

Abstract
This study examines in endothelium-denuded bovine pulmonary arteries the effects of increasing heme oxygenase-1 (HO-1) activity on relaxation and soluble guanylate cyclase (sGC) activation by nitric oxide (NO). A 24 hour organ culture with 0.1 mM cobalt chloride (CoCl2) or 30 μM Co-protoporphyrin IX was developed as a method of increasing HO-1 expression. These treatments increased HO-1 expression and HO activity by ~2–4 fold, and lowered heme levels by 40–45%. Induction of HO-1 was associated with an attenuation of pulmonary arterial relaxation to the NO-donor spermine-NONOate. The presence of a HO-1 inhibitor 30 μM chromium mesoporphyrin during the 24 hour organ culture (but, not acute treatment with this agent) reversed the attenuation of relaxation to NO seen in arteries co-cultured with agents that increased HO-1. Relaxation to isoproterenol, which is thought to be mediated through cAMP, was not altered in arteries with increased HO-1. Inducers of HO-1 did not appear to alter basal sGC activity in arterial homogenates or expression of the β1-subunit of sGC. However, the increase in activity seen in the presence of 1 μM spermine-NONOate was attenuated in homogenates obtained from arteries with increased HO-1. Since arteries with increased HO-1 had decreased levels of superoxide detected by the chemiluminescence of 5 μM lucigenin, superoxide did not appear to be mediating the attenuation of relaxation to NO. These data suggest that increasing HO-1 activity depletes heme, and this is associated with an attenuation of pulmonary artery relaxation and sGC activation responses to NO.
Keywords: cGMP, cobalt protoporphyrin, chromium mesoporphyrin, superoxide
Introduction
The activity of the soluble form of guanylate cyclase (sGC) is a key regulator of vascular smooth muscle contractile function and blood flow as a result of its role in controlling the generation of cGMP, and vascular relaxing mechanisms influenced by the function of this second messenger (25). Control of the activity of sGC by nitric oxide (NO) has a major influence on the function of the neonatal and adult pulmonary circulation (15, 35, 39). In addition, alterations in the regulation of sGC by NO is thought to be a major factor influencing vascular function in multiple diseases, including pulmonary hypertension (39). While the role of superoxide in attenuating the regulation of sGC by NO has been extensively studied in multiple vascular diseases (14, 23), other processes could be contributing factors to alterations in the sensitivity of sGC to regulation by NO.
Early studies on how nitric oxide (NO) regulates the soluble form of guanylate cyclase identified heme as an essential cofactor in mediating the stimulation of cGMP formation (8–11, 18–20). These studies detected evidence for the presence of heme-containing and heme-deficient forms of sGC, where heme was easily lost from sGC as the enzyme was purified. Observations that the iron-free biosynthetic precursor to heme, protoporphyrin IX, activated sGC resulted in a hypothesis that when NO bound to the Fe2+ of heme, it stimulated cGMP production as a result of a loss of coordination of the sGC amino acid that normally bound to the iron this heme group (37). This amino acid was subsequently identified as a histidine (6, 32). Thus, the availability of heme could be a factor which controls the responsiveness of sGC to NO and cGMP-mediated relaxation of vascular tissue in response to NO. In addition, recent studies suggest that sGC heme oxidation and loss could be an important factor in aging and multiple vascular disease models (31)
Heme oxygenase (HO) activity is a key regulator of cellular heme levels (2), and the carbon monoxide (CO) product of heme degradation by this enzyme is also known to bind the heme of sGC in a manner which causes a modest stimulation of cGMP generation (6, 32). The induction of HO-1 in cultured vascular smooth muscle cells was observed to cause an increase in cGMP production through a mechanism that appeared to involve CO generation (7). However, a prolonged exposure of sGC to increased levels of HO-1 in cultured rat pulmonary microvascular endothelial cells was associated with a depletion of heme, a loss of CO production and decreased sGC activity, suggesting heme availability was a factor which controlled sGC activity (3). Vascular tissue appears to relax when exposed to micromolar concentrations of CO through mechanisms that seem to involve stimulation of sGC (16). However, inhibition of NO synthase by CO and the vascular actions of NO (21, 22, 33) can also be a contributing factors to the vasoactive actions of increased HO-1 activity. Although it has been reported that the rat pulmonary circulation appears to show a sGC-mediated vasodilation to CO (29), it has also been observed that porcine pulmonary arteries loose their relaxation to CO in a manner associated with postnatal age (36). While multiple disease processes altering vascular regulation by NO are also associated with increased HO-1 expression (22, 33), little is known about the influence of increased heme metabolizing activity of the regulation of sGC by NO. In this study, organ culture of endothelium-removed bovine pulmonary arteries with agents known to increase HO-1 expression, cobalt protoporphyrin (CoPP) and cobalt chloride (CoCl2) (2, 24, 38), was developed as a method to examine the effects of heme depletion on the sensitivity of pulmonary arteries and sGC to the actions of NO. In addition, a competitive inhibitor of heme metabolism by HO-1, chromium mesoporphyrin (CrMP), which does not alter the sGC activation by NO (4), was used to examine the effects of inhibition of the activity of HO-1.
Experimental Procedure
Materials
Spermine-NONOate, and cGMP Enzyme Immunoassay (ELISA) kits were purchased from Cayman Chemical. All gasses were purchased from Tech Air (White Plains, NY). All salts used were Analyzed Reagent Grade from Baker Chemical Company and all other chemicals were obtained from Sigma Chemical Company (St. Louis, MO). CoPP & CrMP are from Alexis Chemicals and Culture Media is from Gibco Technologies.
Organ Culture Modulation of Heme Metabolism in Bovine Pulmonary Arteries
Intralobar pulmonary arteries from slaughterhouse-derived bovine lungs were isolated and cleaned from surrounding tissue as previously described (26, 27). Arteries were placed directly into in 37°C Dulbecco’s Modified Eagle Medium (DMEM) containing 10% FBS, penicillin, streptomycin, fungizone (complete media). The arteries were isolated and cut into rings ~2 mm in diameter and width, and the endothelium was removed by gentle rubbing of the lumen. Arteries were organ cultured in complete media with various drugs described in the Results (ie. CoCl2, CoPP, CrMP, hemin) for 24 hours at 37°C in a cell culture incubator under an atmosphere of 5% CO2.
Studies on the Regulation of Contractile Function
Following the overnight incubation, arterial rings were mounted on wire hooks attached to Grass (FT03) force displacement transducers for measurement of changes in isometric force. Tension was adjusted to 5 grams, which is the optimal passive force for maximal contraction. Changes in force were recorded on a Grass polygraph (model 7), while vessels were incubated in 10-ml baths (Metro Scientific, Farmingdale, NY), which were individually thermostated (37°C) in Krebs buffer gassed with 21% O2 -5% CO2 (balance N2 ). Arteries were incubated for 1h during which passive tension was adjusted to maintain 5 grams. The vessels were then depolarized with Krebs containing 123 mM KCl (High K+) in place of NaCl, followed by re-equilibration with Krebs, prior to exposure to the experimental protocols.
Protocols for Studies on Mechanisms of NO-Mediated Relaxation in CoCl2 experiments
Following the re-equilibration in Krebs buffer after the High K+ treatment for 30 min, arteries were then contracted with serotonin (5-HT) in a dose-dependent manner. After 5-HT was washed from the tissue baths with Krebs and a 30 min re-equilibration, arteries were then contracted with 1 μM 5-HT and subsequently relaxed with increasing cumulative concentrations of the NO-donor spermine-NONOate (10−8-10−5M) once steady-state force was achieved. In some experiments, arteries precontracted with 0.1 mM U46619 were treated with doses of up to 100 μM of the CO donor CORM-2 prior to exposure to the NO-donor.
Measurements of HO-1 Activity and Heme
Organ cultured pulmonary arteries were homogenized in 20 mM MOPS (pH 7.4) buffer containing 0.25 M sucrose for measurements of HO-1 activity employing an adaptation of methods previously described (1). Measurements of HO-1 activity were made in MOPS-Sucrose Buffer containing 2–3 mg/ml of pulmonary artery homogenate protein in the presence of 1 mM glucose-6-phosphate, 0.2 units/ml glucose-6-phosphate dehydrogenase, 0.8 mM NADPH, and 0.025 mg/ml hemin. After 1 hour incubation at 37°C, bilirubin was removed with a chloroform extraction, and its concentrations were measured using the differences in absorbance at wavelengths from 460 to 530 nm with an absorption coefficient of 40 mM−1 cm−1 with a dual beam spectrophotometer. Heme was quantified in the arterial homogenates diluted to approximately 5 mg protein/ml using a QuantiChrom heme assay kit purchased from BioAssay Systems, based on the manufacture’s instructions, and changes in absorbance at 400 nm were measured in a BIOTEK scanning microplate spectrophotometer. Tissue heme levels were reported as nanomoles/mg protein.
Western Blot Analysis of HO-1 and sGC
Rings from contractile studies were snap-frozen in liquid nitrogen, crushed and homogenized in lysis buffer (50 mM Tris–HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, protease cocktail (Sigma) and phosphatase cocktail (Sigma). Protein assay was preformed and samples were prepared in electrophoresis sample buffer and were separated using a 10% gel SDS-PAGE electrophoresis. Membranes were blocked 1 hour in TBST-5% milk and incubated overnight with respective antibodies. Antibodies for human HO-1 were obtained from Stressgen Biotechnologies, and for the β1-subunit of sGC was obtained from Sigma (Product # G 4405). Membranes were exposed to a secondary horseradish peroxidase linked antibody and visualized with an ECL kit (Amersham). The membranes were subsequently exposed to X-OMAT autoradiography paper (Kodak). Protein loading was normalized to actin.
Guanylate Cyclase Activity
Guanylate cyclase activity of bovine pulmonary artery was measured by enzyme immunoassay (13). Briefly, reaction mixtures (0.2 ml final volume) contained 20 mM MOPS-KOH (pH 7.4), 0.1 mM GTP, 2 mM MgCl2, 0.3 mM of the phosphodiesterase inhibitor IBMX, a GTP-regenerating system consisting of 10 mM phosphocreatine and 150 U/ml creatine phosphokinase, and 0.1 ml of diluted (17) homogenate, and test agents, as indicated. Assays of sGC activity were initiated by the addition of arterial protein. Incubations were conducted for 10 min at 37°C, and they were terminated by the addition of 0.1 ml of preheated 12 mM EDTA. This was followed by boiling the assay mixtures for 10 min and by adding a 5% trichloroacetic acid solution. The precipitate was removed by centrifugation and the supernatant was washed three times with water-saturated diethyl ether. Residual ether was removed by incubation at 70°C for 5 minutes, prior to measurement of cGMP by ELISA, as described in the manual supplied by manufacturer of the ELISA kit.
Measurement of superoxide levels
Organ cultured arteries were placed in plastic scintillation minivials containing 5 μM lucigenin for the detection of O2−by chemiluminescence in a final volume of 1 ml of air-equilibrated Krebs solution buffered with 10 mM HEPES-NaOH (pH 7.4) employing previously described methods (12).
Statistical Analysis
Data are reported as mean ± SEM with n equals the number of arterial segments from different animals, and p < 0.05 was used to determine statistical significance. Statistical analyses were performed by using a Student’s t-test or ANOVA with a post hoc Student’s t-test employing a Bonferroni correction to determine significance between experimental groups.
Results
Effects of Organ Culture of BPA with CoPP and CoCl2 on HO-1 and Heme Levels
Organ culture of BPA with inducers of HO-1 increased the protein expression detected by Western analysis and the heme metabolizing activity of this enzyme, and the increase in HO-1 was associated with the detection of decreased arterial levels of heme. As shown in Figure 1A, HO-1 expression was increased to ~300% of levels seen in organ cultured control arteries when endothelium-denuded bovine pulmonary arteries were organ cultured with 30 μM CoPP or 0.1 mM CoCl2 (See Figure 1A). The activity of HO-1 was increased to >200% of control by CoPP and CoCl2 (See Figure 1B). To determine whether HO-1 induction and expression had any effects on cellular heme content, heme levels were measured from BPA segments which were treated overnight with the inducers CoCl2 and CoPP. The data in figure 1C suggest that these compounds, which significantly increase HO-1 activity, similarly decrease BPA heme levels by 40–45% compared to organ cultured control BPA.
Figure 1.

HO-1 (A) expression (normalized to actin) and (B) activity were increased by CoCl2 (A n=16; B n=12) and CoPP (A n=14; B n=12). (C) Measurements of heme (n=9) showed decreased levels in CoCl2 and CoPP treated vessels, indicative of HO-1 activity.
Effects of HO-1 Inducers on BPA Relaxation to a NO Donor and to Isoproterenol
Spermine-NONOate was used in this study as an NO donor and the β-adrenergic receptor agonist of cAMP-mediated relaxation was used as a sGC-independent relaxing agent. The 24 hr organ cultured BPA control arteries showed relatively normal relaxation responses to spermine-NONOate (Figure 2) and to isoproterenol (figure 3) compared to responses of BPA not exposed to organ culture that have been reported in our previous studies (26). As shown in Figure 2, BPA organ cultured with HO-1 inducers had a response to spermine NONOate which was attenuated compared to organ cultured control arteries. For example, relaxation of BPA to 1 μM NONOate was significantly decreased from control levels of 59±4% to 39±5% (p<0.05, n=11) in CoCl2 treated vessels and to 30±9 (p<0.05 vs. Control) in CoPP treated vessels. As shown in Figure 3, relaxation of BPA to isoproterenol was not altered by either of the treatments used for HO-1 induction in organoid treated vessels.
Figure 2.
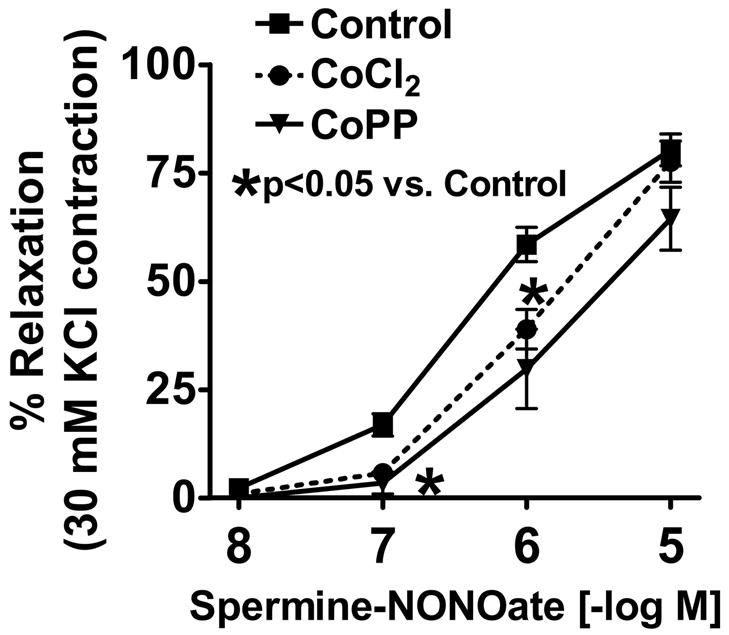
Vascular relaxation of BPA to NONOate was significantly inhibited by organoid treatment with HO-1 inducers CoCl2 and CoPP (p<0.05 vs. Control). n=11
Figure 3.
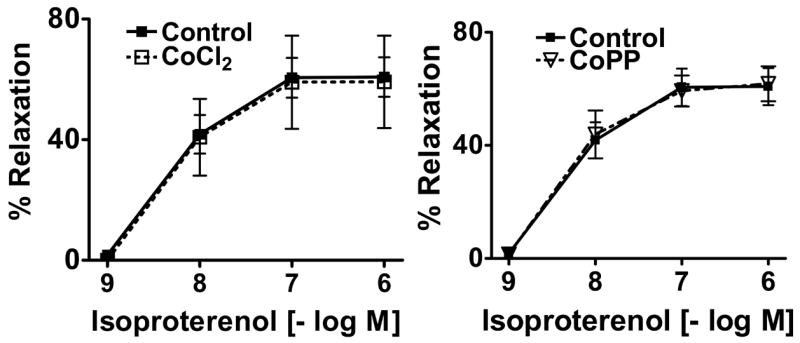
The β-adrenergic receptor agonist isoproterenol induced relaxation was not altered by HO-1 induction. n=4
Effects of Organ Culture and Acute Treatments of BPA with the HO-1 Inhibitor CrMP on Relaxation to NO
Organ culture with the HO-1 inhibitor 30 μM CrMP was used to prevent increased HO-1 activity from influencing vascular function through processes such as depleting heme and regulating gene expression. In addition, a 30 min pretreatment with CrMP was employed to examine to acute vasoactive effects of HO-1 which are potentially associated with its ability to generate CO. As shown in Fig. 4, respectively, the 24 hour or 30 min treatments with CrMP did not alter relaxation to the NO donor spermine NONOate. However, organ culture with CrMP prevented CoCl2 and CoPP from attenuating relaxation to NO. In contrast, acute treatment with CrMP did not alter the ability of inducers of HO-1 from attenuating relaxation to NO. Organ culture with 30 μM hemin (ferriprotoporphyrin IX) to increase cellular heme prevented the attenuating effects of HO-1 induction by CoCl2 on relaxation to NO in a manner similar to CrMP. The relaxation of untreated BPA (n=7–8) precontracted with 1 μM 5-HT to spermine NONOate (e.g., 1 μM = 38±6% relaxation) that was attenuated by 0.1 mM CoCl2 treatment (22±3, p<0.05 vs untreated) was significantly reversed when BPA were organ cultured with CoCl2+hemin (35±5%, p<0.05 vs CoCl2). Hemin did not significantly alter the force generated by 5-HT or relaxation to spermine NONOate (See Table 1 or not shown).
Figure 4.

Relaxation of arteries precontracted with 1 μM serotonin to NONOate was significantly inhibited by CoCl2 and CoPP was reversed by chronic but not acute treatment with HO-1 inhibitor 30 μM CrMP. (n =5–14, p<0.05)
Table 1.
Force generation to 1 μM serotonin (5-HT) was not significantly altered by organ culture with the CoCl2 and CoPP inducers of HO-1 or organ culture (24 hrs) or acute treatment of organ cultured arteries with the HO-1 inhibitor 30 μM CrMP. (n =5–14)
| Condition | Force Generation (g) |
|---|---|
| Control | 8.3±1.0 |
| CrMP (acute) | 7.5±1.5 |
| CrMP (24 hrs) | 7.4±1.2 |
| Hemin (24 hrs) | 9.7±2.0 |
| CoCl2 | 8.5±1.2 |
| CoPP | 6.9±1.4 |
| CoCl2+CrMP (acute) | 9.9±2.2 |
| CoPP+CrMP (acute) | 11.3±1.9 |
| CoCl2+CrMP (24 hrs) | 11.1±1.6 |
| CoPP+CrMP (24 hrs) | 6.4±0.8 |
| CoCl2+Hemin (24 hrs) | 7.1±1.8 |
Effects of Modulators of HO-1 on Contraction to 5-HT
The effects of inducers of HO-1 in the absence and presence of CrMP on contraction to 5-HT was examined to detect if HO-1 was influencing force generation in BPA through mechanisms such as the generation of CO. As shown in the data in Table 1, organ culture in the absence and presence of combinations of with CoPP, CoCl2 and CrMP did not significantly alter force generation to 1 μM 5-HT. In addition, acute treatments of CrMP also did not significantly alter the force generated by 5-HT in organ cultured control or CoPP or CrMP treated BPA (Table 1).
Effects of a CO-Donor on Force Generation by BPA and Relaxation to NO
The actions of tricarbonyldichlororuthenium (II) dimer, a CO-releasing molecule (CORM-2, (28)), as a relaxing agent and its effects on the subsequent response to increasing concentrations of spermine NONOate were examined to determine if CO relaxed BPA or altered relaxation to NO. These studies were examined in the presence of force generated by 0.1 μM U46619, to avoid the time-dependent decay of contractions to 5-HT in the prolonged protocol used in these experiments. Concentrations of the CO donor over the 10 nM –100 μM range did not alter force generation by either 1 μM 5-HT, 0.1 μM U46619 or 30 mM KCl (Fig. 5-left panel or not shown). These BPA were subsequently exposed to increasing concentrations of spermine NONOate. As shown in Figure 5-right panel, the absence or presence of the 100 μM CORM-2 did not alter relaxation to NO.
Figure 5.

The CO-donor CORM-2 (100 μM) does not alter force generation to 0.1 μM U46619 or 30 mM KCl (left) or relaxation to the NONOate donor of NO in arteries precontracted with 30 mM KCl (30K) (right). (n=4)
Effects of Modulators of HO-1 on BPA Levels of Superoxide Detected by Lucigenin Chemiluminescence
The background corrected chemiluminescence originating from 5 μM lucigenin observed in organ cultured control and CoPP and CoCl2 treated BPA was used to detect if these HO-1 inducers attenuated relaxation to NO through increasing the levels of superoxide. As shown by the data in Figure 6, the treatments with either CoPP or CoCl2 has similar effects in decreasing the levels of superoxide that were detected.
Figure 6.
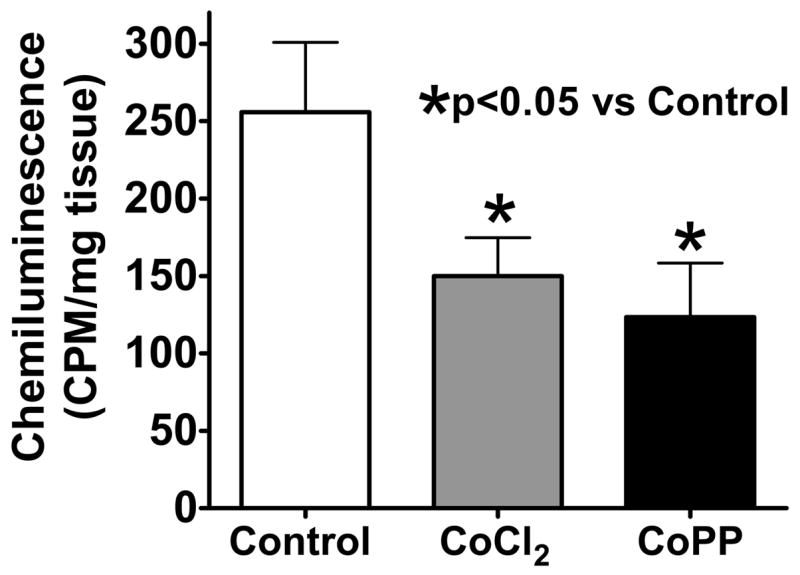
Organoid treatment with HO-1 inducers CoCl2 (n=11) and CoPP (n=7) lowers superoxide in pulmonary arteries which is detected by the chemiluminescence of 5 μM lucigenin.
Effects of CoCl2 Elevated HO-1 Activity on BPA Homogenate sGC Stimulation by NO
The activity of sGC in BPA homogenates was examined to determine if organ culture with CoCl2 altered basal sGC activity and the stimulatory effects of NO. As shown by the data in Figure 7-left panel, basal sGC activity was not altered by organ culture with CoCl2. However, the stimulatory effects of 10 μM spermine NONOate were decreased by ~40% in BPA organ cultured in the presence of CoCl2. Since sGC heme oxidation or depletion has been associated with a degradation of sGC subunits (31), the effect of induction of HO-1 on the expression of the heme-binding β1-subunit was measured by Western analysis. The data in Fig. 7-right panel show that induction of HO-1 with CoCl2 does not appear to alter the levels of the sGC β1-subunit.
Figure 7.
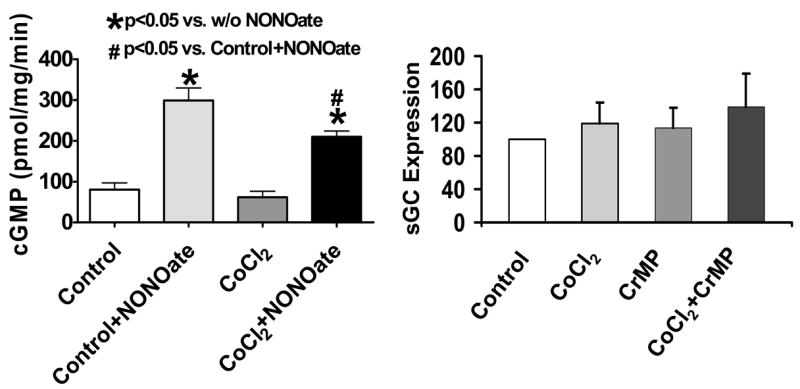
Activity of sGC in BPA homogenates from organoid treated vessels demonstrated an attenuated response to NO due to HO-1 induction by CoCl2 treatment (n=8, left), whereas, HO-1 inducers CoCl2 and CoPP do not alter the expression of the β1-subunit of sGC (n=10, right).
Discussion
The data in this study provide evidence that organ culture of endothelium-removed bovine pulmonary arteries with CoCl2 and CoPP increases HO-1 activity associated with a depletion of heme. The actions of these inducers of HO-1 appeared to selectively attenuate cGMP-associated relaxation to a NO donor, without altering cAMP-associated relaxation to isoproterenol. The model in Figure 8 summarizes some of the potential interactions of HO-1 with the regulation of sGC by NO. Although, increased HO-1 activity could potentially influence cGMP-associated vascular responses through the regulation of sGC as a result of generating CO, heme-depletion would decrease CO generation. In addition, the pulmonary arteries examined in this study did not appear to show altered function in the presence of a CO donor, suggesting CO was not mediating the actions of increased HO-1 activity. The primary effect of increased HO-1 activity observed in the pulmonary arteries studied was an attenuation of relaxation and sGC activation in arterial homogenates in response to a NO donor. Since sGC activation by NO is mediated by the formation of NO-heme at the porphyrin binding site on sGC, increased HO-1 activity could function to inhibit the regulation of sGC and vascular responses to NO by decreasing the availability of heme.
Figure 8.

Model depicting interactions of HO-1, heme and sGC.
Inhibition of vascular relaxation to NO could originate from increased superoxide (14, 23) or by CO binding the heme of sGC preventing further activation by NO (21). Since the cobalt-containing inducers of HO-1 were observed to lower superoxide levels detected with lucigenin, an attenuation of the actions of NO by increased superoxide does not appear to be a contributing factor to the attenuation of the relaxing actions of NO. Increased expression of vascular SOD activity may explain this superoxide lowering effect of HO-1 induction (34). Since there is little evidence that basal superoxide production influences vascular relaxation to NO under physiological conditions, the lowering of superoxide by induction of HO-1 should not effect NO-mediated relaxation. Regulation of sGC by the generation of CO did not appear to be a contributing factor to the actions of the HO-1 inducers. Since increased HO-1 did not detectably alter force generation in arteries contracted with serotonin, endogenous CO was did not appear to be promoting a relaxation associated with activation of sGC. In addition, inhibiting heme oxygenase activity with CrMP did not significantly alter force generation under any of the conditions examined; suggesting CO was not regulating force generation under control conditions or in the presence of elevated levels of HO-1 in the organ cultured bovine calf pulmonary arteries examined in this study. A CO donor did not cause relaxation in the pulmonary arteries studied, further supporting the absence of a detectible relaxation by CO associated with stimulation of the sGC-cGMP system. While organ culture with the CrMP inhibitor of HO-1 attenuated the actions of inducers of HO-1 on relaxation to NO, acute inhibition of heme oxygenase generation of CO by CrMP did not alter relaxation to the NO donor. The presence of CO was also not observed to alter relaxation to the spermine NONOate donor of NO, which suggests that CO does not readily antagonize the actions of NO in the pulmonary arteries examined in this study. While endogenous heme oxygenase-derived CO appears to attenuate vasodilator responses associated endothelium-derived NO, recent studies suggest that CO is functioning as an inhibitor of the generation of NO by NO synthase (22, 33). Thus, the decreased relaxation to NO observed in the presence of pulmonary arteries containing increased HO-1 does not appear to be associated with CO generation by HO-1.
Increased HO-1 activity could be attenuating relaxation to NO through depleting the heme of sGC which controls its cGMP generating activity. Organ culturing BPA with heme (hemin) reversed the attenuation of relaxation to NO under the conditions used to increase HO-1 expression. Early studies on NO regulation of sGC demonstrated that heme was easily lost as sGC was purified (8–11, 18–20), suggesting that heme depletion could be a mechanism of regulating sGC stimulation by NO. Thus, the control of heme availability could be an important factor regulating the responsiveness of sGC and vascular tissue to NO. The data in this study suggest that heme oxygenase could function to attenuate sGC activation by NO under conditions where it lowers basal heme levels in vascular smooth muscle through depleting the heme of sGC which is essential for stimulating cGMP production by NO. Many vascular diseases have recently been shown to be associated with an increased expression of HO-1, but, a major dilemma is that it has been difficult to establish that this increase in endogenous HO-1 was providing a beneficial effect on vascular function (30). Previous work has focused on investigating the role of increased superoxide in causing the attenuation NO-mediated dilator responses that are seen in most vascular disease processes, and it has recently been shown that peroxynitrite generation appears to be a contributing factor to heme oxidation and depletion from sGC (31). The results of this study on sGC heme depletion provide extensive evidence that oxidation and loss of sGC heme is likely to be a major contributor to impaired NO-mediated relaxation that is observed in aging and multiple oxidant stress-associated vascular disease processes, supporting the concept that heme depletion could be an important factor contributing to vascular dysfunction (31). While oxidative stress could contribute to depleting sGC heme, it is not currently known if heme levels in smooth muscle also decrease as a result of increased HO-1 activity and/or a reduced level of heme biosynthesis. Mitochondrial dysfunction is seen in many vascular diseases, including pulmonary hypertension (5), and this could be hypothesized to be a factor in impairing the biosynthesis of heme by this organelle. While it has been observed that a key beneficial effects of HO-1 induction on reversing vascular dysfunction in vivo can originate from its lowering of superoxide levels (34), sGC heme depletion by HO-1 could a factor where vascular disease processes have elevated HO-1 levels and impaired vascular regulation by the NO-sGC system is observed. Thus, many processes regulated by NO such as vasodilation and inhibition of smooth muscle growth could be impaired as a result of depleting the sGC heme essential for the stimulation of cGMP generation by NO, under pathophysiological conditions where this process is potentially a dominant factor controlling the regulation of sGC.
Acknowledgments
We wish to thank Saadet Turkseven for helping adapt measurements of HO-1, and Dr. Xiangmin Zhao for measurements of sGC and HO-1 expression. This study was supported by NIH grants # HL31069, HL43023 and HL66331.
References
- 1.Abraham NG, Lin JH, Dunn MW, Schwartzman ML. Presence of heme oxygenase and NADPH cytochrome P-450 (c) reductase in human corneal epithelium. Invest Ophthalmol Vis Sci. 1987;28:1464–1472. [PubMed] [Google Scholar]
- 2.Abraham NG, Lin JH, Schwartzman ML, Levere RD, Shibahara S. The physiological significance of heme oxygenase. Int J Biochem. 1988;20:543–558. doi: 10.1016/0020-711x(88)90093-6. [DOI] [PubMed] [Google Scholar]
- 3.Abraham NG, Quan S, Mieyal PA, Yang L, Burke-Wolin T, Mingone CJ, Goodman AI, Nasjletti A, Wolin MS. Modulation of cGMP by human HO-1 retrovirus gene transfer in pulmonary microvessel endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2002;283:L1117–1124. doi: 10.1152/ajplung.00365.2001. [DOI] [PubMed] [Google Scholar]
- 4.Appleton SD, Chretien ML, McLaughlin BE, Vreman HJ, Stevenson DK, Brien JF, Nakatsu K, Maurice DH, Marks GS. Selective inhibition of heme oxygenase, without inhibition of nitric oxide synthase or soluble guanylyl cyclase, by metalloporphyrins at low concentrations. Drug Metab Dispos. 1999;27:1214–1219. [PubMed] [Google Scholar]
- 5.Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thebaud B, Bonnet S, Haromy A, Harry G, Moudgil R, McMurtry MS, Weir EK, Archer SL. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation. 2006;113:2630–2641. doi: 10.1161/CIRCULATIONAHA.105.609008. [DOI] [PubMed] [Google Scholar]
- 6.Burstyn JN, Yu AE, Dierks EA, Hawkins BK, Dawson JH. Studies of the heme coordination and ligand binding properties of soluble guanylyl cyclase (sGC): characterization of Fe(II)sGC and Fe(II)sGC(CO) by electronic absorption and magnetic circular dichroism spectroscopies and failure of CO to activate the enzyme. Biochemistry. 1995;34:5896–5903. doi: 10.1021/bi00017a019. [DOI] [PubMed] [Google Scholar]
- 7.Christodoulides N, Durante W, Kroll MH, Schafer AI. Vascular smooth muscle cell heme oxygenases generate guanylyl cyclase-stimulatory carbon monoxide. Circulation. 1995;91:2306–2309. doi: 10.1161/01.cir.91.9.2306. [DOI] [PubMed] [Google Scholar]
- 8.Craven PA, DeRubertis FR. Requirement for heme in the activation of purified guanylate cyclase by nitric oxide. Biochim Biophys Acta. 1983;745:310–321. doi: 10.1016/0167-4838(83)90063-8. [DOI] [PubMed] [Google Scholar]
- 9.Craven PA, DeRubertis FR. Restoration of the responsiveness of purified guanylate cyclase to nitrosoguanidine, nitric oxide, and related activators by heme and hemeproteins. Evidence for involvement of the paramagnetic nitrosyl-heme complex in enzyme activation. J Biol Chem. 1978;253:8433–8443. [PubMed] [Google Scholar]
- 10.Gerzer R, Hofmann F, Schultz G. Purification of a soluble, sodium-nitroprusside-stimulated guanylate cyclase from bovine lung. Eur J Biochem. 1981;116:479–486. doi: 10.1111/j.1432-1033.1981.tb05361.x. [DOI] [PubMed] [Google Scholar]
- 11.Gerzer R, Radany EW, Garbers DL. The separation of the heme and apoheme forms of soluble guanylate cyclase. Biochem Biophys Res Commun. 1982;108:678–686. doi: 10.1016/0006-291x(82)90883-x. [DOI] [PubMed] [Google Scholar]
- 12.Gupte SA, Kaminski PM, Floyd B, Agarwal R, Ali N, Ahmad M, Edwards J, Wolin MS. Cytosolic NADPH may regulate differences in basal Nox oxidase-derived superoxide generation in bovine coronary and pulmonary arteries. American journal of physiology. 2005;288:H13–21. doi: 10.1152/ajpheart.00629.2004. [DOI] [PubMed] [Google Scholar]
- 13.Gupte SA, Rupawalla T, Phillibert D, Jr, Wolin MS. NADPH and heme redox modulate pulmonary artery relaxation and guanylate cyclase activation by NO. Am J Physiol. 1999;277:L1124–1132. doi: 10.1152/ajplung.1999.277.6.L1124. [DOI] [PubMed] [Google Scholar]
- 14.Harrison D, Griendling KK, Landmesser U, Hornig B, Drexler H. Role of oxidative stress in atherosclerosis. Am J Cardiol. 2003;91:7A–11A. doi: 10.1016/s0002-9149(02)03144-2. [DOI] [PubMed] [Google Scholar]
- 15.Hassoun PM, Filippov G, Fogel M, Donaldson C, Kayyali US, Shimoda LA, Bloch KD. Hypoxia decreases expression of soluble guanylate cyclase in cultured rat pulmonary artery smooth muscle cells. Am J Respir Cell Mol Biol. 2004;30:908–913. doi: 10.1165/rcmb.2003-0287OC. [DOI] [PubMed] [Google Scholar]
- 16.Hosein S, Marks GS, Brien JF, McLaughlin BE, Nakatsu K. An extracellular source of heme can induce a significant heme oxygenase mediated relaxation in the rat aorta. Can J Physiol Pharmacol. 2002;80:761–765. doi: 10.1139/y02-086. [DOI] [PubMed] [Google Scholar]
- 17.Iesaki T, Gupte SA, Wolin MS. A flavoprotein mechanism appears to prevent an oxygen-dependent inhibition of cGMP-associated nitric oxide-elicited relaxation of bovine coronary arteries. Circ Res. 1999;85:1027–1031. doi: 10.1161/01.res.85.11.1027. [DOI] [PubMed] [Google Scholar]
- 18.Ignarro LJ, Adams JB, Horwitz PM, Wood KS. Activation of soluble guanylate cyclase by NO-hemoproteins involves NO-heme exchange. Comparison of heme-containing and heme-deficient enzyme forms. J Biol Chem. 1986;261:4997–5002. [PubMed] [Google Scholar]
- 19.Ignarro LJ, Degnan JN, Baricos WH, Kadowitz PJ, Wolin MS. Activation of purified guanylate cyclase by nitric oxide requires heme. Comparison of heme-deficient, heme-reconstituted and heme-containing forms of soluble enzyme from bovine lung. Biochim Biophys Acta. 1982;718:49–59. doi: 10.1016/0304-4165(82)90008-3. [DOI] [PubMed] [Google Scholar]
- 20.Ignarro LJ, Wood KS, Wolin MS. Regulation of purified soluble guanylate cyclase by porphyrins and metalloporphyrins: a unifying concept. Adv Cyclic Nucleotide Protein Phosphorylation Res. 1984;17:267–274. [PubMed] [Google Scholar]
- 21.Imai T, Morita T, Shindo T, Nagai R, Yazaki Y, Kurihara H, Suematsu M, Katayama S. Vascular smooth muscle cell-directed overexpression of heme oxygenase-1 elevates blood pressure through attenuation of nitric oxide-induced vasodilation in mice. Circulation research. 2001;89:55–62. doi: 10.1161/hh1301.092679. [DOI] [PubMed] [Google Scholar]
- 22.Johnson FK, Durante W, Peyton KJ, Johnson RA. Heme oxygenase inhibitor restores arteriolar nitric oxide function in dahl rats. Hypertension. 2003;41:149–155. doi: 10.1161/01.hyp.0000046923.52222.58. [DOI] [PubMed] [Google Scholar]
- 23.Landmesser U, Harrison DG, Drexler H. Oxidant stress-a major cause of reduced endothelial nitric oxide availability in cardiovascular disease. Eur J Clin Pharmacol. 2005:1–7. [Google Scholar]
- 24.Lin JH, Villalon P, Martasek P, Abraham NG. Regulation of heme oxygenase gene expression by cobalt in rat liver and kidney. Eur J Biochem. 1990;192:577–582. doi: 10.1111/j.1432-1033.1990.tb19263.x. [DOI] [PubMed] [Google Scholar]
- 25.Lincoln TM, Dey N, Sellak H. Invited review: cGMP-dependent protein kinase signaling mechanisms in smooth muscle: from the regulation of tone to gene expression. J Appl Physiol. 2001;91:1421–1430. doi: 10.1152/jappl.2001.91.3.1421. [DOI] [PubMed] [Google Scholar]
- 26.Mingone CJ, Gupte SA, Ali N, Oeckler RA, Wolin MS. Thiol Oxidation Inhibits Nitric Oxide-Mediated Pulmonary Artery Relaxation and Guanylate Cyclase Stimulation. Am J Physiol Lung Cell Mol Physiol. 2005:00331.02005. doi: 10.1152/ajplung.00331.2005. [DOI] [PubMed] [Google Scholar]
- 27.Mingone CJ, Gupte SA, Iesaki T, Wolin MS. Hypoxia enhances a cGMP-independent nitric oxide relaxing mechanism in pulmonary arteries. Am J Physiol Lung Cell Mol Physiol. 2003;285:L296–304. doi: 10.1152/ajplung.00362.2002. [DOI] [PubMed] [Google Scholar]
- 28.Motterlini R, Mann BE, Johnson TR, Clark JE, Foresti R, Green CJ. Bioactivity and pharmacological actions of carbon monoxide-releasing molecules. Curr Pharm Des. 2003;9:2525–2539. doi: 10.2174/1381612033453785. [DOI] [PubMed] [Google Scholar]
- 29.Naik JS, Walker BR. Homogeneous segmental profile of carbon monoxide-mediated pulmonary vasodilation in rats. Am J Physiol Lung Cell Mol Physiol. 2001;281:L1436–1443. doi: 10.1152/ajplung.2001.281.6.L1436. [DOI] [PubMed] [Google Scholar]
- 30.Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiological reviews. 2006;86:583–650. doi: 10.1152/physrev.00011.2005. [DOI] [PubMed] [Google Scholar]
- 31.Stasch JP, Schmidt PM, Nedvetsky PI, Nedvetskaya TY, H SA, Meurer S, Deile M, Taye A, Knorr A, Lapp H, Muller H, Turgay Y, Rothkegel C, Tersteegen A, Kemp-Harper B, Muller-Esterl W, Schmidt HH. Targeting the heme-oxidized nitric oxide receptor for selective vasodilatation of diseased blood vessels. The Journal of clinical investigation. 2006;116:2552–2561. doi: 10.1172/JCI28371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stone JR, Marletta MA. Soluble guanylate cyclase from bovine lung: activation with nitric oxide and carbon monoxide and spectral characterization of the ferrous and ferric states. Biochemistry. 1994;33:5636–5640. doi: 10.1021/bi00184a036. [DOI] [PubMed] [Google Scholar]
- 33.Teran FJ, Johnson RA, Stevenson BK, Peyton KJ, Jackson KE, Appleton SD, Durante W, Johnson FK. Heme oxygenase-derived carbon monoxide promotes arteriolar endothelial dysfunction and contributes to salt-induced hypertension in Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol. 2005;288:R615–622. doi: 10.1152/ajpregu.00123.2004. [DOI] [PubMed] [Google Scholar]
- 34.Turkseven S, Kruger A, Mingone CJ, Kaminski P, Inaba M, Rodella LF, Ikehara S, Wolin MS, Abraham NG. Antioxidant mechanism of heme oxygenase-1 involves an increase in superoxide dismutase and catalase in experimental diabetes. American journal of physiology. 2005;289:H701–707. doi: 10.1152/ajpheart.00024.2005. [DOI] [PubMed] [Google Scholar]
- 35.Tzao C, Nickerson PA, Russell JA, Gugino SF, Steinhorn RH. Pulmonary hypertension alters soluble guanylate cyclase activity and expression in pulmonary arteries isolated from fetal lambs. Pediatr Pulmonol. 2001;31:97–105. doi: 10.1002/1099-0496(200102)31:2<97::aid-ppul1016>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 36.Villamor E, Perez-Vizcaino F, Cogolludo AL, Conde-Oviedo J, Zaragoza-Arnaez F, Lopez-Lopez JG, Tamargo J. Relaxant effects of carbon monoxide compared with nitric oxide in pulmonary and systemic vessels of newborn piglets. Pediatr Res. 2000;48:546–553. doi: 10.1203/00006450-200010000-00021. [DOI] [PubMed] [Google Scholar]
- 37.Wolin MS, Wood KS, Ignarro LJ. Guanylate cyclase from bovine lung. A kinetic analysis of the regulation of the purified soluble enzyme by protoporphyrin IX, heme, and nitrosyl-heme. J Biol Chem. 1982;257:13312–13320. [PubMed] [Google Scholar]
- 38.Ye J, Laychock SG. A protective role for heme oxygenase expression in pancreatic islets exposed to interleukin-1beta. Endocrinology. 1998;139:4155–4163. doi: 10.1210/endo.139.10.6244. [DOI] [PubMed] [Google Scholar]
- 39.Zhao L, Mason NA, Morrell NW, Kojonazarov B, Sadykov A, Maripov A, Mirrakhimov MM, Aldashev A, Wilkins MR. Sildenafil inhibits hypoxia-induced pulmonary hypertension. Circulation. 2001;104:424–428. doi: 10.1161/hc2901.093117. [DOI] [PubMed] [Google Scholar]