

Abstract
Background
Hypertrophic (HCM) and dilated (DCM) cardiomyopathies result from sarcomeric protein mutations, including cardiac troponin T (cTnT, TNNT2). We determined whether TNNT2 mutations cause cardiomyopathies by altering cTnT function or quantity; whether the severity of DCM is related to the ratio of mutant to wildtype cTnT; whether Ca2+ desensitization occurs in DCM; and whether absence of cTnT impairs early embryonic cardiogenesis.
Methods and Findings
We ablated Tnnt2 to produce heterozygous Tnnt2 +/− mice, and crossbreeding produced homozygous null Tnnt2 −/− embryos. We also generated transgenic mice overexpressing wildtype (TGWT) or DCM mutant (TGK210Δ) Tnnt2. Crossbreeding produced mice lacking one allele of Tnnt2, but carrying wildtype (Tnnt2 +/−/TGWT) or mutant (Tnnt2 +/−/TGK210Δ) transgenes. Tnnt2 +/− mice relative to wildtype had significantly reduced transcript (0.82±0.06[SD] vs. 1.00±0.12 arbitrary units; p = 0.025), but not protein (1.01±0.20 vs. 1.00±0.13 arbitrary units; p = 0.44). Tnnt2 +/− mice had normal hearts (histology, mass, left ventricular end diastolic diameter [LVEDD], fractional shortening [FS]). Moreover, whereas Tnnt2 +/−/TGK210Δ mice had severe DCM, TGK210Δ mice had only mild DCM (FS 18±4 vs. 29±7%; p<0.01). The difference in severity of DCM may be attributable to a greater ratio of mutant to wildtype Tnnt2 transcript in Tnnt2 +/−/TGK210Δ relative to TGK210Δ mice (2.42±0.08, p = 0.03). Tnnt2 +/−/TGK210Δ muscle showed Ca2+ desensitization (pCa50 = 5.34±0.08 vs. 5.58±0.03 at sarcomere length 1.9 µm, p<0.01), but no difference in maximum force generation. Day 9.5 Tnnt2 −/− embryos had normally looped hearts, but thin ventricular walls, large pericardial effusions, noncontractile hearts, and severely disorganized sarcomeres.
Conclusions
Absence of one Tnnt2 allele leads to a mild deficit in transcript but not protein, leading to a normal cardiac phenotype. DCM results from abnormal function of a mutant protein, which is associated with myocyte Ca2+ desensitization. The severity of DCM depends on the ratio of mutant to wildtype Tnnt2 transcript. cTnT is essential for sarcomere formation, but normal embryonic heart looping occurs without contractile activity.
Introduction
The long-term response of the heart to pathological stimuli is composed of maladaptive remodeling characterized by hypertrophy and/or dilation, leading to heart failure. Individuals in the United States are at a 20% lifetime risk of heart failure, which is the most common cause of death. Cardiomyopathies are primary disorders of the myocardium resulting from heritable mutations in single genes. Familial hypertrophic (HCM) and dilated cardiomyopathy (DCM) are among the most common inherited cardiovascular disorders, with prevalences of 200 and 36.5/100,000, respectively [1]. At least 70% of HCM is caused by mutations in sarcomeric protein genes. The cardiac troponin T protein (cTnT), encoded by the gene TNNT2, is a component of the troponin complex which allows actomyosin interaction and contraction to occur in response to Ca2+. Although TNNT2 is commonly mutated in HCM, surprisingly, it has been found that distinct TNNT2 mutations also lead to DCM [2].
Cardiomyopathies secondary to TNNT2 mutations are inherited as autosomal dominant traits. In autosomal dominant diseases, only one of two alleles of the responsible gene is mutant. In some instances, mutations produce disease by inactivating an allele and reducing the quantity of functional protein (haploinsufficiency). However, in other instances, mutations create a mutant protein (“poison peptide”) which interferes with normal function or assumes a new function. A few mouse models have been reported with ablations of other genes encoding sarcomeric proteins mutated in HCM. In the heterozygous state, in which one allele remains intact, no phenotype abnormalities have been noted for α tropomyosin [3], [4], cardiac myosin binding protein C [5], and cardiac troponin I [6]. These observations suggest that haploinsufficiency of these genes does not lead to HCM. However, no ablations have been studied of a gene mutated with a significant frequency in both HCM and DCM. The differing phenotypes of HCM and DCM resulting from TNNT2 mutations suggests that divergent mechanisms lead from different mutations to either phenotype. To address whether haploinsufficiency of TNNT2 is partially or completely responsible for either HCM or DCM, we ablated Tnnt2 in a mouse model by gene targeting.
Since haploinsufficiency is unlikely to lead to both HCM and DCM, it is probable that abnormal function of a mutated cTnT contributes to at least one phenotype. To assess possible “poison peptide” in vivo effects of the human Lys210 deletion (K210Δ) DCM mutation in TNNT2, we generated transgenic mice with cardiac overexpression of mutant or wildtype cTnT. Moreover, we determined the effect of the relative abundance of mutant cTnT on the severity of DCM. Although previous investigations have been inconsistent, in vitro studies have suggested that HCM mutations in several genes lead to an increase in Ca2+ sensitivity, increases in tension generation, and/or increases in ATPase activity [7]–[12], whereas DCM mutations are associated with Ca2+ desensitization and/or decreased ATPase activity in vitro or in permeabilized rabbit cardiac muscle fibers and isolated myocytes [13]–[18].
In this study, heterozygous mice lacking one allele of Tnnt2 (Tnnt2 +/−) had a mild deficit in transcript, no detectable deficit in protein, and no detectable phenotype abnormalities. In contrast, homozygous null embryos (Tnnt2 −/−) had disorganized sarcomeres and noncontractile hearts leading to death by embryonic day 10.5. Despite the absence of contractile activity, normal cardiac looping occurred in these embryos. Moreover, when a transgene with the human DCM TNNT2 K210Δ mutation was introduced into Tnnt2 +/− mice (Tnnt2 +/−/TGK210Δ), they developed DCM and their papillary muscle fibers showed Ca2+ desensitization. TGK210Δ mice, with two endogenous alleles of Tnnt2 intact, had a lower ratio of mutant to wildtype transcript, and a milder phenotype. These results suggest that TNNT2 mutations lead to cardiomyopathies because of abnormally functioning mutant cTnT rather than haploinsufficiency of the protein. Moreover, the severity of DCM is correlated with the ratio of mutant to wildtype Tnnt2 transcript. Although cTnT is essential for sarcomere formation and contraction, normal looping of the embryonic heart occurs in the absence of contractile activity.
Materials and Methods
All studies were approved by the University of Pittsburgh Institutional Animal Care and Use Committee (IACUC). All mice were studied at 8–10 weeks of age unless otherwise indicated.
Generation of Tnnt2 ablated mice
A murine genomic segment containing the Tnnt2 gene was subcloned from the CitbCJ7 BAC library, clone 353I2 (Invitrogen). A loxP-flanked (floxed) neomycin resistance gene driven by the PGK promoter was inserted in place of the 3′ segment of Tnnt2, including exon 14, with a thymidine kinase gene downstream of the genomic segment (Figure 1) [19]. The construct was electroporated into 129/SvEv strain TC1 ES cells (a kind gift from Philip Leder, M.D., Harvard Medical School). Targeted ES cells were selected with Genetecin/G418 (Invitrogen) and FIAU (Moravek), and homologous recombination confirmed by Southern blotting. ES cells were microinjected into mouse blastocysts to generate chimeras, which were then bred to produce heterozygotes with Tnnt2 ablation (Tnnt2 +/−). Genotypes were confirmed by Southern blotting and multiplex PCR using three primers to amplify the wildtype and mutant alleles (primers F1, 5′-ATGACAACCAGAAAGTGTGAGTGT-3′; R1, 5′-GAGTTGGACAGATACAAGGGTCTT-3′; R3, 5′-CTGGACGTAAACTCCTCTTCAGAC-3′). With Tnnt2 ablation, primers F1 and R3 gave a product size of 260 bp; in the presence of wildtype genomic sequence, F1 and R1 gave a product size of 617 bp. The floxed neomycin cassette was excised by mating with transgenic EIIa-Cre recombinase mice (129/SvEv background). Genotyping was performed by multiplex PCR using three primers designed to amplify the wildtype and mutant alleles (primers F1; F2, 5′-GCTGTATTTCACATCCAAACCATA-3′; R2, 5′-TCCTGGTGACTGATGATAATAACG-3′). With the neomycin cassette excised, primers F1 and R2 gave a product size of 324 bp; in the presence of wildtype genomic sequence, F2 and R2 gave a product size of 506 bp.
Figure 1. Generation of Tnnt2 +/ − mice.

A. A targeting construct containing a neomycin resistance gene (neo/zeo) between loxP sites (triangles) was introduced into the murine Tnnt2 locus by homologous recombination in murine ES cells, ablating the 3′ segment of the gene, including exon 14. ES cells were microinjected into mouse blastocysts to generate chimeras, which were bred with wildtype mice for germline transmission of the Tnnt2 ablation. The neomycin resistance gene was excised using Cre-mediated excision by mating with EIIa-Cre recombinase mice. Horizontal arrows indicate PCR primers (F1, F2, R1, R2, R3) used for genotyping as described in “Materials and Methods.” B. Genomic DNA from mice prior to Cre-mediated excision of the neomycin resistance gene was digested with Hind III and Southern blotted with the probe indicated in panel A to demonstrate homologous recombination. A Hind III restriction site in the neomycin resistance gene produced a smaller restriction product in the presence of homologous recombination. WT, wildtype; TK, thymidine kinase gene.
Generation of Tnnt2 transgenic mice
The Tnnt2 cDNA was amplified from murine cardiac RNA by reverse transcription-polymerase chain reaction (RT-PCR) (Qiagen) with primers F3, 5′- AGACCTGTGTCGACTCCCTGTTCAGAGGGAGAGCCGAGAG-3′, and R4, 5′- AAACAGGAGTAAGCTTTGGGTGCCAAGGAGGACCCAGAGC-3′, and then subjected to PCR mutagenesis to delete nucleotides AAG at positions 628–630, encoding the lysine deletion at codon 210 (K210Δ). The wildtype and K210Δ Tnnt2 cDNA were inserted into a pC126 expression vector, containing a highly active cardiac myocyte specific αMHC promoter, a human growth hormone 3′ untranslated region (UTR), and a polyadenylation terminator (a kind gift from Jeffrey Robbins, Ph.D., Cincinnati Children's Hospital) [20]. The plasmids were linearized, size fractionated, purified (QIAquick Gel Extraction Kit, Qiagen), and microinjected into fertilized FVB mouse oocytes. Presence of the wildtype (TGWT) and K210Δ Tnnt2 (TGK210Δ) transgenes was confirmed by Southern blotting and PCR with primers F4, 5′-CTGAGACAGAGGAGGCCAAC-3′, and R5, 5′-CAGCCTCCAGGTTGTGAATA-3′. Five TGK210Δ and four TGWT founder lines were generated, and lines of each genotype had similar phenotypes. One representative mutant and wildtype transgenic line was backcrossed for at least ten generations into a 129/SvEv background to generate TGK210Δ, TGWT, Tnnt2 +/−/TGK210Δ and Tnnt2 +/−/TGWT mice in a uniform genetic background.
RNA analyses
Hearts were harvested immediately after sacrifice of the mouse, washed in PBS, and flash frozen in liquid nitrogen. RNA was isolated from cardiac tissue by lysis in ice cold Trizol (Invitrogen) and chloroform extraction and treated with 10 U DNase I (Roche) / 10 µg at 37°C for 10 min. RNA quantity was determined by OD measurement at 260 nm. Northern blots were performed with 2 µg RNA per gel lane using the following antisense biotinylated riboprobes (Strip-EZ RNA Kit, Ambion): the Tnnt2 coding sequence, corresponding to nucleotides 169–630 of the published sequence (NM_011619); the Tnnt2 3′ UTR (present only in endogenous gene transcript), corresponding to nucleotides 909–1111 of the published sequence (NM_011619); the human growth hormone 3′ UTR (present only in transgene transcript); or the GAPDH coding sequence. Reverse transcription-PCR was performed and the product sequenced using the BigDye Terminator v3.1 Cycle Sequencing Kit (ABI).
Realtime quantitative reverse transcription-PCR (QPCR) was performed as described [21]. Reverse transcription was carried out with the SuperScript First-Strand Synthesis System as recommended by the manufacturer (Invitrogen). The cDNA was then used as template for QPCR with specific primers on an ABI Prism 7000 Sequence Detection System (Applied Biosystems). Primers amplifying the housekeeping gene cyclophilin were used as a control. Hot-start PCR was performed with the SYBR Green PCR Master Mix (Applied Biosystems). The PCR mixtures were pre-heated at 50°C for 2 min and then at 95°C for 10 min to activate the AmpliTaq Gold DNA polymerase, followed by 40 cycles of amplification (95°C for 15 s; 60°C for 1 min). A final extension step was performed at 60°C for 10 min. Primers were tested on cDNA, reverse transcriptase-negative samples, and 0.1% diethyl pyrocarbonate-treated water to exclude amplification of genomic DNA and primer-primer interactions. Equivalence and efficiency were tested by amplifications on serial dilutions of RNA. Quantification was performed using the comparative Ct method (2−ΔΔCt).
Protein analyses
Cardiac tissue was homogenized in 20 volumes of protein extraction buffer (in mM, 50 Tris at pH 8.0, 200 NaCl, 20 NaF, 20 β-glycerolphosphate, 1 DTT, with 0.5% NP40, 1 protease inhibitor tablet / 7 ml buffer, and 1 phosphatase inhibitor tablet / 10 ml buffer [Roche]). The homogenate was allowed to settle on ice for 10 min, and then centrifuged at 10,000 × g for 10 min. The supernatant was stored at -80°C for protein studies. All protein quantities in the proposed studies were measured by the Bradford method (Bio-Rad). Immunoblots were performed a using 10–20 µg of protein per lane loaded onto an appropriate concentration protein gel (Pierce) and subjected to electrophoresis at 120 V for 45 min. Samples were transferred from the gel to a PVDF membrane. The membrane was blocked and incubated at 4°C overnight with primary antibody recognizing cTnT, troponin C (TnC), troponin I (cTnI), actin, tropomyosin, β-myosin heavy chain (MHC), or GAPDH (Santa Cruz Biotechnology, # sc-8121, #sc-20642, #sc-15368, #sc-1615, #sc-18174, #sc-20641, and RDI Research Diagnostics # RDI-TRK5G4-6C5, respectively) at 1∶200 dilution, followed by the appropriate horseradish peroxidase-conjugated secondary antibody (Santa Cruz or Amersham) at 1∶5000 dilution at room temperature for 45 min. Proteins were visualized and quantified by enhanced chemiluminescence (Pierce). Signal intensity was normalized to protein loading using GAPDH immunoblots or Coomassie blue gel staining.
Echocardiography
Transthoracic echocardiography was performed on a VisualSonics Vevo 770 machine [22]. Mice were sedated with tribromoethanol (125 mg/kg IP). 2D short-axis images of the left ventricle were obtained. Left ventricular end diastolic (LVEDD) and end systolic (LVESD) chamber dimensions and wall thickness (LVWT) were obtained from M-mode tracings based on measurements averaged from three separate cardiac cycles. Left ventricular fractional shortening (FS) was calculated as (LVEDD-LVESD)/ LVEDD × 100%.
Electrocardiography (ECG) and electrophysiological studies
Continuous telemetry electrocardiograms (ECGs) were recorded from conscious mice and analyzed as described [23]. Mice were anesthetized with tribromoethanol, wireless transmitters (Data Sciences International) implanted subcutaneously, and leads tunneled subcutaneously to the right shoulder and the left subcostal areas to simulate a lead II surface ECG. For monitoring, mice were housed in cages over receiver plates connected to a computer where digitized signals were stored. Monitoring was performed for ≥24 hours. Data were analyzed for heart rate, cardiac cycle intervals, and spontaneous atrial or ventricular arrhythmia occurrence.
In vivo electrophysiological studies were performed as described [24]. An ECG was obtained with subcutaneous limb needles. An octapolar catheter (NuMed) was advanced through the right external jugular vein to the right ventricular apex. Baseline cardiac cycle intervals were measured including RR, PR, QRS, QT, AH, and HV intervals. Sinus node function was evaluated by measuring sinus node recovery time (SNRT) after pacing the right atrium at a cycle length of 100 ms for 60 s, and correcting for baseline heart rate (SNRTc = SNRT−RR) (Bloom stimulator, Fisher). Atrioventricular (AV) and VA Wenckebach and 2∶1 cycle lengths were determined. Programmed atrial and ventricular stimulation was performed by delivering a premature stimulus after the eighth stimulus in a drive train. Effective (ERP) and functional refractory periods (FRP) were determined at a cycle length of 100 ms for both AV and VA conduction. Ventricular ERP was determined at a drive cycle length of 100 ms. Programmed ventricular stimulation, consisting of burst pacing at cycle lengths of 100 ms to 50 ms in decrements of 10 ms, and of programmed stimulation with double and triple extrastimuli at a drive cycle length of 100 ms with a coupling interval ≥30 ms, were performed. Inducibility was defined as 10 beats of ventricular tachycardia (VT).
Histopathology
For light microscopy, cardiac tissue was fixed in 4% formaldehyde (Polysciences), mounted in paraffin blocks, and sections stained with hematoxylin and eosin (H&E) or Masson trichrome. For electron microscopy, tissue was fixed with 2.5% gluteraldehyde and 2% paraformaldehyde in 0.11 M cacodylate buffer, postfixed in 1% osmium tetroxide (OsO4), and then 1% uranyl acetate. The fixed tissue was dehydrated in alcohol, rinsed in propylene oxide, and embedded with 1∶1 propylene and Spurr's resin solution. The solution was replaced with 100% Spurr's solution and polymerized at 70°C for 48 hours.
Biomechanical studies
Hearts were excised, placed in ice-cold PBS, and then transferred to relaxing solution (in mM, 40 BES [pH 7.0], 20 KCl, 1 free Mg2+, 5 MgATP, 10 creatine phosphate, 20 EGTA, 1 DTT, 0.01 leupeptin, 0.1 PMSF, 180 ionic strength). Anterior left ventricular papillary muscles were dissected, cut into strips, and skinned overnight in relaxing solution with 1% Triton X-100 at 4°C. Skinned muscle bundles were mounted in a 750 µl bath and were attached to a length controller (model 322B, Aurora Scientific, Toronto, Ontario, Canada) at one end and a force transducer (model 403A, Aurora Scientific, Toronto, Ontario, Canada) at the other end using aluminum T-clips (KEM-MIL-CO, Sunnyvale, CA). Average bundle dimensions were approximately 1 mm long by 180 µm wide. Sarcomere length (SL) was determined using laser diffraction (Spectra-Physics, 10mW HeNe laser, Mountain View, CA) and set at either 1.9 or 2.3 µm. The muscle bundle was then fully activated twice prior to generation of force-pCa curves. Activating solution (pCa 4.33) contained all the ingredients of relaxing solution (pCa 10). Force-pCa data were collected by exposing the skinned fiber to various concentrations of free calcium (pCa range 7.00–4.33) that were generated by mixing relaxing and activating solutions in appropriate proportions.
Normalized force was calculated as the ratio of the measured force at a given pCa and the maximally activated force (i.e., force at pCa = 4.33). Normalized force-pCa data were fitted to a modified Hill equation [25] using a nonlinear regression algorithm (Prism, GraphPad Software, San Diego, CA). Two parameters were estimated from normalized force-pCa data for each fiber: pCa50 (pCa required to produce normalized force of 50%) and Hill coefficient (a measure of the steepness of the normalized force-pCa curve, which characterizes the cooperative phenomena in muscle force generation). All quantitative data are presented in tables as mean ± standard deviation (SD) or mean ± standard error (SE), as indicated. Most of the analysis consisted of comparing two groups. This was accomplished using either Student's t test or chi-squared test. The analysis of force-pCa data consisted of more than two groups: skinned muscle fibers from two types of mice, with measurements made at two sarcomere lengths in each muscle fiber. These data were analyzed using two-way (mouse type and sarcomere length) ANOVA with one repeated measure (sarcomere length). Post hoc pairwise comparisons were made using the Tukey's test.
Embryonic studies
Three to four week old Tnnt2 +/− females were superovulated with pregnant mare serum (5 IU IP), followed two days later by human chorionic gonadotropin (5 IU IP), and then mated with adult Tnnt2 +/− males. Pregnant females were sacrificed and embryos were harvested at 8.5–12.5 days postcoitum (pc). For qualitative studies of contractility, day 9.5 embryos were attached to the experimental chamber on the stage of an Olympus IX71 inverted microscope. The chamber was perfused with 3 ml/min Tyrode solution at 37°C. Cardiac contractile function was indexed by spontaneous or electrically stimulated heartbeats measured with a video edge detector and specialized data acquisition software (SoftEdge Acquisition System and IonWizard, IonOptix).
Data analysis
Data analysis methodologies for biomechanical studies are described above. All other quantitative data are presented in tables as mean ± standard deviation (SD), unless otherwise indicated. Differences between two groups were analyzed by Student's t test or chi-squared test. For comparisons among more than two groups, ANOVA was performed, followed by post hoc Bonferroni correction for multiple comparisons.
Prior presentations of data
This work was presented orally at the 2006 Scientific Sessions of the American Heart Association (November 2006) and the 2007 Keystone Symposium on Molecular Pathways in Cardiac Development and Disease (January 2007).
Results
Tnnt2+/− mice had a mild deficit in transcript and no deficit in protein, with a normal phenotype
Mice lacking one allele of Tnnt2, designated Tnnt2 +/−, were generated by gene targeting (Figure 1). We assessed the effect of loss of one allele on cardiac expression of Tnnt2. Northern blots of cardiac RNA from wildtype and Tnnt2 +/− mice were hybridized to probes complementary to the coding sequence and the 3′ UTR of the transcript (Figure 2A). Signal intensity showed only a mild decrease in Tnnt2 transcript, after correction for loading, with both the coding sequence (wildtype, 1.00±0.12; Tnnt2 +/−, 0.82±0.06; p = 0.029; arbitrary units) and the 3′ UTR probe (wildtype, 1.00±0.13; Tnnt2 +/−, 0.82±0.06; p = 0.025). No bands of unexpected size were observed on northern blots in Tnnt2 +/− mice, indicating that the mutant allele did not give rise to alternatively spliced transcripts and was in effect a null allele. This deficit in transcript was confirmed by QPCR, which showed that Tnnt2 +/− hearts had 0.63±0.01 transcript relative to wildtype (p<0.0001). At the protein level, no detectable deficit in cTnT was observed in Tnnt2 +/− mice. Immunoblots of cardiac protein showed cTnT levels of 1.00±0.13 in wildtype and 1.01±0.20 in Tnnt2 +/− mice (arbitrary units, corrected for total protein loaded, p = 0.44) (Figure 2D).
Figure 2. Tnnt2 gene expression in mouse lines.

A. Northern blots of total cardiac RNA from wildtype (WT) and Tnnt2 heterozygous ablated (Tnnt2 +/−) mice using probes complementary to the coding sequence and the 3′ untranslated region (UTR) of the Tnnt2 transcript, and the GAPDH transcript as a loading control. A mild deficit in Tnnt2 transcript, quantified at 18% by densitometry, was apparent in Tnnt2 +/− mice. B. Northern blots of total cardiac RNA from mice of the indicated genotypes using a probe complementary to the coding sequence of the Tnnt2 transcript, comprising both endogenous and transgene transcript (total); and a probe complementary to the human growth hormone 3′ UTR, specific to the transgene transcript. The intensity of 18S and 28S rRNA bands from ethidium bromide stained agarose gels was used to quantify RNA loading. Significant increases in Tnnt2 transcript were apparent in mice carrying the wildtype (TGWT) or the K210Δ Tnnt2 (TGK210Δ) transgene. C. Reverse transcription-PCR and sequencing of cardiac Tnnt2 mRNA showing deletion of codon AAG encoding lysine at position 210 in hearts from TGK210Δ and Tnnt2 +/−/TGK210Δ, but not wildtype (WT), mice. D. Immunoblots of total protein extracts from the hearts of wildtype (WT) and Tnnt2 +/− mice using antibodies specific for cardiac troponin T (cTnT), troponin C (TnC), troponin I (cTnI), tropomyosin, actin, and α myosin heavy chain (MHC). GAPDH loading control immunoblots are shown corresponding to the membranes used for the immunoblots above. Levels of these sarcomeric proteins were unchanged between genotypes. E. An immunoblot of total protein extracts from the hearts of three wildtype, Tnnt2 +/−, Tnnt2 +/−/TGK210Δ, and Tnnt2 +/−/TGWT mice was performed using an antibody specific for cardiac troponin T (cTnT). cTnT protein levels were unchanged among all genotypes. Electrophoresis of these protein extracts on a polyacrylamide gel, followed by Coomassie blue staining, was used to correct for the relative quantity of protein loaded for the immunoblot.
Consistent with the normal quantity of cTnT observed, no phenotype abnormalities were observed in Tnnt2 +/− mice at age 8–10 weeks. Echocardiography indicated no differences in left ventricular wall thickness (LVWT), left ventricular end diastolic diameter (LVEDD), and fractional shortening (FS) relative to wildtype mice (Table 1). No histopathological abnormalities, including myofibrillar disarray and fibrosis, were present (data not shown). No arrhythmias were observed on continuous ambulatory ECG recordings. Thus, despite the loss of one allele of Tnnt2, Tnnt2 +/− mice had only a mild decrease in transcript, no detectable deficit in protein, and a normal cardiac phenotype.
Table 1. Cardiac morphology and function of mice assessed by echocardiography at nine weeks age.
| Tnnt2 +/+ (WT) | Tnnt2 +/− | TGK210Δ | TGWT | Tnnt2 +/−/TGK210Δ | Tnnt2 +/−/TGWT | |
| No. of mice | 9 | 13 | 7 | 7 | 6 | 8 |
| LVWT (mm) | 0.76±0.10 | 0.77±0.14 | 0.70±0.16 | 0.74±0.07 | 0.67±0.17 | 0.77±0.07 |
| LVEDD (mm) | 3.60±0.27 ║ | 3.62±0.23 ║ | 3.81±0.26 | 3.53±0.31 ║ | 4.21±0.29 *†§# | 3.52±0.30 ║ |
| FS (%) | 37±7 ║ | 37±7 ║ | 29±7 ║# | 39±5 ║ | 18±4 *†‡§# | 41±7 ‡║ |
| HR (bpm) | 422±41 | 374±55 | 390±97 | 398±47 | 408±28 | 397±43 |
LVWT, left ventricular wall thickness at end diastole; LVEDD, left ventricular end diastolic diameter; FS, fractional shortening; HR, heart rate; bpm, beats per minute; WT, wildtype. Significant differences in means for LVEDD and FS were observed between groups by ANOVA (p<0.001). p<0.01 by ANOVA and Bonferroni correction versus *WT, † Tnnt2 +/−, ‡TGK210Δ, §TGWT, ║ Tnnt2 +/−/ TGK210Δ, # Tnnt2 +/−/ TGWT.
Given the close association of cTnT with other components of the sarcomere, we determined whether loss of one allele of Tnnt2 was associated with alterations in these other proteins. Relative to wildtype hearts, no changes in levels of troponin C (TnC), troponin I (cTnI), actin, tropomyosin, or β-myosin heavy chain (MHC) were detected in Tnnt2 +/− hearts by immunoblot (Figure 2D).
TGK210Δ mice had mild DCM
TGK210Δ mice were generated carrying a transgene encoding the Lys210 deletion (K210Δ) in Tnnt2 found in human families with DCM [2]. Similarly, TGWT mice were generated carrying a transgene encoding the wildtype sequence of Tnnt2. The TGK210Δ and TGWT lines studied had greater total Tnnt2 mRNA expression than wildtype mice as assessed by northern blot (Figure 2B). A transgene specific probe showed transgene transcript in only those mice carrying the transgene. Reverse transcription-PCR and sequencing of Tnnt2 mRNA from TGK210Δ hearts showed a deletion of the codon AAG at nucleotide positions 628–630 (K210Δ) (Figure 2C). In the presence of two similar sequences with unequal abundance, direct sequencing is relatively insensitive to the sequence present at lower frequency. Therefore, our failure to identify wildtype sequence by direct sequence in TGK210Δ hearts suggested that a larger proportion of the transcript was mutant. cTnT protein levels remained unchanged in all transgenic lines. Echocardiography at age 9 weeks demonstrated findings consistent with mild DCM in TGK210Δ compared to TGWT mice (Table 1), with trends towards increased LVEDD (3.81±0.26 vs. 3.53±0.31 mm) and decreased FS (29±7 vs. 39±5%) that did not meet statistical significance. No abnormalities were noted on telemetry ECGs, electrophysiological studies, and histological examination (data not shown).
Tnnt2+/−/TGK210Δ mice had severe DCM
When the mutant and wildtype transgenes were introduced into Tnnt2 +/− to generate Tnnt2 +/−/TGK210Δ and Tnnt2 +/−/TGWT mice, the Tnnt2 +/−/TGK210Δ mice recapitulated severe DCM features observed in humans with the K210Δ mutation [2]. As in TGK210Δ mice, reverse transcription-PCR amplification and sequencing of Tnnt2 mRNA from Tnnt2 +/−/TGK210Δ hearts showed only mutant K210Δ sequence (Figure 2C), confirming that a larger proportion of the transcript was mutant. Hearts harvested from Tnnt2 +/−/TGK210Δ mice showed massive dilation (Figure 3). Echocardiography at age 9 weeks showed an increase in LVEDD in Tnnt2 +/−/TGK210Δ relative to Tnnt2 +/−/TGWT mice (4.21±0.29 vs. 3.52±0.30 mm, p<0.01), and a decrease in FS (18±4 vs. 41±7%, p<0.01) (Table 1). Moreover, the phenotype of Tnnt2 +/−/TGK210Δ mice was more severe than that of TGK210Δ mice with significantly greater impairment in contractility (FS, 18±4 vs. 29±7%, p<0.01), along with a trend towards greater left ventricular dilation (LVEDD, 4.21±0.29 vs. 3.81±0.23 mm) that did not meet statistical significance. Electrophysiological studies induced nonsustained ventricular tachycardia at low threshold in two of five Tnnt2 +/−/TGK210Δ mice as contrasted with only one of 10 wildtype mice (p = 0.02) (Figure 4). In addition, Tnnt2 +/−/TGK210Δ mice relative to wildtype mice had a significantly (p<0.05) prolonged QRS duration (24±2 vs. 20±1 ms), HV interval (12±1 vs. 9±1 ms), and QT interval (49±2 vs. 44±2 ms), suggesting conduction delays and repolarization abnormalities.
Figure 3. Hematoxylin and eosin staining of hearts from wildtype and Tnnt2 +/ −/TGK210Δ mice.
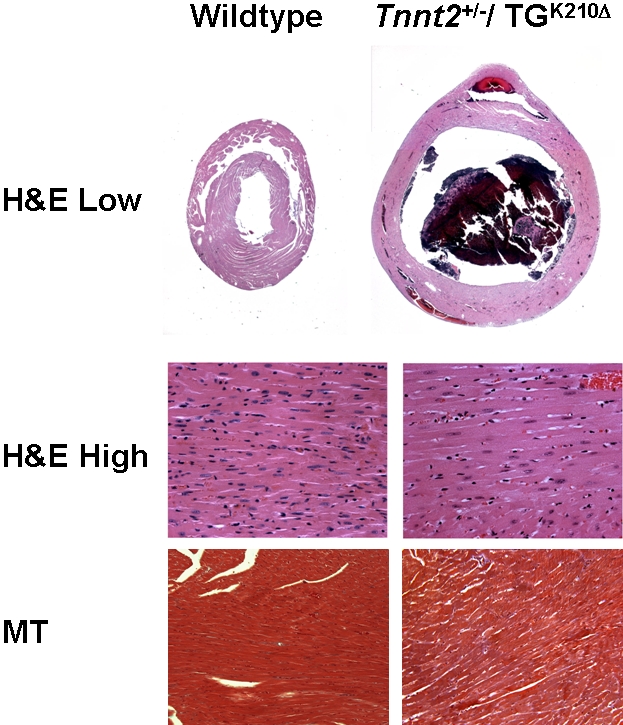
Hematoxylin and eosin (H&E) stained tissue is shown at low (2X) and high (10X) magnification, and Masson's trichrome (MT) stained tissue is shown at high (10X) magnification. Massive dilation of the mutant heart was apparent.
Figure 4. Electrophysiological study in a Tnnt2 +/ −/TGK210Δ mouse.

A surface electrocardiogram (ECG) and an intracardiac electrogram are shown of a Tnnt2 +/−/TGK210Δ mouse which had an inducible arrhythmia. Ventricular tachycardia (VT) was inducible by a drive train at 80 ms cycle length, which self-terminated after approximately four seconds. NSR, normal sinus rhythm.
Molecular changes consistent with heart failure were evident by QPCR at age 50 weeks in Tnnt2 +/−/TGK210Δ relative to Tnnt2 +/−/TGWT mice. Atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), and β-MHC (myosin heavy chain) were all elevated 5.94±1.30, 3.79±1.42, and 9.21±0.68 fold, respectively (p<0.01). Increases in collagen I (2.08±0.03 fold, p<0.01) and matrix metalloproteinase 2 (MMP2) (1.45±0.06 fold, p<0.01) suggested a role for extracellular matrix remodeling in the development of DCM. We observed downregulation of PGC1α 0.13±0.02 (p<0.01) and mitochondrial cytochrome B 0.32±0.03 fold (p<0.05), suggesting impaired mitochondrial biogenesis.
The difference in phenotype severity between TGK210Δ and Tnnt2+/−/TGK210Δ mice correlated with a difference in the proportion of mutant to endogenous wildtype Tnnt2 gene expression
We performed QPCR to determine whether the differences in the severity of the phenotype in TGK210Δ and Tnnt2 +/−/TGK210Δ mice may be related to the relative abundance of mutant Tnnt2 transcript. The relative ratio of mutant to endogenous wildtype transcript was 2.42±0.08 greater in Tnnt2 +/−/TGK210Δ mice relative to TGK210Δ mice (n = 5 each group, p = 0.03), suggesting that the presence of a greater proportion of mutant cTnT in Tnnt2 +/−/TGK210Δ mice leads to a more severe phenotype.
DCM in Tnnt2+/−/TGK210Δ mice is associated with myofiber calcium desensitization
To determine the cellular basis for reduced cardiac contractility, biomechanical studies were performed on skinned papillary muscle fibers from Tnnt2 +/−/TGK210Δ and Tnnt2 +/−/TGWT mice. Tnnt2 +/−/TG K210Δ fibers showed Ca2+ desensitization relative to Tnnt2 +/−/TGWT (pCa50 = 5.34±0.08 vs. 5.58±0.03 at SL = 1.9 µm, p<0.01; 5.46±0.04 vs. 5.71±0.03 at SL = 2.3 µm, p<0.01) (Table 2). The length-dependent increase in Ca2+ sensitivity (Frank-Starling mechanism commonly seen in cardfoiac muscle) was not different between the two groups of mice (ΔpCa50 = 0.13±0.02 for Tnnt2 +/−/TGWT and ΔpCa50 = 0.12±0.05 for Tnnt2 +/−/TGK210Δ, p = 0.86) (Figure 5). Moreover, no difference in maximally activated force was detected (Table 2). There was a tendency for the Hill coefficient to increase in the Tnnt2 +/−/TG K210Δ fibers; however, this increase was not statistically significant. An increase in Hill coefficient would imply increased cooperativity; however, the biological significance of changes in the Hill coefficient in the skinned fiber model remains poorly understood.
Table 2. Characteristics of the force-pCa relationships in skinned papillary muscle fibers.
| pCa50 | Hill Coefficient | Maximal Force (mN mm−2) | pCa50 | Hill Coefficient | Maximal Force (mN mm−2) | |
| Sarcomere Length | 1.9 µm | 1.9 µm | 1.9 µm | 2.3 µm | 2.3 µm | 2.3 µm |
| Tnnt2 +/−/TGWT (n = 10) | 5.58±0.03 | 6.54±0.89 | 45.9±4.6 | 5.71±0.03 | 3.65±0.11 | 62.3±6.5 |
| Tnnt2 +/−/TGK210Δ (n = 7) | 5.34±0.08 | 9.34±2.47 | 34.5±5.3 | 5.46±0.04 | 6.72±1.09 | 63.1±10.9 |
| p | <0.01 | NS | NS | <0.01 | NS | NS |
pCa50, pCa required for generation of 50% of maximal force. n, number of skinned fibers studied, taken from a total of 6 hearts in each group. NS, not significant. Data: mean±SE. p values correspond to the comparison between Tnnt2 +/−/TGWT and Tnnt2 +/−/TGK210Δ fibers at the same sarcomere length.
Figure 5. Normalized force-pCa relationships in Tnnt2 +/ −/TGWT and Tnnt2 +/ −/TGK210Δ skinned papillary muscle fibers.

Normalized force (i.e., ratio of force at a given pCa and maximally activated force at pCa = 4.33) developed at a range of Ca2+ concentrations was assessed at sarcomere lengths 1.9 µm (Tnnt2 +/−/TGWT •, Tnnt2 +/−/TGK210Δ ▪) and 2.3 µm (Tnnt2 +/−/TGWT ○, Tnnt2 +/−/TGK210Δ □). There was a rightward shift of the force-pCa curve in Tnnt2 +/−/TGK210Δ muscle, indicating Ca2+ desensitization. Values are mean±SE (n = 10 for Tnnt2 +/−/TGWT and n = 7 for Tnnt2 +/−/TGK210Δ).
The effect of absence of cTnT on embryonic cardiac development
We studied the consequences of complete loss of Tnnt2 by crossbreeding Tnnt2 +/− males and females to generate Tnnt2 −/− homozygous null embryos. No newborn Tnnt2 −/− pups were observed, suggesting embryonic lethality. Embryos were harvested at 8.5–12.5 days postcoitum (pc). At 11.5–12.5 days pc, 25% of the embryos were dead and in various stages of resorption. All dead embryos which were successfully genotyped were Tnnt2 −/−. All living and grossly normal embryos were wildtype (Tnnt2 +/+) or Tnnt2 +/−. At 9.5 days pc, the genotypes of 76 embryos were 9 Tnnt2 −/− (12%), 46 Tnnt2 +/− (60%), and 21 Tnnt2 +/+ (28%), significantly different from the expected Mendelian ratio (p = 0.03), with an under-representation of Tnnt2 −/− embryos. Thus, some Tnnt2 −/− embryos may be lost even earlier than 9.5 days pc.
A northern blot on pooled RNA from five embryos of each genotype showed the absence of Tnnt2 transcript in Tnnt2 −/− embryos, and a 29% reduction in Tnnt2 transcript in Tnnt2 +/− embryos relative to Tnnt2 +/+ embryos (Figure 6B). Whereas Tnnt2 +/+ and Tnnt2 +/− embryos appeared normal, all Tnnt2 −/− embryos displayed impaired growth and cardiac abnormalities (p = 10−5). Tnnt2 −/− embryos had appropriately looped hearts. However, the ventricular walls appeared much thinner than those of Tnnt2 +/+ and Tnnt2 +/− embryos and large pericardial effusions suggested heart failure (Figure 6A). On electron microscopy, no organized sarcomeres were visible in Tnnt2 −/− hearts. Structures were visible which appeared to be Z bands and possibly disorganized thick filaments. No contractility was observed in any Tnnt2 −/− hearts, either spontaneously or on electrical stimulation.
Figure 6. Morphology of Tnnt2 +/+ (wildtype), Tnnt2 +/ −, and Tnnt2 −/− embryos.

A. Column 1, representative embryos at age 9.5 days postcoitum. Columns 2–3, whole embryos and hearts stained with H&E at low and high power respectively. Columns 4–6, transmission electron microscopy of hearts at increasing magnifications. Whereas Tnnt2 +/− embryos appeared normal, Tnnt2 −/− embryos showed pericardial effusions on gross inspection, thinning of the myocardium on H&E histology, and loss of organized sarcomeres on electron microscopy. B. A northern blot of total pooled RNA from five embryos of each genotype (Tnnt2 +/+, Tnnt2 −/−, and Tnnt2 +/−) hybridized with a probe complementary to the Tnnt2 transcript showed absence of transcript in Tnnt2 −/− embryos and a 29% deficit of transcript in Tnnt2 +/− relative to Tnnt2 +/+ embryos.
Discussion
In this study, we ablated the murine Tnnt2 gene by homologous recombination mediated gene targeting. In the heterozygous state (Tnnt2 +/−), there was a mild decrease in Tnnt2 mRNA, with no detectable decrease in total cTnT protein, and a normal cardiac phenotype. Homozygous null mice (Tnnt2 −/−) were not viable beyond embryonic day 10.5. These embryos demonstrated normal cardiac tube looping, but had thin ventricular walls and apparent heart failure. Electron microscopy of cardiac myocytes showed a lack of organized sarcomeres. When a Tnnt2 cDNA transgene with a human DCM mutation, Lys210 deletion (K210Δ), was introduced into mice retaining two endogenous alleles of Tnnt2 (TGK210Δ), they exhibited mild DCM, trending towards left ventricular dilation and impaired contractility. When this transgene was introduced into heterozygous null Tnnt2 +/− mice lacking one endogenous allele of Tnnt2 (Tnnt2 +/−/TGK210Δ), they displayed findings typical of severe DCM, including significant left ventricular dilation, poor contractility, conduction delays, repolarization abnormalities, and inducible ventricular tachyarrhythmias. Poor contractility was found to correlate with cardiac Ca2+ desensitization in skinned fibers. The difference in severity of DCM in these mice was correlated with the ratio of mutant to wildtype Tnnt2 transcript.
These results confirm and extend previous data suggesting that mutations in genes encoding sarcomeric proteins do not cause cardiomyopathy by means of haploinsufficiency. Since most HCM and DCM mutations in human families are transmitted in an autosomal dominant pattern, affected individuals carry one normal and one mutant allele. Two mechanisms may lead to cardiomyopathy—inactivation of an allele (a null allele), leading to a reduction in transcript and functional protein (haploinsufficiency); or production of a mutant protein (“poison peptide”) which interferes with normal function (dominant negative) or assumes a new function. In the present study, loss of one allele of Tnnt2 did not cause HCM or DCM, suggesting that neither cardiomyopathy results from haploinsufficiency. However, severe DCM developed with the addition of a transgene encoding a DCM mutation, suggesting that the mutant transgene encodes a protein with dominant negative activity.
The ablation of one allele of Tnnt2 in Tnnt2 +/− mice led only to a mild 18–37% deficit in Tnnt2 mRNA, and no detectable decrease in cTnT protein levels. Presumably, the lack of one allele of Tnnt2 is mitigated by increased wildtype allele transcription or decreased degradation of wildtype transcript; similarly, to compensate for the mild deficit in transcript, increased translation and/or decreased protein degradation must be responsible. Moreover, the levels of other sarcomeric proteins closely associated with cTnT, namely troponin C (TnC), troponin I (cTnI), tropomyosin, actin, and α myosin heavy chain (MHC) were unchanged in Tnnt2 +/− mice. In previously reported heterozygous knockout mice lacking single alleles of α-tropomyosin and cardiac myosin binding protein C, mRNA levels were approximately half those of wildtype mice, but protein levels remained unchanged [3]–[5]. Thus, stoichiometric ratios of proteins are tightly regulated in the sarcomere.
The complete absence of cTnT is lethal during embryogenesis. This study is the first to detail the effect of ablation of a sarcomeric protein on mammalian embryonic cardiac development. We have shown that cTnT is critical not only to sarcomere function but also sarcomere assembly and structure. Electron microscopy of Tnnt2 −/−cardiac myocytes demonstrated a complete lack of thin filaments (Figure 6A), although Z bands and thick filaments may possibly be present. Given the lack of organized sarcomeres, it is not surprising that contractile activity was absent in the hearts of Tnnt2 −/− embryos. Null mutations in cardiac troponin T in zebrafish [26] and troponin T in the flight muscle of Drosophila melanogaster [27] similarly cause a lack of organized sarcomeres. In Drosophila, the ablation of myosin heavy chain in the presence of some troponin T mutations restores the normal morphology of the thin filaments and the Z-discs, suggesting that the lack of organized sarcomeres with troponin T mutations alone may be mediated by aberrant actin-myosin interactions.
Whether hemodynamic forces and shear stresses resulting from blood flow are required for normal early cardiac development is controversial. Studies of Na+-Ca2+ exchanger (Ncx1) knock-out embryos, in which some hearts appear not to contract but undergo normal early morphogenesis, suggest that flow is not required. However, a cardiac-specific knock-out [28] and RNA interference [29] of Ncx1 in neonatal cardiac myocytes demonstrated no effect on contraction. Our studies using cTnT, a direct participant in the contractile apparatus, show that early cardiac tube morphogenesis and looping appear to occur normally, in the absence of contractile activity.
When mutant K210Δ cTnT was introduced by means of a transgene, mice developed DCM. Interestingly, the extent of endogenous Tnnt2 transcript correlated with the severity of the phenotype. The mutant transgene led to a milder phenotype when both alleles of the endogenous murine Tnnt2 remained intact, but a more severe phenotype in Tnnt2 +/− mice lacking one allele of endogenous Tnnt2. We have shown that this phenomenon reflects a dose effect of the ratio of mutant to wildtype transcript, so that greater amounts of mutant transcript, and presumably mutant protein, lead to a more severe phenotype. The mutant cTnT protein likely incorporates into the sarcomere along with a smaller amount of wildtype protein and functions abnormally, leading to DCM. Previous studies of other proteins have similarly found that the severity of cardiomyopathy is related to the ratio of mutant to wildtype protein [30]. Interestingly, a similar knock-in murine model of the Tnnt2 K210Δ mutation has just been reported by Du and colleagues [31]. Homozygous mutant knock-in mice were found to have a more severe DCM phenotype than heterozygous knock-in mice, suggesting a gene dosage effect. The mutant to wildtype transcript or protein ratio was not measured in the heterozygous mice. It is interesting that not only the homozygous knock-in mice, but even the heterozygous knock-in mice, reported by Du and colleagues had a more severe phenotype than the TGK210Δ described in this report. It is possible that this difference is related to a greater mutant to wildtype cTnT ratio in the heterozygous knock-in mice than the TGK210Δ mice. Alternatively, strain differences may be responsible for the differences in severity of the phenotype.
The phenotype heterogeneity observed in human cardiomyopathy patients with identical mutations may be related to the ratio of mutant to wildtype transcript and protein, which may be a product of modifier genetic, epigenetic, transcriptional and post-transcriptional processes. However, this hypothesis remains to be tested in human subjects.
The precise mechanisms whereby abnormal cTnT protein leads to DCM will require further study. We have demonstrated Ca2+ desensitization in Tnnt2 +/−/TGK210Δ mice, without any changes in maximally activated force or length-dependent activation characteristics (Frank-Starling mechanism). Although normal maximal force generation was not impaired, it is likely that at physiological intracellular Ca2+ concentrations the observed Ca2+ desensitization leads to decreased force generation in vivo. Ca2+ desensitization has been observed in permeabilized rabbit cardiac muscle fibers and isolated myocytes into which K210Δ or R141W (another DCM mutation) mutant cTnT was introduced [15]–[18]. The knock-in Tnnt2 K210Δ mice reported by Du and colleagues were similarly found to have significantly lower Ca2+ sensitivity in force generation, which was mitigated by a positive inotropic agent, pimobendan, which directly increases myofilament Ca2+ sensitivity [31]. Similar to our findings, these mice retained normal maximal force generation. In mice with DCM mutations in cardiac α-myosin heavy chain (S532P and F764L), we found that contractile function of isolated myocytes was depressed, both mutant myosins exhibited reduced ability to translocate actin but similar force-generating capacities, and actin-activated ATPase activities were reduced [32]. In contrast, some but not all studies have demonstrated an increase in Ca2+ sensitivity resulting in increases in force generation, ATPase activity, and hypercontractility in HCM mutations [1], [33]. These findings suggest a paradigm in which DCM mutations lead to Ca2+ desensitization and impaired motor function, whereas HCM mutations lead to Ca2+ hypersensitization and supernormal motor function.
A potential limitation of this study is that, because the human mutation consists of the deletion of one of four consecutive lysine residues, the mutant and wildtype cTnT proteins could not be differentially quantified in these transgenic mice. Nevertheless, the differences in the relative abundances of mutant and wildtype Tnnt2 transcripts correlated closely with the severity of the phenotype.
Our data lead us to make the following conclusions. Absence of one Tnnt2 allele leads to only a mild deficit in transcript and no detectable deficit in protein, and is associated with a normal phenotype. DCM secondary to the Tnnt2 K210Δ mutation, and likely other sarcomeric protein mutations, results from abnormal function of a mutant protein and is associated with myocyte Ca2+ desensitization in vivo. The severity of DCM is related to the ratio of mutant to wildtype Tnnt2 transcript. Haploinsufficiency of Tnnt2 appears not to be mechanistically related to the development of HCM or DCM. Although cTnT is essential for sarcomere formation and contractility, normal early cardiac morphogenesis and looping of the embryonic heart occurs in the absence of contractile activity.
Acknowledgments
We thank Philip Leder, M.D., Harvard Medical School, for generously providing mouse ES cells; Jeffrey Robbins, Ph.D., Cincinnati Children's Hospital, for generously providing the transgene expression vector; and Dionna Hatch, David Fischer, and Michael Michalec for technical assistance. FA is a Doris Duke Charitable Foundation Clinical Scientist.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This work was supported by an American Heart Association Scientist Development Grant (FA), American Heart Association and Hillgrove Foundation Postdoctoral Fellowships (SKB), the Howard Hughes Medical Institute (FA, DAC, IPM, CES, JGS), and McGinnis Chair Endowment Funds (SGS). We thank Barry London, M.D., Ph.D., Director, Cardiovascular Institute of the University of Pittsburgh, for institutional funds. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Ahmad F, Seidman JG, Seidman CE. The genetic basis for cardiac remodeling. Annu Rev Genomics Hum Genet. 2005;6:185–216. doi: 10.1146/annurev.genom.6.080604.162132. [DOI] [PubMed] [Google Scholar]
- 2.Kamisago M, Sharma SD, DePalma SR, Solomon S, Sharma P, et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N Engl J Med. 2000;343:1688–96. doi: 10.1056/NEJM200012073432304. [DOI] [PubMed] [Google Scholar]
- 3.Blanchard EM, Iizuka K, Christe M, Conner DA, Geisterfer-Lowrance A, et al. Targeted ablation of the murine alpha-tropomyosin gene. Circ Res. 1997;81:1005–10. doi: 10.1161/01.res.81.6.1005. [DOI] [PubMed] [Google Scholar]
- 4.Rethinasamy P, Muthuchamy M, Hewett T, Boivin G, Wolska BM, et al. Molecular and physiological effects of alpha-tropomyosin ablation in the mouse. Circ Res. 1998;82:116–23. doi: 10.1161/01.res.82.1.116. [DOI] [PubMed] [Google Scholar]
- 5.Harris SP, Bartley CR, Hacker TA, McDonald KS, Douglas PS, et al. Hypertrophic cardiomyopathy in cardiac myosin binding protein-C knockout mice. Circ Res. 2002;90:594–601. doi: 10.1161/01.res.0000012222.70819.64. [DOI] [PubMed] [Google Scholar]
- 6.Huang X, Pi Y, Lee KJ, Henkel AS, Gregg RG, et al. Cardiac troponin I gene knockout: a mouse model of myocardial troponin I deficiency. Circ Res. 1999;84:1–8. doi: 10.1161/01.res.84.1.1. [DOI] [PubMed] [Google Scholar]
- 7.Hernandez OM, Szczesna-Cordary D, Knollmann BC, Miller T, Bell M, et al. F110I and R278C troponin T mutations that cause familial hypertrophic cardiomyopathy affect muscle contraction in transgenic mice and reconstituted human cardiac fibers. J Biol Chem. 2005;280:37183–94. doi: 10.1074/jbc.M508114200. [DOI] [PubMed] [Google Scholar]
- 8.Gafurov B, Fredricksen S, Cai A, Brenner B, Chase PB, et al. The Delta 14 mutation of human cardiac troponin T enhances ATPase activity and alters the cooperative binding of S1-ADP to regulated actin. Biochemistry. 2004;43:15276–85. doi: 10.1021/bi048646h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chandra M, Rundell VL, Tardiff JC, Leinwand LA, De Tombe PP, et al. Ca(2+) activation of myofilaments from transgenic mouse hearts expressing R92Q mutant cardiac troponin T. Am J Physiol Heart Circ Physiol. 2001;280:H705–H713. doi: 10.1152/ajpheart.2001.280.2.H705. [DOI] [PubMed] [Google Scholar]
- 10.Montgomery DE, Tardiff JC, Chandra M. Cardiac troponin T mutations: correlation between the type of mutation and the nature of myofilament dysfunction in transgenic mice. J Physiol. 2001;536:583–592. doi: 10.1111/j.1469-7793.2001.0583c.xd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morimoto S, Yanaga F, Minakami R, Ohtsuki I. Ca2+-sensitizing effects of the mutations at Ile-79 and Arg-92 of troponin T in hypertrophic cardiomyopathy. Am J Physiol. 1998;275:C200–7. doi: 10.1152/ajpcell.1998.275.1.C200. [DOI] [PubMed] [Google Scholar]
- 12.Yanaga F, Morimoto S, Ohtsuki I. Ca2+ sensitization and potentiation of the maximum level of myofibrillar ATPase activity caused by mutations of troponin T found in familial hypertrophic cardiomyopathy. J Biol Chem. 1999;274:8806–12. doi: 10.1074/jbc.274.13.8806. [DOI] [PubMed] [Google Scholar]
- 13.Mirza M, Marston S, Willott R, Ashley C, Mogensen J, et al. Dilated cardiomyopathy mutations in three thin filament regulatory proteins result in a common functional phenotype. J Biol Chem. 2005;280:28498–506. doi: 10.1074/jbc.M412281200. [DOI] [PubMed] [Google Scholar]
- 14.Venkatraman G, Gomes AV, Kerrick WG, Potter JD. Characterization of troponin T dilated cardiomyopathy mutations in the fetal troponin isoform. J Biol Chem. 2005;280:17584–92. doi: 10.1074/jbc.M409337200. [DOI] [PubMed] [Google Scholar]
- 15.Morimoto S, Lu QW, Harada K, Takahashi-Yanaga F, Minakami R, et al. Ca(2+)-desensitizing effect of a deletion mutation Delta K210 in cardiac troponin T that causes familial dilated cardiomyopathy. Proc.Natl.Acad.Sci.U.S.A. 2002;99:913–918. doi: 10.1073/pnas.022628899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu QW, Morimoto S, Harada K, Du CK, Takahashi-Yanaga F, et al. Cardiac troponin T mutation R141W found in dilated cardiomyopathy stabilizes the troponin T-tropomyosin interaction and causes a Ca2+ desensitization. J Mol Cell Cardiol. 2003;35:1421–7. doi: 10.1016/j.yjmcc.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 17.Robinson P, Mirza M, Knott A, Abdulrazzak H, Willott R, et al. Alterations in thin filament regulation induced by a human cardiac troponin T mutant that causes dilated cardiomyopathy are distinct from those induced by troponin T mutants that cause hypertrophic cardiomyopathy. J Biol Chem. 2002;277:40710–6. doi: 10.1074/jbc.M203446200. [DOI] [PubMed] [Google Scholar]
- 18.Venkatraman G, Harada K, Gomes AV, Kerrick WG, Potter JD. Different functional properties of troponin T mutants that cause dilated cardiomyopathy. J Biol Chem. 2003;278:41670–6. doi: 10.1074/jbc.M302148200. [DOI] [PubMed] [Google Scholar]
- 19.Bruneau BG, Nemer G, Schmitt JP, Charron F, Robitaille L, et al. A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell. 2001;106:709–21. doi: 10.1016/s0092-8674(01)00493-7. [DOI] [PubMed] [Google Scholar]
- 20.Gulick J, Subramaniam A, Neumann J, Robbins J. Isolation and characterization of the mouse cardiac myosin heavy chain genes. J Biol Chem. 1991;266:9180–5. [PubMed] [Google Scholar]
- 21.Ramani R, Mathier M, Wang P, Gibson G, Togel S, et al. Inhibition of tumor necrosis factor receptor-1-mediated pathways has beneficial effects in a murine model of postischemic remodeling. Am J Physiol Heart Circ Physiol. 2004;287:H1369–77. doi: 10.1152/ajpheart.00641.2003. [DOI] [PubMed] [Google Scholar]
- 22.Ahmad F, Arad M, Musi N, He H, Wolf C, et al. Increased alpha2 subunit-associated AMPK activity and PRKAG2 cardiomyopathy. Circulation. 2005;112:3140–3148. doi: 10.1161/CIRCULATIONAHA.105.550806. [DOI] [PubMed] [Google Scholar]
- 23.Chen HH, Baty CJ, Maeda T, Brooks S, Baker LC, et al. Transcription enhancer factor-1-related factor-transgenic mice develop cardiac conduction defects associated with altered connexin phosphorylation. Circulation. 2004;110:2980–7. doi: 10.1161/01.CIR.0000146902.84099.26. [DOI] [PubMed] [Google Scholar]
- 24.Saba S, Janczewski AM, Baker LC, Shusterman V, Gursoy EC, et al. Atrial contractile dysfunction, fibrosis, and arrhythmias in a mouse model of cardiomyopathy secondary to cardiac-specific overexpression of tumor necrosis factor-{alpha}. Am J Physiol Heart Circ Physiol. 2005;289:H1456–67. doi: 10.1152/ajpheart.00733.2004. [DOI] [PubMed] [Google Scholar]
- 25.Adhikari BB, Regnier M, Rivera AJ, Kreutziger KL, Martyn DA. Cardiac length dependence of force and force redevelopment kinetics with altered cross-bridge cycling. Biophys J. 2004;87:1784–94. doi: 10.1529/biophysj.103.039131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sehnert AJ, Huq A, Weinstein BM, Walker C, Fishman M, et al. Cardiac troponin T is essential in sarcomere assembly and cardiac contractility. Nat Genet. 2002;31:106–10. doi: 10.1038/ng875. [DOI] [PubMed] [Google Scholar]
- 27.Fyrberg E, Fyrberg CC, Beall C, Saville DL. Drosophila melanogaster troponin-T mutations engender three distinct syndromes of myofibrillar abnormalities. J Mol Biol. 1990;216:657–75. doi: 10.1016/0022-2836(90)90390-8. [DOI] [PubMed] [Google Scholar]
- 28.Henderson SA, Goldhaber JI, So JM, Han T, Motter C, et al. Functional adult myocardium in the absence of Na+-Ca2+ exchange: cardiac-specific knockout of NCX1. Circ Res. 2004;95:604–11. doi: 10.1161/01.RES.0000142316.08250.68. [DOI] [PubMed] [Google Scholar]
- 29.Hurtado C, Ander BP, Maddaford TG, Lukas A, Hryshko LV, et al. Adenovirally delivered shRNA strongly inhibits Na+-Ca2+ exchanger expression but does not prevent contraction of neonatal cardiomyocytes. J Mol Cell Cardiol. 2005;38:647–54. doi: 10.1016/j.yjmcc.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 30.James J, Zhang Y, Osinska H, Sanbe A, Klevitsky R, et al. Transgenic modeling of a cardiac troponin I mutation linked to familial hypertrophic cardiomyopathy. Circ Res. 2000;87:805–11. doi: 10.1161/01.res.87.9.805. [DOI] [PubMed] [Google Scholar]
- 31.Du CK, Morimoto S, Nishii K, Minakami R, Ohta M, et al. Knock-in mouse model of dilated cardiomyopathy caused by troponin mutation. Circ Res. 2007;101:185–94. doi: 10.1161/CIRCRESAHA.106.146670. [DOI] [PubMed] [Google Scholar]
- 32.Schmitt JP, Debold EP, Ahmad F, Armstrong A, Frederico A, et al. Cardiac myosin missense mutations cause dilated cardiomyopathy in mouse models and depress molecular motor function. Proc Natl Acad Sci U S A. 2006;103:14525–30. doi: 10.1073/pnas.0606383103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sirenko SG, Potter JD, Knollmann BC. Differential effect of troponin T mutations on the inotropic responsiveness of mouse hearts–role of myofilament Ca2+ sensitivity increase. J Physiol. 2006;575:201–13. doi: 10.1113/jphysiol.2006.107557. [DOI] [PMC free article] [PubMed] [Google Scholar]