

Abstract
Administration of 0.4% clofibrate in the diet stimulated estradiol (E2)-induced mammary carcinogenesis in the August-Copenhagen Irish (ACI) rat without having an effect on serum levels of E2. This treatment stimulated by several-fold the NAD(P)H-dependent oxidative metabolism of E2 and oleyl-CoA-dependent esterification of E2 to 17β-oleyl-estradiol by liver microsomes. Glucuronidation of E2 by microsomal glucuronosyltransferase was increased moderately. In contrast, the activity of NAD(P)H quinone reductase 1 (NQO1), a representative monofunctional phase 2 enzyme, was significantly decreased in liver cytosol of rats fed clofibrate. Decreases in hepatic NQO1 in livers of animals fed clofibrate were noted before the appearance of mammary tumors. E2 was delivered in cholesterol pellets implanted in 7 to 8 week old female ACI rats. The animals received AIN-76A diet containing 0.4% clofibrate for 6, 12 or 28 weeks. Control animals received AIN-76A diet. Dietary clofibrate increased the number and size of palpable mammary tumors but did not alter the histopathology of the E2-induced mammary adenocarcinomas. Collectively, these results suggest that the stimulatory effect of clofibrate on hepatic esterification of E2 with fatty acids coupled with the inhibition of protective phase 2 enzymes, may in part, enhance E2-dependent mammary carcinogenesis in the ACI rat model.
Keywords: beast cancer; clofibrate (2-(4-chlorophenoxy)-2-methyl-propanoic acid, ethyl ester); estradiol
1. Introduction
Xu et al (2001a,b) demonstrated that treatment of overectomized Sprague Dawley rats with dietary clofibrate caused a many-fold increase in the esterification of estradiol (E2) with fatty acids by liver microsomes. This increase in esterification of E2 was accompanied in vivo by E2–induced increases in the formation of lobules in the mammary gland and increased incorporation of bromodeoxyuridine into these lobules (Xu, et al. 2001c). E2 fatty acid esters have very long half-lives in vivo (Hershcopf et al., 1986), and it has been suggested that they are sequestered in fat-rich tissues where they function as a reservoir for the prolonged release of hormonally active E2 (Larner and Hochberg, 1985) (Zielinski et al., 1991). In support of this idea, Xu et al. (2002) found that treatment of rats with clofibrate stimulated the hepatic formation of highly lipophilic fatty acid esters of E2 and their hydrolysis in liver and extrahepatic tissues to the parent steroid hormone by a clofibrate-inducible esterase. Although the chronic administration of clofibrate to Sprague-Dawley rats for 10 days enhanced E2-induced proliferation of rat mammary lobule and ductal cells (Xu et al., 2001c), the effect of clofibrate on the development of mammary cancer is not known.
The object of the present study was to evaluate the influence of dietary clofibrate on the occurence and progression of mammary tumors in female ACI rats treated chronically with E2 and to determine possible correlations between mammary carcinogenesis and hepatic metabolism of E2. We have previously shown that dietary clofibrate markedly inhibits the expression of key antioxidant enzymes in this strain treated chronically with low doses of E2 (Mesia-Vela,et al, 2004) The ovary-intact female ACI rat is a unique and physiologically relevant model to explore mechanisms associated with estrogen-dependent breast cancer because of the high incidence (80–100%) of mammary ductal adenocarcinomas that occur over a relatively short period (median latency 20 weeks) in this strain exposed chronically to low doses of E2 (Stone et al., 1979; Harvel, et al, 2000; Li et al., 2002). Moreover, mammary cancers rarely develop spontaneously in ACI rats in contrast to most other rat strains (Shull et al., 1997). In the present study, we found that administration of dietary clofibrate increases the size and number of E2-dependent mammary tumors in female ACI rats. The onset of mammary tumors is preceded by a significant increase in the esterification and oxidation of E2 by liver microsomes and a marked decline in the activity of the representative phase 2 enzyme NQO1.
2. Material and Methods
2.1. Chemicals
Estradiol, ascorbic acid, NADPH, β-glucuronidase (EC. 3.2.1.3.1), UDP-glucuronic acid, saccharic acid 1,4-lactone, p-nitrophenol, oleoyl coenzyme A and Tris-base were obtained from Sigma Chem. Co. (St. Louis, MO). 35S-PAPS and [2,4,6,7,16,17-3H(N)]-estradiol (s.a.110–170 Ci/mmol) were from New England Nuclear (Boston, MA). AIN-76A diet was purchased from Dyets Inc. (Bethlehem, PA). All other chemicals used were of the highest grade from standard sources.
2.2. Animals and treatments
Female ACI rats (7 to 8 weeks old) were obtained from Harlan Sprague-Dawley Laboratory (Indianapolis, IN). The animals were housed individually in an AAALAC accredited barrier facility under controlled temperature, humidity, and lighting conditions. Animals were fed AIN-76A diet (Dyets Inc., Pennsylvania, PA) with 0.4% clofibrate. This dose was selected based on earlier studies examining the effects of clofibrate on hepatic estrogen metabolizing enzymes in Sprague Dawley rats (Xu S, et al, 2001))This dose is approximately twice the typical dose of clofibrate in humans, which is 2 g daily (Goodman and Gilman, 11th Ed. 2006) or about 30 mg/day/kg. Experimental rats used in our study consumed approximately 15 g of diet containing 4% clofibrate. This corresponds to 60 mg of clofibrate per day. Using the conversion factor of 0.16 (Guidance for Industry and Reviewers: Estmating the safe starting dose in clinical trials for therapeutics in healthy human volunteers, U.S. Food and Drug Administration CDER, 2002), the human equivalent dose consumed by rats in this study is estimated to be 64 mg/day/kg.
Treatment protocols started 4 days after arrival. Rats received food and water ad libitum. A single 20-mg pellet (Hormone Pellets Press, Shawnee Mission, KS) containing E2 was implanted in the shoulder region, as described previously (Li et al., 2002). In experiment 1 (n = 20 rats/group), group A received pellets containing 20 mg cholesterol, group B received pellets containing 1 mg of E2 plus 19 mg of cholesterol pellets, group C received pellets containing 1 mg of E2 plus 19 mg of cholesterol and also 0.4% clofibrate diet, group D received pellets containing 3 mg of E2 plus 17 mg of cholesterol, group E received pellets containing 3 mg of E2 plus 17 mg of cholesterol and also 0.4% clofibrate diet, and group F received pellets containing 20 mg cholesterol and also 0.4% clofibrate diet. Six animals per group were killed after 6 and 12 weeks of treatment. The remaining animals (n= 8/group) were continued until tumors developed, and the animals were killed at 24–36 weeks because of the adverse effects of pituitary tumors and because of the size of mammary tumors. In experiment 2, two groups of animals (n = 15/group) received pellet s containing 3 mg of E2 and 17 mg of cholesterol. One group received AIN76A diet alone whereas the other group received 0.4% clofibrate in AIN-76A diet. Clofibrate-fed and control animals in experiment 2 were sacrificed at the same time point (28 weeks). The rats were palpated for mammary masses twice weekly, and weighed every two weeks for the duration of the experiment.
2.3. Tissue Processing
Trunk blood collected at the time of sacrifice was allowed to clot at 4°C for six hours, and centrifuged. The serum was collected and stored at −80°C. All animals were necropsied and the number, volume and localization of mammary masses recorded. The geometric volume of the tumors was determined using the formula: length × width × height × 0.5326, assuming a hemiellipsoid shape (Shah et al., 1999). The abdominal inguinal mammary glands and the mammary masses were quickly removed and portions were fixed in Carnoy’s solution for 4 h. Fixed tissues were embedded in paraffin. Sections (6 μm) were prepared and stained with hematoxylin and eosin. Selected estrogen target organs were also removed and weighed. Serum levels of E2 were determined by RIA using Coat-A Count® Estradiol RIA kits (Diagnostic Products Corporation, Los Angeles, CA) according to the manufacturer’s instructions.
2.4. Preparation of microsomes and cytosols from rat liver
Liver cytosols and microsomes were prepared by differential centrifugation as described previously (Thomas et al., 1983) and stored at −80°C until used. Protein content of cytosols and microsomes was determined with the BCA™ Protein Assay kit (PIERCE, Rockford, IL) in a 96-well format according to the manufacturer’s recommendations using bovine serum albumin to construct standard curves.
2.5. Enzyme Assays
All assays were performed under conditions yielding linear kinetics with respect to enzyme concentrations and incubation times
2.5.1. NADPH- dependent oxidation of E2
NADPH- dependent oxidation of E2 was carried out using liver microsomes (0.5 mg protein) incubated with 2 mM NADPH, 5 mM ascorbic acid, 3 mM magnesium chloride, 50 μM sodium-phosphate buffer (pH 7.4) and 50 μM [3H]E2 (0.5 μCi) for 20 min at 37 °C. The enzyme reaction was terminated by the addition of 5 ml of ice-cold ethyl acetate and vortexing. The ethyl acetate extracts were evaporated to dryness under nitrogen. The residue was dissolved in methanol and analyzed for metabolite composition by HPLC as described previously (Suchar et al., 1995, Mesia-Vela et al., 2002).
2.5.2. Glucuronosyltransferase activity
Glucuronosyltransferase activity was assayed using E2 as substrate. The reaction mixture contained 1.0 mg of microsomal protein, 2 mM UDPGA, 5mM MgC12, 100 μM [3H]E2 (0.15 μCi) and 50 mM Tris-HCl buffer (pH 8.5) in a final volume of 150 μl. The reaction was initiated by addition of UDPGA. Incubations proceeded at 37°C for 15 min and were terminated by placing them on ice and adding 50-μl ice-cold acetonitrile. The reaction mixtures were then vortexed and centrifuged at 3000 × g for 5 min. Ten μl aliquots of the supernatants were used for the determination of E2 and E2-glucuronides by HPLC. The solvent system consisted of solvent A: 0.1% acetic acid in water and solvent B: containing 20% methanol, 80% acetonitrile and 0.1% acetic acid. E2 and its 3- and 17-hydroxyglucuronides were eluted with a 30 min linear gradient from 25 to 90% B with a flux of 1 ml/min through a Spherisorb column. The glucuronides were identified by their co-elution with authentic standards or by determination of E2-gluucuronides after hydrolysis by β-glucuronidase (200 U/ml) (Mesia-Vela et al., 2006).
2.5.3. Fatty acyl-CoA:estradiol acyltransferase
Fatty acyl-CoA:estradiol acyltransferase was assayed in reaction mixtures containing 50 μM [3H]-E2 (1 μCi), 100 μM fatty acyl-CoA, and 5 mM magnesium chloride in 0.1 M sodium acetate buffer (pH 5.5) in a final volume of 0.5 ml. The reaction was initiated by the addition of liver microsomes (1 mg of protein/ml). After incubation at 37°C for 30 min, the reaction was arrested by placing the tubes on ice, followed by the addition of 0.2 ml of ice-cold sodium acetate buffer (pH 5.5) and extraction with 4 ml of ethyl acetate (HPLC grade from Fisher Scientific, Pittsburgh, PA). Dry extracts were redissolved, and 90-μl aliquots analyzed by HPLC as described previously (Xu et al., 2001b). Metabolite quantification was based on the amount of radioactivity in the metabolite peak compared with the total radioactivity collected from the HPLC column from each sample.
2.5.4. NAD(P)H quinone:oxidoreductase (NQO1)
NAD(P)H quinone:oxidoreductase (NQO1) was measured by the reduction of cytochrome c (50 μM) in the presence of liver cytosol (1–5 μg protein), 10 μM menadione and 1 mM NADPH as described previously (Sanchez et al., 2003). Reactions were carried out in 1 ml of reagent at 25° C and the reduction of cytochrome c (50 μM) was monitored continuously at 550nm.
3. Statistical analysis of data
Data are presented as the mean ± S.E. Differences between means were assessed by ANOVA followed by the Bonferroni post-hoc test with the significance value of p < 0.05.
4. Results
4.1. Tissue wet weights and serum E2 levels
Weights of various organs were measured as potential indices of the sysetemic effects of E2 and clofibrate. For example, the pituitary and uterus are classic target organs for estradiol in intact animals. Treatment of ACI rats with E2 for 6 or 12 weeks did not alter the body weight of the animals or the uterine, kidney or adrenal weights (Table 1). The weights of liver and the ratios of liver to body weight were increased by E2 in both experiments by about 40% (Table 1). E2 administration also increased pituitary weight up to 6-fold and reduced thymus weight by about 50% (Table 1). The pituitary and uterus are classic target organs for estradiol in intact animals. Estradiol induces a rapid and sustained growth of the anterior pituitary in a number of rat strains, including the ACI strain. The grossly enlarged pituitary glands exhibit diffuse lactotroph hyperplasia that result in marked hyperprolactinemia (e.g. Spady et al., 1999, Mol. Carcinogen. 26, 239–253. The failure of E2 to stimulate uterine growth in female ACI rats was expectet (e.g., Turan et al. (2004); Mesia-Vela, et al. (2006)).
Table 1.
Effects of 0.4 % clofibrate diet on body and organ weights and serum estradiol levels after 6, 12 and 28 weeks of treatment of female ACI rats.
| Treatment | E2 (mg)a | Weights | Microsomal protein (mg/liver) | Serum E2 (pg/ml) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Experiment 1 | Body (g) | Uterus (mg) | Liver (g) | Thymus (mg) | Pituitary (mg) | Adrenals (mg) | Kidneys b (g) | |||
| 6weeks (n=6) | ||||||||||
| A.AIN-76A) | 0 | 161 ± 17 | 449 ± 19 | 5.5 ± 0.2 | 240 ± 6 | 9 ± 0.5 | 56 ± 2 | 1.2 ± 0.02 | 81 ± 5 | 30 ± 5 |
| B. AIN-76A | 1 | 172 ± 14 | 551 ± 78 | 7.8 ± 0.5* | 182 ± 7* | 26 ± 3* | 61 ±4 | 1.5 ± 0.14 | 114 ± 9* | 32 ± 3 |
| C. 0.4% clofibrate diet | 1 | 157 ± 3 | 495 ± 44 | 8.5 ± 0.2# | 152 ± 7# | 16 ± 0.6# | 62± 2 | 1.4 ± 0.09 | 180 ± 12* | 35 ± 2 |
| D. AIN-76A | 3 | 175 11 | 547 ± 16 | 7.7 ± 0.2* | 152 ± 8* | 37 ± 3* | 53 ± 1 | 1.6 ± 0.09 | 89 ± 7 | 52 ± 3* |
| E. 0.4% clofibrate diet | 3 | 166 ± 7 | 526 ± 114 | 9.0 ± 0.2# | 122 ± 6# | 31± 3* | 61 ± 1 | 1.5 ± 0.02 | 174 ± 17* | 47 ± 0.7* |
| F. 0.4% clofibrate diet | 0 | 159 ± 13 | 529 ± 37 | 7.6 ± 0.3* | 210 ± 12* | 9.6 ± 0.4 | 61 ± 4 | 1.3 ± 0.08 | 183 ± 10* | 33 ± 1 |
| 12 weeks (n=6) | ||||||||||
| A.AIN-76A) b | 0 | 187 ± 6 | 418 ± 18 | 5.5 ± 0.2 | 199 ± 10 | 9.7 ± 0.6 | 53 ± 2 | 1.2 ± 0.04 | 97 ± 13 | 22 ± 2 |
| B. AIN-76A | 1 | 191 ± 3 | 427 ± 35 | 7.9 ± 0.2* | 121 ± 14* | 33 ± 2* | 54 ± 3 | 1.6 ± 0.06 | 89 ± 8 | 49 ± 5 |
| C. 0.4% clofibrate diet | 1 | 173 ± 8 | 496 ± 24 | 9.4 ± 0.5# | 95 ± 9* | 27 ± 3* | 56 ± 3 | 1.5 ± 0.08 | 205 ± 13* | 53 ± 11 |
| D. AIN-76A | 3 | 185 ± 4 | 494 ± 37 | 7.4 ± 0.3* | 94 ± 5* | 55 ± 4* | 54 ± 3 | 1.6 ± 0.08 | 95 ± 7 | 141 ± 29* |
| E. 0.4% clofibrate diet | 3 | 179 ± 5 | 562 ± 13 | 9.8 ± 0.1# | 95 ± 6* | 49± 3* | 57 ± 2 | 1.5 ± 0.03 | 253 ± 19* | 81 ± 14* |
| F. 0.4% clofibrate diet | 0 | 172 ± 5 | 463 ± 18 | 7.8 ± 0.3* | 146 ± 8* | 9.5 ± 0.9 | 55 ± 3 | 1.4 ± 0.07 | 164 ± 6* | 18 ± 1 |
| Experiment 2 | ||||||||||
| 28 weeks (n=10–11) | ||||||||||
| AIN-76A | 3 | 186 ± 8 | 497 ± 12 | 5.5 ± 0.2 | 103 ± 8 | 187 ± 30 | 51 ± 1 | 1.4 ± 0.02 | n.d. | 103 ± 9 |
| 0.4% clofibrate diet | 3 | 183 ± 24 | 723 ± 29# | 8.6 ± 0.4# | 93 ± 7 | 180 ± 33 | 54 ± 2 | 1.6 ± 0.04 | n.d. | 101 ± 11 |
control animals were implanted with a 20 mg cholesterol pellet (without E2),
Renal unigenic animals were not included in the calculation of this data.
n.d. = not determined. Data is expressed as average ± S.E.
Statistically different from the control group that received only a 20 mg cholesterol pellet,
Statistically different from the group receiving the same amount of E2, P <0.05).
Administration of clofibrate for 6, 12 or 28 weeks did not alter the body weight of control or E2-treated animals (Table 1). An increase in liver size and the induction of microsomal protein/per g liver occurred in rats fed clofibrate (Table 1) and this was reflected by approximately a 2-fold increase of total microsomal protein/liver after 6 or 12 weeks of treatment. Administration of E2 and clofibrate together had a greater effect on liver weight than administration of either compound alone (Table 1).
Table 1 shows serum E2 levels in control and clofibrate-fed animals in trunk blood. Administration of E2 (3 mg) increased the serum level of E2 significantly; however, dietary clofibrate did not alter E2 levels in either control or E2-treated animals (Table 1). Although serum E2 levels were not followed over the course of these experiments, the overall conclusion from these studies is that serum levels of E2 were the same in rats fed control or clofibrate-containing diets. It was necessary to carry out experiments using ovary-intact animals because ovarectomized female ACI rats exposed chronically to low doses of estradiol do not develop mammary tumors (Shull et al., 1997).
4.2. E2-induced mammary carcinogenesis
The cumulative incidence of palpable mammary tumors induced by E2 in ACI rats maintained on normal and clofibrate diets was evaluated in two experiments as described in Methods. The first experiment compared results obtained in groups of rats treated with pellets containing either a low (1 mg) or high dose (3 mg) of E2 and killed at 6, 12 and 36 weeks. Experiment 2, in which the incidence of mammary tumors was conducted with a second group of animals implanted with pellets containing 3 mg E2 and maintained on either normal or 0.4% clofibrate diets for 28 weeks.
4.2.2. Experiment 1
The cumulative incidence of palpable mammary tumors detected ex vivo was lower in the group of rats treated with pellets containing 1 mg E2 then in the group implanted with pellets containing 3 mg.E2; however, addition of clofibrate to diets did not significantly alter the incidence of mammary tumors in either group of rats (Fig. 1). A 50% incidence of mammary tumors in animals treated with E2 (3mg) occurred at 16 weeks, whereas a 50% incidence in rats treated with the low dose of E2 (1 mg) was noted at about 36 weeks. This dose dependency of the incidence of palpable mammary tumors is comparable to that reported earlier for ACI rats implanted with cholesterol pellets containing 1 or 3 mg E2 (Turan, et al.,2004). With the exeception of one rat treated with the high dose of E2 and fed the clofibrate-containing diet, there were no detectable palpable mammary tumors before 15 weeks.
Fig. 1. Effect of dietary clofibrate on the cumulative incidence of palpable mammary tumors induced by E2 in the ACI rat.
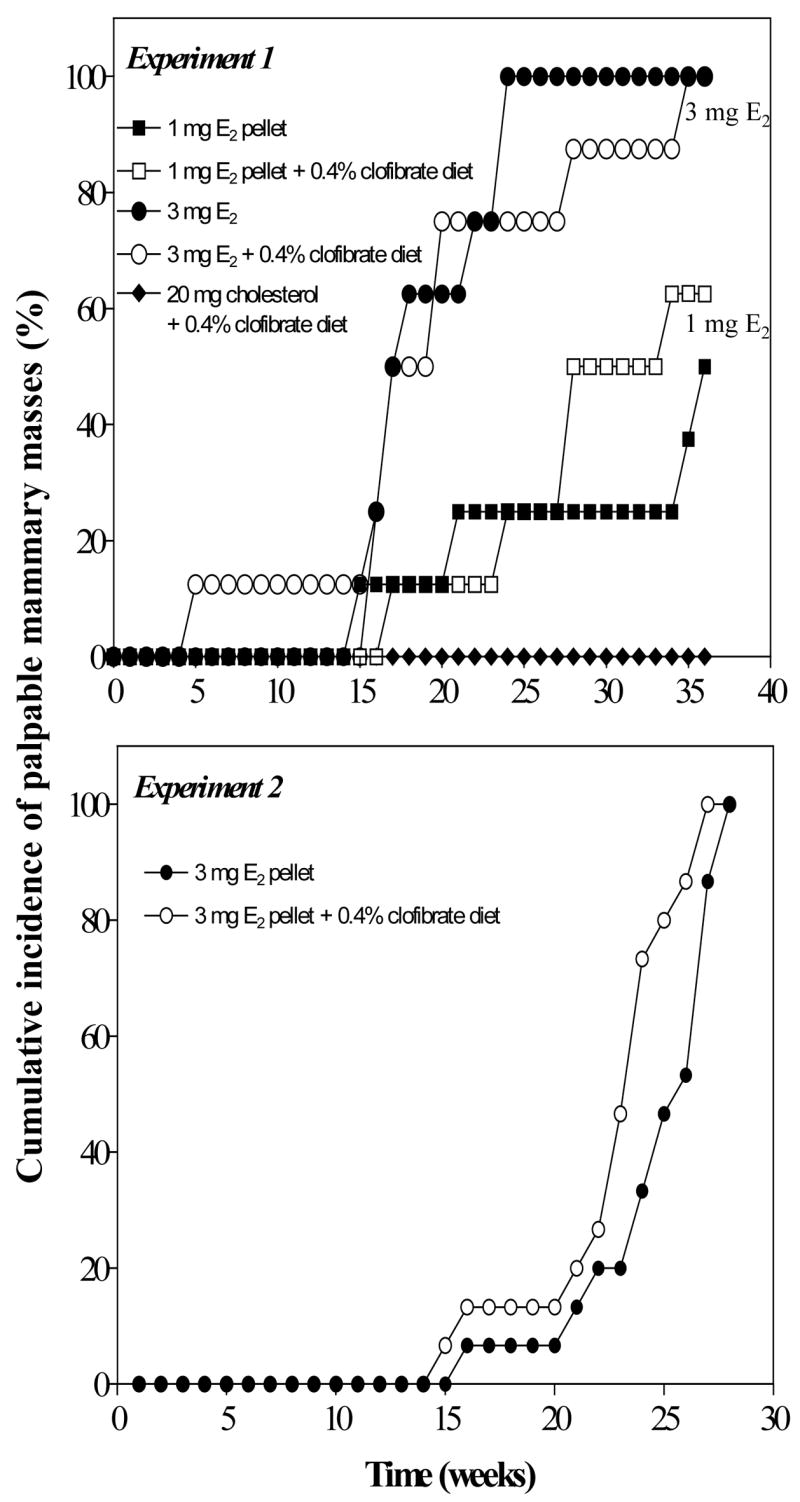
In Experiment 1, E2 (0, 1 or 3 mg) was administered in cholesterol-containing pellets to animals maintained on control or 0.4% clofibrate-contianing AIN76A diets as described in Methods. Six animals from each group at 6 and 12 weeks were killed for necroscopy and tissue sampling. The 8 remaining animals, in each group, were followed for development of mammary tumors and were sacrificed at 36 weeks. In Experiment 2, eleven control and ten clofibrated-fed animals received 3 mg of E2 contained in 17 mg cholesterol pellets and were sacrificed at the same time point (28 weeks). Throughout both experiments, all animals were examined twice ex vivo a week for palpable mammary tumors.
4.2.2. Experiment 2
In experiment 2, 15 rats were implanted E2-containing (3 mg) pellets and fed the control diet. An additional 15 rats were implanted with E2-containing pellets (3 mg) and fed the 0.4% clofibrate diet. Animals that received E2 (3 mg) together with dietary 0.4% clofibrate tended to develop tumors slightly earlier than animals fed the control diet treated with E2 (Fig. 1). Although some animals with moribund pituitary and mammary tumors were sacrificed early (5 animals from the clofibrate group and 4 from the control group), all other animals from both groups were killed at 28 weeks of treatment. In surviving animals, we found that concurrent administration of clofibrate with E2 (3 mg) increased the number of E2-induced mammary tumors per rat by about 2-fold (Fig. 2A) and the size per tumor by 4- to 5-fold (Fig. 2B).
Fig 2. Effects of dietary clofibrate on the number and the size of mammary tumors induced by E2 in the ACI rat.
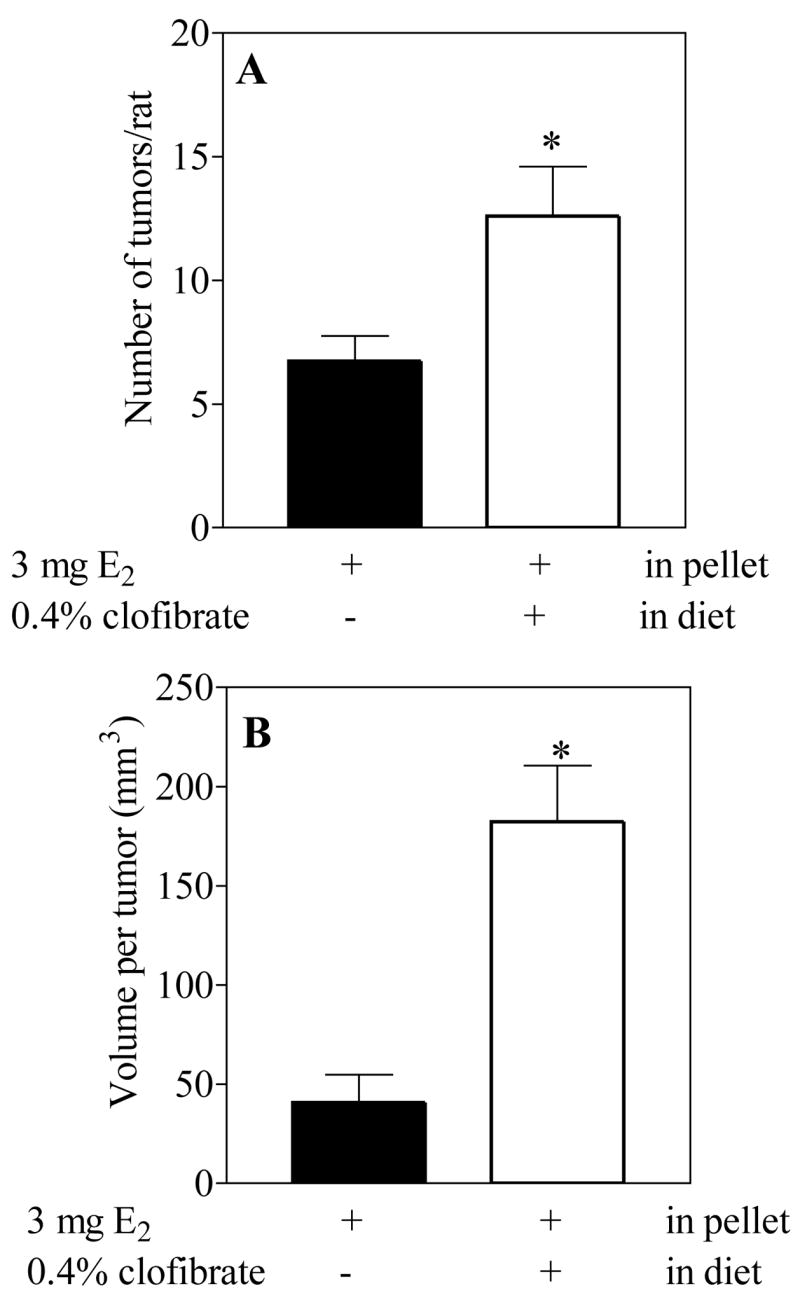
Data were obtained from animals described in Experiment 2 sacrificed at 28 weeks. Each value is the average ± S.E. from 10 control 11 clofibrate-fed animals.
* (p <0.05) control vs E2 treated group.
4.3. Tissue Histopathology
Microscopic examination of mammary tissues from rats treated with E2, maintained on normal or clofibrate-containing diets and killed at 6 or at 12 weeks revealed essentially identical alterations (Fig. 3). Mammary glands displayed pronounced ductal hyperplasia as early as six weeks after the beginning of the treatments with E2 and no quantitative differences could be observed in the extent of hyperplasia induced by E2 in relation to time of exposure. Administration of clofibrate alone caused a relatively high mitotic index in liver without altering mammary ductal cellular histology. Co-treatment with clofibrate and E2 resulted in the same extent of mammary hyperplasia as induced by E2 alone. Mammary neoplasms were qualitatively similar in both groups (Fig. 3) and most were classified as adenocarcinomas. The tumor architecture showed a mixture of solid and glandular phases of densely packed, moderately pleomorphic cells displaying considerable nuclear atypia. Mitoses, including tripolar mitotic figures, were abundant throughout the tumors, and the larger lesions contained zones of tumor necrosis. Invasion of the underlying skeletal muscle by tumor occurred in rats from both treatment groups.
Fig. 3. Histopathology of mammary tumors from a normal rat (A, C) or clofibrate-fed rat (B, D) treated with estradiol (3mg) for 12 weeks.
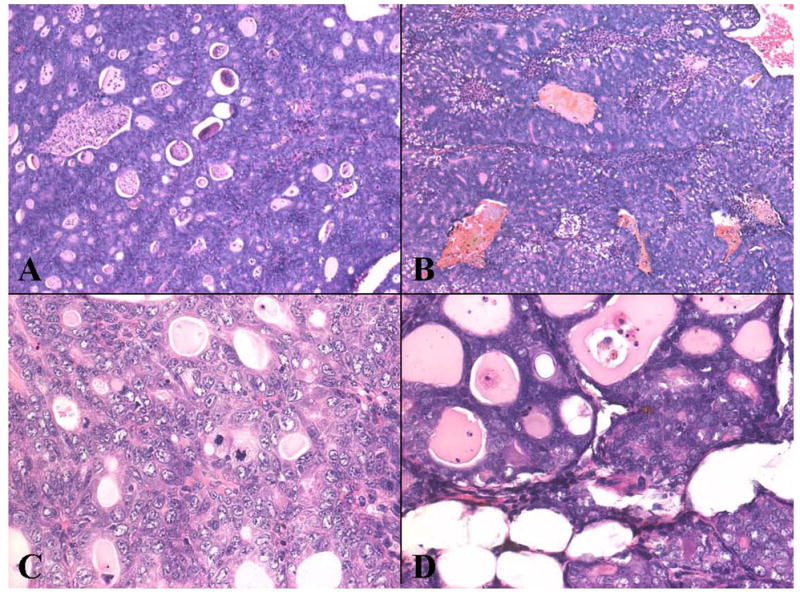
The tumors were composed of solid nests of neoplastic cells and well-differentiated glands. Both histological forms showed considerable atypia and a high mitotic index. Tumor necrosis was pronounced in most tumors. Original magnification 5X (A, C) and 20X (C, D).
Peroxisomes were not examined in liver because we did not utilize electromicroscopy; however, catalase, as expected, was elevated significantly in groups of control and estradiol-treated ACI rats fed diets containing clofibrate under conditions identical to those used in the current report (Mesia-Vela et al., 2004). Hypertrophy of hepatocytes was minimal. Mitotic index was high relative to historic age matched control ACI rats implanted with cholesterol pellets alone.
4.4. NADPH-dependent oxidative metabolism of E2 by rat liver microsomes
Incubation of E2 with liver microsomes from ACI rats and NADPH resulted in the formation of 2-OH E2, 4-OH E2, 2-OH E1 and E1 as well as unidentified metabolites; 2-OH E2 and E1 were the major metabolites observed (Table 2). Treatment with dietary clofibrate alone or together with an E2 stimulated liver microsomal metabolism of E2 to 2-OH E2, 4-OH E2 and 2-OH E1 by several fold, and this corresponded to an increase in the total amount of E2 metabolized (Table 2). Treatment of the rats with E2 stimulated the metabolism of E2 to E1, but this treatment had no effect on the hydroxylation of E2 in the 2- or 4-position (Table 2).
Table 2.
Effect of dietary clofibrate on the NADPH-dependent oxidation of estradiol by liver microsomes from ACI rats
| Treatment in diet | E2 (mg) pellet | Estradiol metabolites formed (pmol/mg protein/min)
|
E2 metabolized pmol/mg protein/min (%)a | E2 metabolized nmol/liver/min | |||
|---|---|---|---|---|---|---|---|
| 4-OH- E2 | 2 -OH- E2 | 2-OH- E1 | E1 | ||||
| 6 weeks | |||||||
| A. AIN-76A | 0 | 13 ± 2 | 154 ± 5 | 20 ± 1 | 83 ± 11 | 387 ± 27 (16%) | 32 ± 3 |
| B. AIN-76A | 1 | 18 ± 4 | 195 ± 22 | 18 ± 2 | 121 ± 8* | 526 ± 11 (21%) | 56 ± 15 |
| C. 0.4% clofibrate | 1 | 27 ± 3# | 402 ± | 42 ± 3# | 140 ± | 825 ± 54# (33%) | 147 ± 19# |
| D. AIN-76A | 3 | 14 ± 4 | 191 ± 18 | 21 ± 4 | 125 ± | 487 ± 26 (17%) | 42 ± 5 |
| E. 0.4% clofibrate | 3 | 34 ± 7# | 427 ± | 34 ± 6# | 115 ± 18 | 835 ± 59# (33%) | 155 ±18# |
| F. 0.4% clofibrate | 0 | 34 ± 7* | 547 ± | 46 ± 6* | 137 ± | 989 ± 35* (40%) | 181 ±12* |
| 12 weeks | |||||||
| A. AIN-76A | 0 | 13 ±2 | 154 ± 12 | 17 ± 3 | 93 ± 7 | 384 ± 24 (14%) | 36 ± 9 |
| B. AIN-76A | 1 | 9 ± 1 | 150 ± 29 | 18 ± 3 | 150 ± | 413 ± 30 (14%) | 31 ± 3 |
| C. 0.4% clofibrate | 1 | 9 ± 2 | 368 ± | 16 ± 3 | 97 ± 17# | 638 ± 46# (22%) | 113 ± 6# |
| D. AIN-76A | 3 | 12 ± 2 | 182 ± 78 | 27 ± 6 | 157 ± 25* | 404 ± 64 (16%) | 40 ± 5 |
| E. 0.4% clofibrate | 3 | 16 ± 5 | 278 ± 36# | 19 ± 4 | 66 ± 14# | 606 ± 56# (16%) | 95 ± 10# |
| F. 0.4% clofibrate | 0 | 18 ± 5 | 246 ± 18* | 23 ± 4 | 127 ± 13* | 507 ± 37* (20%) | 81 ± 4* |
| D. AIN-76A | 3 | 12 ± 2 | 182 ± 78 | 27 ± 6 | 157 ± 25* | 404 ± 64 (16%) | 40 ± 5 |
| E. 0.4% clofibrate | 3 | 16 ± 5 | 278 ± 36# | 19 ± 4 | 66 ± 14# | 606 ± 56# (16%) | 95 ± 10# |
| F. 0.4% clofibrate | 0 | 18 ± 5 | 246 ± 18* | 23 ± 4 | 127 ± 13* | 507 ± 37* (20%) | 81 ± 4* |
Liver microsomes (0.5 mg protein) were incubated at 37 °C for 20 min with 50 μM [3H]-estradiol (E2), 2 mM NADPH and 5 mM ascorbic acid. Each value is mean ± S.E. obtained with liver microsomes from 5 rats.
Percentage of E2 metabolized is indicated in parenthesis.
Statistically different from control group receiving a 20 mg cholesterol pellet,
Statistically different from group receiving the same amount of E2, P <0.05). (ANOVA followed by Bonferroni hoc post test)
4.5. Esterification of E2 by rat liver microsomes
Incubation of [3H]E2 with liver microsomes in the presence of oleoyl-CoA as a cofactor resulted in the formation of a single radioactive peak less polar than E2, and this corresponded to the previously reported E2-17β-oleoyl ester (Xu et al., 2001b). Formation of E2-oleoyl ester was not altered by E2 administration but was increased many fold after treatment with clofibrate for 6 or 12 weeks (Table 3).
Table 3.
Esterification of E2 by liver microsomes from control and clofibrate-fed ACI rats
| Treatment | E2 mg in pellet | E2-oleoyl-ester formed (pmol/mg protein/min) | E2-oleoyl-ester formed (nmol/liver/min) |
|---|---|---|---|
| 6 weeks | |||
| A. AIN-76A | 0 | 12 ± 2 | 1.10 ± 0.17 |
| B. AIN-76A | 1 | 9 ± 2 | 1.08 ± 0.05 |
| C. 0.4% Clofibrate | 1 | 85 ± 3# | 14.32 ± 1.67# |
| D. AIN-76A | 3 | 8 ± 2 | 0.66 ± 0.15 |
| E. 0.4% Clofibrate | 3 | 74 ± 8# | 13.21 ± 1.91# |
| F. 0.4% Clofibrate | 0 | 72 ± 7* | 12.50 ± 0.69* |
| 12 weeks | |||
| A. AIN-76A | 0 | 15 ± 2 | 1.30 ± 0.08 |
| B. AIN-76A | 1 | 14 ± 2 | 1.23 ± 0.12 |
| C. 0.4% Clofibrate | 1 | 55 ± 9# | 10.53 ± 1.30# |
| D. AIN-76A | 3 | 7 ± 2* | 0.61 ± 0.12* |
| E. 0.4% Clofibrate | 3 | 20 ± 3# | 5.22 ± 1.26# |
| F. 0.4% Clofibrate | 0 | 28 ± 5* | 4.65 ± 0.91* |
Liver microsomes (1 mg protein/ml) were incubated at 37 °C for 30 min with 50 μM [3H]-E2 (3 μCi), 100 μM oleoyl-CoA. Data are presented as the mean ± S.E. of five animals per group.
Statistically different from the control group receiving only a 20 mg cholesterol pellet,
Statistically different from the group receiving same amount of E2, P <0.05). (ANOVA followed by Bonferroni hoc post test)
4.6. Glucuronidation of E2 by rat liver microsomes
Similar amounts of E2-3-glucuronide and E2-17-glucuronide were formed by liver microsomes from control animals incubated with 0.1 mM [3H]E2. Rates of formation of each glucuronide by microsomes isolated from animals after 6 and 12 weeks were essentially the same: (221 ± 33 and 239 ± 33 μmol/min/liver) vs (233 ± 29 and 237 ± 93 μmol/min/liver), respectively. Formation of glucuronides was not modified by treatment with either 1 or 3mg E2-containing pellets alone for 6 or 12 weeks. Clofibrate treatment alone increased the formation of E2-3-glucuronide by about 60% (p<0.01). This modest stimulatory effect was not observed when clofibrate was co-administered with E2 (data not presented).
4.7. Hepatic NQO1 Activity
NQO1 activity in livers from fed control and 0.4% clofibrate-containing diets is shown in Figure 4. Activity of NQO1 was reduced significantly (25–37%) in hepatic cytosol of rats fed clofibrate-containg diets for 4 weeks and remained depressed for at least 12 weeks. It is noteworthy that reduction of this enzyme occurred prior to the detection of palpable mammary tumors (Fig. 4). We cannot predict that other compounds that are PPAR alpha agonist would produce similar effects on tumor development and NQO1 activity to those noted with clofibrate in the ACI rat model. PPAR alpha agonists effects variable and may be strain-dependent (e.g., Wy 14643 treatment decreases NQO1 activity in rats but only sporadically affected hamsters. Gemfibrozil induced an increase in NQO1 activity in rats and hamsters (O’Brein et al., 2001, Tox.icological Sciences, 60:271–278).
Fig. 4. Effect of dietary clofibrate on hepatic NQO1 activity.
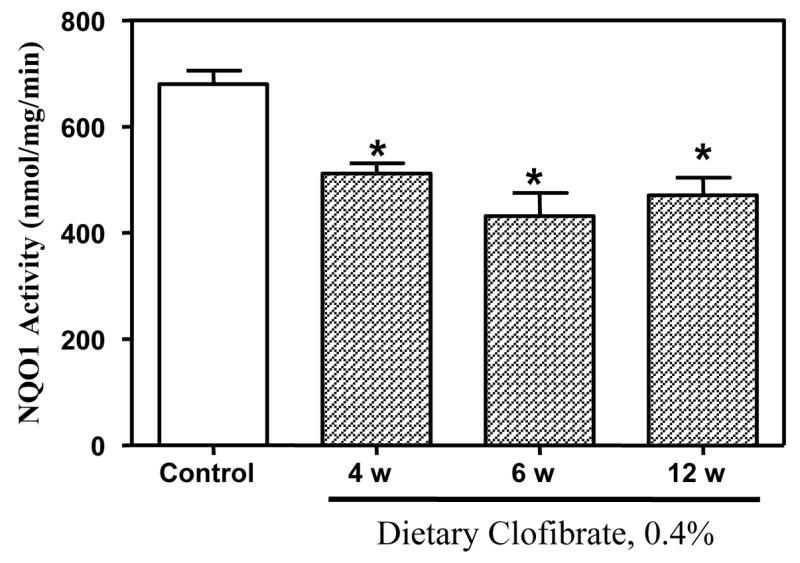
Activities of NQO1 were determined using 1–5 mg of cytosolic protein at 25°C as described in Methods. Activities measured in cytosols from control rats at 4, 6 and 12 weeks were pooled (N=4 at each period) since were not different. Activities in rats fed clofibrate-containing diets were determined in groups (n=4) at 4, 6, and 12 weeks. The height of the bar is the average S.E.M of 12 control and 4 clobrate-fed rats at each time period. *p < 0.05
5. Discussion and Conclusions
5.1. Mammary Carcinogenesis
Administration of dietary clofibrate enhanced E2-induced mammary carcinogenesis as indicated by increased number and size of palpable mammary tumors. Hispathology of E2-induced mammary tumors ACI rats fed clofibrate was the same as that in E2-treated rats fed control diets suggesting that tumor type was not altered by clofibrate treatment. The mechanism of tumor induction does not appear to be related to alterations in circulating levels of E2 since these were basically the same in control and clofibrate-fed rats. Clofibrate is a well known peroxisome proliferator (Hess et al., 1965; Reddy and Lalwani, 1983) that has non-genotoxic hepatocarcinogenic activity in rodents (Reddy et al., 1980; Nilakantan et al., 1998). Induction of peroxisomes leads to increased expression of genes encoding enzymes of the peroxisomal β-oxidation pathway (Yeldandi et al., 2000). Among these enzymes, acyl-coenzyme A oxidase generates excessive amounts of free radicals that in turn initiate oxidative stress in the cell. Sustained oxidative stress is thought to play a key role in oncogenesis (Rao and Reddy, 1996). The hepatocarcinogenicity of clofibrate in rats is well established and is correlated to ROS-mediated impairment of mitochondrial function (Qu et al., 2001). Future studies directed at determining if clofibrate induces oxidative stress in mammary tissue and whether this contributes to enhanced mammary carcinogenesis would be valuable.
5.2. Hydroxylation of estradiol
The possibility that clofibrate stimulates the formation of catechol estrogen quinones, which promote breast tumorigenesis has been suggested by Cavalieri and his colleagues (Cavalieri et al., 1997; Cavalieri et al., 2002). This mechanism, however, seems unlikely because administration of 2-OH E2, 4-OH E1 and 4-OH E2 via implanted cholesterol pellets did not cause mammary tumors in the ACI rat (Turan et al., 2005), and the ortho-quinone derived from 4-OH E1 does not cause mammary tumors when injected into the mammary gland of rats (el-Bayoumy et al., 1996). Results of a recent parallel study carried out in ACI rats treated with phenobarbital in drinking water (Mesia-Vela et al., 2006) indicated that phenobarbital had a large stimulatory effect on the conversion of E2 to catechol estrogens by liver microsomes, yet phenobarbital treatment markedly inhibited E2-induced mammary carcinogenesis (Mesia-Vela et al., 2006). Further, the inhibitory action of phenobarbital on E2-induced carcinogenesis was accompanied by a marked stimulation of NQO1 activity and a significant inhibition of E2 esterification. These results are in agreement with the present findings in which stimulation of mammary carcinogenesis by dietary clofibrate was accompanied by a marked induction of E2 esterification coupled with inhibition of NQO1.
5.3. Esterification of Estradiol
Fatty acid esters of estrogens are a unique family of extremely hydrophobic hormonal derivatives that are transported in blood exclusively in lipoprotein particles Miilunpohja, et al., 2006). Esters of estradiol formed largely in liver have prolonged activity in vivo because they are slowly metabolized (Larner and Hochberg, 1985) and large amounts accumulate in extrahepatic fatty tissues (Larner et al., 1992). Thus, in theory, treatment of ACI rats with clofibrate would enhance the formation of fatty acid esters in the liver that would subsequently be transported to and stored in fat rich tissues such as mammary gland. Subsequent hydrolysis of sequestered fatty acid esters of estradiol by esterases present in mammary tissue would release biologically active hormone. In accord with this idea, administration of clofibrate to ovarectomized rats infused with estradiol resulted in selective hormonal stimulation of the mammary gland but not the uterus (Xu, et al., 2001c). The biological significance of the fall in esterase activity from 6 to 12 weeks is unknown. Future studies directed at measuring circulating amounts of estradiol fatty acid esters, as well as other conjugates of estradiol, in the ACI rat model would be valuable in addressing this question.
5.4 NQO1
NQO1 is considered a prototype of phase 2 enzymes that are up-regulated by a number of oxidants and electrophiles acting via the Keap1-Nrf2 signaling pathway and antioxidant response elements (Dinkova-Kostova et al., 2005). Studies by Talalay and his co-workers over a number of years have established a strong relationship between the induction of NQO1 and protection against chemically-induced carcinogenesis in a number of animal models (Talalay, 2005). For example, these investigators found that sulforaphane, an isothiocyanate isolated from broccoli sprouts, was a very potent inducer of NQO1 in murine hepatoma cells in culture and also markedly inhibited the formation of mammary tumors in Sprague-Dawley rats treated with 9, 10-dimethyl-1,2 benzanthracene (DMBA) (Zhang et al., 1994). More than 10 different classes of chemicals that react chemically with thiol groups by alkylation, oxidation or reduction have been found to be inducers of NQO1 and other phase 2 enzymes (Talalay, 2005). Based on a recent survey of NQO1-inducing activity in extracts of approximately 3000 plants and marine organisms, and the finding that one of these compounds also inhibited DMBA-induced mammary tumors in Sprague-Dawley rats, it was suggested that NQO1 induction serves as a biomarker of cancer chemoprevention (Cuendet, et al. 2006). Dietary clofibrate reduced the activity of NQO1 activity and enhanced mammary tumor formation in ACI rats treated chronically with E2. Moreover, E2-induced increases in this activity are completely inhibited in ACI rats fed clofibrate-containing diets under identical conditions to those used in the present study (Mesia-Vela et al., 2004). In contrast, chronic phenobarbital treatment increases hepatic NQO1 activity and stimulates mammary tumor formation in this model (Mesia-Vela, et al., 2006). These findings are in accord with the idea that induction of NQO1 may serve as a useful index of anticarcinogenic activity.
NQO1 has been suggested to detoxify catechol estrogen quinones and to reduce damage to DNA that may arise from reactive electrophiles and reactive oxygen species generated redox cycling in estrogen sensitive tissues (Cavelieri, et al., 1997, 2002) Recent studies on the functional role of ER β-mediated upregulation of NQO1 in the ACI rat in vivo and in mammary epithelial cells in vitro suggest that increases in NQO1 are associated with the protection against estrogen-induced mammary tumorigenesis secondary to inhibition of oxygen dependent DNA damage (Montano et al., 2006). Data reported above, and previously (Mesia-Vela, et al., 2004), indicating that both constituitive and E2-induced activities of hepatic NQO1 and glutathione S-transferase are markedly decreased by dietary clofibrate, argue strongly that this important antioxidant mechanism of protection against estrogen-dependent mammary tumorigenesis has been inactivated by clofibrate in the ACI rat.
6. Conclusion
Results of the present study indicate a stimulatory effect of dietary clofibrate on E2-induced mammary tumorigenesis in female ACI rats in the absence of detectable effects of clofibrate on serum levels of E2. Stimulation of mammary tumogenesis was accompanied by enhanced hepatic fatty acid esterification of E2 and inhibition of NQO1. Although the mechanism(s) underlying this stimulatory effect of dietary clofibrate on E2-induced mammary carcinogenesis remains unclear, it is plausible that enhanced hepatic fatty acid esterification leading to increased levels of E2 fatty acid esters in fatty tissues such as the mammary gland, induction of esterase activity that liberates estradiol in the target tissue, and inhibition of protective phase 2 enzyme activities contribute to the formation of E2-dependent mammary tumors. Future studies exploring the relationship, if any, between hormone-dependent mammary carcinogenesis in female ACI rats and elevated hepatic fatty acid esterification accompanied by inhibition of antioxidant phase-2 enzymes should be directed at determining profiles of E2 metabolites in the circulation and target tissues as well as measuring activities of key enzymes associated with esterification/de-esterification and anti-oxidation in mammary tissue.
Acknowledgments
This work was supported in part by NIH/NIEHS ES05022.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Cavalieri EL, Li K, Balu N, Saeed M, Devanesan P, Higginbotham S, Zhao J, Gross LM, Rogan EG. Catechol ortho-quinones: the electrophilic compounds that form depurinating DNA adducts and could initiate cancer and other diseases. Carcinogenesis. 2002;23:1071–1077. doi: 10.1093/carcin/23.6.1071. [DOI] [PubMed] [Google Scholar]
- Cavalieri EL, Stack DE, Devanesan PD, Todorovic R, Dwivedy I, Higginbotham S, Johansson SL, Patil KD, Gross ML, Gooden JK, Ramanathan R, Cerny RL, Rogan EG. Molecular origin of cancer: catechol estrogen-3,4 quinones as endogenous tumor initiators. Proc Natl Acad Sci USA. 1997;99:10937–10942. doi: 10.1073/pnas.94.20.10937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuendet M, Oteham CP, Moon RC, Pezzuto JM. Quinone reductase induction as a biomarker for cancer chemoprotection. J Nat Prod. 2006;69:460–463. doi: 10.1021/np050362q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el-Bayoumy K, Ji BY, Upadhyaya P, Chae YH, Kurtzke C, Rivenson A, Reddy BS, Amin S, Hecht SS. Lack of tumorigenicity of cholesterol epoxides and estrone-3,4-quinone in the rat mammary gland. Cancer Res. 1996;56:1970–1973. [PubMed] [Google Scholar]
- Dinkova-Kostova AT, Holtzclaw WD, Kensler TW. The role of Keap1 in cellular protective responses. Chem Res Toxicol. 2005;18:1779–1791. doi: 10.1021/tx050217c. [DOI] [PubMed] [Google Scholar]
- Harvell DM, Strecker TE, Tochacek M, Xie B, Pennington KL, McComb RD, Roy SK, Shull JD. Rat strain-specific actions of 17 beta-estradiol in the mammary gland: correlation between estrogen-induced lobuloalveolar hyperplasia and susceptibility to estrogen-induced mammary cancers. Proc Natl Acad Sci USA. 2000;97:2779–2784. doi: 10.1073/pnas.050569097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess R, Staubli W, Riess W. Nature of the hepatomegalic effect produced by ethyl-chlorophenoxy-isobutyrate in the rat. Nature. 1965;208:856–858. doi: 10.1038/208856a0. [DOI] [PubMed] [Google Scholar]
- Hershcopf RJ, Bradlow HL, Fishman J. Differential hydroxylations of estrone and estradiol in man. J Clin Endocrinol Metab. 1986;62:170–173. doi: 10.1210/jcem-62-1-170. [DOI] [PubMed] [Google Scholar]
- Larner JM, Hochberg RB. The clearance and metabolism of estradiol-17 esters in the rat. Endocrinol. 1985;117:1209–1214. doi: 10.1210/endo-117-3-1209. [DOI] [PubMed] [Google Scholar]
- Larner JM, Shackleton CHL, Roitman E, Schwartz PE, Hochberg RB. Measurement of estradiol-17-esters in human tissues. J Clin Endocrinol Metab. 1992;75:195–200. doi: 10.1210/jcem.75.1.1619010. [DOI] [PubMed] [Google Scholar]
- Li SA, Weroha SJ, Tawfik O, Li JJ. Prevention of solely estrogen-induced mammary tumors by Tamoxifen: Evidence for estrogen receptor mediation. J Endocrinol. 2002;175(2):297–305. doi: 10.1677/joe.0.1750297. [DOI] [PubMed] [Google Scholar]
- Mesia-Vela S, Sanchez RI, Reuhl KR, Conney AH, Kauffman FC. Phenobarbital treatment inhibits the formation of estradiol-dependent mammary tumors in the august-copenhagen irish rat. J Pharmacol Exp Ther. 2006;317(2):590–7. doi: 10.1124/jpet.105.096867. [DOI] [PubMed] [Google Scholar]
- Mesia-Vela S, Sanchez RI, Reuhl KR, Conney AH, Kauffman FC. Dietary clofibrate inhibits induction of hepatic antioxidant enzymes by chronic estradiol in female ACI rats. Toxicology. 2004;200(2–3):103–11. doi: 10.1016/j.tox.2004.03.009. [DOI] [PubMed] [Google Scholar]
- Mesia-Vela S, Sanchez RI, Li JJ, Li SA, Conney AH, Kauffman FC. Catechol estrogen formation in liver microsomes from female ACI and Sprague-Dawley rats: comparison of 2-and 4-hydroxylation revisited. Carcinogenesis. 2002;23(8):1369–72. doi: 10.1093/carcin/23.8.1369. [DOI] [PubMed] [Google Scholar]
- Miilunpohja M, Uphoff A, Somerharju P, Tiitinen A, Wahala, Tikkanen MJ. Fatty acid esterification of lipoprotein-associated estrone in human plasma and follicular fluid. J Steroid Biochem Mol Biol. 2006;100(2):59–66. doi: 10.1016/j.jsbmb.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Montano MM, Chaplin LJ, Deng H, Mesia-Vela S, Gaikwad N, Zahid M, Rogan E. Protective roles of quinone reductase and tamoxifen against estrogen-induced mammary tumorigenesis. Oncogene. 2006:1–4. doi: 10.1038/sj.onc.1210144. [DOI] [PubMed] [Google Scholar]
- Nilakantan V, Apear BT, Glauert HP. Effect of the peroxisome proliferator ciprofibrate on lipid peroxidation and 8-hydroxydeoxyguanosine formation in transgenic mice with elevated hepatic catalase activity. Free Radic Biol Med. 1998;24(9):1430–6. doi: 10.1016/s0891-5849(98)00007-0. [DOI] [PubMed] [Google Scholar]
- O’Brien ML, Twaroski TP, Cunningham ML, Glauert HP, Spear BT. Effects of peroxisome proliferators on antioxidant enzymes and antioxidant vitamins in rats and hamsters. Toxicol Sci. 2001;60:271–278. doi: 10.1093/toxsci/60.2.271. [DOI] [PubMed] [Google Scholar]
- Qu B, Li OT, Wong KP, Tan TM, Halliwell B. Mechanism of clofibrate hepatotoxicity: mitochondrial damage and oxidative stress in hepatocytes. Free Radic Biol Med. 2001;31(5):659–69. doi: 10.1016/s0891-5849(01)00632-3. [DOI] [PubMed] [Google Scholar]
- Rao MS, Reddy JK. Hepatocarcinogenesis of peroxisome proliferators. Ann NY Acad Sci. 1996;804:573–87. doi: 10.1111/j.1749-6632.1996.tb18646.x. [DOI] [PubMed] [Google Scholar]
- Reddy JK, Lalwal ND. Carcinogenesis by hepatic peroxisome proliferators, evaluation of the risk of hypolipidemic drugs and industrial plasticizers to humans. Crit Rev Toxicol. 1983;12:1–58. doi: 10.3109/10408448309029317. [DOI] [PubMed] [Google Scholar]
- Reddy JK, Azarnoff DL, Hignite CE. Hypolipidaemic hepatic peroxisome proliferators form a novel class of chemical carcinogens. Nature. 1980;283:397–398. doi: 10.1038/283397a0. [DOI] [PubMed] [Google Scholar]
- Sanchez RI, Mesia-Vela S, Kauffman FC. Induction of NADP(H) quinone oxidoreductase and glutathione S-transferase activities in livers of female August-Copenhagen Irish rats treated chronically with estradiol: comparison with the Sprague-Dawley rat. J Steroid Biochem Mol Biol. 2003;87(2–3):199–206. doi: 10.1016/j.jsbmb.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Shah N, Antony T, Haddad S, Amenta P, Shirahata A, Thomas TJ, Thomas T. Antitumor effects of bis(ethyl)polyamine analogs on mammary tumor development in FVB/NTgN (MMTVneu) transgenic mice. Cancer Lett. 1999;146(1):15–23. doi: 10.1016/s0304-3835(99)00215-3. [DOI] [PubMed] [Google Scholar]
- Shull JD, Spady TJ, Snyder MC, Johansson SL, Pennington KL. Ovary-intact, but not ovarectomized female ACI rats treated with 17b-estradiol develop mammary carcinomas. Carcinogenesis. 1997;15(8):1595–1601. doi: 10.1093/carcin/18.8.1595. [DOI] [PubMed] [Google Scholar]
- Spady TJ, Pennington KL, McComb RD, Shull JD. Genetic bases of estrogen-induced pituitary growth in an intercross between the ACI and Copenhagen rat strains: dominant mendelian inheritance of the ACI phenotype. Endocrinology. 1999 Jun;140(6):2828–35. doi: 10.1210/endo.140.6.6757. [DOI] [PubMed] [Google Scholar]
- Stone JP, Holtzman S, Shellabarger CJ. Neoplastic responses and correlated plasma prolactin levels in diethylstilbestrol-treated ACI and Spraque-Dawley rats. Cancer Res. 1979;39:773–778. [PubMed] [Google Scholar]
- Suchar LA, Chang RL, Rosen RT, Lech J, Conney AH. High-performance liquid chromatography separation of hydroxylated estradiol metabolites: formation of estradiol metabolites by liver microsomes from male and female rats. J Pharmacol Exp Ther. 1995;272:197–206. [PubMed] [Google Scholar]
- Talalay P. A fascination with enzymes: the journey not the arrival matters. J Biol Chem. 2005;280:28829–28847. doi: 10.1074/jbc.X500004200. [DOI] [PubMed] [Google Scholar]
- Thomas PE, Reick LM, Ryan DE, Levin W. Induction of two immunochemically related rat liver cytochrome P450 isozymes cytochromes P450c and P450d, by structurally diverse xenobiotics. J Biol Chem. 1983;258:4590–4598. [PubMed] [Google Scholar]
- Turan VK, Sanchez RI, Li JJ, Li SA, Reuhl KR, Thomas PE, Conney AH, Gallo MA, Kauffman FC, Mesia-Vela S. The effects of steroidal estrogens in ACI rat mammary carcinogenesis: 17beta-estradiol, 2-hydroxyestradiol, 4-hydroxyestradiol, 16alpha-hydroxyestradiol, and 4-hydroxyestrone. J Endocrinol. 2004;183(1):91–9. doi: 10.1677/joe.1.05802. [DOI] [PubMed] [Google Scholar]
- Xu S, Zhu BT, Conney AH. Effect of clofibrate administration on the esterification and deesterification of steroid hormones by liver and extrahepatic tissues in rats. Biochem Pharmacol. 2002;63(5):985–92. doi: 10.1016/s0006-2952(01)00921-2. [DOI] [PubMed] [Google Scholar]
- Xu S, Zhu BT, Turan V, Rusyn I, Thurman R, Peters JM, Gonzalez FJ, Conney AH. PPARalpha-dependent induction of liver microsomal esterification of estradiol and testosterone by a prototypical peroxisome proliferator. Endocrinol. 2001a;142(8):3554–7. doi: 10.1210/endo.142.8.8330. [DOI] [PubMed] [Google Scholar]
- Xu S, Zhu BT, Conney AH. Stimulatory effect of clofibrate and gemfibrozil administration on the formation of fatty acid esters of estradiol by rat liver microsomes. J Pharmacol Exp Ther. 2001b;296:188–197. [PubMed] [Google Scholar]
- Xu S, Zhu BT, Cai MX, Conney AH. Stimulatory effect of clofibrate on the action of estradiol in the mammary gland but not in the uterus of rats. J Pharmacol Exp Ther. 2001c;297(1):50–6. [PubMed] [Google Scholar]
- Yeldandi AV, Rao MS, Reddy JK. Hydrogen peroxide generation in peroxisome proliferator-induced oncogenesis. Mutat Res. 2000;448(2):159–77. doi: 10.1016/s0027-5107(99)00234-1. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Kensler TW, Cho CG, Posner GH, Talalay P. Anticarcinogenic activities of sulforaphane and structurally related synthetic norbornyl isothiocyanates. Proc Natl Acad Sci USA. 1994;91:3147–3150. doi: 10.1073/pnas.91.8.3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielinski JE, Pahuja SL, Larner JM, Hochberg RB. Estrogenic action of estriol fatty acid esters. J Steroid Biochem Molec Biol. 1991;38:399–405. doi: 10.1016/0960-0760(91)90327-2. [DOI] [PubMed] [Google Scholar]