

Abstract
Prior work has shown that d-amphetamine (AMPH) treatment or voluntary exercise improves cognitive functions after traumatic brain injury (TBI). In addition, voluntary exercise increases levels of brain-derived neurotrophic factor (BDNF). The current study was conducted to determine how AMPH and exercise treatments, either alone or in combination, affect molecular events that may underlie recovery following controlled cortical impact (CCI) injury in rats. We also determined if these treatments reduced injury-induced oxidative stress. Following a CCI or sham injury, rats received AMPH (1 mg/kg/day) or saline treatment via an ALZET® pump and were housed with or without access to a running wheel for 7 days. CCI rats ran significantly less than sham controls, but exercise level was not altered by drug treatment. On day 7 the hippocampus ipsilateral to injury was harvested and BDNF, synapsin I and phosphorylated (P) -synapsin I proteins were quantified. Exercise or AMPH alone significantly increased BDNF protein in sham and CCI rats, but this effect was lost with the combined treatment. In sham-injured rats synapsin I increased significantly after AMPH or exercise, but did not increase after combined treatment. Synapsin levels, including the P-synapsin/total synapsin ratio, were reduced from sham controls in the saline-treated CCI groups, with or without exercise. AMPH treatment significantly increased the P-synapsin/total synapsin ratio after CCI, an effect that was attenuated by combining AMPH with exercise. Exercise or AMPH treatment alone significantly decreased hippocampal carbonyl groups on oxidized proteins in the CCI rats, compared with saline-treated sedentary counterparts, but this reduction in a marker of oxidative stress was not found with the combination of exercise and AMPH treatment. These results indicate that, whereas exercise or AMPH treatment alone may induce plasticity and reduce oxidative stress after TBI, combining these treatments may cancel each other’s therapeutic effects.
Keywords: controlled cortical impact, norepinephrine, oxidized proteins, running wheel, traumatic brain injury
Traumatic brain injury (TBI) continues to be one of the leading causes of mortality and morbidity, with approximately 5.3 million Americans suffering from enduring disabilities resulting from TBI (Binder et al., 2005). Despite many experimental and clinical efforts using numerous therapeutic approaches, there are currently no proven treatments to alleviate the sequelae of TBI or to enhance recovery of function.
Our laboratories have been investigating treatment strategies that may facilitate endogenous repair mechanisms and enhance functional recovery after experimental TBI in the rat. One approach has been to use spontaneous or voluntary exercise, obtained via running wheel (RW) exposure. Exercise is known to increase key molecules involved in neuroplasticity, such as brain-derived neurotrophic factor (BDNF) and synapsin I (Neeper et al., 1995; van Praag et al., 1999; Cotman and Berchtold, 2002), as well as reduce markers of oxidative stress (Navarro et al., 2004; Wu et al., 2004; Pan et al., 2007). While experimental TBI can acutely decrease expression of BDNF mRNA within the hippocampus (Hellmich et al., 2005), most studies have reported hippocampal increases in BDNF mRNA within the first 24 h following TBI (Oyesiku et al., 1999; Truettner et al., 1999; Morrison et al., 2000). At later post-injury times, neither hippocampal BDNF mRNA (Hicks et al., 2002) nor BDNF protein differs from that of controls (Griesbach et al., 2004b, 2007; Chen et al., 2005). However, spontaneous exercise in rats with TBI can increase the expression and/or levels of BDNF and synapsin I within the hippocampus (Hicks et al., 1998; Griesbach et al., 2004b, 2007), and this effect is associated with an improvement in cognitive performance (Griesbach et al., 2004b). The timing of this post-injury exercise has been shown to play an important role after experimental fluid percussion injury (FPI) to the brain. We previously reported that voluntary exercise during the first week after sham injury increased hippocampal BDNF and synapsin I protein, but exercise did not increase these molecules after a FPI (Griesbach et al., 2004a, 2007).
The mechanisms underlying the inability of acute exercise to up-regulate BDNF after TBI are not known, but may be related to alterations in neurotransmitter systems during this acute period following injury. Transmitter depletion studies have shown that norepinephrine (NE) is necessary for voluntary wheel-running to up-regulate BDNF mRNA in the hippocampus (Garcia et al., 2003). Anti-depressant drugs, which increase synaptic levels of NE and 5-HT, have been reported to increase levels of hippocampal BDNF mRNA (Nibuya et al., 1995) and protein (Peng et al., 2008; Kozisek et al., 2008). In addition, anti-depressant drugs strengthen BDNF mRNA up-regulation when given in combination with exercise (Russo-Neustadt et al., 1999, 2000). Repeated treatments with d-amphetamine (AMPH) have also been reported to increase BDNF levels in multiple brain regions (Meredith et al., 2002). Brain injury induced by experimental stroke or cortical ablation reduces brain levels and/or turnover rates of NE, dopamine (DA) and/or 5-HT for days or weeks after injury (Feeney and Sutton, 1987). Experimental TBI in the rat produces early and widespread reductions in brain NE levels and turnover (Dunn-Meynell et al., 1994; Krobert et al., 1994; Levin et al., 1995). Hence, although exercise in non-injured rats is known to increase NE release (Dunn et al., 1996; Dishman et al., 2000; Garcia et al., 2003), a TBI-induced reduction of NE could conceivably prevent exercise-dependent increases in NE and BDNF during the acute period after injury.
Another approach for the treatment of TBI has been to administer treatments that increase central levels of monoamines. Single or multiple treatments with AMPH have been frequently reported to accelerate functional recovery in various brain injury models (Feeney and Sutton, 1988; Sutton and Feeney, 1994), including cortical contusion (Sutton et al., 1987) or FPI (Prasad et al., 1994; Hovda, 1996). While AMPH increases levels of all the mono-amines (Fleckenstein et al., 2007), the beneficial effects of post-injury AMPH treatment appear to be mediated by NE release since intraventricular NE, but not DA (Boyeson and Feeney, 1990) or 5-HT (Boyeson et al., 1994), mimics the AMPH induced recovery of function in ablation models. In addition, drugs causing central NE release induce recovery similar to AMPH (Sutton and Feeney, 1992; Goldstein, 1993), and drugs that interfere with NE release or synaptic transmission can retard recovery or reinstate deficits in recovered animals (Feeney and Westerberg, 1990; Goldstein and Davis, 1990; Dunn-Meynell et al., 1997). In addition to AMPH, multiple treatments with methylphenidate (Kline et al., 2000), a D2 receptor agonist (Kline et al., 2002) or a 5- HT(1A) receptor agonist (Kline et al., 2004) are reported to improve outcome after experimental TBI. More recently, chronic intermittent vagus nerve stimulation, which increases NE release (Roosevelt et al., 2006) and BDNF mRNA levels (Follesa et al., 2007) in cortex and hippocampus, has been shown to improve outcome after experimental TBI (Smith et al., 2005, 2006; Clough et al., 2007). Given these preceding reports on the effects of TBI and the apparent links between AMPH, NE, exercise and BDNF, the current study was designed to determine if AMPH administration during the acute period after TBI would enable or facilitate spontaneous exercise-induced increases of hippocampal BDNF and synapsin I. Because oxidative stress plays an important role in neuronal injury after TBI (Shohami et al., 1997; Sullivan et al., 1998; Hall et al., 2004), we also determined if these treatments would alter oxidative stress after TBI. We employed the controlled cortical impact (CCI) model of TBI for this study, as this injury model is known to produce molecular changes and cellular damage in the hippocampus (Dash et al., 1995; Colicos et al., 1996; Oyesiku et al., 1999; McCullers et al., 2002; Saatman et al., 2006), induce hippocampal-dependent learning deficits (Hamm et al., 1992; Lindner et al., 1998; Dixon et al., 1999), and to reduce brain NE levels and turnover (Dunn-Meynell et al., 1994; Levin et al., 1995). While increased expression or production of hippocampal BDNF and synapsin I has been reported to occur after exercise in both intact rats and in rats with FPI (Timmusk et al., 1993; Neeper et al., 1996; Molteni et al., 2002; Griesbach et al., 2004b, 2007), exercise-induced effects of these markers of plasticity have not yet been evaluated in the CCI model. Based on prior studies we hypothesized that: 1) acute exercise after CCI would have detrimental or neutral effects on outcome measures, 2) low-dose AMPH treatment would improve these outcome measures, and 3) AMPH treatment combined with RW exposure would enable acute exercise to up-regulate BDNF and synapsin I and reduce oxidative stress.
EXPERIMENTAL PROCEDURES
Subjects
A total of 48 male Sprague–Dawley rats (mean weight: 285 g) were utilized in these experiments. Rats underwent surgery to induce either sham injury (Sham) or CCI injury followed by AMPH or saline (S) treatment. Rats were single-housed with a RW or under sedentary (Sed) conditions for 7 days, thus resulting in the following groups (n=6 rats per group): Sham-S-Sed, Sham-AMPH-Sed, Sham-S-RW, Sham-AMPH-RW, CCI-S-Sed, CCI-AMPH-Sed, CCI-S-RW, CCI-AMPH-RW. All animals were monitored and cared for by veterinary care staff upon arrival to University of California at Los Angeles (UCLA). During the experiments, rats were housed in opaque plastic bins (50.8×25.4×25.4 cm), which were lined with bedding material. All procedures were performed in accordance with the United States National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80–23) revised 1996. Procedures were approved by the UCLA Chancellor’s Animal Research Committee and can be provided upon request. The suffering and number of animals used were minimized.
CCI injury
The previously characterized CCI injury device (Sutton et al., 1993; Lindner et al., 1998; Kelly et al., 2000) was used to generate a cortical contusion to the left hemisphere. Each animal was placed under inhalation anesthesia with isoflurane (4% for induction, 2.0% for maintenance, in 100% O2 at 1.5 l/min). The level of anesthesia was monitored by level of respiration, muscular relaxation and the corneal and pedal reflexes. After loss of corneal and pedal reflexes the scalp and scapular regions were shaved, the animal was secured in a stereotaxic head frame, and the scalp was cleansed with ethanol and Betadine. Rectal temperature was monitored and maintained between 36.5–38.0 °C with a thermostatically controlled heating pad (Braintree Scientific Inc., Brain-tree, MA, USA). A midline sagittal incision was made, the scalp and temporal muscle were reflected and a 6-mm-diameter circular craniotomy was made over the left parietal cortex, centered at 3 mm posterior and 3.5 mm lateral to bregma. The bone flap was removed and the dura left intact in all animals to receive CCI. An electronically controlled pneumatic piston cylinder (Hydraulics Control Inc., Emeryville, CA, USA) mounted onto a stereotaxic micromanipulator (Kopf Instruments, Tujunga, CA, USA) was used to allow for precise localization and control of the impact (Sutton et al., 1993). The piston cylinder was angled 19° away from vertical to allow the flat (5 mm diameter) impactor tip to make contact perpendicular to the brain’s surface. A moderate CCI was induced using a 2 mm compression of tissue under the exposed dura (250 ms, 1.9 m/s velocity). After controlling for any mild bleeding following the injury, the scalp incision was sutured closed. Bupivacaine (0.25 mg) was injected into the margins of the scalp incision and triple antibiotic ointment was applied over the incision. Sham-injured animals underwent all surgical procedures, except for the craniotomies and CCI delivery.
Drug administration
Immediately after scalp closure the skin over the scapular region and nape of the neck were cleansed with Betadine and 70% ethanol. After making a small (0.5 cm) incision an s.c. pocket was created over the scapular region, a pre-filled osmotic minipump (ALZET® Model 2001; DURECT Corporation, Cupertino, CA, USA) was placed under the skin, and the incision was sutured closed. After injection of bupivacaine (0.25 mg) and application of triple antibiotic ointment to this incision, anesthesia was discontinued and animals were placed into a heated recovery cage until ambulatory. The osmotic pumps were filled with sterile S (0.9%) or freshly prepared AMPH sulfate (Sigma, St. Louis, MO, USA) dissolved in S (12 mg/ml, filtered via a 0.2 μm syringe filter) prior to each surgery session. Pumps and drug concentration were chosen to infuse (to rats weighing on average 285 g) 1 mg/kg/day of AMPH or 1.0 μl/h of the vehicle (S) over 7 days. This drug administration method was chosen since our prior studies of RW exposure after TBI have left animals undisturbed for the 1 week exercise period (Vaynman et al., 2003; Griesbach et al., 2004b, 2007). The dose was chosen based on the recent report that 1 mg/kg/day of AMPH combined with rehabilitation training facilitates recovery after ischemic brain injury (Adkins and Jones, 2005).
Voluntary wheel exercise
Rats were individually caged with or without access to a RW from post-surgery day 0–6 (a total of 7 days). The time length for exercise was based on previous studies indicating increases in BDNF and synapsin I after 7 days of voluntary exercise (Vaynman et al., 2003; Griesbach et al., 2004b, 2007). Rats had ad libitum access to food and water and were maintained on a 12-h light/dark cycle. Animals provided the opportunity for spontaneous exercise were placed in standard cages equipped with a RW (diameter=31.8 cm, width=10 cm; Nalge Nunc International, Rochester, NY, USA) that rotated against a resistance of 100 g. Non-exercised (Sed) animals were left undisturbed in their home-cages. Exercise was quantified by recording the number of wheel revolutions per hour using VitalView Data Acquisition System software (Mini Mitter Company Inc., Sunriver, OR, USA). The mean number of revolutions was calculated for each night (7 PM to 7 AM), given that this was the most active period.
Protein immunoassay and Western blot analysis
Rats were killed by decapitation at post-injury day 7 for molecular studies. Hippocampal tissue within the injured hemisphere was dissected and homogenized in homogenization buffer (137 mM NaCl, 20 mM Tris–HCl pH 8.0, 1% NP40, 10% glycerol, 1 mM PMSF, 10 μg/ml aprotinin, 0.1 mM benzethonium, 0.5 mM sodium vanadate). After centrifuging at 12,500×g for 15 min, supernatants were collected and immediately processed for total protein concentration determination according to the Micro BCA procedure (Pierce, Rockford, IL, USA), using bovine serum albumin as standard. All chemicals were obtained from Sigma unless otherwise noted.
BDNF protein was quantified using an enzyme-linked immunosorbent assay (ELISA) and standard protocols (BDNF Emax ImmunoAssay System kit; Promega Inc., Madison, WI, USA). Briefly, 96 wells plates were coated with 0.1 ml of a monoclonal antibody against BDNF in a buffer containing 0.025 M sodium bicarbonate and 0.025 M sodium carbonate (pH 9.7) for 16 h at 4 °C. After washing in TBST [20 mM Tris–HCl (pH 7.6), 150 mM NaCl, 0.05% Tween 20], wells were incubated with 0.2 ml of a blocking buffer at room temperature for 1 h and then washed in TBST again. Samples, six serial dilutions of a BDNF standard (500 pg/ml), and a blank (no BDNF) were added in triplicate into separate wells. Plates were incubated for 2 h at room temperature and washed five times in TBST. A polyclonal antibody against BDNF (1:500 dilution) was added into each well and plates were incubated for 2 h at room temperature. After five washes in TBST, 0.1 ml of a secondary anti-IgY antibody with a horseradish peroxidase conjugate was added to each well, and plates were incubated for 1 h at room temperature. Wells were washed five times with TBST. A hydrogen peroxidase solution with a peroxidase substrate was added and incubated for 10 min at room temperature. Reactions were stopped with 1 M phosphoric acid, and absorbance at 450 nm was measured using an automated microplate reader (HTS7000 Plus Bioassay Reader, Perkin Elmer, Waltham, MA, USA). Standard curves were plotted for each plate. Triplicate measures were averaged and values were corrected for total amount of protein in the sample to derive the pg of BDNF protein/mg of total protein. For each plate, the final BDNF data were expressed as the percent change from the mean Sham-S-Sed value.
Total synapsin and phosphorylated (P) -synapsin was analyzed by Western blot. Separate gels, each including samples from one experimental group and control rats (Sham-S-Sed), were processed. Actin was utilized as an internal control, and each blot was standardized to its corresponding actin value. Protein samples were separated by electrophoresis on a 10% polyacrylamide gel and electrotransferred to a polyvinylidene fluoride membrane (Millipore, Bedford, MA, USA). Non-specific binding sites were blocked in TBS with 5% nonfat milk and with 0.1% Tween-20 for 1 h at room temperature. Membranes were incubated at 4 °C overnight, with anti-actin, anti-synapsin (1:1000, Santa Cruz Bio-technology Inc., Santa Cruz, CA, USA), or anti-P-synapsin (1: 2000, Cell Signaling, Beverly, MA, USA). After rinsing in buffer four times for 10 min, membranes were incubated with anti-goat IgG horseradish peroxidase-conjugate (1:10,000 Santa Cruz Bio-technology Inc.). Immunocomplexes were analyzed by chemiluminescence using the ECL Plus kit (Amersham Pharmacia Bio-tech Inc., Piscataway, NJ, USA), according to manufacturer’s instructions. Optical densities for Sham-S-Sed blots were normalized across all gels for total synapsin and P-synapsin, and blots for each experimental group were normalized to Sham-S-Sed values within the same gel. The final total synapsin and P-synapsin data were expressed as the percent change from the mean Sham-S-Sed values. To aid in the interpretation of the synapsin data, ratios of P-synapsin to total synapsin for each rat were obtained and group means were calculated for this measure of synapsin activation.
Measurement of oxidized proteins
Because an elevation of oxidative stress, which could be caused by neuronal activation (either drug or exercise-induced), leads to neuronal damage after CCI injury (Sullivan et al., 1998; Hall et al., 2004), we measured oxidized proteins as an outcome measure of exercise and AMPH treatments after CCI. The oxidized proteins containing carbonyl groups, as a marker of cellular protein oxidation, were measured using an OxyBlot kit (Intergen, Purchase, NY, USA). Separate gels, each including samples from a CCI group and control rats (Sham-S-Sed), were processed. Briefly, carbonyl groups in the protein side chains are derivatized to 2,4-dinitrophenylhydrazone (DNPH). Protein samples were reacted with 1×DNPH for 15 min, followed by neutralization with a solution containing glycerol and β-mercaptoethanol. Samples were then electrophoresed on an 8% polyacrylamide gel and electrotransferred to a nitrocellulose membrane. After blocking and overnight incubation with a rabbit DNPH antibody (1:150) at 4 °C, membranes were incubated in goat anti-rabbit (1:300) for 1 h at room temperature and rinsed with buffer. Immunocomplexes were visualized by chemiluminescence using the ECL kit (Amersham Pharmacia Biotech Inc.), according to the manufacturer’s instructions. Optical densities for the oxidized protein bands in each CCI group were normalized to Sham-S-Sed values within the same gel, and the final oxidized protein data were expressed as the percent change from the mean Sham-S-Sed values.
Statistical analysis
All data are expressed as the mean±standard error of the mean (S.E.M.). The amount of exercise was analyzed through a two-way repeated measures analysis of variance (ANOVA) [(injury: Sham vs. CCI) and (drug treatment: S vs. AMPH)], with the number of nightly wheel revolutions being the dependent variables. Hippocampal BDNF, total synapsin I, P-synapsin I, and the P-synapsin/total synapsin ratio data were analyzed using a three-way ANOVA [(injury: Sham vs. CCI), (drug treatment: S vs. AMPH) and (exercise: Sed vs. RW)]. Interaction effects were further analyzed by performing means comparisons, and desired contrast weights were specified. Analysis of correlation (linear regression) was performed between the amount of exercise (mean nightly wheel revolutions) and BDNF protein levels for each exercised rat. Oxidized protein data were analyzed using a one-way ANOVA. Post hoc analyses were conducted using Bonferroni comparisons. For all analyses, P<0.05 was considered significant in this study.
RESULTS
Amount of exercise
Although gross motor impairments of ambulatory ability in home cages were not observed in any of the injured rats, the CCI groups exercised significantly less across all days compared with the Sham groups [F1,20=26.44, P<0.0005]. Drug treatment did not significantly affect RW performance in Sham or CCI rats. The number of nightly revolutions increased over time during the RW caging conditions [F6,120=26.75, P<0.0005] (Fig. 1).
Fig. 1.
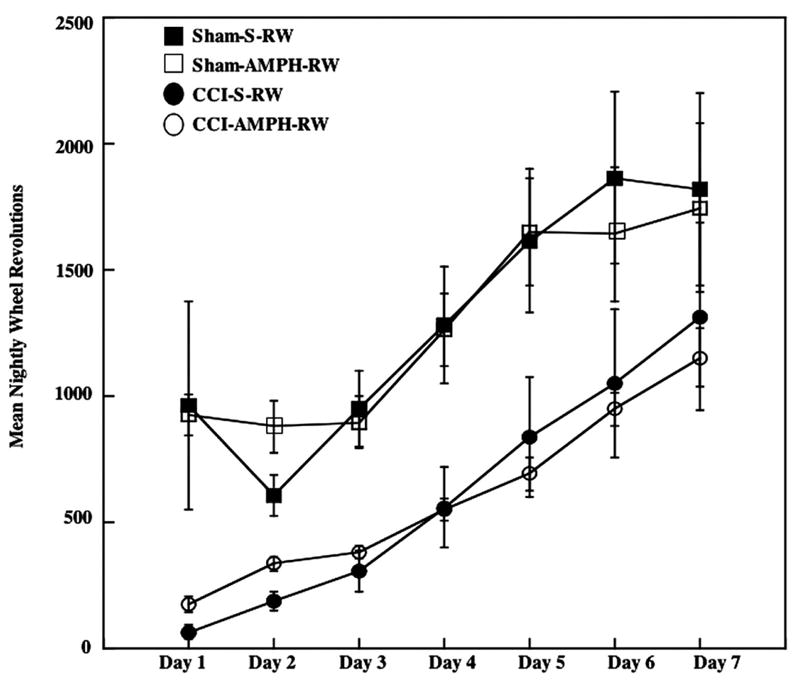
Exercise levels assessed using nightly RW activity in the week following surgery. Rats with CCI injury exercised significantly less than rats with a Sham injury. Drug treatment (AMPH vs. S) did not affect RW activity. Group means±S.E.M.
Hippocampal BDNF protein
A three-way ANOVA indicated no significant main effects, but revealed a significant drug treatment by exercise interaction [F1,40=10.14, P<0.005] for hippocampal BDNF levels. As can be seen in Fig. 2, voluntary exercise alone increased levels of BDNF in Sham and CCI groups. Means comparisons analysis indicated that exercise significantly increased BDNF levels in Sham-S-RW and CCI-S-RW rats compared with the Sham-S-Sed and CCI-S-Sed groups (F=5.76, P<0.05).
Fig. 2.
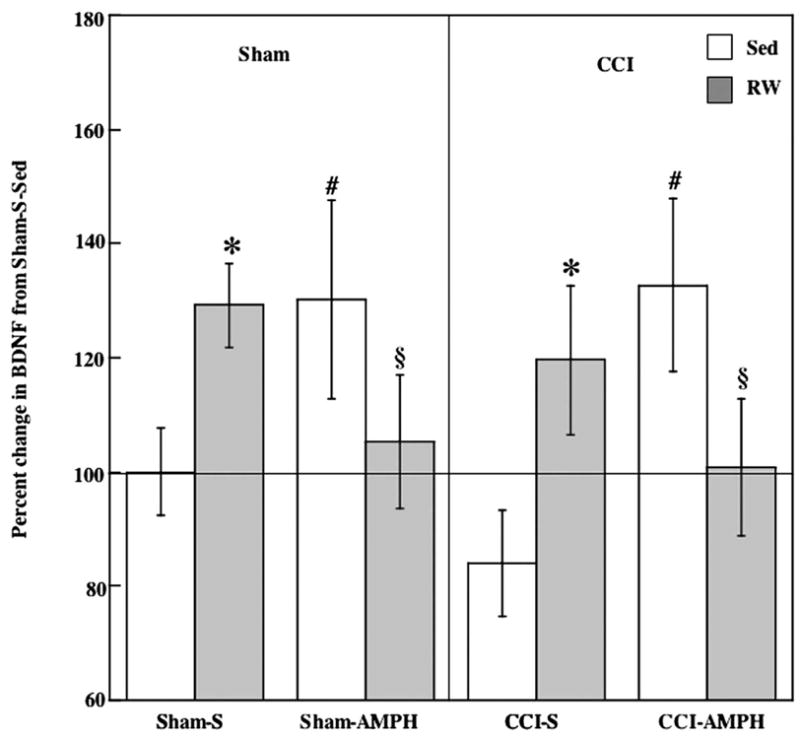
Effects of AMPH treatment and RW exposure on BDNF protein. Exercise or AMPH treatment alone significantly increased levels of BDNF in Sham and CCI rats. This increase was not found when AMPH and exercise were combined. Group means±S.E.M. * P<0.05 comparing Sham-S-RW and CCI-S-RW against Sham-S-Sed and CCI-S-Sed respectively; # P<0.05 comparing Sham-AMPH-Sed and CCI-AMPH-Sed against Sham-S-Sed and CCI-S-Sed respectively; § P<0.05 comparing Sham-AMPH-Sed and CCI-AMPH-Sed against Sham-AMPH-RW and CCI-AMPH-RW.
AMPH treatment alone also increased levels of BDNF in Sham and CCI groups (Fig. 2). Hippocampal BDNF levels in both Sham-AMPH-Sed and CCI-AMPH-Sed significantly increased compared with the Sham-S-Sed and CCI-S-Sed groups (F=8.51, P<0.05).
Increases in BDNF were not observed in Sham or CCI rats when exercise and AMPH were combined (Fig. 2). Although not decreased below Sham-S-Sed levels, BDNF levels in Sham-AMPH-RW and CCI-AMPH-RW were decreased compared with the Sham-AMPH-Sed and CCI-AMPH-Sed groups (F=4.42, P<0.05).
Exercise-induced increases in hippocampal BDNF protein have been reported to be proportional to the amount of exercise in sham-injured and FPI rats (Griesbach et al., 2004b). This relationship was again observed in the ShamS-RW animals, where there was a positive correlation (r=0.74) between the amount of exercise (total nightly revolutions over 7 days) and the hippocampal BDNF levels. However the correlation was not as high when sham rats were treated with AMPH (Sham-AMPH-RW: r=0.47) (Fig. 3A). A positive correlation between the amount of exercise and BDNF was not found for CCI groups, in either the S or AMPH treatment conditions (Fig. 3B).
Fig. 3.
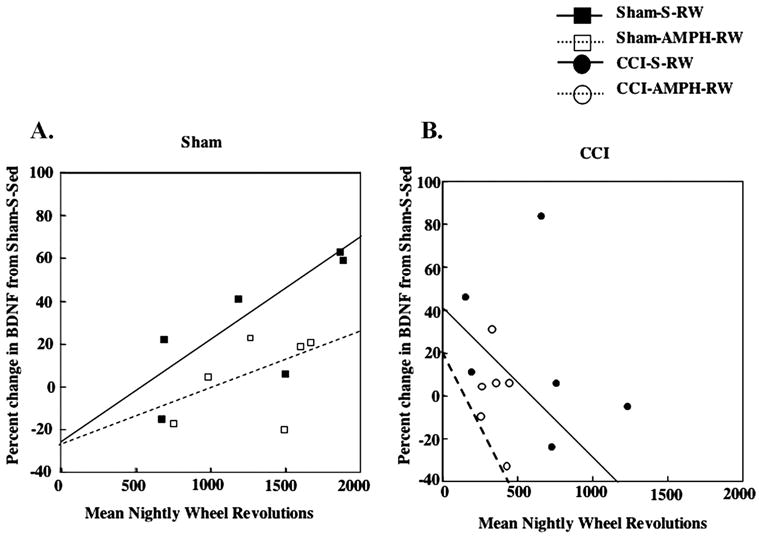
Exercise-dependent increases in hippocampal BDNF. (A) A positive relationship between the amount of exercise (mean nightly RW revolutions) and BDNF was observed in Sham-S-RW rats (r=0.74). A smaller positive correlation was seen in the Sham-AMPH-RW group (r=0.47). (B) A positive correlation between the amount of exercise and BDNF was not observed after CCI injury.
Hippocampal synapsin I
Tissue samples for one Sham-S-RW animal were lost subsequent to BDNF assays, thus reducing the sample size for synapsin I assays to five rats in this group. A three-way ANOVA on total synapsin I data (Fig. 4A) indicated significant effects for injury [F1,39=57.92, P<0.001], the injury×drug treatment interaction [F1,39=10.04, P<0.005] and the three-way interaction [F2,39=8.18, P<0.005]. The exercise-induced increase (28%) in total synapsin in Sham-S-RW rats was not significant, whereas the increase in Sham-AMPH-Sed rats was significant (P<0.001), compared with levels in Sham-S-Sed controls as well as in the combined treatment (Sham-AMPH-RW) group. The CCI-induced decrease (39%) in total synapsin I in the CCI-S-Sed group was not significant compared with Sham-S-Sed controls (Fig. 4A). AMPH treatment or RW exposure after CCI further decreased total synapsin, and levels in the CCI-AMPH-Sed group were significantly decreased compared with Sham-S-Sed (P<0.001) or CCI-S-Sed (P<0.05) conditions.
Fig. 4.
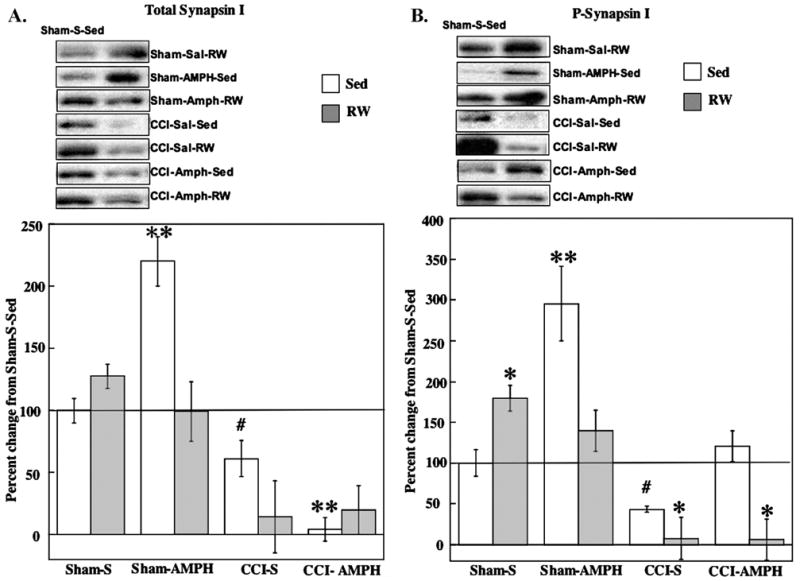
Effects of AMPH treatment and RW exposure on total and P-synapsin I. Panels contain data for all group means±S.E.M. (A) Total synapsin I increased in Sham-AMPH-Sed rats compared with Sham-S-Sed controls. Total synapsin I levels were decreased after CCI injury, with the greatest reductions occurring after AMPH or RW treatments. ** P<0.005 compared with Sham-S-Sed; # P<0.05 compared with CCI-AMPH-Sed. (B) P-synapsin I increased after RW or AMPH treatment alone in Shams compared with Sham-S-Sed. P-synapsin I levels were decreased in CCI-S-RW and CCI-AMPH-RW groups compared with Sham-S-Sed. The CCI-AMPH-Sed treatments increased P-synapsin compared with CCI-S-Sed counterparts. * P<0.05 compared with Sham-S-Sed; ** P<0.005 compared with Sham-S-Sed; # P<0.05 compared with CCI-AMPH-Sed. Representative blots are shown for each experimental group compared with Sham-S-Sed controls (left side blots).
The three-way ANOVA on the P-synapsin data (Fig. 4 B) indicated significant effects for injury [F1,39=53.97,P<0.001], drug treatment [F1,39=12.69, P<0.005] and the injury×drug treatment×exercise interaction [F2,39=11.62, P<0.001]. Compared with the Sham-S-Sed controls, P-synapsin I was increased significantly with exercise in the Sham-S-RW group (P<0.05) as well as by AMPH in the Sham-AMPH-Sed group (P<0.005). The smaller increase (40%) in P-synapsin after combined treatment (Sham-AMPH-RW) was not significant compared with Shams-S-Sed (Fig. 4B). Although CCI-S-Sed treatment did not significantly decrease P-synapsin I, the P-synapsin I levels were significantly decreased in the CCI-S-RW (P<0.05) and CCI-AMPH-RW (P<0.05) groups compared with Sham-S-Sed (Fig. 4B). AMPH treatment alone increased levels of P-synapsin I in the CCI-AMPH-Sed group, and the P-synapsin in this group did not differ from Sham-S-Sed controls and was also significantly increased above levels in the CCI-S-Sed group (P<0.05).
The three-way ANOVA on P to total synapsin levels (Fig. 5) indicated significant effects for injury [F1,35=23.65, P<0.005], drug treatment [F1,35=31.46, P<0.005], injury×drug treatment interaction [F1,35=32.52, P<0.001], injury×exercise interaction [F1,35=14.85, P<0.005] and the drug treatment×exercise interaction [F1,35=39.06, P<0.005]. Compared with the Sham-S-Sed controls, synapsin activation was increased significantly with exercise in the Sham-S-RW group (P<0.0005) as well as by AMPH in the Sham-AMPH-Sed group (P<0.005). There was no increase in activation of synapsin after combined treatment (Sham-AMPH-RW) compared with Sham-S-Sed. Synapsin activation was significantly decreased in CCI-S-Sed and CCI-S-RW compared with Sham-S-Sed (P<0.005). Increases in synapsin activation were observed in the CCI-AMPH-Sed (P<0.0005) and CCI-AMPH-RW (P<0.05) compared with CCI-S-Sed group.
Fig. 5.
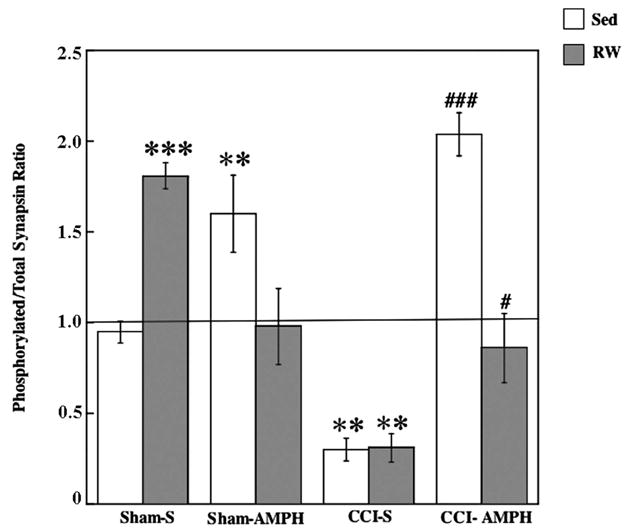
Ratio of P to total synapsin levels. Increases in activation were observed after AMPH or RW treatment alone in the Shams. The CCI-S-Sed and CCI-S-RW groups had a decrease in activation. The CCI-induced decrease was not observed after AMPH treatment alone or when combined with exercise. Data show group means±S.E.M. ** P<0.005 compared with Sham-S-Sed; *** P<0.0005 compared with Sham-S-Sed; # P<0.05 compared with CCI-S-Sed; ## P<0.005 compared with CCI-S-Sed.
Hippocampal oxidized proteins
One-way ANOVA indicated significant differences between the five groups included in the oxidized protein analysis [F4,25=9.97, P<0.001]. As illustrated in Fig. 6, the slight increase (17%) in protein carbonyl levels for CCI-S-Sed rats was not significant compared with Sham-S-Sed levels. CCI rats allowed to exercise (CCI-S-RW) had significant reductions in protein carbonyl levels compared with either Sham-S-Sed controls (P<0.001) or CCI-S-Sed rats (P<0.001). CCI-AMPH-Sed treatments significantly reduced oxidized proteins compared with CCI-S-Sed conditions (P<0.005), although these CCI rats with AMPH treatment did not differ from Sham-S-Sed controls and they had significantly greater protein oxidation than the CCI-S-RW group (P < 0.05). No significant effects of combined treatment (CCI-AMPH-RW) were found compared with Sham-S-Sed or CCI-S-Sed (P=0.052) conditions, but this combined treatment group had increased protein oxidation compared with the CCI-S-RW group (P<0.01) (Fig. 6).
Fig. 6.
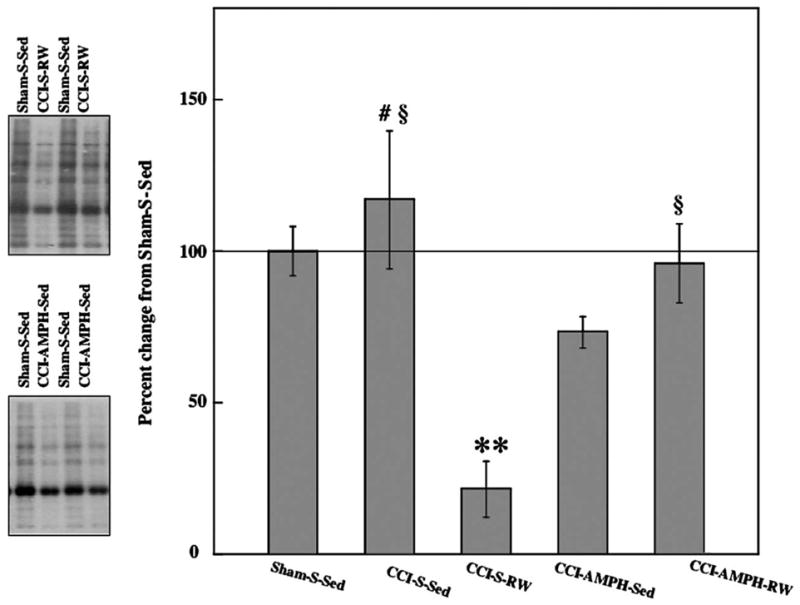
Effects of AMPH treatment or RW exposure on oxidized protein levels after CCI injury. Representative blots for the Sham controls and two CCI groups are shown next to the graph containing data for all group means±S.E.M. Protein carbonyl contents were significantly decreased after exercise or AMPH treatment alone, but not the combined treatment. ** P<0.001 compared with Sham-S-Sed, CCI-S-Sed or CCI-AMPH-RW; # P<0.005 compared with CCI-S-Sed; § P<0.05 compared with CCI-S-RW.
DISCUSSION
These studies tested several hypotheses based on previous findings. First, we hypothesized that acute exercise would not increase BDNF and synapsin I following CCI injury. Second, we hypothesized that AMPH treatment would increase BDNF and synapsin I. Finally, and foremost, we hypothesized that AMPH treatment would enable acute exercise to increase BDNF and synapsin I in the injured hippocampus. The results showed that AMPH treatment alone increased BDNF and P-synapsin I protein levels after CCI injury. To our surprise, acute exercise alone also increased BDNF levels after CCI, but the combination of exercise and AMPH did not. The current results also indicate that exercise or AMPH treatment, but not the combined treatments, reduces oxidative stress after CCI.
Effects of voluntary exercise
The current study found that voluntary exercise increased levels of BDNF protein in the hippocampus of Sham-S-Sed rats, with increases being proportional to the amount of exercise, as has been previously reported (Griesbach et al., 2004b). In addition, exercise in these S-treated sham injury controls produced a significant increase in P-synapsin I protein.
A fact worthy of noting was that the CCI rats did show an increase in BDNF, accompanied by a decrease in protein carbonyl contents, when exposed to the RW during the first post-injury week. These findings led us to reject our initial hypothesis that “acute exercise after CCI would have detrimental or neutral effects on outcome measures.” Previous work from this laboratory had indicated that exercise dependent increases in BDNF are not found when rats with mild or moderate FPI are exposed to exercise during post-injury days 0–6, but occur at later post-injury time periods (Griesbach et al., 2004b, 2007). Thus, it appears that the effects that TBI has on voluntary running activity affect the BDNF outcome. These differential effects may be related to the RW activity in FPI compared with CCI animals. Rats receiving a FPI exercised as much as sham rats during the first week post-injury (Griesbach et al., 2004b). However, CCI rats engaged in very little spontaneous exercise over the first 3 days post-injury, and continued to exercise less than sham animals throughout the 7 day RW exposure. Thus, due to CCI rats engaging in most of their spontaneous exercise from days 4–7 compared with days 1–3 post-injury, the non-responsive period to exercise induced up-regulation of BDNF previously observed after FPI may have passed. These findings also highlight the importance in determining appropriate exercise regimens after TBI. An exercise regimen that is too intensive may be counterproductive during the early post-injury time period.
Although BDNF increased with exercise after CCI, molecules downstream to BDNF were still non-responsive to exercise. A reduction in synapsin activation, in the hippocampus ipsilateral to CCI, was not alleviated by acute exercise. This inability of exercise to up-regulate synapsin I after CCI is similar to our previous findings where an exercise-induced increase in synapsin I mRNA or protein was absent after FPI (Griesbach et al., 2004a,b).
Effects of amphetamine
Although multiple injections of high-dose AMPH (5 mg/kg) have been reported to increase BDNF expression and production in multiple brain regions (Meredith et al., 2002), to our knowledge this is the first report that low-dose AMPH treatment increases levels of BDNF protein. A 1-week infusion of AMPH (1 mg/kg/day) significantly increased hippocampal BDNF levels in non-exercising sham and CCI groups. This may be due to AMPH increasing levels of NE, by indirectly promoting its release as well as blocking NE reuptake (Florin et al., 1994). In turn, NE can produce elevations of BDNF mRNA in astrocytes (Zafra et al., 1992) and, more recently, NE has been found to lead to BDNF expression in hippocampal neurons in a cyclic pathway (Chen et al., 2007). However, BDNF elevations with AMPH treatment could also be related to drug-induced increases of 5-HT and DA. Monoaminergic activity, which is elevated with AMPH, has been found to enhance the expression of BDNF (Fumagalli et al., 2003; Mattson et al., 2004; Miklic et al., 2004; Juric et al., 2006; Luellen et al., 2007). A third putative mechanism for AMPH to increase BDNF is through the cocaine- and amphetamine-regulated transcript (CART). CART is abundantly expressed in the hippocampus and has been proposed for therapeutic use, in part due to its ability to up-regulate BDNF (Douglass and Daoud, 1996; Wu et al., 2006; Pae et al., 2007).
In contrast to exercise alone, AMPH treatment significantly increased synapsin activation, in the hippocampus of non-exercising sham operates. Synapsin I responses to AMPH treatment in the non-exercised CCI rats differed from patterns seen in sham injury conditions. Total synapsin was significantly decreased from Sham-S-Sed and CCI-S-Sed controls only in the non-exercised CCI group treated with AMPH. However, AMPH treatment prevented the reduction in P-synapsin observed in the CCI-S-Sed rats, and raised hippocampal levels of P-synapsin I back to at least Sham-S-Sed control levels. Thus, AMPH treatment after CCI significantly increased synapsin activation.
The current results also indicate that AMPH treatment alone can decrease protein oxidation after CCI. While levels of carbonyl proteins were significantly higher in hippocampal tissue from CCI-AMPH-Sed compared with CCI-S-RW rats, oxidized proteins in this AMPH-treated group were significantly reduced compared with CCI-S-Sed controls.
Effects of combined treatment of voluntary exercise and amphetamine
Interestingly, although exercise or AMPH treatment alone elevated levels of BDNF, this effect was lost when treatments were combined in either sham operates of CCI rats. However, CCI-induced decreases in BDNF and synapsin activation were not observed when AMPH and exercise were combined. This pattern of molecular changes is consistent with synapsin’s downstream nature to BDNF. The ability of exercise or AMPH treatment to individually reduce levels of protein oxidation after CCI was also attenuated in the combined treatment group. Given that combined treatment effects were encountered in both CCI and sham rats, it is unlikely they are due to brain injury.
The current findings appear to contrast with prior reports that some antidepressants not only rapidly increase levels of BDNF mRNA, but also potentiate an exercise-induced increase of BDNF mRNA within the adult rat hippocampus (Russo-Neustadt et al., 2000, 2001, 2004). However, these studies did not assess protein levels, and chronic treatment with antidepressant drugs is required to up-regulate BDNF protein in adult mice or rats (Kozisek et al., 2008; Peng et al., 2008). It is possible that AMPH plus exercise increased mRNA for BDNF, but translation to the BDNF protein did not occur with the combined treatment. On the other hand, it is also possible that sensitization properties of AMPH prevent it from acting synergistically with exercise in the same manner as antidepressants. AMPH treatment can lead to dissociation in the mutual regulation of noradrenergic and serotonergic neurons, whereas antidepressants which block NE and 5-HT re-uptake do not produce this sensitization (Lanteri et al., 2007; Salomon et al., 2006). Activation of autoregulatory responses with combined treatments may have altered monoamine release, which could affect BDNF levels since BDNF-catecholamine regulation is bidirectional. For example, NE increases the levels of BDNF, as indicated above, and in turn BDNF regulates noradrenergic neuron plasticity (Akbarian et al., 2002). In addition, exercise alone increases levels of NE in brain and spinal cord (Dunn et al., 1996; Dishman et al., 2000), as does AMPH. A “dose-response” effect with the combination of exercise and AMPH treatments may have resulted in an enhanced autoregulatory response (feedback inhibition) for NE release. AMPH was administered continuously in the current study, and it is possible that single or intermittent AMPH treatment(s), where autoregulatory responses and tolerance mechanisms are not as strong, would act synergistically with exercise to enhance markers of plasticity. In such a scenario, AMPH might potentiate an exercise-induced BDNF increase, such as those mentioned above for anti-depressants paired with exercise. Further studies will be needed to determine if AMPH increases BDNF mRNA, or if antidepressants increase BDNF protein, when these drug treatments and exercise are paired. In particular, further studies exploring the effects that TBI has on BDNF translation will aid in interpreting these findings.
The current results did not support our original hypothesis that AMPH treatment would augment an exercise-induced increase in BDNF. Consequently, this result has specific implications for future proposals to treat TBI using a combination of treatments. This complication of combining treatments is not novel. There have been similar findings where two potentially therapeutic TBI treatments cancel each other out when combined or are even counterproductive (Guluma et al., 1999; Kline et al., 2004). Particularly relevant is a recent study combining a behavioral manipulation with a pharmacological agent. Exposure to an enriched environment and treatment with a 5-HT receptor agonist improved recovery after a CCI when given individually, but had a neutral effect when combined (Kline et al., 2007). The current findings that combined exercise and AMPH treatments did not lead to elevated BDNF or synapsin suggest these two therapies should not be combined, at least in the acute post-injury phase. The CCI animals with combined therapy had levels of BDNF, P-synapsin/total synapsin ratios and oxidized proteins that were similar to Sed, S-treated, sham-injured and CCI controls, which does not indicate any deleterious effect of the combined treatments. However, the P-synapsin results in the CCI-AMPH-RW group may indicate effects of combined treatments that could impact negatively on neuronal recovery and functional outcomes after CCI.
The current results for molecular effects of combined treatments within the hippocampus are somewhat difficult to reconcile with prior studies on combined treatment effects on improved motor functions after brain injury. For example, it has been frequently noted that the pairing of AMPH treatment with task-relevant experience or rehabilitation can enhance neuronal plasticity and recovery of motor functions after brain injury (Feeney and Sutton, 1988; Stroemer et al., 1998; Butefisch et al., 2002; Adkins and Jones, 2005; Ramic et al., 2006). Thus, it is possible that spontaneous exercise in combination with AMPH may enhance molecular changes underlying neuroplasticity within motor pathways after TBI. Conversely, combining AMPH treatment with cognitive therapies or task demands, rather than with a RW task, might facilitate molecular changes underlying neuroplasticity within the hippocampus. Further studies will be needed to evaluate these hypotheses.
Potential neuroprotective effects
In spite of the negative findings of the combined treatment, this work indicates that either exercise or AMPH may be beneficial for the treatment of TBI. In addition to increasing neuroplasticity through BDNF-mediated pathways, these treatments appear to offer protection from oxidative damage. Oxidative stress is one of the key factors in neuronal degeneration after TBI (Shohami et al., 1997; Sullivan et al., 1998), where an increase in free radicals leads to protein oxidation, lipid peroxidation and DNA damage. Both exercise and AMPH-treated CCI groups showed decreases in protein carbonyl contents, indicating that these individual treatments reduced the metal-catalyzed oxidation reactions on hippocampal proteins.
Acknowledgments
This study was supported by NINDS awards NSO48535, NS27544 and the UCLA Brain Injury Research Center.
Abbreviations
- AMPH
d-amphetamine
- ANOVA
analysis of variance
- BDNF
brain-derived neurotrophic factor
- CART
cocaine- and amphetamine-regulated transcript
- CCI
controlled cortical impact
- DA
dopamine
- DNPH
2,4-dinitrophenylhydrazone
- FPI
fluid percussion injury
- NE
norepinephrine
- P
phosphorylated
- RW
running wheel
- S
saline
- Sed
sedentary
- TBI
traumatic brain injury
- UCLA
University of California at Los Angeles
References
- Adkins DL, Jones TA. D-amphetamine enhances skilled reaching after ischemic cortical lesions in rats. Neurosci Lett. 2005;380:214–218. doi: 10.1016/j.neulet.2005.01.036. [DOI] [PubMed] [Google Scholar]
- Akbarian S, Rios M, Liu RJ, Gold SJ, Fong HF, Zeiler S, Coppola V, Tessarollo L, Jones KR, Nestler EJ, Aghajanian GK, Jaenisch R. Brain-derived neurotrophic factor is essential for opiate-induced plasticity of noradrenergic neurons. J Neurosci. 2002;22:4153–4162. doi: 10.1523/JNEUROSCI.22-10-04153.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder S, Corrigan JD, Langlois JA. The public health approach to traumatic brain injury: an overview of CDC’s research and programs. J Head Trauma Rehabil. 2005;20:189–195. doi: 10.1097/00001199-200505000-00002. [DOI] [PubMed] [Google Scholar]
- Boyeson MG, Feeney DM. Intraventricular norepinephrine facilitates motor recovery following sensorimotor cortex injury. Pharmacol Biochem Behav. 1990;35:497–501. doi: 10.1016/0091-3057(90)90279-q. [DOI] [PubMed] [Google Scholar]
- Boyeson MG, Harmon RL, Jones JL. Comparative effects of fluoxetine, amitriptyline and serotonin on functional motor recovery after sensorimotor cortex injury. Am J Phys Med Rehabil. 1994;72:76–83. doi: 10.1097/00002060-199404000-00002. [DOI] [PubMed] [Google Scholar]
- Butefisch CM, Davis BC, Sawaki L, Waldvogel D, Classen J, Kopylev L, Cohen LG. Modulation of use-dependent plasticity by d-amphetamine. Ann Neurol. 2002;51:59–68. doi: 10.1002/ana.10056. [DOI] [PubMed] [Google Scholar]
- Chen MJ, Nguyen TV, Pike CJ, Russo-Neustadt AA. Norepinephrine induces BDNF and activates the PI-3K and MAPK cascades in embryonic hippocampal neurons. Cell Signal. 2007;19:114–128. doi: 10.1016/j.cellsig.2006.05.028. [DOI] [PubMed] [Google Scholar]
- Chen X, Li Y, Kline AE, Dixon CE, Zafonte RD, Wagner AK. Gender and environmental effects on regional brain-derived neurotrophic factor expression after experimental traumatic brain injury. Neuroscience. 2005;135:11–17. doi: 10.1016/j.neuroscience.2005.05.041. [DOI] [PubMed] [Google Scholar]
- Clough RW, Neese SL, Sherill LK, Tan AA, Duke A, Roosevelt RW, Browning RA, Smith DC. Cortical edema in moderate fluid percussion brain injury is attenuated by vagus nerve stimulation. Neuroscience. 2007;147:286–293. doi: 10.1016/j.neuroscience.2007.04.043. [DOI] [PubMed] [Google Scholar]
- Colicos MA, Dixon CE, Dash PK. Delayed, selective neuronal death following experimental cortical impact injury in rats: possible role in memory deficits. Brain Res. 1996;739:111–119. doi: 10.1016/s0006-8993(96)00819-0. [DOI] [PubMed] [Google Scholar]
- Cotman CW, Berchtold NC. Exercise: a behavioral intervention to enhance brain health and plasticity. Trends Neurosci. 2002;25:295–301. doi: 10.1016/s0166-2236(02)02143-4. [DOI] [PubMed] [Google Scholar]
- Dash PK, Moore AN, Dixon CE. Spatial memory deficits, increased phosphorylation of the transcription factor CREB, and induction of the AP-1 complex following experimental brain injury. J Neurosci. 1995;15:2030–2039. doi: 10.1523/JNEUROSCI.15-03-02030.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dishman RK, Renner KJ, White-Welkley JE, Burke KA, Bunnell BN. Treadmill exercise training augments brain norepinephrine response to familiar and novel stress. Brain Res Bull. 2000;52:337–342. doi: 10.1016/s0361-9230(00)00271-9. [DOI] [PubMed] [Google Scholar]
- Dixon CE, Kochanek PM, Yan HQ, Schiding JK, Griffith RG, Baum E, Marion DW, Dekosky ST. One-year study of spatial memory performance, brain morphology, and cholinergic markers after moderate controlled cortical impact in rats. J Neurotrauma. 1999;16:109–122. doi: 10.1089/neu.1999.16.109. [DOI] [PubMed] [Google Scholar]
- Douglass J, Daoud S. Characterization of the human cDNA and genomic DNA encoding CART: a cocaine- and amphetamine-regulated transcript. Gene. 1996;169:241–245. doi: 10.1016/0378-1119(96)88651-3. [DOI] [PubMed] [Google Scholar]
- Dunn-Meynell A, Pan S, Levin BE. Focal traumatic brain injury causes widespread reductions in rat brain norepinephrine turnover from 6 to 24 h. Brain Res. 1994;660:88–95. doi: 10.1016/0006-8993(94)90842-7. [DOI] [PubMed] [Google Scholar]
- Dunn-Meynell AA, Yarlagadda Y, Levin BE. Alpha 1-adrenoceptor blockade increases behavioral deficits in traumatic brain injury. J Neurotrauma. 1997;14:43–52. doi: 10.1089/neu.1997.14.43. [DOI] [PubMed] [Google Scholar]
- Dunn AL, Reigle TG, Youngstedt SD, Armstrong RB, Dishman RK. Brain norepinephrine and metabolites after treadmill training and wheel running in rats. Med Sci Sports Exerc. 1996;28:204–209. doi: 10.1097/00005768-199602000-00008. [DOI] [PubMed] [Google Scholar]
- Feeney DM, Sutton RL. Pharmacotherapy for recovery of function after brain injury. CRC Crit Rev Neurobiol. 1987;3:135–197. [PubMed] [Google Scholar]
- Feeney DM, Sutton RL. Catecholamines and recovery of function after brain damage. In: Stein DG, Sabel BA, editors. Pharmacological approaches to the treatment of brain and spinal cord injury. New York: Plenum Press; 1988. pp. 121–142. [Google Scholar]
- Feeney DM, Westerberg VS. Norepinephrine and brain damage: alpha noradrenergic pharmacology alters functional recovery after cortical trauma. Can J Psychol. 1990;44:233–252. doi: 10.1037/h0084243. [DOI] [PubMed] [Google Scholar]
- Fleckenstein AE, Volz TJ, Riddle EL, Gibb JW, Hanson GR. New insights into the mechanism of action of amphetamines. Annu Rev Pharmacol Toxicol. 2007;47:681–698. doi: 10.1146/annurev.pharmtox.47.120505.105140. [DOI] [PubMed] [Google Scholar]
- Florin SM, Kuczenski R, Segal DS. Regional extracellular norepinephrine responses to amphetamine and cocaine and effects of clonidine pretreatment. Brain Res. 1994;654:53–62. doi: 10.1016/0006-8993(94)91570-9. [DOI] [PubMed] [Google Scholar]
- Follesa P, Biggio F, Gorini G, Caria S, Talani G, Dazzi L, Puligheddu M, Marrosu F, Biggio G. Vagus nerve stimulation increases norepinephrine concentration and the gene expression of BDNF and bFGF in the rat brain. Brain Res. 2007;1179:28–34. doi: 10.1016/j.brainres.2007.08.045. [DOI] [PubMed] [Google Scholar]
- Fumagalli F, Racagni G, Colombo E, Riva MA. BDNF gene expression is reduced in the frontal cortex of dopamine transporter knockout mice. Mol Psychiatry. 2003;8:898–899. doi: 10.1038/sj.mp.4001370. [DOI] [PubMed] [Google Scholar]
- Garcia C, Chen MJ, Garza AA, Cotman CW, Russo-Neustadt A. The influence of specific noradrenergic and serotonergic lesions on the expression of hippocampal brain-derived neurotrophic factor transcripts following voluntary physical activity. Neuroscience. 2003;119:721–732. doi: 10.1016/s0306-4522(03)00192-1. [DOI] [PubMed] [Google Scholar]
- Goldstein LB, Davis JN. Clonidine impairs recovery of beam-walking after sensorimotor cortex lesion in the rat. Brain Res. 1990;508:305–309. doi: 10.1016/0006-8993(90)90413-6. [DOI] [PubMed] [Google Scholar]
- Goldstein LB. Basic and clinical studies of pharmacologic effects on recovery from brain injury. J Neural Transplant Plast. 1993;4:175–192. doi: 10.1155/NP.1993.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesbach GS, Gomez-Pinilla F, Hovda DA. The upregulation of plasticity related proteins following TBI is disrupted with acute voluntary exercise. Brain Res. 2004a;1016:154–162. doi: 10.1016/j.brainres.2004.04.079. [DOI] [PubMed] [Google Scholar]
- Griesbach GS, Hovda DA, Molteni R, Wu A, Gomez-Pinilla F. Voluntary exercise following traumatic brain injury: brain-derived neurotrophic factor upregulation and recovery of function. Neuroscience. 2004b;125:129–139. doi: 10.1016/j.neuroscience.2004.01.030. [DOI] [PubMed] [Google Scholar]
- Griesbach GS, Gomez-Pinilla F, Hovda DA. Time window for voluntary exercise-induced increases in hippocampal neuroplasticity molecules after traumatic brain injury is severity dependent. J Neurotrauma. 2007;24:1161–1171. doi: 10.1089/neu.2006.0255. [DOI] [PubMed] [Google Scholar]
- Guluma KZ, Saatman KE, Brown A, Raghupathi R, McIntosh TK. Sequential pharmacotherapy with magnesium chloride and basic fibroblast growth factor after fluid percussion brain injury results in less neuromotor efficacy than that achieved with magnesium alone. J Neurotrauma. 1999;16:311–321. doi: 10.1089/neu.1999.16.311. [DOI] [PubMed] [Google Scholar]
- Hall ED, Detloff MR, Johnson K, Kupina NC. Peroxynitrite-mediated protein nitration and lipid peroxidation in a mouse model of traumatic brain injury. J Neurotrauma. 2004;21:9–20. doi: 10.1089/089771504772695904. [DOI] [PubMed] [Google Scholar]
- Hamm RJ, Dixon CE, Gbadebo DM, Singha AK, Jenkins LW, Lyeth BG, Hayes RL. Cognitive deficits following traumatic brain injury produced by controlled cortical impact. J Neurotrauma. 1992;9:11–20. doi: 10.1089/neu.1992.9.11. [DOI] [PubMed] [Google Scholar]
- Hellmich HL, Garcia JM, Shimamura M, Shah SA, Avila MA, Uchida T, Parsley MA, Capra BA, Eidson KA, Kennedy DR, Winston JH, Dewitt DS, Prough DS. Traumatic brain injury and hemorrhagic hypotension suppress neuroprotective gene expression in injured hippocampal neurons. Anesthesiology. 2005;102:806–814. doi: 10.1097/00000542-200504000-00017. [DOI] [PubMed] [Google Scholar]
- Hicks RR, Boggs A, Leider D, Kraemer P, Brown R, Scheff SW, Serrogy KB. Effects of exercise following lateral fluid percussion brain injury in rats. Restor Neurol Neurosci. 1998;12:41–47. [PubMed] [Google Scholar]
- Hicks RR, Zhang L, Atkinson A, Stevenon M, Veneracion M, Seroogy KB. Environmental enrichment attenuates cognitive deficits, but does not alter neurotrophin gene expression in the hippocampus following lateral fluid percussion brain injury. Neuroscience. 2002;112:631–637. doi: 10.1016/s0306-4522(02)00104-5. [DOI] [PubMed] [Google Scholar]
- Hovda DA. Metabolic dysfunction. In: Narayan RK, Wilberger JE, Povlishock JT, editors. Neurotrauma. New York: McGraw-Hill Inc; 1996. pp. 1459–1478. [Google Scholar]
- Juric DM, Miklic S, Carman-Krzan M. Monoaminergic neuronal activity upregulates BDNF synthesis in cultured neonatal rat astrocytes. Brain Res. 2006;1108:54–62. doi: 10.1016/j.brainres.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Kelly DF, Kozlowski DA, Haddad E, Echiverri A, Hovda DA, Lee SM. Ethanol reduces metabolic uncoupling following experimental head injury. J Neurotrauma. 2000;17:261–272. doi: 10.1089/neu.2000.17.261. [DOI] [PubMed] [Google Scholar]
- Kline AE, Yan HQ, Bao J, Marion DW, Dixon CE. Chronic methylphenidate treatment enhances water maze performance following traumatic brain injury in rats. Neurosci Lett. 2000;280:163–166. doi: 10.1016/s0304-3940(00)00797-7. [DOI] [PubMed] [Google Scholar]
- Kline AE, Massucci JL, Marion DW, Dixon CE. Attenuation of working memory and spatial acquisition deficits after a delayed and chronic bromocriptine treatment regimen in rats subjected to traumatic brain injury by controlled cortical impact. J Neurotrauma. 2002;19:415–425. doi: 10.1089/08977150252932370. [DOI] [PubMed] [Google Scholar]
- Kline AE, Massucci JL, Dixon CE, Zafonte RD, Bolinger BD. The therapeutic efficacy conferred by the 5-HT(1A) receptor agonist 8-Hydroxy-2-(di-npropylamino) tetralin (8-OH-DPAT) after experimental traumatic brain injury is not mediated by concomitant hypothermia. J Neurotrauma. 2004;21:175–185. doi: 10.1089/089771504322778631. [DOI] [PubMed] [Google Scholar]
- Kline AE, Wagner AK, Westergom BP, Malena RR, Zafonte RD, Olsen AS, Sozda CN, Luthra P, Panda M, Cheng JP, Aslam HA. Acute treatment with the 5- HT(1A) receptor agonist 8-OH-DPAT and chronic environmental enrichment confer neurobehavioral benefit after experimental brain trauma. Behav Brain Res. 2007;177:186–194. doi: 10.1016/j.bbr.2006.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozisek ME, Middlemas D, Bylund DB. The differential regulation of BDNF and TrkB levels in juvenile rats after four days of escitalopram and desipramine treatment. Neuropharmacology. 2008;54:251–257. doi: 10.1016/j.neuropharm.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Krobert KA, Sutton RL, Feeney DM. Spontaneous and amphetamine-evoked release of cerebellar noradrenaline after sensorimotor cortex contusion: an in vivo microdialysis study in the awake rat. J Neurochem. 1994;62:2233–2240. doi: 10.1046/j.1471-4159.1994.62062233.x. [DOI] [PubMed] [Google Scholar]
- Lanteri C, Salomon L, Torrens Y, Glowinski J, Tassin JP. Drugs of abuse specifically sensitize noradrenergic and serotonergic neurons via a nondopaminergic mechanism. Neuropsychopharmacology. 2007 doi: 10.1038/sj.npp.1301548. advance online publication, 5 September:1–11. [DOI] [PubMed] [Google Scholar]
- Levin BE, Brown KL, Pawar G, Dunn-Meynell A. Widespread and lateralized effects of acute traumatic brain injury on norepinephrine turnover in the rat brain. Brain Res. 1995;674:307–313. doi: 10.1016/0006-8993(95)00032-l. [DOI] [PubMed] [Google Scholar]
- Lindner MD, Plone MA, Cain CK, Frydel B, Francis JM, Emerich DF, Sutton RL. Dissociable long-term cognitive deficits after frontal versus sensorimotor cortical contusions. J Neurotrauma. 1998;15:199–216. doi: 10.1089/neu.1998.15.199. [DOI] [PubMed] [Google Scholar]
- Liu QR, Lu L, Zhu XG, Gong JP, Shaham Y, Uhl GR. Rodent BDNF genes, novel promoters, novel splice variants, and regulation by cocaine. Brain Res. 2006;1067:1–12. doi: 10.1016/j.brainres.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Luellen BA, Bianco LE, Schneider LM, Andrews AM. Reduced brain-derived neurotrophic factor is associated with a loss of serotonergic innervation in the hippocampus of aging mice. Genes Brain Behav. 2007;6:482–490. doi: 10.1111/j.1601-183X.2006.00279.x. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Maudsley S, Martin B. BDNF and 5-HT: a dynamic duo in age-related neuronal plasticity and neurodegenerative disorders. Trends Neurosci. 2004;27:589–594. doi: 10.1016/j.tins.2004.08.001. [DOI] [PubMed] [Google Scholar]
- McCullers DL, Sullivan PG, Scheff SW, Herman JP. Mifepristone protects CA1 hippocampal neurons following traumatic brain injury in rat. Neuroscience. 2002;109:219–230. doi: 10.1016/s0306-4522(01)00477-8. [DOI] [PubMed] [Google Scholar]
- Meredith GA, Callen S, Scheuer DA. Brain-derived neurotrophic factor expression is increased in the rat amygdala, piriform cortex and hypothalamus following repeated amphetamine administration. Brain Res. 2002;949:218–227. doi: 10.1016/s0006-8993(02)03160-8. [DOI] [PubMed] [Google Scholar]
- Miklic S, Juric DM, Carman-Krzan M. Differences in the regulation of BDNF and NGF synthesis in cultured neonatal rat astrocytes. Int J Dev Neurosci. 2004;22:119–130. doi: 10.1016/j.ijdevneu.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Molteni R, Ying Z, Gomez-Pinilla F. Differential effects of acute and chronic exercise on plasticity-related genes in the rat hippocampus revealed by microarray. Eur J Neurosci. 2002;16:1107–1116. doi: 10.1046/j.1460-9568.2002.02158.x. [DOI] [PubMed] [Google Scholar]
- Morrison B, Meaney DF, McIntosh TK. Dynamic mechanical stretch of organotypic brain slice cultures induces differential genomic expression: Relationship to mechanical parameters. J Biomech Eng. 2000;122:224–230. doi: 10.1115/1.429650. [DOI] [PubMed] [Google Scholar]
- Navarro A, Gomez C, Lopez-Cepero JM, Boveris A. Beneficial effects of moderate exercise on mice aging: survival, behavior, oxidative stress, and mitochondrial electron transfer. Am J Physiol Regul Integr Comp Physiol. 2004;286:R505–R511. doi: 10.1152/ajpregu.00208.2003. [DOI] [PubMed] [Google Scholar]
- Neeper SA, Gomez-Pinilla F, Choi F, Cotman CW. Exercise and brain neurotrophins. Nature. 1995;373:109. doi: 10.1038/373109a0. [DOI] [PubMed] [Google Scholar]
- Neeper SA, Gomez-Pinilla F, Choi F, Cotman CW. Physical activity increases mRNA for brain-derived neurotrophic factor and nerve growth factor in rat brain. Brain Res. 1996;726:49–56. [PubMed] [Google Scholar]
- Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci. 1995;15:7539–7547. doi: 10.1523/JNEUROSCI.15-11-07539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyesiku NM, Evans CO, Houston S, Darrell RS, Smith JS, Fulop ZL, Dixon CE, Stein DG. Regional changes in the expression of neurotrophic factors and their receptors following acute traumatic brain injury in the adult rat brain. Brain Res. 1999;833:161–172. doi: 10.1016/s0006-8993(99)01501-2. [DOI] [PubMed] [Google Scholar]
- Pae CU, Lee C, Paik IH. Therapeutic implication of cocaine- and amphetamine-regulated transcript (CART) in the treatment of depression. Med Hypotheses. 2007;69:132–135. doi: 10.1016/j.mehy.2006.11.009. [DOI] [PubMed] [Google Scholar]
- Pan YX, Gao L, Wang WZ, Zheng H, Liu D, Patel KP, Zucker IH, Wang W. Exercise training prevents arterial baroreflex dysfunction in rats treated with central angiotensin II. Hypertension. 2007;49:519–527. doi: 10.1161/01.HYP.0000256955.74461.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Q, Masuda N, Jiang M, Li Q, Zhao M, Ross CA, Duan W. The antidepressant sertraline improves the phenotype, promotes neurogenesis and increases BDNF levels in the R6/2 Huntington’s disease mouse model. Exp Neurol. 2008;210:154–163. doi: 10.1016/j.expneurol.2007.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad RM, Dose JM, Dhillon HS, Carbary T, Kraemer PJ. Amphetamine affects the behavioral outcome of lateral fluid percussion brain injury in the rat. Restor Neurol Neurosci. 1995;9:65–75. doi: 10.3233/RNN-1995-9201. [DOI] [PubMed] [Google Scholar]
- Ramic M, Emerick AJ, Bollnow MR, O’Brien TE, Tsai SY, Kartje GL. Axonal plasticity is associated with motor recovery following amphetamine treatment combined with rehabilitation after brain injury in the adult rat. Brain Res. 2006;1111:176–186. doi: 10.1016/j.brainres.2006.06.063. [DOI] [PubMed] [Google Scholar]
- Roosevelt RW, Smith DC, Clough RW, Jensen RA, Browning RA. Increased extracellular concentrations of norepinephrine in cortex and hippocampus following vagus nerve stimulation in the rat. Brain Res. 2006;1119:124–132. doi: 10.1016/j.brainres.2006.08.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo-Neustadt A, Beard RC, Cotman CW. Exercise, antide-pressant medications, and enhanced brain derived neurotrophic factor expression. Neuropsychopharmacology. 1999;21:679–682. doi: 10.1016/S0893-133X(99)00059-7. [DOI] [PubMed] [Google Scholar]
- Russo-Neustadt A, Ha T, Ramirez R, Kesslak JP. Physical activity-antidepressant treatment combination: impact on brain-derived neurotrophic factor and behavior in an animal model. Behav Brain Res. 2001;120:87–95. doi: 10.1016/s0166-4328(00)00364-8. [DOI] [PubMed] [Google Scholar]
- Russo-Neustadt AA, Beard RC, Huang YM, Cotman CW. Physical activity and antidepressant treatment potentiate the expression of specific brain-derived neurotrophic factor transcripts in the rat hippocampus. Neuroscience. 2000;101:305–312. doi: 10.1016/s0306-4522(00)00349-3. [DOI] [PubMed] [Google Scholar]
- Russo-Neustadt AA, Alejandre H, Garcia C, Ivy AS, Chen MJ. Hippocampal brain-derived neurotrophic factor expression following treatment with reboxetine, citalopram, and physical exercise. Neuropsychopharmacology. 2004;29:2189–2199. doi: 10.1038/sj.npp.1300514. [DOI] [PubMed] [Google Scholar]
- Saatman KE, Feeko KJ, Pape RL, Raghupathi R. Differential behavioral and histopathological responses to graded cortical impact injury in mice. J Neurotrauma. 2006;23:1241–1253. doi: 10.1089/neu.2006.23.1241. [DOI] [PubMed] [Google Scholar]
- Salomon L, Lanteri C, Glowinski J, Tassin JP. Behavioral sensitization to amphetamine results from an uncoupling between noradrenergic and serotonergic neurons. Proc Natl Acad Sci U S A. 2006;103:7476–7481. doi: 10.1073/pnas.0600839103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shohami E, Beit-Yannai E, Horowitz M, Kohen R. Oxidative stress in closed-head injury: brain antioxidant capacity as an indicator of functional outcome. J Cereb Blood Flow Metab. 1997;17:1007–1019. doi: 10.1097/00004647-199710000-00002. [DOI] [PubMed] [Google Scholar]
- Smith DC, Modglin AA, Roosevelt RW, Neese SL, Jensen RA, Browning RA, Clough RW. Electrical stimulation of the vagus nerve enhances cognitive and motor recovery following moderate fluid percussion injury in the rat. J Neurotrauma. 2005;22:1485–1502. doi: 10.1089/neu.2005.22.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DC, Tan AA, Duke A, Neese SL, Clough RW, Browning RA, Jensen RA. Recovery of function after vagus nerve stimulation initiated 24 hours after fluid percussion brain injury. J Neurotrauma. 2006;23:1549–1560. doi: 10.1089/neu.2006.23.1549. [DOI] [PubMed] [Google Scholar]
- Stroemer RP, Kent TA, Hulsebosch CE. Enhanced neocortical neural sprouting, synaptogenesis, and behavioral recovery with D-amphetamine therapy after neocortical infarction in rats. Stroke. 1998;29:2381–2393. doi: 10.1161/01.str.29.11.2381. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Keller JN, Mattson MP, Scheff SW. Traumatic brain injury alters synaptic homeostasis: implications for impaired mitochondrial and transport function. J Neurotrauma. 1998;15:789–798. doi: 10.1089/neu.1998.15.789. [DOI] [PubMed] [Google Scholar]
- Sutton RL, Weaver MS, Feeney DM. Drug-induced modifications of behavioral recovery following cortical trauma. J Head Trauma Rehabil. 1987;2:50–58. [Google Scholar]
- Sutton RL, Feeney DM. a-Noradrenergic agonists and antagonists affect recovery and maintenance of beam-walking ability after sensorimotor cortex ablation in the rat. Restor Neurol Neurosci. 1992;4:1–11. doi: 10.3233/RNN-1992-4101. [DOI] [PubMed] [Google Scholar]
- Sutton RL, Lescaudron L, Stein DG. Unilateral cortical contusion injury in the rat: Vascular disruption and temporal development of cortical necrosis. J Neurotrauma. 1993;10:135–149. doi: 10.1089/neu.1993.10.135. [DOI] [PubMed] [Google Scholar]
- Sutton RL, Feeney DM. Pharmacological approaches to rehabilitation: Noradrenergic pharmacotherapy and functional recovery after cortical injury. In: Illis LS, editor. Neurological rehabilitation. Oxford: Blackwell Scientific Publications; 1994. pp. 469–481. [Google Scholar]
- Timmusk T, Palm K, Metsis M, Reintam T, Paalme V, Saarma M, Persson H. Multiple promoters direct tissue-specific expression of the rat BDNF gene. Neuron. 1993;10:475–489. doi: 10.1016/0896-6273(93)90335-o. [DOI] [PubMed] [Google Scholar]
- Truettner J, Schmidt-Kastner R, Busto R, Alonso OF, Loor JY, Dietrich WD, Ginsberg MD. Expression of brain-derived neurotrophic factor, nerve growth factor, and heat shock protein HSP70 following fluid percussion brain injury in rats. J Neurotrauma. 1999;16:471– 486. doi: 10.1089/neu.1999.16.471. [DOI] [PubMed] [Google Scholar]
- van Praag H, Christie BR, Sejnowski TJ, Gage FH. Running enhances neurogenesis, learning, and long-term potentiation in mice. Proc Natl Acad Sci U S A. 1999;96:13427–13431. doi: 10.1073/pnas.96.23.13427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaynman S, Ying Z, Gomez-Pinilla F. Interplay between brain-derived neurotrophic factor and signal transduction modulators in the regulation of the effects of exercise on synaptic-plasticity. Neuroscience. 2003;122:647–657. doi: 10.1016/j.neuroscience.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Wu A, Ying Z, Gomez-Pinilla F. The interplay between oxidative stress and brain-derived neurotrophic factor modulates the outcome of a saturated fat diet on synaptic plasticity and cognition. Eur J Neurosci. 2004;19:1699–1707. doi: 10.1111/j.1460-9568.2004.03246.x. [DOI] [PubMed] [Google Scholar]
- Wu B, Hu S, Yang M, Pan H, Zhu S. CART peptide promotes the survival of hippocampal neurons by upregulating brain-derived neurotrophic factor. Biochem Biophys Res Commun. 2006;347:656–661. doi: 10.1016/j.bbrc.2006.06.117. [DOI] [PubMed] [Google Scholar]
- Zafra F, Lindholm D, Castren E, Hartikka J, Thoenen H. Regulation of brain-derived neurotrophic factor and nerve growth factor mRNA in primary cultures of hippocampal neurons and astrocytes. J Neurosci. 1992;12:4793–4799. doi: 10.1523/JNEUROSCI.12-12-04793.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]