

Abstract
The biology and pathogenesis of hepatitis E virus are poorly understood due to the lack of an in vitro culture or infection models. The viral Orf3 protein activates the cellular mitogen-activated protein kinase pathway and is likely to modulate the host cell environment for efficient viral replication. We screened for cellular genes whose transcription was differentially up-regulated in an Orf3-expressing stable cell line (ORF3/4). The gene for mitochondrial voltage-dependent anion channel (VDAC) was one such candidate. The up-regulation of VDAC in ORF3/4 cells was confirmed by Northern and Western blotting in various cell lines. Transfection of ORF3/4 cells with an ORF3-specific small interfering RNA led to a reduction in VDAC protein levels. VDAC is a critical mitochondrial outer membrane protein, and its overexpression results in apoptosis. Surprisingly, Orf3-expressing cells were protected against staurosporine-induced cell death by preservation of mitochondrial potential and membrane integrity. A small interfering RNA-mediated reduction in Orf3 and VDAC levels also made cells sensitive to staurosporine. Chemical cross-linking showed Orf3-expressing cells to contain higher levels of oligomeric VDAC. These cells also contained higher levels of hexokinase I that directly interacted with VDAC. This interaction is known to preserve mitochondrial potential and prevent cytochrome c release. We report here the first instance of a viral protein promoting cell survival through such a mechanism.
Hepatitis E virus (HEV)4 is the causative agent of hepatitis E, a major form of viral hepatitis in developing countries (1-3). It is a waterborne pathogen that is transmitted primarily through the feco-oral route, causing rampant sporadic infections and large outbreaks in endemic areas (1-3). Although the infection is self-limited and never proceeds to chronicity, fulminant hepatitis with high rates of mortality is known to occur in a small fraction of patients, especially in pregnant women (4, 5). The HEV was recently classified as the only member of Hepevirus in the family Hepeviradae (6). It is a nonenveloped virus with a single-stranded positive sense RNA genome of ∼7.2 kb and contains three open reading frames (ORFs) (7). The orf1 encodes the major nonstructural polyprotein, orf2 codes for the major viral capsid protein, and orf3 codes for a small protein whose functions are not fully understood. Due to the lack of reliable cell culture systems or small animal models of infection and disease, subgenomic expression strategies have been employed to functionally characterize HEV-encoded proteins (8-10).
The orf3 overlaps the other two ORFs and encodes a protein of 123 amino acids (Orf3). A recent report proposes that Orf2 and Orf3 are translated from the same subgenomic RNA and that the latter protein was 9 amino acids shorter at its N-terminal end than previously reported (11). The Orf3 protein is phosphorylated at a single serine residue by the cellular mitogen-activated protein kinase (9). In its N-terminal half, Orf3 contains two hydrophobic domains, the first shown to be responsible for its cytoskeletal association (9). In its C-terminal half, the protein contains two proline-rich regions, of which one carries the phosphorylated serine residue (9). The other region was shown to contain a PXXPXXP motif and to bind several proteins that contain Src homology 3 domains (12). Such PXXP motifs are part of polyproline helices found in a number of viral and cellular proteins involved in signal transduction and bind the Src homology 3 domains found in a diverse group of signal-transducing molecules (13, 14). Previously, we have also shown Orf3 to activate the extracellularly regulated kinase (ERK), a member of the mitogen-activated protein kinase superfamily, supporting a prosurvival role for it. The Orf3 protein binds Pyst1, an ERK-specific member of dual specificity mitogen-activated protein kinase phosphatases; this interaction inhibits ERK dephosphorylation, thereby prolonging its activated state (15).
In the present study, we found increased expression of the voltage-dependent anion channel (VDAC) protein in Orf3-expressing cells. The VDAC protein, also known as porin for its pore forming ability, is a resident outer mitochondrial membrane (OMM) protein of 30-35 kDa that regulates the transport of solutes such as Ca2+ and ATP across the OMM (16, 17). The mitochondrion plays an important and fateful role in the apoptotic death of mammalian cells by releasing apoptogenic proteins into the cytoplasm (18). The release of cytochrome c from mitochondria into the cytoplasm activates caspases, setting the cell on an apoptotic path (19). The VDAC protein regulates OMM permeability and is in turn regulated by direct interaction with Bcl-2 family members as well as other proteins, such as hexokinase and mitochondrial creatine kinase (20). Further, VDAC is known to be up-regulated in cancer cells (21) and exhibits elevated binding to hexokinase I and/or hexokinase II. Due to their higher energy requirements, cancer cells are characterized by a high rate of glycolysis (22, 23). This in turn requires various genetic and biochemical adaptations, including increased expression of mitochondria-bound isoforms of hexokinase (HK I and HK II) (23-25). Purified HK I has been shown to interact directly with purified VDAC reconstituted into a planar lipid bilayer, leading to channel closure. This prevents opening of the permeability transition pore and release of cytochrome c, thus inhibiting the mitochondrial phase of apoptosis (26). A dynamic equilibrium exists between monomeric and oligomeric forms of VDAC that determines its interactions with other proteins and the regulated formation of pores large enough to allow the release of apoptogenic factors from mitochondria (27).
We show here a prosurvival effect of the HEV Orf3 protein and relate this to its ability to modulate VDAC expression and oligomerization. We also show that VDAC oligomerization in Orf3-expressing cells protects against an apoptotic insult and is accompanied by enhanced hexokinase I expression and its interaction with VDAC. These findings are discussed in the context of HEV pathogenesis.
EXPERIMENTAL PROCEDURES
Plasmids, Cell Lines, and Antibodies
The expression vectors for orf3, pSG-ORF3 (8), and pORF3-ECFP (15) have been described earlier. The orf3 and vector control stable cell lines (12) as well as polyclonal antibodies to Orf3 (8) have also been described earlier. The VDAC1, VDAC2, and VDAC3 genes were generously provided by Dr. Michael Forte (Vollum Institute for Advanced Biomedical Research, Oregon Health Sciences University, Portland, OR). The VDAC antibody was purchased from Cell Signaling Technology (Beverly, MA), whereas the Alexa dye-conjugated secondary antibodies were procured from Molecular Probes, Inc. (Eugene, OR). The EGFP-cytochrome c expression vector was kindly provided by Dr. Israrul Haq Ansari (University of Nebraska, Lincoln, NE).
Suppressive Subtractive Hybridization
Suppressive subtractive hybridization was performed using orf3 and vector control stable cell lines according to Mishra et al. (28). Briefly, total RNA was isolated from the cells and mRNA prepared by magnetic separation after annealing with biotinylated oligo(dT) primer and immobilizing it onto streptavidin-linked paramagnetic beads. Five μg of mRNA from each cell line was used to synthesize first strand cDNA by separately priming with P1-dT and P2-dT oligonucleotide primers (28). After reverse transcription, mRNA and excess primers were removed before ligating the P1-3′ and P2-3′ adaptor oligonucleotides to the 3′-ends of first strand cDNAs with T4 RNA ligase (28). The first strand cDNA population from control samples was PCR-amplified using 5′ biotinylated P1 forward and nonbiotinylated P1 reverse primers (28). The PCR conditions included 30 cycles of 94 °C for 1 min, 55 °C for 1 min, and 72 °C for 3 min. The biotinylated double-stranded cDNAs thus obtained were immobilized onto streptavidin-linked paramagnetic beads, whereas the nonbiotinylated complementary antisense strands were denatured with 0.15 m NaOH for 10 min at room temperature and separated out from the beads with a magnet. The beads were washed twice with 0.15 m NaOH and used for the first round of cDNA subtraction. The unamplified first strand cDNAs that were made from ORF3/4 cells were hybridized for 5 h at 65 °C with PCR-amplified sense strand cDNAs of control cells that were immobilized onto the magnetic beads. The hybrids between the sense strand and the common complementary antisense cDNA strands, as well as the beads, were magnetically separated. Differentially up-regulated genes in ORF3/4 cells remained in the hybridization solution and were PCR-amplified in a sequence-independent manner with P2 forward and P2 reverse primers. The PCR conditions included 30 cycles of 94 °C for 1 min, 55 °C for 1 min, and 72 °C for 3 min. The PCR-amplified differentially expressed cDNA population was then cloned into the pGEMT vector (Promega, Madison, WI). The recombinant plasmids thus obtained were digested with NotI; depending on insert size, six different clones were selected and sequenced, and the genes were identified by BLAST analysis.
Northern Blotting
For probe preparation, the VDAC1 clone was digested with AccI-BglI to give a 230-bp fragment, the VDAC2 clone was digested with AccI-HindIII for a 530-bp fragment, and the VDAC3 clone was digested with HindIII for a 470-bp fragment. The probe fragments corresponded to the 3′-ends of the VDAC genes and showed very low nucleotide homology, enabling specific detection of VDAC RNAs by Northern blotting. Total RNA was isolated with Trizol (Invitrogen) as per the manufacturer’s guidelines. Samples of total RNA (20 μg/lane) were denatured in 17.4% (v/v) formaldehyde, 50% (v/v) formamide, 20 mm 3-(N-morpholino) propanesulfonic acid, 5 mm sodium acetate, and 1 mm EDTA (pH 7.0) for 5 min at 65 °C and separated on a 1% (w/v) agarose, 0.66 m formaldehyde gel. Following electrophoresis, the RNA was transferred to a nylon membrane (HyBond-N+; Amersham Biosciences) by capillary blotting for 24 h in 10× SSC (1× SSC: 150 mm NaCl and 15 mm sodium citrate, pH 7.0) and UV-cross-linked to the nylon membrane. The blot was stained with methylene blue for RNA integrity and equivalence by determining transfer of 28 and 18 S rRNA from the gel. Membranes were prehybridized at 42 °C for 5 h in hybridization solution containing 50% (v/v) deionized formamide, 50 mm sodium phosphate, 0.8 m NaCl, 2% (w/v) SDS, 100 μg salmon sperm DNA/ml, 20 μg of transfer RNA/ml, and 1× Denhardt’s solution (50× Denhardt’s solution: 1% each of bovine serum albumin, Ficoll, and polyvinylpyrrolidone). The VDAC probes were labeled with [α-32P]dCTP using a random priming labeling kit (Hexalabel DNA labeling kit; Fermentas). Hybridization was carried out at 42 °C for 20 h in hybridization solution containing the radiolabeled VDAC isoform-specific probe. Membranes were washed sequentially once in 2× SSC at room temperature for 10 min and twice with 0.5× SSC, 0.1% SDS at 65 °C for 30 min. The bands were visualized by autoradiography.
Immunoprecipitation and Western Blotting
Cells were lysed with a buffer containing 20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, and a protease inhibitor mixture (Roche Applied Science). The cell lysates were normalized for protein content, and 1 mg of total proteins in 500 μl of lysis buffer were incubated with 10 μl of Protein G-agarose (Amersham Biosciences) beads for 1 hat 4 °C. The precleared lysate was then incubated with 2 μg of anti-hexokinase I polyclonal antibody (sc-6518; Santa Cruz Biotechnology, Inc., Santa Cruz, CA) overnight at 4 °C. This was followed by incubation with 10 μl of Protein G-agarose beads for 2 h at 4 °C. After five washings in lysis buffer, the beads were boiled in Laemmli buffer, and the proteins were separated by SDS-PAGE. For Western blotting, the separated proteins were transferred to a nitrocellulose membrane (MDI, Advanced Microdevices Pvt. Ltd., Ambala, India). After blocking with Tris-buffered saline (TBS) containing 5% nonfat milk (Nestle) for 1-2 h at room temperature, the membrane was washed with TBST (TBS containing 0.1% Tween 20) and incubated overnight at 4 °C with the primary antibody appropriately diluted in TBST containing 5% bovine serum albumin. The blot was then washed three times for 10 min each with TBST and then incubated with horseradish peroxidase-linked anti-rabbit or anti-goat IgG diluted in TBST containing 5% nonfat milk. Chemiluminescent detection of proteins was carried out using the Phototope horseradish peroxidase Western blot detection system (Cell Signaling Technology, Beverly, MA) according to the supplier’s protocol. In all Western blotting experiments, total ERK levels were evaluated as loading controls.
Cell Viability Assay
For the cell survival assay, cells were treated with 1 μm staurosporine (STS) for 10 h, followed by two washes with PBS. The treated cells were then stained with 0.25% Coomassie Brilliant Blue R-250 in 10% acetic acid, 50% methanol (v/v) and washed, and the stained colonies of live cells were counted on an Elispot Reader (Bioreader 4000; Biosys). The cell viability was also evaluated in attached cells using a colorimetric assay based on 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) staining (29) that measures mitochondrial function (30, 31). The cells were either mock-treated or treated with various concentrations of STS for the indicated time(s) followed by the MTT assay. All data are expressed as percentages with respect to mock-treated cells and represent the mean ± S.E. of at least three independent experiments.
Mitochondrial Membrane Potential Measurement
The change in mitochondrial membrane potential (ΔΨm) was analyzed by flow cytometry using the ΔΨm-sensitive dye 5,5′,6,6′-tetrachloro-1,1′3,3′-tetraethyl benzimidazolcarbocyanine iodide (JC-1; Sigma) (32). Briefly, cells were harvested, washed once in PBS, and then resuspended in complete culture medium containing 1 μm JC-1 at 37 °C for the indicated time(s) in the presence or absence of 2 μm STS. Stained cells were then washed with PBS and analyzed by flow cytometry. The emission maxima of JC-1 monomers and aggregates are 527 nm (FL-1 channel) and 590 nm (FL-2 channel), respectively.
Microscopy
For immunofluorescence staining and colocalization experiments, Huh-7 cells were seeded at about 30% confluence on coverslips in 12-well plates, grown for 18 h, and then cotransfected with Ds-Red Mito and EGFP-cytc, either with or without ECFP-ORF3. Also, pCN and ORF3/4 cells were cotransfected to express Ds-Red Mito and EGFP-cytc. At 48 h post-transfection, the cells were treated with 2 μm STS for 2 h, washed with PBS, fixed in 4% paraformaldehyde for 15 min at room temperature, and washed once again in phosphate-buffered saline. The cells were then mounted using Antifade (Bio-Rad) and sealed with a synthetic rubber-based adhesive, Fevibond (Fevicol; Pidilite Industries). Confocal images were collected using a 60× planapo objective on a Bio-Rad Radiance 2100 system attached to a Nikon inverted microscope. Multiple randomly selected cells from each group were analyzed, and the amount (percentage) of EGFP-cytochrome c that colocalized with Ds-Red Mito was quantitated with the LaserPix software (Bio-Rad). Such colocalization calculations are independent of the relative intensities in the green and red channels.
Cross-linking Experiments
The pCN and ORF3/4 cells were treated with or without 5 μm STS for 30 min followed by a PBS wash. Cells were lysed by five or six repeated freeze-thaw cycles in liquid nitrogen and at room temperature. The unclarified lysate was then incubated with 1 mm DSP (Pierce) dissolved in Me2SO for 30 min at 30 °C according to manufacturer’s protocol. The Me2SO concentration in control and reagent-containing samples was up to 2.5% (v/v). The reaction was stopped by adding 50 mm Tris, pH 7.5, and the samples were analyzed on nonreducing as well as reducing SDS-PAGE.
siRNA-mediated Knockdown of Orf3 and VDAC
The orf3 siRNA 5′-UCACGUCGUAGACCUACCAUU-3′ was designed using the “Dharmacon siDESIGN Center” program available on the World Wide Web and was obtained from Dharmacon (Lafayette, CO) as a duplex. The VDAC1-specific and GFP siRNAs were obtained as siGENOME SMARTpool reagent from Dharmacon. Cells were transfected using RNAiFect (Qiagen, Hilden, Germany) with 200 nm VDAC siRNA (si-V), 300 nm orf3 siRNA (si-3), and 200 nm GFP siRNA (si-G). All siRNA-related experiments were performed in 12-well culture plates, and GFP-specific siRNA was used as nonspecific control. For siRNA-based survival experiments, cells were transfected, with either the orf3 siRNA for 48 h or the VDAC siRNA pool for 36 h followed by treatment with 2 μm STS for 2 h. The cell viability assays were then performed as described above.
Reverse Transcription-PCR Analysis
Total RNA was isolated from pCN and ORF3/4 cells using the Trizol reagent (Invitrogen). Four μg of RNA in a 25-μl reaction mixture was used for cDNA synthesis with Reverse Transcriptase (Promega, Madison, WI) according to the supplier’s protocol. Of this, 0.5 μl of cDNA was used as a template for PCR amplification of target genes. The PCRs were performed in a 50-μl volume containing 1× reaction buffer, 200 μm dNTPs, 10 pmol of each primer, and 1.25 unit of TaqDNA polymerase (Real Biotech Corp., Taipei, Taiwan), for 35 cycles of 94 °C for 30 s, 55 °C for 30 s, 72 °C for 30 s, and a final extension at 72 °C for 2 min. The primer sequences for hexokinase I and II were retrieved from the World Wide Web and custom-synthesized. Primer pairs HK1-F (5′-TCCTGGCCTATTACTTCACGG-3′) and HK1-R (5′-GGACCTTACGAATGTTGGCAA-3′) and HK2-F (5′-GAGCCACCACTCACCCTACT-3′) and HK2-R (5′-ACCCAAAGCACACGGAAGTT-3′) were used to amplify hexokinase I and II, respectively. The histone H4 gene was amplified as a reference control using the primers HISTH4-F (5′-TGAGAGACAACATTCAGGGCATCAC-3′) and HISTH4-R (5′-CGCTTGAGCGCGTACACCACATCCAT-3′). The amplified products were resolved on a 2% agarose gel and visualized following staining with ethidium bromide. The sizes of the amplified fragments were as follows: hexokinase I, 194 bp; hexokinase II, 130 bp; histone H4, 211 bp.
RESULTS
Suppression subtractive hybridization was performed using control (pCN) and Orf3-expressing (ORF3/4) stable cell lines as described under “Experimental Procedures.” The differentially expressed cDNAs were PCR-amplified and cloned. A number of clones were randomly picked and analyzed by restriction digestion. Clones with varying sizes of inserts were identified and sequenced to determine the identity of the up-regulated genes they represented. One such up-regulated gene was VDAC1.
To validate Orf3-mediated increased expression of VDAC, we carried out Northern and Western blot analyses. The ORF3/4 cell line showed higher levels of VDAC1 RNA (Fig. 1A). Using probes from the 3′-end of the VDAC gene, where minimal homology was observed between VDAC1 and its two other isoforms, VDAC2 and VDAC3, increased expression of all three VDAC isoforms was observed in Orf3-expressing cells (Fig. 1A). To confirm that the higher RNA levels were also reflected in higher levels of the VDAC protein, Western blotting was performed on lysates prepared from pCN and two independent Orf3-expressing cell lines, ORF3/1 and ORF3/4. Western blotting using anti-VDAC antibodies showed higher expression of the VDAC protein in Orf3-expressing cells (Fig. 1B, top, lanes 1-3). Since the anti-VDAC antibodies reacted with all three isoforms of the protein, it was not possible to study the effects on individual isoforms. In another experiment, human hepatoma Huh-7 cells that were transiently transfected with an orf3 expression vector (pSG-ORF3) also showed higher levels of the VDAC protein compared with Huh-7 cells transfected with the empty vector (Fig. 1B, top, lanes 4-6). We checked a HeLa/Tet-OFF-inducible stable cell line5 to show that compared with control cells, the Orf3-expressing cells showed higher levels of the VDAC protein (Fig. 1B, bottom, lanes 1-3). These cells showed leaky expression of Orf3 even in the presence of tetracycline (lane 2); this was also reflected in VDAC expression in HeLa/Tet-OFF/ORF3 cells in the presence (lane 2) and absence of tetracycline (lane 3). Thus, using multiple cell line systems, either transiently transfected or stably expressing Orf3, we show that increased VDAC expression correlated with Orf3 expression.
FIGURE 1. Analysis of VDAC expression in cell lines.

A, total RNA extracted from pCN (−) and ORF3/4 (+) cells was Northern blotted with orf3-, VDAC1-, VDAC2-, and VDAC3-specific probes. The 18 and 28 S rRNA stained with methylene blue served as a control for equal loading and RNA integrity. B, Western blot analysis for VDAC expression was carried out on lysates prepared from various cell lines. These included the following: pCN (lane 1), ORF3/1 (lane 2), ORF3/4 (lane 3), vector-transfected Huh-7 cells (lane 4), and pSG-orf3-transfected Huh-7 cells (lanes 5 and 6). Western blotting of cell lysates with anti- Orf3 and anti-ERK antibodies served as expression and loading controls, respectively. The bottom panel shows results from HeLa/Tet-OFF cells (lane 1) or HeLa/Tet-OFF-ORF3 cells in the presence (lane 2) or absence (lane 3) of tetracycline. A nonspecific band detected with anti-VDAC antibodies served as a loading control; Western blotting for Orf3 showed its expression in these cells.
The VDAC protein is the principal mitochondrial channel protein, and its regulated expression is important for cell survival. A higher VDAC expression level in cells results in cell death by apoptosis (33). For insight into VDAC overexpression in Orf3-stable cell lines, we treated the cells with STS, a potent apoptogenic stimulus. Apoptosis mediated by STS occurs through a mitochondrial pathway and leads to loss of mitochondrial transmembrane potential. Cells that survived STS-initiated death were initially scored by staining with Coomassie Blue and counting. Surprisingly, significantly reduced cell death was observed in ORF3/4 cells compared with the control pCN cells (Fig. 2A), which was in contrast to previous reports where higher VDAC expression levels resulted in enhanced apoptosis and cell death. Although treatment with 1 μm STS for 10 h killed about 75% of the pCN cells, about 50% of the ORF3/4 cells survived this treatment (Fig. 2A). The cell viability was also evaluated by using a colorimetric assay based on MTT staining. The pCN and ORF3/4 cells were treated with various concentrations of STS for different times. Although pCN cells showed greater sensitivity toward STS-mediated death, ORF3/4 cells resisted this effect. An optimum 2 μm STS treatment for 2 h resulted in ∼40% cell death in pCN cells, whereas only about 20% death was observed in ORF3/4 cells (Fig. 2B).
FIGURE 2. The Orf3 protein protects cells from staurosporine-induced death.

A, control (pCN) and ORF3/4 cells were treated with 1 μm STS for 10 h followed by staining with Coomassie Brilliant Blue R-250 and quantitation of surviving cells as described under “Experimental Procedures.” The picture on the left is representative of a survival assay in a 12-well plate. The bar graph on the right shows results from three independent experiments. The dark and light bars show results for pCN and ORF3/4 cells, respectively. B, cell viability was also evaluated by using a colorimetric assay based on the MTT uptake. The pCN and ORF3/4 cells were treated with various concentrations of STS for 30 min, 60 min, 1 h, and 5 h, followed by the MTT assay. Data are expressed as percentage of cell viability with respect to control and represent an average of three independent experiments.
Mitochondria are critical integrators of apoptosis, the initiator event being the loss of mitochondrial potential. We therefore compared mitochondrial depolarization in control and Orf3-expressing cells following STS treatment. To detect changes in the mitochondrial membrane potential, we used JC-1, a cationic dye that detects mitochondrial depolarization in apoptotic cells by a shift in its fluorescence emission. Treatment with STS and JC-1 staining was followed by flow cytometric analysis of the cells. Although the pCN cells showed a quantitative loss of mitochondrial potential characterized by a large increase in fluorescence (Fig. 3A), only minimal shifts were observed in ORF3/4 cells (Fig. 3B).
FIGURE 3. The Orf3 protein prevents mitochondrial depolarization.

The pCN (A) and ORF3/4 (B) cells were either mock-treated or treated with 2 μm STS for 30 min, 1 h, and 2 h followed by JC-1 staining as described under “Experimental Procedures.” The JC-1-stained cells were then subjected to flow cytometry, and results were analyzed for the FL-1 channel that scores JC-1 monomers. The solid and dotted lines show JC-1 staining without or with STS treatment, respectively.
The retention of mitochondrial potential in Orf3-expressing cells in response to STS suggested the involvement of other players in controlling mitochondrial membrane integrity. The mitochondrial VDAC acts as a docking site for various pro- and antiapoptotic proteins that affect its oligomerization status. Monomeric VDAC is reported to form the functional channel whose conductance decreases following VDAC oligomerization (27). To evaluate the oligomeric status of VDAC in Orf3-expressing cells, we used the thiol-cleavable bifunctional chemical cross-linker DSP. Chemical cross-linking of lysates from pCN and ORF3/4 cells with this membrane-permeable cross-linker was performed following STS treatment. The lysates from cross-linked pCN and ORF3/4 cells were separated by SDS-PAGE under nonreducing and reducing conditions followed by Western blotting with anti-VDAC antibodies. Substantially higher levels of VDAC oligomers were observed in ORF3/4 cells compared with pCN cells (Fig. 4, lanes 1-8). These oligomers were formed specifically by cross-linking VDAC monomers as evidenced by their disappearance following thiol-mediated cleavage of the DSP spacer arm (Fig. 4, lanes 9-12).
FIGURE 4. VDAC oligomerization status.
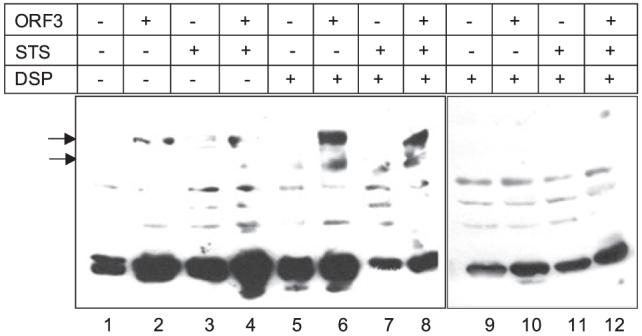
Control and ORF3/4 cells were either mock-treated or treated with 5 μm STS for 30 min. The cells were then lysed by freeze-thaw, and the unclarified lysates were treated with the membrane-permeable bifunctional cross-linker DSP at a concentration of 1 mm. As a control, cells were treated with an equivalent volume of Me2SO. The lysates were then mixed with Laemmli SDS dye loading buffer (β-mercaptoethanol-free for nonreducing conditions), boiled, and resolved on nonreducing (lanes 1-8) as well as reducing (lanes 9 -12) SDS-polyacrylamide gels. Western blotting was performed using anti-VDAC antibodies. The arrows indicate oligomeric forms of VDAC.
Hexokinase (HK), the first enzyme in the glycolytic pathway, is reported to bind to the mitochondrial membrane due to its interaction with VDAC. This interaction is important for the integration of glycolysis with mitochondrial energy metabolism and the antiapoptotic role of HK in cells (34). Hexokinase binding cross-links VDAC and suppresses the release of intermembrane space proteins, thus preventing apoptosis (35). To evaluate the status of HK in Orf3-expressing cells, we carried out reverse transcription-PCR analysis of RNA prepared from pCN and ORF3/4 cells. Although there was a large increase in HK I expression in ORF3/4 cells, there was no change in the expression of HK II in these cells compared with the pCN cells (Fig. 5A). The Western blot analysis of these cell lines using antibodies specific for HK I and HK II also revealed higher levels of HK I in Orf3-expressing cells, whereas the HK II levels remained unchanged (Fig. 5B). Human hepatoma Huh-7 cells transiently transfected with pSG-ORF3 also exhibited higher levels of HK I (Fig. 5C). A direct interaction between HK I and VDAC was tested by immunoprecipitation of cell lysates with anti-HK I antibodies followed by Western blotting with anti-VDAC antibodies. Anti-HK I antibodies co-immunoprecipitated VDAC from pCN and ORF3/4 cells. However, a larger amount of the VDAC protein was immunoprecipitated from Orf3-expressing cells compared with control cells (Fig. 5D).
FIGURE 5. Hexokinase expression and its interaction with VDAC.

A, reverse transcription-PCR analysis was carried out to estimate the expression of histone H4 (reference control), hexokinase I (HK I) and hexokinase II (HK II) genes in pCN (lanes 1, 3, and 5) and ORF3/4 (lanes 2, 4, and 6) cells. The molecular size markers are shown. B, Western blot analysis of control and ORF3/4 cells was performed using anti-HK I or HK II antibodies. Western blotting for ORF3 and ERK served as expression and loading controls. C, lysates prepared from Huh-7 cells transiently transfected with either control vector or pSG-orf3 were subjected to Western analysis using anti-HK I antibody. Levels of total ERK served as loading control. D, the lysates prepared from control and ORF3/4 cells were immunoprecipitated with anti-HK I antibodies followed by Western blotting with anti-VDAC antibodies.
If Orf3 conferred resistance against a strong apoptotic stimulus (STS) and if it was indeed mediated through the up-regulation of VDAC and HK I and their enhanced interaction, an siRNA-mediated knockdown of Orf3 should affect this survival advantage. To confirm the protective effects of Orf3, we transfected ORF3/4 cells with an orf3-specific siRNA and 48 h later treated the transfected cells with 2 μm STS for 2 h, followed by MTT staining. Although a majority of GFP siRNA-transfected ORF3/4 cells survived the STS treatment, this number was reduced in the orf3 siRNA-transfected cells (Fig. 6A). Although this reduction was not dramatic, it was reproducible. This pool is likely to contain a mixture of cells that received the orf3 siRNA following transfection and those that did not, thus potentially reducing the overall siRNA effect. The siRNA against orf3 also led to a decrease in HK I and VDAC levels (Fig. 6B). To demonstrate a direct role for VDAC in protection from cell death, we transfected pCN or ORF3/4 cells with an siRNA pool directed against VDAC. This reduced endogenous VDAC protein levels by about 80-90% in both cell lines at 36 h post-transfection (Fig. 6C). At this time, cells were treated with STS, and cell viability was scored by the MTT assay. Reduced cell survival was found in VDAC siRNA-transfected pCN cells compared with mock-transfected cells following STS treatment (Fig. 6D). Similarly, ORF3/4 cells also showed reduced survival following siRNA-mediated VDAC knockdown (Fig. 6D); the effects were, however, not as pronounced as those observed for pCN cells.
FIGURE 6. Orf3-, VDAC-, and STS-mediated cell death.

A, the ORF3/4 cells were transfected with either control siRNA for GFP (si-G) or directed against orf3 (si-3), and 48 h later, the cells were treated with 2 μm STS for 2 h. Cell viability was then measured by the MTT assay. Controls included mock transfection and no STS treatment. The values shown are means ± S.E. of three independent experiments. B, the lysates from mock-, control (si-G)-, or orf3 siRNA (si-3)-transfected ORF3/4 cells were Western blotted with HK I, VDAC, Orf3, and ERK antibodies. C, Western blot analysis of lysates from mock-, control (si-G)-, or VDAC (si-V) siRNA-transfected cells 36 h post-transfection to estimate levels of VDAC and ERK (loading control) proteins. D, pCN (dark bars) and ORF3/4 (light bars) cells were either mock-transfected or transfected with GFP siRNA (si-G) or VDAC siRNA (si-V). After 36 h, these cells were treated with 2 μm STS for 2 h and then assayed for cell viability by the MTT method. Mock-treated cells were taken as 100% survival. The results are presented as mean ± S.E. of three independent experiments.
The release of cytochrome c from mitochondria is the first irreversible step toward the commitment of cells to apoptosis. We therefore assessed the release of cytochrome c from mitochondria. Control and ORF3/4 cells were transfected to express EGFP-cytc together with a mitochondrial marker, DsRed-mito. Huh-7 cells were also transiently transfected with EGFP-cytochrome c and DsRed-mito in the absence or presence of ECFP-tagged ORF3. After 48 h, the cells were treated with 2 μm STS for 2 h and analyzed by confocal microscopy. Control (pCN and Huh-7) cells showed extensive mitochondrial disintegration and release of cytochrome c in the cytosol following STS treatment (Fig. 7A). In mock-treated cells, EGFP-cytc was found to quantitatively localize to the mitochondria (not shown). However, in ORF3/4 cells and ECFP-ORF3-expressing Huh-7 cells, EGFP-cytochrome c was clearly retained in the mitochondria following STS treatment, as evidenced by its colocalization with the DsRed-mito marker (Fig. 7A). The degree of extramitochondrial EGFP-cytc was determined using the LaserPix colocalization module and plotted in Fig. 7B. Although control cells (pCN and Huh-7) showed about 75-80% of cytc to be released from mitochondria, only about 20-25% of it was released in Orf3-expressing cells. In the same cells, the Orf3 protein was found not to localize to the mitochondria; thus, the protective effect was not due to a direct association of Orf3 with the mitochondria. Together, the JC-1 and cytc results showed the Orf3 prosurvival effect to be due to its ability to preserve mitochondrial integrity in the face of an apoptotic insult.
FIGURE 7. Effect of Orf3 on mitochondrial cytochrome c release.

A, control (pCN) and ORF3/4 cells were cotransfected with pDs-Red-Mito and EGFP-cytc. Huh-7 cells were cotransfected to express Ds-Red Mito and EGFP-cytochrome c either without or with plasmid ECFP-orf3. After 48 h, the cells were treated with 2 μm STS for 2 h, fixed, and visualized by confocal microscopy. The individual images and their merged pictures are shown. B, the amount of cytochrome c released from the mitochondria to the cytoplasm in these cells was determined by LaserPix software and plotted. Average cytochrome c release values derived from at least six randomly selected cells are shown.
DISCUSSION
The Orf3 protein appears to modulate the host cell environment for efficient replication and propagation of HEV. We have shown earlier that this viral protein up-regulates ERK activity (12) and that this is due to its ability to bind and inhibit mitogen-activated protein kinase phosphatases that negatively regulate ERK activation (15). The Orf3 protein also binds α-1-microglobulin, a protein with immunosuppressive properties, and promotes its secretion into the extracellular medium (36). Although not directly demonstrated, this is likely to have an impact on the host immune response, at least in the milieu of an infected liver. The prolonged activation of ERK would have a prosurvival effect, protecting the infected cell from premature cell death and increasing the efficiency of viral replication. Thus, Orf3 appears to be a regulatory protein that is likely to influence multiple pathways that promote establishment and propagation of HEV infection. Indeed, Orf3 was shown to be required for viral infection following intrahepatic inoculation of HEV genomic RNA directly into monkey liver (37) but was found to be dispensable when HEV genomic RNA was tested by transfection of cells in vitro (38, 39). Since no efficient cell culture system, small animal models, or in vitro genomic replicon systems are available for studying HEV biology, our strategy is based on subgenomic expression to study the characteristics and possible functions of the Orf3 protein.
We used suppression-subtractive hybridization (28), a PCR-based technique for cloning differentially expressed genes, to search for cellular genes whose expression was increased in Orf3-expressing cells. For this, we used a cell line that stably and constitutively expressed Orf3; another cell line made with the empty expression vector served as a control. At least six different up-regulated genes were cloned and sequenced. One of these matched the gene for VDAC, and this was verified by Northern blotting. The RNAs for all three isoforms of VDAC were found to be up-regulated in Orf3-expressing cells. Increased levels of the VDAC protein in these cells were confirmed using multiple cell lines that included transient as well as stable expression of Orf3. Further, siRNA-mediated silencing of Orf3 expression led to reduced VDAC expression, supporting a direct causal relationship between these proteins.
Mitochondria are organelles that coordinate cellular energy metabolism and integrate signals for cell survival and apoptosis (35, 40). Exchange of metabolites across the inner and outer mitochondrial membranes and the maintenance of a transmembrane potential are critical for normal cell physiology. The VDAC is an important mitochondrial protein whose expression levels are regulated and critical for cell survival. An imbalance in VDAC expression disturbs cellular metabolism, with lower levels resulting in slow growth and up-regulation, leading to apoptosis. A preliminary analysis was performed by scoring cell viability of Orf3-expressing cells in response to STS, a potent apoptotic agent that acts at the level of mitochondria. Interestingly, Orf3-expressing cells showed better survival to STS-mediated death when assayed by two different methods, although higher VDAC expression levels are correlated with increased apoptosis. Since STS is known to cause mitochondrial depolarization, the mitochondrial membrane potential (ΔΨ)-sensitive dye JC-1 was used to assess this as well. This dye accumulates as aggregates in the mitochondria, resulting in red fluorescence, the brightness of which is proportional to ΔΨ and varies among different cell types. However, in apoptotic and necrotic cells, JC-1 exists in its monomeric form in the cytoplasm and stains cells green. Although the control cells showed quantitative mitochondrial depolarization following STS treatment, the ORF3/4 cells were again better protected. Increased VDAC expression on one hand and cell survival and mitochondrial depolarization results on the other suggested the involvement of other interacting partners and compensatory mechanisms in the fate of Orf3-expressing cells.
There are two main models to explain the release of apoptogenic factors from mitochondria. One is based on increased Ca2+ uptake, swelling of the matrix, and mitochondrial rupture (41-44). The other model supports formation of a pore in the OMM that allows passage of large molecules without damage to the organelle (43, 45-48). Recent reports suggest that VDAC forms this pore whose opening is regulated by association with proteins of the Bcl-2 family as well as other proteins like hexokinase and mitochondrial creatine kinase. Microinjected VDAC-specific antibodies inhibit apoptosis induced by microinjected Bax, demonstrating the importance of VDAC cross-linking in cells (48).
To understand the mechanism(s) underlying the maintenance of mitochondrial potential in ORF3/4 cells, we assessed the oligomeric nature of VDAC in these cells through chemical cross-linking studies. The membrane-permeable but thiol-cleavable cross-linker, DSP, showed the presence of oligomeric VDAC in ORF3/4 cells when the lysates were analyzed under nonreducing conditions but not under reducing conditions. It is known that monomeric VDAC serves as the functional channel, and a relatively intimate contact exists between monomers in the VDAC, whereas cross-linked VDAC results in decreased channel conductance and voltage-independent channel activity (27). It has been shown that cross-linking of VDAC prevents the movement of cytochrome c from the mitochondria to the cytoplasm (27). A dynamic equilibrium exists between monomeric and oligomeric VDAC that upon cross-linking shifts in favor of the latter. Oligomeric VDAC is also a binding site for the hexokinase I tetramer (26, 49), this binding leading to closure of the VDAC pore, preventing the release of proapoptotic molecules.
Our hexokinase experiments further strengthened the correlation between up-regulated VDAC and increased cell survival. Although HK I and HK II are functionally similar, they differ in their tissue distribution (30). Hexokinase binding to VDAC suppresses the release of intermembrane space proteins and inhibits apoptosis, thereby contributing to the survival advantage of cells (31, 50). This is achieved by the binding of tetrameric HK to VDAC tetramers (26, 51-53). Both isoforms interact through a hydrophobic 15-amino acid sequence in their N-terminal region with mitochondrial VDAC (51, 54). In ORF3/4 cells, we observed higher expression levels of HK I, the isoform predominantly present in hepatocytes; there was no effect on the levels of HK II, an isoform known to be weakly expressed in liver cells (50, 55). Transient expression of Orf3 in Huh-7 cells also had a similar effect on HK I levels. In an independent microarray analysis, VDAC and HK I, but not HK II, genes were found to be up-regulated in ORF3/4 cells compared with pCN cells (not shown). Co-immunoprecipitation of HK I and VDAC showed a direct interaction between these proteins. Further, higher amounts of VDAC were pulled down with HK I in Orf3-expressing cells compared with pCN cells. This interaction between HK I and VDAC would prevent opening of the pore and release of the proapoptotic protein cytochrome c, thereby suppressing the initiation of apoptosis.
The VDAC channel controls the release of solutes, including cytochrome c and other proapoptotic molecules residing in the intermembrane space of mitochondria. We tested this in Huh-7 cells transiently transfected with EGFP-cytc, either alone or together with ECFP-ORF3. Following STS treatment, large amounts of EGFP-cytc were released from the mitochondria; this effect was missing in Huh-7 cells that also expressed ECFP-ORF3. Similar results were obtained with cells stably expressing Orf3. Reducing VDAC expression through transfection of its specific siRNA made cells more susceptible to STS; this effect was more pronounced in control compared with Orf3-expressing cells. A similar effect was also observed when orf3 siRNA was used. In that case, although the effects on Orf3 or VDAC expression were not dramatic, this down-regulation still made orf3 siRNA-transfected cells more prone to STS-mediated death as seen in the cell viability assay. Together, these results provided an explanation for Orf3-mediated protection of cells from STS-induced mitochondrial depolarization, cytochrome c release, and death. In related work, we have also observed that Orf3 protects cells from death induced by U0126 and LY294002, which are pharmacological inhibitors of the ERK and phosphatidylinositol 3-kinase/Akt signaling pathways.6
A number of viral proteins target mitochondria for the suppression or induction of apoptosis (55, 57). Importantly, it appears that such viral proteins have little or no structural similarity and that they target very different mitochondrial receptors (58). Of the mitochondrial proteins that interact with viral proteins, several are also involved in the physiological regulation of mitochondrial membrane potential in response to endogenous regulators of apoptosis, such as the Bcl-2 family proteins, ANT, VDAC, and PBR (59). Many viruses also encode death-inhibitory Bcl-2 analogs that preferentially localize to mitochondria and may interact with the proapoptotic Bax protein. Examples of this include the Epstein-Barr virus BORFB2F and BALF1 proteins, the African swine fever virus 5-HL/A179L protein, the herpesvirus saimiri HVS-Bcl-2 protein, the Kaposi sarcoma-associated herpesvirus 8 KSBcl-2 protein, the bovine herpesvirus 4 BHRF-1 protein, and the murine herpesvirus-68 M11 protein (55). The hepatitis B virus X protein shares 31% homology with human VDAC3 and colocalizes with it to the mitochondria (60). Thus, it appears that viruses exploit several distinct apoptosis-regulatory pathways at the level of mitochondria.
Based on the current study, we propose that the Orf3 protein of HEV protects cells from mitochondrial depolarization and death following apoptotic insult. This is achieved by overexpression of the mitochondrial VDAC, a key channel protein that integrates cellular energy metabolism with the mitochondrial phase of apoptosis (61). Although this up-regulation would lead to enhanced mitochondrial energy metabolism, it would also make cells susceptible to apoptosis. Up-regulation of another glycolytic protein hexokinase by Orf3 negates the proapoptotic function of VDAC. By directly interacting with and cross-linking VDAC, hexokinase would serve in stabilizing the mitochondrial permeability transition pore in its closed state and prevent mitochondrial depolarization, thereby maintaining mitochondrial membrane integrity and preventing the subsequent release of cytochrome c into the cytoplasm. Hexokinase is also known to compete with the proapoptotic Bax protein in binding to mitochondria; this would further suppress Bax-induced cytochrome c release and promote cell survival (50, 56). Further, being the first enzyme in glycolysis, higher levels of hexokinase could be an adaptation to maintain efficient ATP transfer for glucose metabolism to fuel the increased energy demands of virus-infected cells.
In this work, we have demonstrated a relationship between expression of the HEV Orf3 protein and the ability of the cell to cope with apoptotic stress. Results from intrahepatic inoculation of a genomic HEV RNA in monkeys showed orf3 to be important for infection and disease progression (37). Similar experiments involving cells in culture show orf3 to be dispensable for the viral life cycle (38). These observations indicate that Orf3 could be a viral accessory protein. A number of viruses are known to encode such proteins to optimize the host cell environment for efficient viral replication (62). For example, HIV-1 encodes at least four accessory proteins that are dispensable for viral replication in cell culture models (63, 64) but are required for establishment of infection, high titer viral replication, and AIDS progression in the host (65).
In the present study, we have delineated a biochemical pathway through which an acute, noncytopathic virus, such as HEV, can protect infected cells from apoptotic death. This is the first report that correlates prosurvival effects of a viral protein to enhanced VDAC and hexokinase expression. An intracellular survival signal thus would shift the balance toward viral replication and gene expression, and this would contribute to pathogenesis.
Acknowledgments
We thank Dr. Michael Forte for the VDAC genes, Dr. Israrul Haq Ansari for the EGFP-cytochrome c plasmid, Rahul Arya for the reverse transcription-PCR analysis, Maitreyi Rajala for the ORF3-HeLa-Tet/OFF cells, Ravinder Kumar for overall cell culture support, and Anindita Kar-Roy for helpful discussions.
Footnotes
- HEV
- hepatitis E virus
- ORF
- open reading frame
- ERK
- extracellular signal-regulated kinase
- VDAC
- voltage-dependent anion channel
- OMM
- outer mitochondrial membrane
- EGFP
- enhanced green fluorescence protein
- TBS
- Tris-buffered saline
- STS
- staurosporine
- MTT
- 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide
- HK
- hexokinase
- siRNA
- small interfering RNA
- JC-1
- 5,5′,6,6′-tetrachloro-1,1′3,3′-tetraethyl benzimidazolcarbocyanine iodide
- cytc
- cytochrome c
M. Rajala and S. Jameel, unpublished observations.
R. Arya and S. Jameel, unpublished observations.
This work was supported by an International Senior Research Fellowship in Biomedical Sciences of the Wellcome Trust (to S. J.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Purcell RH, Ticehurst JR. In: Viral Hepatitis and Liver Disease. Zuckerman AJ, editor. New York: Alan R. Liss; 1988. pp. 131–137. [Google Scholar]
- 2.Ramalingaswami V, Purcell RH. Lancet. 1988;1:571–573. doi: 10.1016/s0140-6736(88)91362-1. [DOI] [PubMed] [Google Scholar]
- 3.Krawczynski K. Hepatology. 1993;17:932–941. [PubMed] [Google Scholar]
- 4.Khuroo MS, Teli MR, Skidmore S, Sofi MA, Khuroo M. Am. J. Med. 1981;70:252–255. doi: 10.1016/0002-9343(81)90758-0. [DOI] [PubMed] [Google Scholar]
- 5.Nayak NC, Panda SK, Datta R, Zuckerman AJ, Guha DK, Madanagopalan N, Buckshee K. J. Gastroenterol. Hepatol. 1989;4:345–352. doi: 10.1111/j.1440-1746.1989.tb00846.x. [DOI] [PubMed] [Google Scholar]
- 6.Emerson SU, Anderson D, Arankalle A, Meng XJ, Purdy M, Schlauder GG, Tsarev SA. In: Virus Taxonomy, VIIIth Report of the ICTV. Fauquet CM, Mayo MA, Maniloff J, Desselberger U, Ball LA, editors. London: Elsevier/Academic Press; 2004. pp. 851–855. [Google Scholar]
- 7.Tam AW, Smith MM, Guerra ME, Huang CC, Bradley DW, Fry KE, Reyes GR. Virology. 1991;185:120–131. doi: 10.1016/0042-6822(91)90760-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jameel S, Zafrullah M, Ozdener MH, Panda SK. J. Virol. 1996;70:207–216. doi: 10.1128/jvi.70.1.207-216.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zafrullah M, Ozdener MH, Panda SK, Jameel S. J. Virol. 1997;71:9045–9053. doi: 10.1128/jvi.71.12.9045-9053.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zafrullah M, Ozdener MH, Kumar R, Panda SK, Jameel S. J. Virol. 1999;73:4074–4082. doi: 10.1128/jvi.73.5.4074-4082.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Graff J, Torian U, Nguyen H, Emerson SU. J. Virol. 2006;80:5919–5926. doi: 10.1128/JVI.00046-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Korkaya H, Jameel S, Gupta D, Tyagi S, Kumar R, Zafrullah M, Mazumdar M, Lal SK, Xiaofang L, Sehgal D, Das SR, Sahal D. J. Biol. Chem. 2001;276:42389–42400. doi: 10.1074/jbc.M101546200. [DOI] [PubMed] [Google Scholar]
- 13.Pawson T. Nature. 1995;373:573–580. doi: 10.1038/373573a0. [DOI] [PubMed] [Google Scholar]
- 14.Cohen GB, Ren R, Baltimore D. Cell. 1995;80:237–248. doi: 10.1016/0092-8674(95)90406-9. [DOI] [PubMed] [Google Scholar]
- 15.Kar-Roy A, Korkaya H, Oberoi R, Lal SK, Jameel S. J. Biol. Chem. 2004;279:28345–28357. doi: 10.1074/jbc.M400457200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Colombini M. J. Membr. Biol. 1989;111:103–111. doi: 10.1007/BF01871775. [DOI] [PubMed] [Google Scholar]
- 17.Bernardi P. Physiol. Rev. 1999;79:1127–1155. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- 18.Halestrap AP, Doran E, Gillespie JP, O’Toole A. Biochem. Soc. Trans. 2000;28:170–177. doi: 10.1042/bst0280170. [DOI] [PubMed] [Google Scholar]
- 19.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 20.Tsujimoto Y, Shimizu S. Biochimie (Paris) 2002;84:187–193. doi: 10.1016/s0300-9084(02)01370-6. [DOI] [PubMed] [Google Scholar]
- 21.Shinohara Y, Ishida T, Hino M, Yamazaki N, Baba Y, Terada H. Eur. J. Biochem. 2000;267:6067–6073. doi: 10.1046/j.1432-1327.2000.01687.x. [DOI] [PubMed] [Google Scholar]
- 22.Arora KK, Pedersen PL. J. Biol. Chem. 1988;263:17422–17428. [PubMed] [Google Scholar]
- 23.Pedersen PL, Mathupala S, Rempel A, Geschwind JF, Ko YH. Biochim. Biophys. Acta. 2002;1555:14–20. doi: 10.1016/s0005-2728(02)00248-7. [DOI] [PubMed] [Google Scholar]
- 24.Rempel A, Bannasch P, Mayer D. Biochim. Biophys. Acta. 1994;1219:660–668. doi: 10.1016/0167-4781(94)90225-9. [DOI] [PubMed] [Google Scholar]
- 25.Sebastian S, Kenkare UW. Biochem. Biophys. Res. Commun. 1997;235:389–393. doi: 10.1006/bbrc.1997.6797. [DOI] [PubMed] [Google Scholar]
- 26.Azoulay-Zohar H, Israelson A, Abu-Hamad S, Shoshan-Barmatz V. Biochem. J. 2004;377:347–355. doi: 10.1042/BJ20031465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zalk R, Israelson A, Garty ES, Azoulay-Zohar H, Shoshan-Barmatz V. Biochem. J. 2005;386:73–83. doi: 10.1042/BJ20041356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mishra RN, Ramesha A, Kaul T, Nair S, Sopory SK, Reddy MK. Anal. Biochem. 2005;345:149–157. doi: 10.1016/j.ab.2005.07.029. [DOI] [PubMed] [Google Scholar]
- 29.Mosmann T. J. Immunol. Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 30.Carmichael J, DeGraff WG, Gazdar AF, Minna JD, Mitchell JB. Cancer Res. 1987;47:936–942. [PubMed] [Google Scholar]
- 31.Vistica DT, Skehan P, Scudiero D, Monks A, Pittman A, Boyd MR. Cancer Res. 1991;51:2515–2520. [PubMed] [Google Scholar]
- 32.Cossarizza A, Baccarani-Contri M, Kalashnikova G, Franceschi C. Biochem. Biophys. Res. Commun. 1993;197:40–45. doi: 10.1006/bbrc.1993.2438. [DOI] [PubMed] [Google Scholar]
- 33.Abu-Hamad S, Sivan S, Shoshan-Barmatz V. Proc. Natl. Acad. Sci. U. S. A. 2006;103:5787–5792. doi: 10.1073/pnas.0600103103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilson JE. Rev. Physiol. Biochem. Pharmacol. 1995;126:65–198. doi: 10.1007/BFb0049776. [DOI] [PubMed] [Google Scholar]
- 35.Pastorino JG, Hoek JB. Curr. Med. Chem. 2003;10:1535–1551. doi: 10.2174/0929867033457269. [DOI] [PubMed] [Google Scholar]
- 36.Tyagi S, Surjit M, Roy AK, Jameel S, Lal SK. J. Biol. Chem. 2004;279:29308–29319. doi: 10.1074/jbc.M402017200. [DOI] [PubMed] [Google Scholar]
- 37.Graff J, Nguyen H, Yu C, Elkins WR, St Claire M, Purcell RH, Emerson SU. J. Virol. 2005;79:6680–6689. doi: 10.1128/JVI.79.11.6680-6689.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Emerson SU, Nguyen H, Torian U, Purcell RH. J. Virol. 2006;80:10457–10464. doi: 10.1128/JVI.00892-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang YW, Opriessnig T, Halbur PG, Meng XJ. J. Virol. 2007;81:3018–3026. doi: 10.1128/JVI.02259-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brown GC. Science. 2003;299:838–839. doi: 10.1126/science.1082028. [DOI] [PubMed] [Google Scholar]
- 41.Skulachev VP. FEBS Lett. 1996;397:7–10. doi: 10.1016/0014-5793(96)00989-1. [DOI] [PubMed] [Google Scholar]
- 42.Vander Heiden MG, Chandel NS, Williamson EK, Schumacker PT, Thompson CB. Cell. 1997;91:627–637. doi: 10.1016/s0092-8674(00)80450-x. [DOI] [PubMed] [Google Scholar]
- 43.Martinou JC, Desagher S, Antonsson B. Nat. Cell Biol. 2000:E41–E43. doi: 10.1038/35004069. [DOI] [PubMed] [Google Scholar]
- 44.Doran E, Halestrap AP. Biochem. J. 2000;348:343–350. [PMC free article] [PubMed] [Google Scholar]
- 45.Schlesinger PH, Gross A, Yin XM, Yamamoto K, Saito M, Waksman G, Korsmeyer SJ. Proc. Natl. Acad. Sci. U. S. A. 1997;94:11357–11362. doi: 10.1073/pnas.94.21.11357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Antonsson B, Conti F, Ciavatta A, Montessuit S, Lewis S, Martinou I, Bernasconi L, Bernard A, Mermod JJ, Mazzei G, Maundrell K, Gambale F, Sadoul R, Martinou JC. Science. 1997;277:370–372. doi: 10.1126/science.277.5324.370. [DOI] [PubMed] [Google Scholar]
- 47.Zoratti M, Szabo I. Biochim. Biophys. Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]
- 48.Ferri KF, Jacotot E, Blanco J, Este JA, Zamzami N, Susin SA, Xie Z, Brothers G, Reed JC, Penninger JM, Kroemer G. J. Exp. Med. 2000;192:1081–1092. doi: 10.1084/jem.192.8.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aflalo C, Azoulay H. J. Bioenerg. Biomembr. 1998;30:245–255. doi: 10.1023/a:1020544803475. [DOI] [PubMed] [Google Scholar]
- 50.Pastorino JG, Shulga N, Hoek JB. J. Biol. Chem. 2002;277:7610–7618. doi: 10.1074/jbc.M109950200. [DOI] [PubMed] [Google Scholar]
- 51.Xie GC, Wilson JE. Arch. Biochem. Biophys. 1988;267:803–810. doi: 10.1016/0003-9861(88)90090-2. [DOI] [PubMed] [Google Scholar]
- 52.Xie G, Wilson JE. Arch. Biochem. Biophys. 1990;276:285–293. doi: 10.1016/0003-9861(90)90040-6. [DOI] [PubMed] [Google Scholar]
- 53.Azoulay-Zohar H, Aflalo C. Eur. J. Biochem. 2000;267:2973–2980. doi: 10.1046/j.1432-1033.2000.01313.x. [DOI] [PubMed] [Google Scholar]
- 54.Beutner G, Ruck A, Riede B, Brdiczka D. Biochim. Biophys. Acta. 1998;1368:7–18. doi: 10.1016/s0005-2736(97)00175-2. [DOI] [PubMed] [Google Scholar]
- 55.Boya P, Roques B, Kroemer G. EMBO J. 2001;20:4325–4331. doi: 10.1093/emboj/20.16.4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bryson JM, Coy PE, Gottlob K, Hay N, Robey RB. J. Biol. Chem. 2002;277:11392–11400. doi: 10.1074/jbc.M110927200. [DOI] [PubMed] [Google Scholar]
- 57.Tschopp J, Thome M, Hofmann K, Meinl E. Curr. Opin. Genet. Dev. 1998;8:82–87. doi: 10.1016/s0959-437x(98)80066-x. [DOI] [PubMed] [Google Scholar]
- 58.Boya P, Roumier T, Andreau K, Gonzalez-Polo RA, Zamzami N, Castedo M, Kroemer G. Biochem. Biophys. Res. Commun. 2003;304:575–581. doi: 10.1016/s0006-291x(03)00630-2. [DOI] [PubMed] [Google Scholar]
- 59.Boya P, Pauleau AL, Poncet D, Gonzalez-Polo RA, Zamzami N, Kroemer G. Biochim. Biophys. Acta. 2004;1659:178–189. doi: 10.1016/j.bbabio.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 60.Rahmani Z, Huh KW, Lasher R, Siddiqui A. J. Virol. 2000;74:2840–2846. doi: 10.1128/jvi.74.6.2840-2846.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Downward J. Nature. 2003;424:896–897. doi: 10.1038/424896a. [DOI] [PubMed] [Google Scholar]
- 62.Agrawal N, Korkaya H, Jameel S. Curr. Sci. 2000;79:711–724. [Google Scholar]
- 63.Steffens CM, Hope TJ. AIDS. 2001;15(Suppl 5):21–26. doi: 10.1097/00002030-200100005-00004. [DOI] [PubMed] [Google Scholar]
- 64.Bour S, Strebel K. Adv. Pharmacol. 2000;48:75–120. doi: 10.1016/s1054-3589(00)48004-x. [DOI] [PubMed] [Google Scholar]
- 65.Rucker E, Grivel JC, Munch J, Kirchhoff F, Margolis L. J. Virol. 2004;78:12689–12693. doi: 10.1128/JVI.78.22.12689-12693.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]