteases and their role as regulators of the inflammatory process and immune response.
Neutrophil serine protease biosynthesis and secretion
Neutrophil serine proteases CG, NE, and PR3 are encoded by highly homologous genes in humans and mice. NE and PR3 are tightly linked in a cluster found on chromosome 19 (Zimmer et al., 1992) and chromosome 10 (Belaaouaj et al., 1997) in humans and mice, respectively. In humans the gene for AZU is also encoded on chromosome 19. CG on the other hand is found in a separate cluster on syntenic regions of human and mouse chromosome 14 that include genes encoding for granzymes and mast cell chymases (Caughey et al., 1993; Heusel et al., 1993). The neutrophil granule-associated serine protease genes are tightly regulated and expressed only during the promyelocytic stage of myeloid development (Garwicz et al., 2005). Despite their distinct chromosomal localization, the expression of these proteases appear to be controlled at the transcriptional level by similar factors that bind to similar motifs in their proximal promoter regions (Faurschou and Borregaard, 2003; Lennartsson et al., 2005). The mRNAs of these serine proteases are expressed at the highest levels during the promyelocytic stage and then subsequently downregulated as the neutrophils mature.
Neutrophils contain several subsets of granules that are formed sequentially during neutrophil differentiation in the bone marrow. The granules found in neutrophils are azurophil granules (also known as primary granules), specific granules (also known as secondary granules), gelatinase granules (also known as tertiary granules), and secretory granules (Faurschou and Borregaard, 2003). The segregation of proteins into different subsets of granules is determined solely by the time of their biosynthesis and does not depend on individual sorting information present in the proteins (Le Cabec et al., 1996). The maturation-specific expression of serine proteases expression therefore explains why these enzymes are only present in azurophil granules, which are formed during the promyelocytic stage, and absent in granules formed at later stages of neutrophil differentiation. However, the mechanism by which these proteases are targeted to the granules instead of being constitutively secreted is still unknown. No sorting motifs have been found for these proteases. And although the major mechanism for sorting proteins to granules involve mannose-6-phosphate receptors, this pathway is not implicated in the sorting and targeting of proteins in neutrophils. Instead, adaptor protein 3 (AP3) seems to play an important role in targeting serine proteases to azurophil granules. AP3 is a cargo protein responsible for shuttling proteins from the trans-Golgi network to the granule compartment. The role of AP3 in granule sorting is seen in the Hermansky-Pudlak syndrome and in canine cyclic neutropenia where mutations in AP3 disrupt the normal trafficking of NE (Benson et al., 2003). Recent evidence also indicates that serglycin, the major intracellular proteoglycan, is required for NE localization to granules (Lemansky et al., 2007; Niemann et al., 2007). Serglycin has previously been shown to complex with the highly related cytotoxic T lymphocyte granule-associated serine proteases granzymes and may also act as a chaperone for these secreted proteases (Raja et al., 2002). The targeting of CG and PR3 to azurophil granules however is mediated by an unknown mechanism.
Serine proteases are synthesized as inactive zymogens that require two N-terminal proteolytic modifications to become active. After the signal peptide removal, the proenzyme is further processed by the lysosomal cysteine protease dipeptidyl peptidase I (DPPI, also known as cathepsin C) en route to the granules where they are stored as active enzymes (Adkison et al., 2002). Processing by DPPI is required for targeting and stable storage in granules, as most of the proform is either constitutively secreted (Garwicz et al., 2005) or degraded, as has been shown for the proform of granzyme A (Pham and Ley, 1999). In addition, neutrophil serine proteases also undergo C-terminal processing. Although this proteolytic modification is not required for enzymatic activity, the removal of this C-terminal sequence reveals a docking site that allows NE to interact with AP3 (Benson et al., 2003). The retention of this C-terminus apparently results in mistrafficking of NE and its relocation to the surface membrane (Benson et al., 2003). Because of this tightly regulated expression and storage, perturbation of the intracellular trafficking of NE to granules results characteristic disorders that will be discussed below. Whether C-terminal truncation is also required for the proper trafficking of CG and PR3 to granules is still unknown.
Although neutrophil serine proteases function mainly as intracellular antimicrobial agents, they are also released following limited granule exocytosis. It is well established that a hierarchy of mobilization exists for the exocytosis of different granule subsets, specifically regarding the extent to which the granule content is released in response to stimuli. Specific and tertiary granules are readily exocytosed upon cell activation with a variety of stimuli while azurophil granules mainly fuse with the phagosome and only release a limited amount of their content extracellularly (Borregaard and Cowland, 1997; Mollinedo et al., 1999). The molecular basis for this hierarchy in exocytosis is still poorly understood but recent data suggest that different combinations of soluble N-ethylaleimide-sensitive factor attachment protein receptors (SNAREs), vesicle-associated membrane proteins (VAMPs), 23-kDa synaptosome-associated protein (SNAP-23), and syntaxin 4 control the differential mobilization of neutrophil granule subsets (Mollinedo et al., 2006). Thus, a higher ability to form diverse SNARE complexes renders the granules more prone to exocytosis. In addition, the actin cytoskeleton also regulates exocytosis of neutrophil granules. The actin cytoskeleton acts to limit the rate and extent of granule exocytosis for secretory, gelatinase, and specific granules and blocks fusion of azurophil granules to the plasma membrane under basal and certain stimulated conditions (Jog et al., 2007). Thus, these different mechanisms act to prevent uncontrolled secretion of destructive proteases from intracellular granules unless neutrophils are appropriately activated.
Once released from the neutrophils these proteases remain active although the extracellular environment is replete with highly specific endogenous inhibitors. Under normal circumstances, α1-proteinase inhibitor and elafin (also known as skin-derived antileukoprotease) antagonize the excessive extracellular activity of NE and PR3 whereas secretory leukocyte protease inhibitor (SLPI) is a potent inhibitor of CG and NE. In addition, CG can also be inhibited by α1-antichymotrypsin (Fitch et al., 2006; Reid and Sallenave, 2001; Rest, 1988). However, there are several mechanisms by which neutrophil serine proteases evade endogenous inhibitors. Some studies suggest that the high local concentration of proteases at the sites of inflammation can overwhelm the action of protease inhibitors (Owen and Campbell, 1995). Others suggest that extracellular proteases remain bound to neutrophil cell surface through their interactions with chondroitin sulfate and heparan sulfate proteoglycans (Campbell and Owen, 2007) and this interaction may render the proteases inaccessible to circulating, high-molecular weight inhibitors (Campbell et al., 2000; Owen et al., 1995). More recent studies however indicate that cell membrane-bound NE can be fully inhibited by α1-proteinase inhibitor when the cells are in suspension (Korkmaz et al., 2005) and perhaps it its the tight adhesion between neutrophils and their substrates that leads to the compartmentalization of the membrane-bound proteases, thus excluding natural circulating inhibitors.
Neutrophil serine proteases in infections
Studies using loss-of-function mutations in neutrophil serine proteases indicate that mice deficient in CG are more susceptible to infections with Gram-positive Staphylococcus aureus (Reeves et al., 2002). NE-deficient mice on the other hand have impaired host defense against infections with Escherichia coli and Klebsiella pneumoniae (Belaaouaj et al., 1998). NE kills E. coli by degrading their outer membrane protein A (OmpA) (Belaaouaj et al., 2000). In addition, NE also cleaves the virulence factors of enterobacteria Salmonella enterica, Shigella flexneri, and Yersinia enterocolitica (Weinrauch et al., 2002). Moreover NE and CG are important for the antifungal activity against Candida albicans and Aspergillus fumigatus (Reeves et al., 2002; Tkalcevic et al., 2000). Recently, a novel mechanism has been described where stimulated neutrophils release serine proteases and chromatin to form structures called neutrophil extracellular traps (NETs) that capture and kill pathogens (Brinkmann et al., 2004; Brinkmann and Zychlinsky, 2007). In contrast, several studies suggest that neutrophil serine proteases may also interfere with the host normal defense and promote viral infection. Indeed CG has been shown to inhibit the host’s ability to clear Pseudomonas aeruginosa from the lung, as indicated by a reduction in bacterial load recovered from infected CG-deficient mice (Sedor et al., 2007). NE on the other hand facilitates reovirus infection in U937 cells (Golden and Schiff, 2005) and CG increases the susceptibility of macrophages to acute human immunodeficiency virus type 1 (Moriuchi et al., 2000), thus raising the possibility that neutrophil serine protease release during inflammation may actually exacerbate certain infections by promoting viral spread. In summary, loss-of-function mutations in the mouse system reveal a prominent role for neutrophil serine proteases in the clearance of microorganisms. However, direct comparisons between mice and men are difficult, as murine neutrophils do not contain the broad-spectrum antimicrobial peptides defensins that are abundant in the human system (Eisenhauer and Lehrer, 1992).
Neutrophil serine proteases in human diseases
Although deficiencies in specific neutrophil serine proteases have not been described in humans, relative deficiencies of these enzymes are associated with distinct disorders. Chediak-Higashi syndrome is a disease characterized by recurrent bacterial infections, bleeding defects, and partial albinism (Holt et al., 2006). Mutations in these patients have been mapped to the CHS or “Lyst” protein that is believed to function in membrane interactions and vesicle transport (Ward et al., 2000). Neutrophils from patients and the beige mice with the same mutation form giant granules with a severe defect in exocytosis and significantly reduced serine protease activity. It is believed that the defect in granule secretion and the reduced protease activity may in part contribute to the increased susceptibility to infections. Loss-of-function mutations in DPPI leading to defects in neutrophil serine proteases have also been identified in patients with Papillon-Lefevre syndrome (PLS) (Hart et al., 2000; Toomes et al., 1999). Neutrophils from these patients have minimal residual activities in all three serine proteases (de Haar et al., 2004; Pham et al., 2004). Loss of neutrophil serine protease activity may also affect the processing of human 18-kDa cationic antimicrobial protein (hCAP18) to the antimicrobial peptide LL-37 (Sorensen et al., 2001), which may also potentially explain the susceptibility of PLS patients to periodontal diseases (de Haar et al., 2006). However, not all patients with PLS have defective antimicrobial activity against common microorganisms (Pham et al., 2004), indicating that human neutrophils do not rely solely on serine proteases to kill invading pathogens.
Antineutrophil cytoplasmic antibodies (ANCAs) are diagnostic markers found in several forms of small vessel vasculitis, including Wegener’s granulomatosis, microscopic polyangiitis, and Churg-Strauss syndrome. ANCAs are directed primarily against myeloperoxidase (MPO, another major protein found in azurophil granules) and PR3 (Bosch et al., 2006; Kallenberg et al., 2006). ANCAs have been shown to activate cytokine-primed neutrophils to release ROS and pro-inflammatory cytokines in vitro and anti-MPO ANCAs directly induce pauci-immune glomerulonephritis and systemic vasculitis in vivo (Xiao et al., 2002). Although anti-PR3 ANCAs are found in 90% of the cases of Wegener’s granulomatosis and they activate neutrophils in vitro, it is unclear whether they are directly pathogenic in vivo (Pfister et al., 2004).
Other disorders mediated by neutrophil serine proteases includde the hereditary neutropenia. Cyclic neutropenia (CN) and severe congenital neutropenia (SCN) are diseases characterized by maturation arrest of myelopoiesis leading to neutropenia. The molecular basis of CN and SCN has been identified as heterozygous mutations in the gene ELA2 encoding NE (Horwitz et al., 2007). Recent observations indicate that mutations in ELA2 disrupt normal intracellular trafficking of NE, leading to the accumulation of mutant NE protein intracellularly and resulting in apoptosis through the induction of the unfolded protein response (Kollner et al., 2006).
Neutrophil serine proteases in the modulation of chemokines and cytokines
Growing evidence suggests that neutrophil serine proteases are closely involved in the regulation of chemokine and cytokine bioavailability. Indeed several studies have shown that neutrophil serine proteases proteolytically modify the activity of several chemokines and provide an alternative mechanism to convert cytokine precursors into their active forms. Chemokines are a family of small chemoattractant proteins that play an important role in recruiting leukocytes to the sites of inflammation. Studies have shown that limited truncation of chemokines by enzymatic proteolysis can enhance their activity and stability. In fact, N-terminal proteolytic modification of CXC-chemokine ligand 8 (CXCL8, also known as IL-8) by PR3 (Padrines et al., 1994) and CXCL5 (also known as ENA-78) by CG (Nufer et al., 1999) regulate their biological activity as the truncated variants have higher potency and efficiency for neutrophil chemotaxis than the full-length forms. Likewise, the N-terminal processing of CCL15 (also known as MIP-1δ) by CG leads to significant increased potency in its ability to induce calcium fluxes and chemotactic activity on monocytes (Richter et al., 2005). CG also converts connective tissue-activating peptide III (CTAP-III) into CXCL7 (also known as neutrophil-activating peptide 2 or NAP-2) by limited proteolysis (Schiemann et al., 2006). In addition, CG and NE convert prochemerin to active chemerin, a novel chemoattractant factor that specifically attracts antigen-presenting cells, through the removal of a C-terminal peptide (Wittamer et al., 2005). Human NE and CG cleave the alternatively spliced form of CCL23 by both N-terminal and C-terminal processing, producing a CC chemokine “body” that can engage two distinct chemokine receptors with high affinity (Miao et al., 2007). In contrast, several studies have also shown that proteolytic processing of chemokines by serine proteases can result in decreased activity. CG digests CCL5 (also known as RANTES) into a variant lacking three N-terminal residues and exhibiting lower chemotactic activity (Lim et al., 2006). Proteolysis of CCL3 (also known as MIP-1α) by all three neutrophil serine proteases (Ryu et al., 2005) and proteolysis of CXCL12 (also known as SDF-1α) and its cognate receptor CXCR4 by NE and CG (Delgado et al., 2001; Rao et al., 2004; Valenzuela-Fernandez et al., 2002) abrogate their chemotactic activity. In addition to proteolytic modifications, studies also indicate that neutrophil serine proteases control the release of chemokines from neutrophils. Neutrophils themselves are a source of chemokines (Cassatella, 1995; Scapini et al., 2000) and the release of CXCL2 (also known as MIP-2) from neutrophils depends on the activity of CG (Raptis et al., 2005). The ability of neutrophil serine proteases to control chemokine release and bioactivity therefore suggests that they may play a crucial role in orchestrating the subsequent recruitment of different leukocyte subsets to the inflamed site.
Likewise, most of the important cytokines produced at the sites inflammation also require proteolytic processing to be released from an inactive precursor form. Interleukin-18 (IL-18), originally named interferon-gamma (IFN-γ)-inducing factor, is constitutively secreted by a variety of cells as a pro-IL-18 precursor that is activated by caspase 1 (Ghayur et al., 1997; Gu et al., 1997). However, stimulation of epithelial cells with IFN-γ in the presence of PR3 and lipopolysaccharide (LPS) leads to the release of biologically active mature IL-18, independent of caspase 1 (Sugawara et al., 2001). In contrast, it has been shown that mature IL-18 released by neutrophils can be proteolysed by NE leading to diminished biological activity (Robertson et al., 2006).
Tumor necrosis factor alpha (TNF-α) exists as a proform bound to the surface membrane and is proteolytically activated by the membrane-bound metalloproteinase TNF-α converting enzyme (TACE) (Black et al., 1997). Similarly, pro-IL-1β is converted to its active, soluble form by caspase 1 (also known as interleukin-1 converting enzyme or ICE) (Thornberry et al., 1992). However, studies indicate that serine proteases may alternatively process TNF-α and IL-1β. It has been shown that serine protease inhibitors suppress the release of TNF-α from the surface of activated macrophages (Armstrong et al., 2006; Scuderi, 1989). Moreover, pre-treatment with a serine protease inhibitor leads to suppression of TNF-α release in vivo and protects mice against the effects of LPS (Wendel, 1990). In vitro studies confirm that PR3 processes the cytokine precursor to generate a biologically active form of TNF-α (Coeshott et al., 1999; Robache-Gallea et al., 1995). In contrast, the effect of CG and NE on TNF-α processing is controversial. Some studies indicate that CG and NE degrade TNF-α, resulting in a loss of activity (Scuderi et al., 1991). Others suggest that at the appropriate concentrations CG and NE can process the membrane-bound TNF-α into a soluble and biologically active form (Mezyk-Kopec et al., 2005). Although the enzymatic activity of PR3 closely resembles that of NE, only PR3 has been shown to convert the precursor of IL-1β into its biologically active form (Coeshott et al., 1999). The fact that there are converting-enzyme-independent pathways for TNF-α and IL-1β release and that IL-1β production is not entirely abolished in the absence caspase 1 (Fantuzzi et al., 1997) suggests that alternative processing of these cytokines by neutrophil serine proteases may be operative at sites of inflammation.
IL-32 is a pro-inflammatory cytokine originally found as a transcript termed NK4 in activated natural killer cells and T lymphocytes (Dahl et al., 1992). IL-32 expression is increased in a variety of inflammatory diseases, including rheumatoid arthritis, crohn’s disease, psoriasis, and chronic obstructive pulmonary disease (Dinarello and Kim, 2006). It has been shown that PR3 is a binding protein for IL-32 and limited proteolysis of IL-32 by PR3 enhances the activity of the cytokine (Novick et al., 2006). In contrast CG, NE, and PR3 have all been shown to degrade and inactivate IL-6 at sites of inflammation (Bank et al., 2000). Since IL-6 has both pro- and anti-inflammatory effects (Tilg et al., 1997), it is unclear whether IL-6 inactivation has limiting or pro-inflammatory consequences. Thus, neutrophil serine proteases can fine-tune the local inflammatory response by modulating the biological activity of chemokines and cytokines through limited proteolysis. In some cases proteolysis enhances biological activity. In others, proteolysis leads to inactivation and may provide a negative feedback mechanism to terminate immunostimulating signals.
Neutrophil serine proteases in growth factor regulation
Mucus hypersecretion is a prominent feature of chronic inflammatory lung diseases and stimulation of the epidermal growth factor receptor (EGFR) has been shown to be a major cause of mucus secretion (Nadel, 2007). NE promotes mucous cell hyperplasia and MUC5AC mRNA expression by modulating the cleavage of the membrane-bound transforming growth factor alpha (TGF-α), releasing a soluble form that binds to and activates EGFR (Kohri et al., 2002; Voynow et al., 2004). The TGF-α soluble form can also be released from the surface membrane by TACE, which is activated by ROS (Nadel, 2007). NE is also thought to activate EGFR through an indirect pathway involving protein kinase C (PKC) activation and ROS production that in turn activate TACE, leading to the cleavage of pro-TGF-α into its active soluble form (Shao and Nadel, 2005). In addition, TGF-α liberated by NE also induces keratinocyte proliferation via EGFR activation, suggesting that NE may play a role in epidermal proliferation (Meyer-Hoffert et al., 2004). The fact that NE activity is elevated in psoriatic lesions suggests that it may be a relevant stimulus for epidermal proliferation in this inflammatory disease (Wiedow et al., 1995). Transforming growth factors, such as TGF-β, exist in latent form complexed with different binding proteins. The latent form of TGF-β consists of the mature TGF-β homodimer, the latency-associated protein (LAP), and a latent TGF-beta-binding protein (LTBP), which mediates the association of TGF-β to the extracellular matrix. Through the proteolysis of LTBP, NE releases the active growth factor and therefore contributes to the tissue remodeling that accompanies inflammation in the lung (Taipale et al., 1995). NE has also been shown to stimulate the release of TGF-β through a myeloid differentiation primary-response gene 88 (MyD88)/IL-1 receptor-associated kinase (IRAK)/nuclear factor κB (NF-κB) pathway from airway smooth muscle cells, suggesting another mechanism by which NE can cause airway mucus hypersecretion (Lee et al., 2006). The role of NE in growth factor release was also confirmed in vivo where intra-tracheal instillation of elastase into mice leads to a time-dependent increase in the active form of TGF-β1 in the bronchoalveolar lavage (Buczek-Thomas et al., 2004).
Neutrophil serine proteases in the activation of cell surface receptors
In addition to the proteolytic modulation and release of cytokines and growth factors, neutrophil serine proteases also contribute to the inflammatory response through the specific activation of cell surface receptors. Integrins are a large family of cell surface receptors that mediate the interaction between cells and the environment. NE has previously been shown to proteolytically activate the platelet integrin alphaIIbbeta3 through limited cleavage of the carboxyl terminus of the alphaIIb subunit heavy chain, a process that enhances platelet aggregation (Si-Tahar et al., 1997). Recently the presence of neutrophil serine protease CG on the surface of cells has been shown to modulate neutrophil spreading, clustering of integrins, which lead to optimal neutrophil effector functions (Raptis et al., 2005). The action of CG requires its proteolytic activity but does not involve direct cleavage of integrins. The mechanism by which CG modulates neutrophil spreading is unclear; however, it has been shown that shedding of CD43 (also known as leukosialin) is required for neutrophils to fully spread (Nathan et al., 1993). Furthermore, it has been shown that NE cleaves the ectodomain of CD43 (Remold-O'Donnell and Parent, 1995). Whether CG also cleaves CD43 remains to be studied.
On the other hand, studies indicate that integrins may provide binding sites for neutrophil serine proteases on the cell surface. Cai and colleagues have shown that NE binds directly to integrin CD11b/CD18 (CR3, Mac-1) and regulates integrin-mediated cellular attachment and detachment (Cai and Wright, 1996). Others also report that PR3 can co-immunoprecipitate with CD11b and Fc gamma receptor IIIb (FcγRIIIb) (David et al., 2005; David et al., 2003). However, recent evidence indicates that neutrophil serine proteases bind to proteoglycans on the surface of cells in a saturable and reversible manner (Campbell and Owen, 2007). Rather than being contradictory, these studies suggest that serine proteases likely bind to proteoglycans on the cell surface and can be found in the same complex that includes Fc gamma receptor and CD11b/CD18 (Fridlich et al., 2006).
Protease-activated receptors (PARs) belong to a family of G-protein-coupled receptors that undergo N-terminal cleavage to reveal a tethered ligand. The role of PARs in inflammation is well documented (Coughlin and Camerer, 2003). It has been shown that CG directly activates PAR4 on platelets and this activation may mediate neutrophil-platelet interactions at the sites of inflammation (Sambrano et al., 2000). In contrast, the role of neutrophil serine proteases in the activation of other PARs is more controversial. Activation of PAR2 has been shown to be both pro- and anti-inflammatory (Chignard and Pidard, 2006) and neutrophil serine proteases can simultaneously activate and disarm this receptor. Uehera et al. have shown that in vitro, all three neutrophil serine proteases activate PAR2, which then induces the release of the chemokines CXCL8 and CCL2 (Uehara et al., 2002; Uehara et al., 2003; Uehara et al., 2004). On the other hand, these proteases have also been shown to disarm PAR2 through different cleavage sites (Dulon et al., 2003). Likewise, NE, CG, and PR3 cleave PAR1 at distinct sites from thrombin and inhibit subsequent thrombin-induced activation of the receptor (Renesto et al., 1997). Thus it is clear that in vitro neutrophil serine proteases can display opposite effects on PARs but the precise role of these enzymes in the activation of these receptors in vivo requires further studies.
Previous studies have shown that subcutaneous injection of CG into mice induces the recruitment of monocytes and neutrophils, suggesting that CG is a potent chemoattractant in vivo (Chertov et al., 1997). Neutrophil serine protease inhibitors abolish the chemotactic activity of CG, indicating a requirement for its enzymatic activity. In addition, alpha-1-antichymotrypsin and antibodies to CG inhibit the bacterial chemotactic peptide N-formyl-L-methionyl-L-leucyl-L-phenylalanine (fMLP) and C5a-induced chemotaxis, indicating that CG also modulates leukocyte chemotaxis in vitro (Lomas et al., 1995). The fact that CG inhibits the action of fMLP suggests that the two molecules may share a common receptor. Indeed, Sun et al. recently show that the chemotactic activity of CG is mediated through the G-protein-coupled formyl peptide receptor (FPR). CG binds to FPR, inducing calcium fluxes and translocation of protein kinase C-ζ(PKCζ), a step that is essential for CG-mediated chemotactic activity (Sun et al., 2004).
NE has been shown to induce the release of IL-8 (Devaney et al., 2003), cathepsin B, and matrix metalloproteinase 2 (MMP2) (Geraghty et al., 2007) through a MyD88/IRAK/TNF-receptor-associated factor 6 (TRAF-6)-dependent pathway that also implicates toll-like receptor 4 (TLR4) (Devaney et al., 2003; Walsh et al., 2001). Neutralizing antibodies to TLR4 and a dominant negative IRAK-4 abrogate the NE-induced upregulation of cathepsin B and MMP2 (Geraghty et al., 2007), further confirming the involvement of this pathway; but how NE activates TLR4 is unknown.
Neutrophil serine proteases in the shedding of surface receptors
Proteolytic shedding of cell surface receptors provides a mechanism for controlling cytokine bioactivity at the sites of inflammation. Shedding of receptors renders the cells less responsive to the cytokine effects and provides a source of soluble proteins in the body fluids. Porteu et al. show that in vitro stimulation of neutprophils with chemotactic factors, such as fMLP and C5a, and pharmacological agonists, such as calcium ionophores, leads to the loss of TNF receptors from the cell surface (Porteu and Nathan, 1990). This TNF receptor sheddase was identified as NE (Porteu et al., 1991). NE acts on the p75 TNF receptor, releasing a 32-kDa soluble fragment, while the 55-kDa TNF receptor is completely resistant to the activity of NE. The shedding of TNF receptor by NE may provide an additional mechanism for the control of cellular response to TNF at sites of inflammation.
Urokinase-type plasminogen activator receptor (CD87) is a glycophosphatidylinositol (GPI)-anchored protein that participates in cell migration and tissue remodeling. Cleavage of CD87 by CG and NE reduces the ability of the cell to bind urokinase, leading to reduced pericellular proteolysis and thus affecting cellular migration in the context of tissue remodeling. Moreover, proteolysis of CD87 generates soluble chemotactic fragments that can influence the inflammatory response (Beaufort et al., 2004).
CD14 is the main bacterial LPS cell surface receptor. Recognition of LPS occurs mainly through the TLR4-MD2-CD14 complex (Miller et al., 2005). CG and NE released by neutrophils cleave CD14 from the cell surface in a time- and concentration-dependent manner (Le-Barillec et al., 1999; Le-Barillec et al., 2000; Nemoto et al., 2000). Proteolysis of CD14 suppresses LPS-induced cell activation and CXCL8 production, suggesting that these neutrophil serine proteases might provide a negative feedback mechanism to downmodulate the inflammatory response. On the other hand, cleavage of CD14 by NE has been shown to reduce the ability of macrophages to recognize apoptotic cells (Henriksen et al., 2004). Moreover, NE can also disrupt apoptotic cell phagocytosis by cleaving the phosphatidylserine receptor (Vandivier et al., 2002). Since the resolution of inflammation requires the recognition and phagocytic removal of apoptotic cells by macrophages, impaired apoptotic cell recognition may contribute to prolonged inflammation.
Neutrophils and complement are crucial components of the innate immunity and the interaction of complement and neutrophils modulates the inflammatory response. Complement receptor 1 (CR1, also known as CD35 or C3b/C4b receptor) is a transmembrane protein of many hematopoietic cells. CR1 is cleaved from the surface of neutrophils by NE and NE-like activity, releasing the functional soluble fragment sCR1 that inhibits complement activation and prevents further complement-mediated injury (Hamacher et al., 1998; Sadallah et al., 1999). In addition, previous studies also show that membrane bound CG can generate active fragments of C3 (Maison et al., 1991) and NE cleaves C5 in the presence of C6, leading to the formation of an active C5b6-like complex that lyse non-sensitized cells upon the addition of the terminal components C7, C8, and C9 (Vogt, 2000). In addition, the cleavage of C5 by NE modulates the relative yield of C5a-like fragment, a chemotactic peptide. In contrast, NE and CG have been shown to inhibit the responsiveness of neutrophils to C5a, thus potentially down-regulating the acute inflammatory response (Tralau et al., 2004).
Neutrophil serine proteases have also been implicated in the release of the cell surface-bound ligand-binding chains of IL-6 receptor (Bank et al., 2000). The soluble IL-6 receptor subunits retain ligand-binding capacity and therefore may influence cellular responsiveness to IL-6. In addition, CG and NE can release soluble CD23 fragments that stimulate monocytes to produce oxidative burst and pro-inflammatory cytokines without any co-stimulatory signals (Brignone et al., 2001). In summary, these studies suggest that neutrophil serine proteases have the potential to activate and shed many receptors. However, the consequences of these activities in vivo remain a matter of further investigation.
Neutrophil serine proteases in cell adhesion and transmigration
Recruitment of neutrophils to sites of inflammation requires firm adhesion and transmigration of the cells across the endothelium. Studies have previously shown that inhibition of NE alone was sufficient to reduce neutrophil infiltration (without affecting neutrophil adherence) after ischemia/reperfusion in skeletal muscle (Carden and Korthuis, 1996). Also, incubation of neutrophils with a combined inhibitor of CG and NE markedly reduces the adhesion of neutrophils to various surfaces (Delyani et al., 1996; Murohara et al., 1995). Due to their proteolytic properties, it was originally thought that neutrophil serine proteases degrade components of the extracellular matrix, thus allowing cells to crawl through the gaps. Studies using serine protease inhibitors subsequently show that neutrophil transendothelial migration in vitro does not require serine proteases (Mackarel et al., 1999). The generation of serine protease-deficient mice further confirms the fact that neutrophil serine proteases are not required for in vitro chemotaxis and certain in vivo inflammatory models. (Hirche et al., 2004; MacIvor et al., 1999; Raptis et al., 2005). However, more recently Young et al., using NE-deficient mice, demonstrate that NE in fact plays a non-redundant role in zymosan-induced leukocyte firm adhesion and transmigration (Young et al., 2004). The defect in transmigration in NE-deficient mice is accompanied by reduction in the levels of pro-inflammatory chemokines and cytokines. Furthermore the authors show that NE cooperates with platelet/endothelial –cell adhesion molecule 1 (PECAM-1) and alpha6 integrins in mediating neutrophil migration through the perivascular environment (Wang et al., 2005). CG and NE have also been shown to cleave vascular endothelial cadherins (E-cadherins) (Hermant et al., 2003; Mayerle et al., 2005), intercellular adhesion molecule 1 (ICAM-1) (Champagne et al., 1998; Robledo et al., 2003), and vascular cell adhesion molecule 1 (VCAM-1) (Levesque et al., 2001; Xu et al., 2005). Since these adhesion molecules play important role in cell-cell contact formation, their extracellular cleavage may induce formation of gaps through which neutrophils transmigrate. Thus how neutrophil serine proteases actually participate in leukocyte adhesion and transmigration remains controversial.
Neutrophil serine proteases and apoptosis
The role of granzymes in the induction of apoptosis is well established. However, emerging data suggest that neutrophil serine proteases also have caspase-like activity and can induce cellular apoptosis at the sites of inflammation. NE and PR3 entry into endothelial cells coincides with the sustained activation of proapoptotic-signaling events through extracellular signal-regulated kinase (ERK), Jun N-terminal kinase (JNK), and p38 mitogen-activated protein kinase (MAPK) pathways (Preston et al., 2002). Inhibition of JNK blocks PR3-induced apoptosis; likewise, inhibition of p38 MAPK blocks PR3- and NE-induced apoptosis, indicating a direct role for serine proteases in controlling cell viability. NE and PR3 directly cleave NF-κB (Preston et al., 2002) and PR3 can cleave p21 (Waf1/Cip1/Sdi1) to induce endothelial cell apoptosis (Pendergraft et al., 2004) and modulate differentiation of monocytic cell line (Dublet et al., 2005). In addition, incubation of epithelial cells with NE leads to decreased mitochondrial membrane potential, release of cytochrome c to the cytosol, and cleavage of caspases 9 and 3, indicating that neutrophil serine proteases may induce apoptosis through the alterations of mitochondrial membrane permeability (Ginzberg et al., 2004). These studies suggest that neutrophil serine proteases might play specific functions in cell survival during the course of inflammation.
Neutrophil serine proteases in lymphocyte activation
CG has been shown to bind to and activate lymphocytes and modulate antigen-specific humoral responses in mice (Hase-Yamazaki and Aoki, 1995; Tani et al., 2001; Yamazaki and Aoki, 1997). Co-injection of CG into mice immunized with an antigen results in significant increase in antibody production. In vitro restimulation of lymph node cells from immunized mice indicates that CG induces a marked increase in IFN-γ production and antigen-specific production of IL-4. In addition, CG enhances the cytotoxicity of natural killer cells in a dose-dependent manner (Yamazaki and Aoki, 1998). Thus CG may act as an adjuvant to modulate the immune response.
Concluding remarks
Neutrophil serine proteases have emerged as important regulators of the immune response and the generation of protease-deficient mice confirms their critical role in the perpetuation of inflammation. Studies over the last several years demonstrate that, rather than being simply degradative intracellular enzymes, neutrophil serine proteases maintain their enzymatic activity in the extracellular environment and may participate in the regulation of various biological pathways (Table 1 and Figure 1). However, most of the regulatory pathways summarized in this review come from in vitro studies. The true extent of neutrophil serine proteases involvement in these regulatory networks in specific diseases and under pathological conditions requires further studies. Nonetheless, these studies suggest that in vivo antagonism of serine protease activity may provide new therapeutic strategies in inflammatory diseases. Indeed, attempts have been made to suppress the activity of NE in hereditary emphysema (Dirksen et al., 1999; Doring, 1999), chronic obstructive pulmonary disease (COPD) (Luisetti et al., 1996; Ohbayashi, 2002), cystic fibrosis (Cantin et al., 2006; Doring, 1999; Martin et al., 2006), and the management of acute lung injury (Hoshi et al., 2005; Ono et al., 2007; Zeiher et al., 2004), all with varying results. To date, no inhibitors of serine proteases have been approved for use in patients as many practical problems associated with the synthesis and delivery of these agents still remain [Reviewed in (Chughtai and O'Riordan, 2004)].
Table 1.
Effects of neutrophil serine proteases on selected biological targets
| Targets | Proteases | Potential biological effects |
|---|---|---|
| Chemokines | ||
| CCL3 | CG, NE, PR3 | Abrogation of chemotactic activity |
| CCL5 | CG | N-terminal truncation and ↓ chemotactic activity |
| CCL15 | CG | N-terminal truncation and ↑ chemotactic activity |
| CCL23 | CG, NE | N- and C-terminal truncation and ↑ affinity to receptors |
| CXCL2 | CG | ↑ release from neutrophils |
| CXCL5 | CG | N-terminal truncation and ↑ chemotactic activity |
| CXCL7 | CG | Conversion of CTAP-III to CXCL7 |
| CXCL8 | PR3 | N-terminal truncation and ↑ chemotactic activity |
| CXCL12 | CG, NE | Abrogation of chemotactic activity |
| Chemerin | CG, NE | Conversion of prochemerin to chemerin |
| Cytokines | ||
| IL-1β | PR3 | Conversion of pro-IL-1β to soluble, active IL-1β |
| IL-6 | CG, NE, PR3 | Degradation and inactivation |
| IL-18 | PR3, NE | Conversion of pro-IL-18 to mature IL-8 by PR3; proteolysis and abrogation of activity by NE |
| IL-32 | PR3 | Increase in activity |
| TNF-α | PR3, CG, NE | Activation of membrane-bound pro-TNF-α by CG, NE, and PR3; degradation by CG and NE |
| Growth factors | ||
| TGF-β | NE | Release of membrane-bound TGF-β to activate EGFR; cleavage of LTBP to release latent TGF-β |
| Antimicrobial peptides | ||
| LL-37 | PR3 | Conversion of hCAP18 to LL-37 |
| Receptors | ||
| αIIβ3 integrin | NE | ↑ platelet aggregation |
| CD14 | CG, NE | ↓ LPS-induced cell activation; ↓ ability of macrophages to recognize apoptotic cells |
| CD43 | NE | ↑ cell spreading |
| CD87 | CG, NE | ↓ urokinase binding; generation of chemotactic fragments |
| CR1 | NE | Release of sCR1 and inhibition of complement activation |
| CXCR4 | NE | N-terminal proteolysis, ↓ CXCL12 binding |
| FPR | CG | Induction of PKCζ translocation, Ca+ flux, and chemotaxis |
| IL-6R | CG | Inactivation |
| PAR1 | CG, NE, PR3 | Inactivation |
| PAR2 | CG, NE, PR3 | Activation and inactivation of the receptor through distinct cleavage sites |
| PAR4 | CG | Activation |
| TLR4 | NE | Activation through a MyD88/IRAK/TRAF-6-dependent pathway |
| TNFR | NE | Inactivation of the p75 receptor |
| Adhesion molecules | ||
| E-cadherins | CG, NE | ↑ neutrophil transmigration |
| ICAM-1 | CG, NE | ↑ neutrophil infiltration |
| VCAM-1 | CG, NE | ↑ mobilization of hematopoietic cells |
| Apoptotic molecules | ||
| NF-κB | NE, PR3 | Induction of apoptosis |
| p21 | PR3 | Induction of apoptosis |
Figure 1.
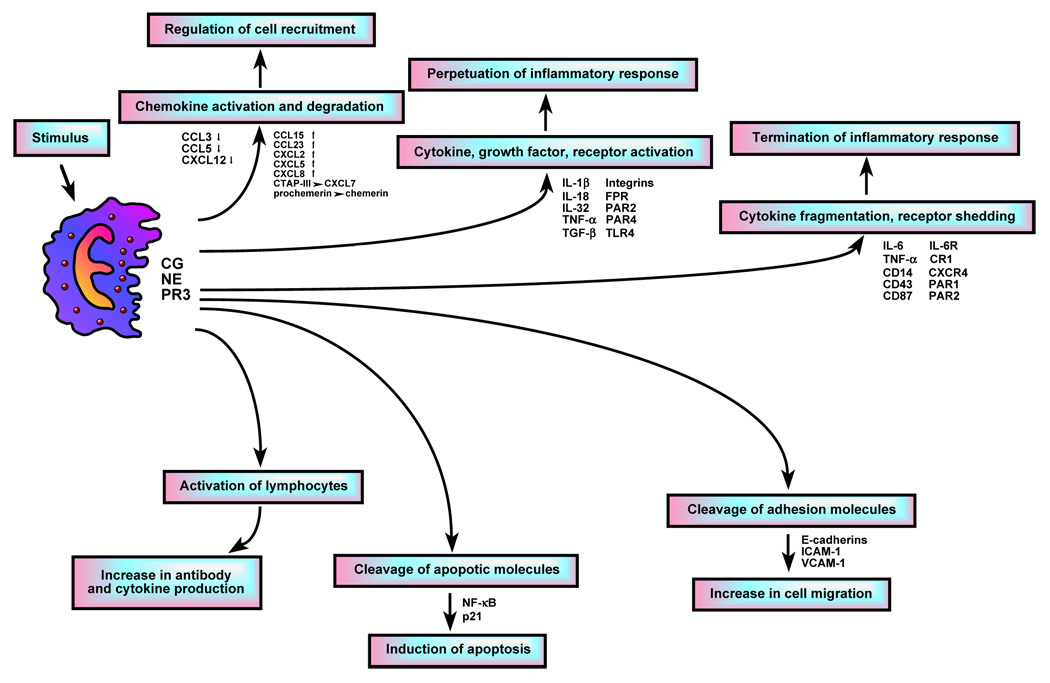
Potential mechanisms by which extracellular neutrophil serine proteases may be involved in the regulation of the inflammatory process and the immune response.
Acknowledgements
This work is supported by grants from the Sandler Program for Asthma Research and the National Institutes of Health.
Abbreviations
- CG
cathepsin G
- NE
neutrophil elastase
- PR3
proteinase 3
- AZU
azurocidin
- AP3
adaptor protein 3
- DPPI
dipeptidyl peptidase I
- SNAREs
N-ethylaleimide-sensitive factor attachment protein receptors
- VAMP
vesicle-associated membrane protein
- SNAP
synaptosome-associated protein
- SLPI
secretory leukocyte protease inhibitor
- NET
neutrophil extracellular traps
- CHS
Chediak-Higashi syndrome
- PLS
Papillon-Lefevre syndrome
- ANCA
antineutrophil cytoplasmic antibody
- ROS
reactive oxygen species
- CN
cyclic neutropenia
- SCN
severe congenital neutropenia
- MPO
myeloperoxidase
- ENA78
epithelial neutrophil activating peptide 78
- MIP
macrophage inflammatory protein
- CTAP
connective tissue-activating peptide
- NAP
neutrophil-activating peptide
- RANTES
regulated upon activation, normal T cell expressed and secreted
- SDF1
stromal cell derived factor 1
- IFN-γ
interferon gamma
- TNF
tumor necrosis factor
- TACE
TNF-α converting enzyme
- ICE
IL-1 converting enzyme
- LPS
lipopolysaccharide
- IL
interleukin
- EGFR
epidermal growth factor receptor
- TGF
transforming growth factor
- PKC
protein kinase C
- LAP
latency-associated protein
- LTBP
latent-TGF-beta-binding protein
- IRAK
IL-1 receptor-associated kinase
- MyD88
myeloid differentiation primary-response gene 88
- NF-κB
nuclear factor κB
- TRAF
TNF-receptor-associated factor
- PAR
protease-acivated receptor
- FPR
formyl peptide receptor
- fMLP
N-formyl-L-methionyl-L-leucyl-L-phenylalanine
- TLR
toll-like receptor
- PECAM
platelet/endothelial cell adhesion molecule
- ICAM
intercellular adhesion molecule
- VCAM
vascular cell adhesion molecule
- ERK
extracellular signal-regulated kinase
- JNK
Jun N-terminal kinase
- MAPK
mitogenactivated protein kinase
- APC
antigen-presenting cell
- GPI
glycophosphatidylinositol
- hCAP18
human 18-kDa cationic antimicrobial protein
- COPD
chronic obstructive pulmonary disease
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adkison AM, Raptis SZ, Kelley DG, Pham CT. Dipeptidyl peptidase I activates neutrophil-derived serine proteases and regulates the development of acute experimental arthritis. The Journal of Clinical Investigation. 2002;109:363–371. doi: 10.1172/JCI13462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida RP, Vanet A, Witko-Sarsat V, Melchior M, McCabe D, Gabay JE. Azurocidin, a natural antibiotic from human neutrophils: expression, antimicrobial activity, and secretion. Protein Expression and Purification. 1996;7:355–366. doi: 10.1006/prep.1996.0053. [DOI] [PubMed] [Google Scholar]
- Armstrong L, Godinho SI, Uppington KM, Whittington HA, Millar AB. Contribution of TNF-alpha converting enzyme and proteinase-3 to TNF-alpha processing in human alveolar macrophages. American Journal of Respiratory Cell and Molecular Biology. 2006;34:219–225. doi: 10.1165/rcmb.2005-0087OC. [DOI] [PubMed] [Google Scholar]
- Bank U, Kupper B, Ansorge S. Inactivation of interleukin-6 by neutrophil proteases at sites of inflammation. Protective effects of soluble IL-6 receptor chains. Advances in Experimental Medicine and Biology. 2000;477:431–437. doi: 10.1007/0-306-46826-3_43. [DOI] [PubMed] [Google Scholar]
- Beaufort N, Leduc D, Rousselle JC, Magdolen V, Luther T, Namane A, Chignard M, Pidard D. Proteolytic regulation of the urokinase receptor/CD87 on monocytic cells by neutrophil elastase and cathepsin G. Journal of Immunology. 2004;172:540–549. doi: 10.4049/jimmunol.172.1.540. [DOI] [PubMed] [Google Scholar]
- Belaaouaj A, Kim KS, Shapiro SD. Degradation of outer membrane protein A in Escherichia coli killing by neutrophil elastase. Science. 2000;289:1185–1188. doi: 10.1126/science.289.5482.1185. [DOI] [PubMed] [Google Scholar]
- Belaaouaj A, McCarthy R, Baumann M, Gao Z, Ley TJ, Abraham SN, Shapiro SD. Mice lacking neutrophil elastase reveal impaired host defense against gram negative bacterial sepsis. Nature Medicine. 1998;4:615–618. doi: 10.1038/nm0598-615. [DOI] [PubMed] [Google Scholar]
- Belaaouaj A, Walsh BC, Jenkins NA, Copeland NG, Shapiro SD. Characterization of the mouse neutrophil elastase gene and localization to chromosome 10. Mammalian Genome. 1997;8:5–8. doi: 10.1007/s003359900337. [DOI] [PubMed] [Google Scholar]
- Benson KF, Li FQ, Person RE, Albani D, Duan Z, Wechsler J, Meade-White K, Williams K, Acland GM, Niemeyer G, et al. Mutations associated with neutropenia in dogs and humans disrupt intracellular transport of neutrophil elastase. Nature Genetics. 2003;35:90–96. doi: 10.1038/ng1224. [DOI] [PubMed] [Google Scholar]
- Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385:729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- Borregaard N, Cowland JB. Granules of the human neutrophilic polymorphonuclear leukocyte. Blood. 1997;89:3503–3521. [PubMed] [Google Scholar]
- Bosch X, Guilabert A, Font J. Antineutrophil cytoplasmic antibodies. Lancet. 2006;368:404–418. doi: 10.1016/S0140-6736(06)69114-9. [DOI] [PubMed] [Google Scholar]
- Brignone C, Munoz O, Batoz M, Rouquette-Jazdanian A, Cousin JL. Proteases produced by activated neutrophils are able to release soluble CD23 fragments endowed with proinflammatory effects. Faseb Journal. 2001;15:2027–2029. doi: 10.1096/fj.00-0773fje. [DOI] [PubMed] [Google Scholar]
- Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- Brinkmann V, Zychlinsky A. Beneficial suicide: why neutrophils die to make NETs. Nature Reviews Microbiology. 2007;5:577–582. doi: 10.1038/nrmicro1710. [DOI] [PubMed] [Google Scholar]
- Buczek-Thomas JA, Lucey EC, Stone PJ, Chu CL, Rich CB, Carreras I, Goldstein RH, Foster JA, Nugent MA. Elastase mediates the release of growth factors from lung in vivo. American Journal of Respiratory Cell and Molecular Biology. 2004;31:344–350. doi: 10.1165/rcmb.2003-0420OC. [DOI] [PubMed] [Google Scholar]
- Cai TQ, Wright SD. Human leukocyte elastase is an endogenous ligand for the integrin CR3 (CD11b/CD18, Mac-1, alpha M beta 2) and modulates polymorphonuclear leukocyte adhesion. The Journal of Experimental Medicine. 1996;184:1213–1223. doi: 10.1084/jem.184.4.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell EJ, Campbell MA, Owen CA. Bioactive proteinase 3 on the cell surface of human neutrophils: quantification, catalytic activity, and susceptibility to inhibition. Journal of Immunology. 2000;165:3366–3374. doi: 10.4049/jimmunol.165.6.3366. [DOI] [PubMed] [Google Scholar]
- Campbell EJ, Owen CA. The sulfate groups of chondroitin sulfate- and heparan sulfate-containing proteoglycans in neutrophil plasma membranes are novel binding sites for human leukocyte elastase and cathepsin G. The Journal of Biological Chemistry. 2007;282:14645–14654. doi: 10.1074/jbc.M608346200. [DOI] [PubMed] [Google Scholar]
- Cantin AM, Berthiaume Y, Cloutier D, Martel M. Prolastin aerosol therapy and sputum taurine in cystic fibrosis. Clinical and Investigative Medicine. 2006;29:201–207. [PubMed] [Google Scholar]
- Carden DL, Korthuis RJ. Protease inhibition attenuates microvascular dysfunction in postischemic skeletal muscle. The American Journal of Physiology. 1996;271:H1947–H1952. doi: 10.1152/ajpheart.1996.271.5.H1947. [DOI] [PubMed] [Google Scholar]
- Cassatella MA. The production of cytokines by polymorphonuclear neutrophils. Immunology Today. 1995;16:21–26. doi: 10.1016/0167-5699(95)80066-2. [DOI] [PubMed] [Google Scholar]
- Caughey GH, Schaumberg TH, Zerweck EH, Butterfield JH, Hanson RD, Silverman GA, Ley TJ. The human mast cell chymase gene (CMA1): mapping to the cathepsin G/granzyme gene cluster and lineage-restricted expression. Genomics. 1993;15:614–620. doi: 10.1006/geno.1993.1115. [DOI] [PubMed] [Google Scholar]
- Champagne B, Tremblay P, Cantin A, St Pierre Y. Proteolytic cleavage of ICAM-1 by human neutrophil elastase. Journal of Immunology. 1998;161:6398–6405. [PubMed] [Google Scholar]
- Chertov O, Ueda H, Xu LL, Tani K, Murphy WJ, Wang JM, Howard OM, Sayers TJ, Oppenheim JJ. Identification of human neutrophil-derived cathepsin G and azurocidin/CAP37 as chemoattractants for mononuclear cells and neutrophils. The Journal of Experimental Medicine. 1997;186:739–747. doi: 10.1084/jem.186.5.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chignard M, Pidard D. Neutrophil and pathogen proteinases versus proteinase-activated receptor-2 lung epithelial cells: more terminators than activators. American Journal of Respiratory Cell and Molecular Biology. 2006;34:394–398. doi: 10.1165/rcmb.2005-0250TR. [DOI] [PubMed] [Google Scholar]
- Chua F, Dunsmore SE, Clingen PH, Mutsaers SE, Shapiro SD, Segal AW, Roes J, Laurent GJ. Mice lacking neutrophil elastase are resistant to bleomycin-induced pulmonary fibrosis. The American Journal of Pathology. 2007;170:65–74. doi: 10.2353/ajpath.2007.060352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chughtai B, O'Riordan TG. Potential role of inhibitors of neutrophil elastase in treating diseases of the airway. Journal of Aerosol Medicine. 2004;17:289–298. doi: 10.1089/jam.2004.17.289. [DOI] [PubMed] [Google Scholar]
- Coeshott C, Ohnemus C, Pilyavskaya A, Ross S, Wieczorek M, Kroona H, Leimer AH, Cheronis J. Converting enzyme-independent release of tumor necrosis factor alpha and IL-1beta from a stimulated human monocytic cell line in the presence of activated neutrophils or purified proteinase 3. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:6261–6266. doi: 10.1073/pnas.96.11.6261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlin SR, Camerer E. PARticipation in inflammation. The Journal of Clinical Investigation. 2003;111:25–27. doi: 10.1172/JCI17564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crinnion JN, Homer-Vanniasinkam S, Hatton R, Parkin SM, Gough MJ. Role of neutrophil depletion and elastase inhibition in modifying skeletal muscle reperfusion injury. Cardiovascular Surgery. 1994;2:749–753. [PubMed] [Google Scholar]
- Dahl CA, Schall RP, He HL, Cairns JS. Identification of a novel gene expressed in activated natural killer cells and T cells. Journal of Immunology. 1992;148:597–603. [PubMed] [Google Scholar]
- David A, Fridlich R, Aviram I. The presence of membrane Proteinase 3 in neutrophil lipid rafts and its colocalization with FcgammaRIIIb and cytochrome b558. Experimental Cell Research. 2005;308:156–165. doi: 10.1016/j.yexcr.2005.03.034. [DOI] [PubMed] [Google Scholar]
- David A, Kacher Y, Specks U, Aviram I. Interaction of proteinase 3 with CD11b/CD18 (beta2 integrin) on the cell membrane of human neutrophils. Journal of Leukocyte Biology. 2003;74:551–557. doi: 10.1189/jlb.1202624. [DOI] [PubMed] [Google Scholar]
- de Haar SF, Jansen DC, Schoenmaker T, De Vree H, Everts V, Beertsen W. Loss-of-function mutations in cathepsin C in two families with Papillon-Lefevre syndrome are associated with deficiency of serine proteinases in PMNs. Human Mutation. 2004;23:524–530. doi: 10.1002/humu.9243. [DOI] [PubMed] [Google Scholar]
- de Haar SF, Hiemstra PS, van Steenbergen MT, Everts V, Beertsen W. Role of polymorphonuclear leukocyte-derived serine proteases in defense against Actinobacillus actinomycetemcomitans. Infection and Immunity. 2006;74:5284–5291. doi: 10.1128/IAI.02016-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado MB, Clark-Lewis I, Loetscher P, Langen H, Thelen M, Baggiolini M, Wolf M. Rapid inactivation of stromal cell-derived factor-1 by cathepsin G associated with lymphocytes. European Journal of Immunology. 2001;31:699–707. doi: 10.1002/1521-4141(200103)31:3<699::aid-immu699>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Delyani JA, Murohara T, Lefer AM. Novel recombinant serpin, LEX-032, attenuates myocardial reperfusion injury in cats. The American Journal of Physiology. 1996;270:H881–H887. doi: 10.1152/ajpheart.1996.270.3.H881. [DOI] [PubMed] [Google Scholar]
- Devaney JM, Greene CM, Taggart CC, Carroll TP, O'Neill SJ, McElvaney NG. Neutrophil elastase up-regulates interleukin-8 via toll-like receptor 4. FEBS Letters. 2003;544:129–132. doi: 10.1016/s0014-5793(03)00482-4. [DOI] [PubMed] [Google Scholar]
- Dinarello CA, Kim SH. IL-32, a novel cytokine with a possible role in disease. Annals of the Rheumatic Diseases. 2006;65 Suppl 3:iii61–iii64. doi: 10.1136/ard.2006.058511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirksen A, Dijkman JH, Madsen F, Stoel B, Hutchison DC, Ulrik CS, Skovgaard LT, Kok-Jensen A, Rudolphus A, Seersholm N, et al. A randomized clinical trial of alpha(1)-antitrypsin augmentation therapy. American Journal of Respiratory and Critical Care Medicine. 1999;160:1468–1472. doi: 10.1164/ajrccm.160.5.9901055. [DOI] [PubMed] [Google Scholar]
- Doring G. Serine proteinase inhibitor therapy in alpha(1)-antitrypsin inhibitor deficiency and cystic fibrosis. Pediatric Pulmonology. 1999;28:363–375. doi: 10.1002/(sici)1099-0496(199911)28:5<363::aid-ppul9>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Dublet B, Ruello A, Pederzoli M, Hajjar E, Courbebaisse M, Canteloup S, Reuter N, Witko-Sarsat V. Cleavage of p21/WAF1/CIP1 by proteinase 3 modulates differentiation of a monocytic cell line. Molecular analysis of the cleavage site. The Journal of Biological Chemistry. 2005;280:30242–30253. doi: 10.1074/jbc.M414609200. [DOI] [PubMed] [Google Scholar]
- Dulon S, Cande C, Bunnett NW, Hollenberg MD, Chignard M, Pidard D. Proteinase-activated receptor-2 and human lung epithelial cells: disarming by neutrophil serine proteinases. American Journal of Respiratory Cell and Molecular Biology. 2003;28:339–346. doi: 10.1165/rcmb.4908. [DOI] [PubMed] [Google Scholar]
- Eisenhauer PB, Lehrer RI. Mouse neutrophils lack defensins. Infection and Immunity. 1992;60:3446–3447. doi: 10.1128/iai.60.8.3446-3447.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyles JL, Roberts AW, Metcalf D, Wicks IP. Granulocyte colony-stimulating factor and neutrophils--forgotten mediators of inflammatory disease. Nature clinical practice. 2006;2:500–510. doi: 10.1038/ncprheum0291. [DOI] [PubMed] [Google Scholar]
- Fantuzzi G, Ku G, Harding MW, Livingston DJ, Sipe JD, Kuida K, Flavell RA, Dinarello CA. Response to local inflammation of IL-1 beta-converting enzyme-deficient mice. Journal of Immunology. 1997;158:1818–1824. [PubMed] [Google Scholar]
- Faurschou M, Borregaard N. Neutrophil granules and secretory vesicles in inflammation. Microbes and Infection. 2003;5:1317–1327. doi: 10.1016/j.micinf.2003.09.008. [DOI] [PubMed] [Google Scholar]
- Fitch PM, Roghanian A, Howie SE, Sallenave JM. Human neutrophil elastase inhibitors in innate and adaptive immunity. Biochemical Society Transactions. 2006;34:279–282. doi: 10.1042/BST20060279. [DOI] [PubMed] [Google Scholar]
- Fridlich R, David A, Aviram I. Membrane proteinase 3 and its interactions within microdomains of neutrophil membranes. Journal of Cellular Biochemistry. 2006;99:117–125. doi: 10.1002/jcb.20901. [DOI] [PubMed] [Google Scholar]
- Garwicz D, Lennartsson A, Jacobsen SE, Gullberg U, Lindmark A. Biosynthetic profiles of neutrophil serine proteases in a human bone marrow-derived cellular myeloid differentiation model. Haematologica. 2005;90:38–44. [PubMed] [Google Scholar]
- Geraghty P, Rogan MP, Greene CM, Boxio RM, Poiriert T, O'Mahony M, Belaaouaj A, O'Neill SJ, Taggart CC, McElvaney NG. Neutrophil elastase up-regulates cathepsin B and matrix metalloprotease-2 expression. Journal of Immunology. 2007;178:5871–5878. doi: 10.4049/jimmunol.178.9.5871. [DOI] [PubMed] [Google Scholar]
- Ghayur T, Banerjee S, Hugunin M, Butler D, Herzog L, Carter A, Quintal L, Sekut L, Talanian R, Paskind M, et al. Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature. 1997;386:619–623. doi: 10.1038/386619a0. [DOI] [PubMed] [Google Scholar]
- Ginzberg HH, Shannon PT, Suzuki T, Hong O, Vachon E, Moraes T, Abreu MT, Cherepanov V, Wang X, Chow CW, et al. Leukocyte elastase induces epithelial apoptosis: role of mitochondial permeability changes and Akt. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2004;287:G286–G298. doi: 10.1152/ajpgi.00350.2003. [DOI] [PubMed] [Google Scholar]
- Golden JW, Schiff LA. Neutrophil elastase, an acid-independent serine protease, facilitates reovirus uncoating and infection in U937 promonocyte cells. Virology Journal. 2005;2:48–61. doi: 10.1186/1743-422X-2-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Kuida K, Tsutsui H, Ku G, Hsiao K, Fleming MA, Hayashi N, Higashino K, Okamura H, Nakanishi K, et al. Activation of interferon-gamma inducing factor mediated by interleukin-1beta converting enzyme. Science. 1997;275:206–209. doi: 10.1126/science.275.5297.206. [DOI] [PubMed] [Google Scholar]
- Hamacher J, Sadallah S, Schifferli JA, Villard J, Nicod LP. Soluble complement receptor type 1 (CD35) in bronchoalveolar lavage of inflammatory lung diseases. European Respiratory Journal. 1998;11:112–119. doi: 10.1183/09031936.98.11010112. [DOI] [PubMed] [Google Scholar]
- Hart PS, Zhang Y, Firatli E, Uygur C, Lotfazar M, Michalec MD, Marks JJ, Lu X, Coates BJ, Seow WK, et al. Identification of cathepsin C mutations in ethnically diverse papillon-Lefevre syndrome patients. Journal of Medical Genetics. 2000;37:927–932. doi: 10.1136/jmg.37.12.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hase-Yamazaki T, Aoki Y. Stimulation of human lymphocytes by cathepsin G. Cellular Immunology. 1995;160:24–32. doi: 10.1016/0008-8749(95)80005-4. [DOI] [PubMed] [Google Scholar]
- Henriksen PA, Devitt A, Kotelevtsev Y, Sallenave JM. Gene delivery of the elastase inhibitor elafin protects macrophages from neutrophil elastase-mediated impairment of apoptotic cell recognition. FEBS Letters. 2004;574:80–84. doi: 10.1016/j.febslet.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Hermant B, Bibert S, Concord E, Dublet B, Weidenhaupt M, Vernet T, Gulino-Debrac D. Identification of proteases involved in the proteolysis of vascular endothelium cadherin during neutrophil transmigration. The Journal of Biological Chemistry. 2003;278:14002–14012. doi: 10.1074/jbc.M300351200. [DOI] [PubMed] [Google Scholar]
- Heusel JW, Scarpati EM, Jenkins NA, Gilbert DJ, Copeland NG, Shapiro SD, Ley TJ. Molecular cloning, chromosomal location, and tissue-specific expression of the murine cathepsin G gene. Blood. 1993;81:1614–1623. [PubMed] [Google Scholar]
- Hirche TO, Atkinson JJ, Bahr S, Belaaouaj A. Deficiency in neutrophil elastase does not impair neutrophil recruitment to inflamed sites. American Journal of Respiratory Cell and Molecular Biology. 2004;30:576–584. doi: 10.1165/rcmb.2003-0253OC. [DOI] [PubMed] [Google Scholar]
- Holt OJ, Gallo F, Griffiths GM. Regulating secretory lysosomes. Journal of Biochemistry. 2006;140:7–12. doi: 10.1093/jb/mvj126. [DOI] [PubMed] [Google Scholar]
- Horwitz MS, Duan Z, Korkmaz B, Lee HH, Mealiffe ME, Salipante SJ. Neutrophil elastase in cyclic and severe congenital neutropenia. Blood. 2007;109:1817–1824. doi: 10.1182/blood-2006-08-019166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi K, Kurosawa S, Kato M, Andoh K, Satoh D, Kaise A. Sivelestat, a neutrophil elastase inhibitor, reduces mortality rate of critically ill patients. The Tohoku Journal of Experimental Medicine. 2005;207:143–148. doi: 10.1620/tjem.207.143. [DOI] [PubMed] [Google Scholar]
- Jog NR, Rane MJ, Lominadze G, Luerman GC, Ward RA, McLeish KR. The actin cytoskeleton regulates exocytosis of all neutrophil granule subsets. American Journal of Physiology. 2007;292:C1690–C1700. doi: 10.1152/ajpcell.00384.2006. [DOI] [PubMed] [Google Scholar]
- Kallenberg CG, Heeringa P, Stegeman CA. Mechanisms of Disease: pathogenesis and treatment of ANCA-associated vasculitides. Nature Clinical Practice. 2006;2:661–670. doi: 10.1038/ncprheum0355. [DOI] [PubMed] [Google Scholar]
- Kohri K, Ueki IF, Nadel JA. Neutrophil elastase induces mucin production by ligand-dependent epidermal growth factor receptor activation. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2002;283:L531–L540. doi: 10.1152/ajplung.00455.2001. [DOI] [PubMed] [Google Scholar]
- Kollner I, Sodeik B, Schreek S, Heyn H, von Neuhoff N, Germeshausen M, Zeidler C, Kruger M, Schlegelberger B, Welte K, et al. Mutations in neutrophil elastase causing congenital neutropenia lead to cytoplasmic protein accumulation and induction of the unfolded protein response. Blood. 2006;108:493–500. doi: 10.1182/blood-2005-11-4689. [DOI] [PubMed] [Google Scholar]
- Korkmaz B, Attucci S, Jourdan ML, Juliano L, Gauthier F. Inhibition of neutrophil elastase by alpha1-protease inhibitor at the surface of human polymorphonuclear neutrophils. Journal of Immunology. 2005;175:3329–3338. doi: 10.4049/jimmunol.175.5.3329. [DOI] [PubMed] [Google Scholar]
- Kyriakides C, Austen WG, Jr, Wang Y, Favuzza J, Moore FD, Jr, Hechtman HB. Neutrophil mediated remote organ injury after lower torso ischemia and reperfusion is selectin and complement dependent. The Journal of Trauma. 2000;48:32–38. doi: 10.1097/00005373-200001000-00006. [DOI] [PubMed] [Google Scholar]
- Le Cabec V, Cowland JB, Calafat J, Borregaard N. Targeting of proteins to granule subsets is determined by timing and not by sorting: The specific granule protein NGAL is localized to azurophil granules when expressed in HL-60 cells. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:6454–6457. doi: 10.1073/pnas.93.13.6454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le-Barillec K, Si-Tahar M, Balloy V, Chignard M. Proteolysis of monocyte CD14 by human leukocyte elastase inhibits lipopolysaccharide-mediated cell activation. The Journal of Clinical Investigation. 1999;103:1039–1046. doi: 10.1172/JCI5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le-Barillec K, Pidard D, Balloy V, Chignard M. Human neutrophil cathepsin G down-regulates LPS-mediated monocyte activation through CD14 proteolysis. Journal of Leukocyte Biology. 2000;68:209–215. [PubMed] [Google Scholar]
- Lee KY, Ho SC, Lin HC, Lin SM, Liu CY, Huang CD, Wang CH, Chung KF, Kuo HP. Neutrophil-derived elastase induces TGF-beta1 secretion in human airway smooth muscle via NF-kappaB pathway. American Journal of Respiratory Cell and Molecular Biology. 2006;35:407–414. doi: 10.1165/rcmb.2006-0012OC. [DOI] [PubMed] [Google Scholar]
- Lemansky P, Smolenova E, Wrocklage C, Hasilik A. Neutrophil elastase is associated with serglycin on its way to lysosomes in U937 cells. Cellular Immunology. 2007;246:1–7. doi: 10.1016/j.cellimm.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Lennartsson A, Garwicz D, Lindmark A, Gullberg U. The proximal promoter of the human cathepsin G gene conferring myeloid-specific expression includes C/EBP, c-myb and PU.1 binding sites. Gene. 2005;356:193–202. doi: 10.1016/j.gene.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Levesque JP, Takamatsu Y, Nilsson SK, Haylock DN, Simmons PJ. Vascular cell adhesion molecule-1 (CD106) is cleaved by neutrophil proteases in the bone marrow following hematopoietic progenitor cell mobilization by granulocyte colony-stimulating factor. Blood. 2001;98:1289–1297. doi: 10.1182/blood.v98.5.1289. [DOI] [PubMed] [Google Scholar]
- Lim JK, Lu W, Hartley O, DeVico AL. N-terminal proteolytic processing by cathepsin G converts RANTES/CCL5 and related analogs into a truncated 4–68 variant. Journal of Leukocyte Biology. 2006;80:1395–1404. doi: 10.1189/jlb.0406290. [DOI] [PubMed] [Google Scholar]
- Liu Z, Shapiro SD, Zhou X, Twining SS, Senior RM, Giudice GJ, Fairley JA, Diaz LA. A critical role for neutrophil elastase in experimental bullous pemphigoid. The Journal of Clinical Investigation. 2000;105:113–123. doi: 10.1172/JCI3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z. Bullous pemphigoid: using animal models to study the immunopathology. The Journal of Investigative Dermatology. 2004;9:41–46. doi: 10.1111/j.1087-0024.2004.00841.x. [DOI] [PubMed] [Google Scholar]
- Lomas DA, Stone SR, Llewellyn-Jones C, Keogan MT, Wang ZM, Rubin H, Carrell RW, Stockley RA. The control of neutrophil chemotaxis by inhibitors of cathepsin G and chymotrypsin. The Journal of Biological Chemistry. 1995;270:23437–23443. doi: 10.1074/jbc.270.40.23437. [DOI] [PubMed] [Google Scholar]
- Luisetti M, Sturani C, Sella D, Madonini E, Galavotti V, Bruno G, Peona V, Kucich U, Dagnino G, Rosenbloom J, et al. MR889, a neutrophil elastase inhibitor, in patients with chronic obstructive pulmonary disease: a double-blind, randomized, placebo-controlled clinical trial. European Respiratory Journal. 1996;9:1482–1486. doi: 10.1183/09031936.96.09071482. [DOI] [PubMed] [Google Scholar]
- MacIvor DM, Shapiro SD, Pham CT, Belaaouaj A, Abraham SN, Ley TJ. Normal neutrophil function in cathepsin G-deficient mice. Blood. 1999;94:4282–4293. [PubMed] [Google Scholar]
- Mackarel AJ, Cottell DC, Russell KJ, FitzGerald MX, O'Connor CM. Migration of neutrophils across human pulmonary endothelial cells is not blocked by matrix metalloproteinase or serine protease inhibitors. American Journal of Respiratory Cell and Molecular Biology. 1999;20:1209–1219. doi: 10.1165/ajrcmb.20.6.3539. [DOI] [PubMed] [Google Scholar]
- Maison CM, Villiers CL, Colomb MG. Proteolysis of C3 on U937 cell plasma membranes. Purification of cathepsin G. Journal of Immunology. 1991;147:921–926. [PubMed] [Google Scholar]
- Martin SL, Downey D, Bilton D, Keogan MT, Edgar J, Elborn JS. Safety and efficacy of recombinant alpha(1)-antitrypsin therapy in cystic fibrosis. Pediatric Pulmonology. 2006;41:177–183. doi: 10.1002/ppul.20345. [DOI] [PubMed] [Google Scholar]
- Mayerle J, Schnekenburger J, Kruger B, Kellermann J, Ruthenburger M, Weiss FU, Nalli A, Domschke W, Lerch MM. Extracellular cleavage of E-cadherin by leukocyte elastase during acute experimental pancreatitis in rats. Gastroenterology. 2005;129:1251–1267. doi: 10.1053/j.gastro.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Meyer-Hoffert U, Wingertszahn J, Wiedow O. Human leukocyte elastase induces keratinocyte proliferation by epidermal growth factor receptor activation. The Journal of Investigative Dermatology. 2004;123:338–345. doi: 10.1111/j.0022-202X.2004.23202.x. [DOI] [PubMed] [Google Scholar]
- Mezyk-Kopec R, Bzowska M, Bzowska M, Mickowska B, Mak P, Potempa J, Bereta J. Effects of elastase and cathepsin G on the levels of membrane and soluble TNFalpha. Biological Chemistry. 2005;386:801–811. doi: 10.1515/BC.2005.094. [DOI] [PubMed] [Google Scholar]
- Miao Z, Premack BA, Wei Z, Wang Y, Gerard C, Showell H, Howard M, Schall TJ, Berahovich R. Proinflammatory proteases liberate a discrete high-affinity functional FPRL1 (CCR12) ligand from CCL23. Journal of Immunology. 2007;178:7395–7404. doi: 10.4049/jimmunol.178.11.7395. [DOI] [PubMed] [Google Scholar]
- Miller SI, Ernst RK, Bader MW. LPS, TLR4 and infectious disease diversity. Nature Reviews Microbiology. 2005;3:36–46. doi: 10.1038/nrmicro1068. [DOI] [PubMed] [Google Scholar]
- Mollinedo F, Borregaard N, Boxer LA. Novel trends in neutrophil structure, function and development. Immunology Today. 1999;20:535–537. doi: 10.1016/s0167-5699(99)01500-5. [DOI] [PubMed] [Google Scholar]
- Mollinedo F, Calafat J, Janssen H, Martin-Martin B, Canchado J, Nabokina SM, Gajate C. Combinatorial SNARE complexes modulate the secretion of cytoplasmic granules in human neutrophils. Journal of Immunology. 2006;177:2831–2841. doi: 10.4049/jimmunol.177.5.2831. [DOI] [PubMed] [Google Scholar]
- Moriuchi H, Moriuchi M, Fauci AS. Cathepsin G, a neutrophil-derived serine protease, increases susceptibility of macrophages to acute human immunodeficiency virus type 1 infection. Journal of Virology. 2000;74:6849–6855. doi: 10.1128/jvi.74.15.6849-6855.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murohara T, Guo JP, Lefer AM. Cardioprotection by a novel recombinant serine protease inhibitor in myocardial ischemia and reperfusion injury. The Journal of Pharmacology and Experimental Therapeutics. 1995;274:1246–1253. [PubMed] [Google Scholar]
- Nadel JA. Innate immune mucin production via epithelial cell surface signaling: relationship to allergic disease. Current opinion in allergy and clinical immunology. 2007;7:57–62. doi: 10.1097/ACI.0b013e328012ce22. [DOI] [PubMed] [Google Scholar]
- Nathan C. Neutrophils and immunity: challenges and opportunities. Nature Reviews Immunolgy. 2006;6:173–182. doi: 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- Nathan C, Xie QW, Halbwachs-Mecarelli L, Jin WW. Albumin inhibits neutrophil spreading and hydrogen peroxide release by blocking the shedding of CD43 (sialophorin, leukosialin) The Journal of Cell Biology. 1993;122:243–256. doi: 10.1083/jcb.122.1.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemoto E, Sugawara S, Tada H, Takada H, Shimauchi H, Horiuchi H. Cleavage of CD14 on human gingival fibroblasts cocultured with activated neutrophils is mediated by human leukocyte elastase resulting in down-regulation of lipopolysaccharide-induced IL-8 production. Journal of Immunology. 2000;165:5807–5813. doi: 10.4049/jimmunol.165.10.5807. [DOI] [PubMed] [Google Scholar]
- Newburger PE. Disorders of neutrophil number and function. Hematology / The Education Program of the American Society of Hematology American Society of Hematology. 2006:104–110. doi: 10.1182/asheducation-2006.1.104. [DOI] [PubMed] [Google Scholar]
- Niemann CU, Abrink M, Pejler G, Fischer RL, Christensen EI, Knight SD, Borregaard N. Neutrophil elastase depends on serglycin proteoglycan for localization in granules. Blood. 2007;109:4478–4486. doi: 10.1182/blood-2006-02-001719. [DOI] [PubMed] [Google Scholar]
- Novick D, Rubinstein M, Azam T, Rabinkov A, Dinarello CA, Kim SH. Proteinase 3 is an IL-32 binding protein. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:3316–3321. doi: 10.1073/pnas.0511206103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nufer O, Corbett M, Walz A. Amino-terminal processing of chemokine ENA-78 regulates biological activity. Biochemistry. 1999;38:636–642. doi: 10.1021/bi981294s. [DOI] [PubMed] [Google Scholar]
- Ohbayashi H. Neutrophil elastase inhibitors as treatment for COPD. Expert Opinion on Investigational Drugs. 2002;11:965–980. doi: 10.1517/13543784.11.7.965. [DOI] [PubMed] [Google Scholar]
- Ono S, Tsujimoto H, Hiraki S, Takahata R, Kimura A, Kinoshita M, Ichikura T, Mochizuki H. Effects of neutrophil elastase inhibitor on progression of acute lung injury following esophagectomy. World Journal of Surgery. 2007;31:1996–2001. doi: 10.1007/s00268-007-9172-6. [DOI] [PubMed] [Google Scholar]
- Owen CA, Campbell EJ. Neutrophil proteinases and matrix degradation. The cell biology of pericellular proteolysis. Seminars in Cell Biology. 1995;6:367–376. doi: 10.1016/s1043-4682(05)80007-8. [DOI] [PubMed] [Google Scholar]
- Owen CA, Campbell MA, Sannes PL, Boukedes SS, Campbell EJ. Cell surface-bound elastase and cathepsin G on human neutrophils: a novel, non-oxidative mechanism by which neutrophils focus and preserve catalytic activity of serine proteinases. The Journal of Cell Biology. 1995;131:775–789. doi: 10.1083/jcb.131.3.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padrines M, Wolf M, Walz A, Baggiolini M. Interleukin-8 processing by neutrophil elastase, cathepsin G and proteinase-3. FEBS Letters. 1994;352:231–235. doi: 10.1016/0014-5793(94)00952-x. [DOI] [PubMed] [Google Scholar]
- Park SJ, Wiekowski MT, Lira SA, Mehrad B. Neutrophils regulate airway responses in a model of fungal allergic airways disease. Journal of Immunology. 2006;176:2538–2545. doi: 10.4049/jimmunol.176.4.2538. [DOI] [PubMed] [Google Scholar]
- Pendergraft WF, 3rd, Rudolph EH, Falk RJ, Jahn JE, Grimmler M, Hengst L, Jennette JC, Preston GA. Proteinase 3 sidesteps caspases and cleaves p21(Waf1/Cip1/Sdi1) to induce endothelial cell apoptosis. Kidney International. 2004;65:75–84. doi: 10.1111/j.1523-1755.2004.00364.x. [DOI] [PubMed] [Google Scholar]
- Pfister H, Ollert M, Frohlich LF, Quintanilla-Martinez L, Colby TV, Specks U, Jenne DE. Antineutrophil cytoplasmic autoantibodies against the murine homolog of proteinase 3 (Wegener autoantigen) are pathogenic in vivo. Blood. 2004;104:1411–1418. doi: 10.1182/blood-2004-01-0267. [DOI] [PubMed] [Google Scholar]
- Pham CT, Ivanovich JL, Raptis SZ, Zehnbauer B, Ley TJ. Papillon-Lefevre syndrome: correlating the molecular, cellular, and clinical consequences of cathepsin C/dipeptidyl peptidase I deficiency in humans. Journal of Immunology. 2004;173:7277–7281. doi: 10.4049/jimmunol.173.12.7277. [DOI] [PubMed] [Google Scholar]
- Pham CT, Ley TJ. Dipeptidyl peptidase I is required for the processing and activation of granzymes A and B in vivo. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:8627–8632. doi: 10.1073/pnas.96.15.8627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porteu F, Nathan C. Shedding of tumor necrosis factor receptors by activated human neutrophils. The Journal of Experimental Medicine. 1990;172:599–607. doi: 10.1084/jem.172.2.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porteu F, Brockhaus M, Wallach D, Engelmann H, Nathan CF. Human neutrophil elastase releases a ligand-binding fragment from the 75-kDa tumor necrosis factor (TNF) receptor. Comparison with the proteolytic activity responsible for shedding of TNF receptors from stimulated neutrophils. The Journal of Biological Chemistry. 1991;266:18846–18853. [PubMed] [Google Scholar]
- Preston GA, Zarella CS, Pendergraft WF, 3rd, Rudolph EH, Yang JJ, Sekura SB, Jennette JC, Falk RJ. Novel effects of neutrophil-derived proteinase 3 and elastase on the vascular endothelium involve in vivo cleavage of NF-kappaB and proapoptotic changes in JNK, ERK, and p38 MAPK signaling pathways. Journal of the American Society of Nephrology. 2002;13:2840–2849. doi: 10.1097/01.asn.0000034911.03334.c3. [DOI] [PubMed] [Google Scholar]
- Qualls JE, Kaplan AM, van Rooijen N, Cohen DA. Suppression of experimental colitis by intestinal mononuclear phagocytes. Journal of Leukocyte Biology. 2006;80:802–815. doi: 10.1189/jlb.1205734. [DOI] [PubMed] [Google Scholar]
- Raja SM, Wang B, Dantuluri M, Desai UR, Demeler B, Spiegel K, Metkar SS, Froelich CJ. Cytotoxic cell granule-mediated apoptosis. Characterization of the macromolecular complex of granzyme B with serglycin. The Journal of Biological Chemistry. 2002;277:49523–49530. doi: 10.1074/jbc.M209607200. [DOI] [PubMed] [Google Scholar]
- Rao RM, Betz TV, Lamont DJ, Kim MB, Shaw SK, Froio RM, Baleux F, Arenzana-Seisdedos F, Alon R, Luscinskas FW. Elastase release by transmigrating neutrophils deactivates endothelial-bound SDF-1alpha and attenuates subsequent T lymphocyte transendothelial migration. The Journal of Experimental Medicine. 2004;200:713–724. doi: 10.1084/jem.20040499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raptis SZ, Shapiro SD, Simmons PM, Cheng AM, Pham CT. Serine protease cathepsin G regulates adhesion-dependent neutrophil effector functions by modulating integrin clustering. Immunity. 2005;22:679–691. doi: 10.1016/j.immuni.2005.03.015. [DOI] [PubMed] [Google Scholar]
- Reeves EP, Lu H, Jacobs HL, Messina CG, Bolsover S, Gabella G, Potma EO, Warley A, Roes J, Segal AW. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature. 2002;416:291–297. doi: 10.1038/416291a. [DOI] [PubMed] [Google Scholar]
- Reid PT, Sallenave JM. Neutrophil-derived elastases and their inhibitors: potential role in the pathogenesis of lung disease. Current Opinion in Investigational Drugs. 2001;2:59–67. [PubMed] [Google Scholar]
- Remold-O'Donnell E, Parent D. Specific sensitivity of CD43 to neutrophil elastase. Blood. 1995;86:2395–2402. [PubMed] [Google Scholar]
- Renesto P, Si-Tahar M, Moniatte M, Balloy V, Van Dorsselaer A, Pidard D, Chignard M. Specific inhibition of thrombin-induced cell activation by the neutrophil proteinases elastase, cathepsin G, and proteinase 3: evidence for distinct cleavage sites within the aminoterminal domain of the thrombin receptor. Blood. 1997;89:1944–1953. [PubMed] [Google Scholar]
- Rest RF. Human neutrophil and mast cell proteases implicated in inflammation. Methods in Enzymology. 1988;163:309–327. doi: 10.1016/0076-6879(88)63030-8. [DOI] [PubMed] [Google Scholar]
- Richter R, Bistrian R, Escher S, Forssmann WG, Vakili J, Henschler R, Spodsberg N, Frimpong-Boateng A, Forssmann U. Quantum proteolytic activation of chemokine CCL15 by neutrophil granulocytes modulates mononuclear cell adhesiveness. Journal of Immunology. 2005;175:1599–1608. doi: 10.4049/jimmunol.175.3.1599. [DOI] [PubMed] [Google Scholar]
- Robache-Gallea S, Morand V, Bruneau JM, Schoot B, Tagat E, Realo E, Chouaib S, Roman-Roman S. In vitro processing of human tumor necrosis factor-alpha. The Journal of Biological Chemistry. 1995;270:23688–23692. doi: 10.1074/jbc.270.40.23688. [DOI] [PubMed] [Google Scholar]
- Robertson SE, Young JD, Kitson S, Pitt A, Evans J, Roes J, Karaoglu D, Santora L, Ghayur T, Liew FY, et al. Expression and alternative processing of IL-18 in human neutrophils. European Journal of Immunology. 2006;36:722–731. doi: 10.1002/eji.200535402. [DOI] [PubMed] [Google Scholar]
- Robledo O, Papaioannou A, Ochietti B, Beauchemin C, Legault D, Cantin A, King PD, Daniel C, Alakhov VY, Potworowski EF, et al. ICAM-1 isoforms: specific activity and sensitivity to cleavage by leukocyte elastase and cathepsin G. European Journal of Immunology. 2003;33:1351–1360. doi: 10.1002/eji.200323195. [DOI] [PubMed] [Google Scholar]
- Ryu OH, Choi SJ, Firatli E, Choi SW, Hart PS, Shen RF, Wang G, Wu WW, Hart TC. Proteolysis of macrophage inflammatory protein-1alpha isoforms LD78beta and LD78alpha by neutrophil-derived serine proteases. The Journal of Biological Chemistry. 2005;280:17415–17421. doi: 10.1074/jbc.M500340200. [DOI] [PubMed] [Google Scholar]
- Sadallah S, Hess C, Miot S, Spertini O, Lutz H, Schifferli JA. Elastase and metalloproteinase activities regulate soluble complement receptor 1 release. European Journal of Immunology. 1999;29:3754–3761. doi: 10.1002/(SICI)1521-4141(199911)29:11<3754::AID-IMMU3754>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Sambrano GR, Huang W, Faruqi T, Mahrus S, Craik C, Coughlin SR. Cathepsin G activates protease-activated receptor-4 in human platelets. The Journal of Biological Chemistry. 2000;275:6819–6823. doi: 10.1074/jbc.275.10.6819. [DOI] [PubMed] [Google Scholar]
- Scapini P, Lapinet-Vera JA, Gasperini S, Calzetti F, Bazzoni F, Cassatella MA. The neutrophil as a cellular source of chemokines. Immunological Reviews. 2000;177:195–203. doi: 10.1034/j.1600-065x.2000.17706.x. [DOI] [PubMed] [Google Scholar]
- Schiemann F, Grimm TA, Hoch J, Gross R, Lindner B, Petersen F, Bulfone-Paus S, Brandt E. Mast cells and neutrophils proteolytically activate chemokine precursor CTAP-III and are subject to counterregulation by PF-4 through inhibition of chymase and cathepsin G. Blood. 2006;107:2234–2242. doi: 10.1182/blood-2005-06-2424. [DOI] [PubMed] [Google Scholar]
- Scuderi P. Suppression of human leukocyte tumor necrosis factor secretion by the serine protease inhibitor p-toluenesulfonyl-L-arginine methyl ester (TAME) Journal of Immunology. 1989;143:168–173. [PubMed] [Google Scholar]
- Scuderi P, Nez PA, Duerr ML, Wong BJ, Valdez CM. Cathepsin-G and leukocyte elastase inactivate human tumor necrosis factor and lymphotoxin. Cellular Immunology. 1991;135:299–313. doi: 10.1016/0008-8749(91)90275-g. [DOI] [PubMed] [Google Scholar]
- Sedor J, Hogue L, Akers K, Boslaugh S, Schreiber J, Ferkol T. Cathepsin-G interferes with clearance of Pseudomonas aeruginosa from mouse lungs. Pediatric Research. 2007;61:26–31. doi: 10.1203/01.pdr.0000250043.90468.c2. [DOI] [PubMed] [Google Scholar]
- Shao MX, Nadel JA. Neutrophil elastase induces MUC5AC mucin production in human airway epithelial cells via a cascade involving protein kinase C, reactive oxygen species, and TNF-alpha-converting enzyme. Journal of Immunology. 2005;175:4009–4016. doi: 10.4049/jimmunol.175.6.4009. [DOI] [PubMed] [Google Scholar]
- Shapiro SD, Goldstein NM, Houghton AM, Kobayashi DK, Kelley D, Belaaouaj A. Neutrophil elastase contributes to cigarette smoke-induced emphysema in mice. The American Journal of Pathology. 2003;163:2329–2335. doi: 10.1016/S0002-9440(10)63589-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoda N, Fukazawa N, Nonomura K, Fairchild RL. Cathepsin g is required for sustained inflammation and tissue injury after reperfusion of ischemic kidneys. The American Journal of Pathology. 2007;170:930–940. doi: 10.2353/ajpath.2007.060486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si-Tahar M, Pidard D, Balloy V, Moniatte M, Kieffer N, Van Dorsselaer A, Chignard M. Human neutrophil elastase proteolytically activates the platelet integrin alphaIIbbeta3 through cleavage of the carboxyl terminus of the alphaIIb subunit heavy chain. Involvement in the potentiation of platelet aggregation. The Journal of Biological Chemistry. 1997;272:11636–11647. doi: 10.1074/jbc.272.17.11636. [DOI] [PubMed] [Google Scholar]
- Sisley AC, Desai T, Harig JM, Gewertz BL. Neutrophil depletion attenuates human intestinal reperfusion injury. The Journal of Surgical Research. 1994;57:192–196. doi: 10.1006/jsre.1994.1130. [DOI] [PubMed] [Google Scholar]
- Sorensen OE, Follin P, Johnsen AH, Calafat J, Tjabringa GS, Hiemstra PS, Borregaard N. Human cathelicidin, hCAP-18, is processed to the antimicrobial peptide LL-37 by extracellular cleavage with proteinase 3. Blood. 2001;97:3951–3959. doi: 10.1182/blood.v97.12.3951. [DOI] [PubMed] [Google Scholar]
- Stuart LM, Ezekowitz RA. Phagocytosis: elegant complexity. Immunity. 2005;22:539–550. doi: 10.1016/j.immuni.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Sugawara S, Uehara A, Nochi T, Yamaguchi T, Ueda H, Sugiyama A, Hanzawa K, Kumagai K, Okamura H, Takada H. Neutrophil proteinase 3-mediated induction of bioactive IL-18 secretion by human oral epithelial cells. Journal of Immunology. 2001;167:6568–6575. doi: 10.4049/jimmunol.167.11.6568. [DOI] [PubMed] [Google Scholar]
- Sun R, Iribarren P, Zhang N, Zhou Y, Gong W, Cho EH, Lockett S, Chertov O, Bednar F, Rogers TJ, et al. Identification of neutrophil granule protein cathepsin G as a novel chemotactic agonist for the G protein-coupled formyl peptide receptor. Journal of Immunology. 2004;173:428–436. doi: 10.4049/jimmunol.173.1.428. [DOI] [PubMed] [Google Scholar]
- Taipale J, Lohi J, Saarinen J, Kovanen PT, Keski-Oja J. Human mast cell chymase and leukocyte elastase release latent transforming growth factor-beta 1 from the extracellular matrix of cultured human epithelial and endothelial cells. The Journal of Biological Chemistry. 1995;270:4689–4696. doi: 10.1074/jbc.270.9.4689. [DOI] [PubMed] [Google Scholar]
- Tanaka D, Kagari T, Doi H, Shimozato T. Essential role of neutrophils in antitype II collagen antibody and lipopolysaccharide-induced arthritis. Immunology. 2006;119:195–202. doi: 10.1111/j.1365-2567.2006.02424.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tani K, Murphy WJ, Chertov O, Oppenheim JJ, Wang JM. The neutrophil granule protein cathepsin G activates murine T lymphocytes and upregulates antigen-specific IG production in mice. Biochemical and Biophysical Research Communications. 2001;282:971–976. doi: 10.1006/bbrc.2001.4676. [DOI] [PubMed] [Google Scholar]
- Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, Miller DK, Molineaux SM, Weidner JR, Aunins J, et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature. 1992;356:768–774. doi: 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- Tilg H, Dinarello CA, Mier JW. IL-6 and APPs: anti-inflammatory and immunosuppressive mediators. Immunology Today. 1997;18:428–432. doi: 10.1016/s0167-5699(97)01103-1. [DOI] [PubMed] [Google Scholar]
- Tkalcevic J, Novelli M, Phylactides M, Iredale JP, Segal AW, Roes J. Impaired immunity and enhanced resistance to endotoxin in the absence of neutrophil elastase and cathepsin G. Immunity. 2000;12:201–210. doi: 10.1016/s1074-7613(00)80173-9. [DOI] [PubMed] [Google Scholar]
- Toomes C, James J, Wood AJ, Wu CL, McCormick D, Lench N, Hewitt C, Moynihan L, Roberts E, Woods CG, et al. Loss-of-function mutations in the cathepsin C gene result in periodontal disease and palmoplantar keratosis. Nature Genetics. 1999;23:421–424. doi: 10.1038/70525. [DOI] [PubMed] [Google Scholar]
- Tralau T, Meyer-Hoffert U, Schroder JM, Wiedow O. Human leukocyte elastase and cathepsin G are specific inhibitors of C5a-dependent neutrophil enzyme release and chemotaxis. Experimental Dermatology. 2004;13:316–325. doi: 10.1111/j.0906-6705.2004.00145.x. [DOI] [PubMed] [Google Scholar]
- Uehara A, Sugawara S, Muramoto K, Takada H. Activation of human oral epithelial cells by neutrophil proteinase 3 through protease-activated receptor-2. Journal of Immunology. 2002;169:4594–4603. doi: 10.4049/jimmunol.169.8.4594. [DOI] [PubMed] [Google Scholar]
- Uehara A, Muramoto K, Takada H, Sugawara S. Neutrophil serine proteinases activate human nonepithelial cells to produce inflammatory cytokines through protease-activated receptor 2. Journal of Immunology. 2003;170:5690–5696. doi: 10.4049/jimmunol.170.11.5690. [DOI] [PubMed] [Google Scholar]
- Uehara A, Sugawara Y, Sasano T, Takada H, Sugawara S. Proinflammatory cytokines induce proteinase 3 as membrane-bound and secretory forms in human oral epithelial cells and antibodies to proteinase 3 activate the cells through protease-activated receptor-2. Journal of Immunology. 2004;173:4179–4189. doi: 10.4049/jimmunol.173.6.4179. [DOI] [PubMed] [Google Scholar]
- Valenzuela-Fernandez A, Planchenault T, Baleux F, Staropoli I, Le-Barillec K, Leduc D, Delaunay T, Lazarini F, Virelizier JL, Chignard M, et al. Leukocyte elastase negatively regulates stromal cell-derived factor-1 (SDF-1)/CXCR4 binding and functions by amino-terminal processing of SDF-1 and CXCR4. Journal of Biological Chemistry. 2002;277:15677–15689. doi: 10.1074/jbc.M111388200. [DOI] [PubMed] [Google Scholar]
- Vandivier RW, Fadok VA, Hoffmann PR, Bratton DL, Penvari C, Brown KK, Brain JD, Accurso FJ, Henson PM. Elastase-mediated phosphatidylserine receptor cleavage impairs apoptotic cell clearance in cystic fibrosis and bronchiectasis. The Journal of Clinical Investigation. 2002;109:661–670. doi: 10.1172/JCI13572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt W. Cleavage of the fifth component of complement and generation of a functionally active C5b6-like complex by human leukocyte elastase. Immunobiology. 2000;201:470–477. doi: 10.1016/S0171-2985(00)80099-6. [DOI] [PubMed] [Google Scholar]
- Voynow JA, Fischer BM, Malarkey DE, Burch LH, Wong T, Longphre M, Ho SB, Foster WM. Neutrophil elastase induces mucus cell metaplasia in mouse lung. American Journal of Physiology- Lung Cellular and Molecular Physiology. 2004;287:L1293–L1302. doi: 10.1152/ajplung.00140.2004. [DOI] [PubMed] [Google Scholar]
- Walsh DE, Greene CM, Carroll TP, Taggart CC, Gallagher PM, O'Neill SJ, McElvaney NG. Interleukin-8 up-regulation by neutrophil elastase is mediated by MyD88/IRAK/TRAF-6 in human bronchial epithelium. The Journal of Biological Chemistry. 2001;276:35494–35499. doi: 10.1074/jbc.M103543200. [DOI] [PubMed] [Google Scholar]
- Wang S, Dangerfield JP, Young RE, Nourshargh S. PECAM-1, alpha6 integrins and neutrophil elastase cooperate in mediating neutrophil transmigration. Journal of Cell Science. 2005;118:2067–2076. doi: 10.1242/jcs.02340. [DOI] [PubMed] [Google Scholar]
- Ward DM, Griffiths GM, Stinchcombe JC, Kaplan J. Analysis of the lysosomal storage disease Chediak-Higashi syndrome. Traffic. 2000;1:816–822. doi: 10.1034/j.1600-0854.2000.011102.x. [DOI] [PubMed] [Google Scholar]
- Weinrauch Y, Drujan D, Shapiro SD, Weiss J, Zychlinsky A. Neutrophil elastase targets virulence factors of enterobacteria. Nature. 2002;417:91–94. doi: 10.1038/417091a. [DOI] [PubMed] [Google Scholar]
- Weiss SJ. Tissue destruction by neutrophils. The New England Journal of Medicine. 1989;320:365–376. doi: 10.1056/NEJM198902093200606. [DOI] [PubMed] [Google Scholar]
- Wendel A. Biochemical pharmacology of inflammatory liver injury in mice. Methods in Enzymology. 1990;186:675–680. doi: 10.1016/0076-6879(90)86166-s. [DOI] [PubMed] [Google Scholar]
- Wiedow O, Wiese F, Christophers E. Lesional elastase activity in psoriasis. Diagnostic and prognostic significance. Archives of Dermatological Research. 1995;287:632–635. doi: 10.1007/BF00371734. [DOI] [PubMed] [Google Scholar]
- Wipke BT, Allen PM. Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. Journal of Immunology. 2001;167:1601–1608. doi: 10.4049/jimmunol.167.3.1601. [DOI] [PubMed] [Google Scholar]
- Wittamer V, Bondue B, Guillabert A, Vassart G, Parmentier M, Communi D. Neutrophil-mediated maturation of chemerin: a link between innate and adaptive immunity. Journal of Immunology. 2005;175:487–493. doi: 10.4049/jimmunol.175.1.487. [DOI] [PubMed] [Google Scholar]
- Xiao H, Heeringa P, Hu P, Liu Z, Zhao M, Aratani Y, Maeda N, Falk RJ, Jennette JC. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. The Journal of Clinical Investigation. 2002;110:955–963. doi: 10.1172/JCI15918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Bruno E, Chao J, Huang S, Finazzi G, Fruchtman SM, Popat U, Prchal JT, Barosi G, Hoffman R. Constitutive mobilization of CD34+ cells into the peripheral blood in idiopathic myelofibrosis may be due to the action of a number of proteases. Blood. 2005;105:4508–4515. doi: 10.1182/blood-2004-08-3238. [DOI] [PubMed] [Google Scholar]
- Yamazaki T, Aoki Y. Cathepsin G binds to human lymphocytes. Journal of Leukocyte Biology. 1997;61:73–79. doi: 10.1002/jlb.61.1.73. [DOI] [PubMed] [Google Scholar]
- Yamazaki T, Aoki Y. Cathepsin G enhances human natural killer cytotoxicity. Immunology. 1998;93:115–121. doi: 10.1046/j.1365-2567.1998.00397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RE, Thompson RD, Larbi KY, La M, Roberts CE, Shapiro SD, Perretti M, Nourshargh S. Neutrophil elastase (NE)-deficient mice demonstrate a nonredundant role for NE in neutrophil migration, generation of proinflammatory mediators, and phagocytosis in response to zymosan particles in vivo. Journal of Immunology. 2004;172:4493–4502. doi: 10.4049/jimmunol.172.7.4493. [DOI] [PubMed] [Google Scholar]
- Zeiher BG, Artigas A, Vincent JL, Dmitrienko A, Jackson K, Thompson BT, Bernard G. Neutrophil elastase inhibition in acute lung injury: results of the STRIVE study. Critical Care Medicine. 2004;32:1695–1702. doi: 10.1097/01.ccm.0000133332.48386.85. [DOI] [PubMed] [Google Scholar]
- Zimmer M, Medcalf RL, Fink TM, Mattmann C, Lichter P, Jenne DE. Three human elastase-like genes coordinately expressed in the myelomonocyte lineage are organized as a single genetic locus on 19pter. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:8215–8219. doi: 10.1073/pnas.89.17.8215. [DOI] [PMC free article] [PubMed] [Google Scholar]