

Abstract
Neuroprotection is increasingly considered as a promising therapy for preventing and treating temporal lobe epilepsy (TLE). The development of chronic TLE, also termed as epileptogenesis, is a dynamic process. An initial precipitating injury (IPI) such as the status epilepticus (SE) leads to neurodegeneration, abnormal reorganization of the brain circuitry and a significant loss of functional inhibition. All of these changes likely contribute to the development of chronic epilepsy, characterized by spontaneous recurrent motor seizures (SRMS) and learning and memory deficits. The purpose of this review is to discuss the current state of knowledge pertaining to neuroprotection in epileptic conditions, and to highlight the efficacy of distinct neuroprotective strategies for preventing or treating chronic TLE. Although the administration of certain conventional and new generation antiepileptic drugs is effective for primary neuroprotection such as reduced neurodegeneration after acute seizures or the SE, their competence for preventing the development of chronic epilepsy after an IPI is either unknown or not promising. On the other hand, alternative strategies such as the ketogenic diet therapy, administration of distinct neurotrophic factors, hormones or antioxidants seem useful for preventing and treating chronic TLE. However, long term studies on the efficacy of these approaches introduced at different time-points after the SE or an IPI are lacking. Additionally, grafting of fetal hippocampal cells at early time-points after an IPI holds considerable promise for preventing TLE, though issues regarding availability of donor cells, ethical concerns, timing of grafting after SE, and durability of graft-mediated seizure suppression need to be resolved for further advances with this approach. Overall, from the studies performed so far, there is consensus that neuroprotective strategies need to be employed as quickly as possible after the onset of the SE or an IPI for considerable beneficial effects. Nevertheless, ideal strategies that are capable of facilitating repair and functional recovery of the brain after an IPI and preventing the evolution of IPI into chronic epilepsy are still hard to pin down.
Keywords: aberrant synaptic reorganization, antiepileptic drugs, antioxidants, dentate gyrus, dentate neurogenesis, GABA-ergic interneurons, head injury, hippocampus, hippocampal cell grafts, hormones, ketogenic diet, learning and memory deficits, mossy fiber sprouting, neural cell transplants, neural stem cells, neurodegeneration, neurotrophic factors, spontaneous seizures, status epilepticus, synaptic plasticity, temporal lobe epilepsy
1. Introduction
Epilepsy is one of the oldest neurological conditions known to humankind. The term “epilepsy” is derived from Greek word “epilambanein”, which means “to seize upon” or “to attack”. In this modern era, epilepsy is the most frequent neurodegenerative disease after stroke. It afflicts more than 2 million Americans and 50 million people worldwide (Strine et al., 2005), and the temporal lobe epilepsy (TLE) is among the most frequent types of drug-resistant epilepsy (Engel, 2001; Litt et al., 2001; Mckeown and McNamara, 2001). In a population of new patients presented with epilepsy, almost 30% of them have seizures originating from the temporal lobe of the brain (Manford et al., 1992). Individuals affected with TLE typically have comparable clinical description, including an initial precipitating injury (IPI) such as the status epilepticus (SE), head trauma, encephalitis or childhood febrile seizures (Harvey et al., 1997; Fisher et al., 1998; Cendes, 2004). There is usually a latent period of several years between this injury and the emergence of the chronic TLE characterized by spontaneous recurrent motor seizures (SRMS) originating from temporal lobe foci, and learning and memory impairments (Devinsky, 2004; Detour et al., 2005). Further, the TLE is frequently associated with hippocampal sclerosis, mainly exemplified by significant neurodegeneration in the dentate hilus (DH), and the CA1 and CA3c sub regions (Sloviter, 2005). Human studies suggest that the hippocampal sclerosis likely initiates or contributes to the generation of most TLEs (Engel, 1996). A significant number of people (~25%) afflicted with epilepsy have seizures that cannot be controlled by antiepileptic drugs (Litt et al., 2001; McKeown and McNamara, 2001). Moreover, antiepileptic drugs (AEDs) merely provide symptomatic treatment without having any influence on the course of the disease. Thus, there is a pressing need to develop alternative therapeutic approaches that prevent the epileptogenesis after the SE or an IPI. From this perspective, identification of compounds or approaches that are efficacious for providing neuroprotection to the hippocampus after the onset of SE has great significance.
2. Definition, causes and consequences of seizures
A seizure is a convulsive episode, which starts of as atypical, excessive hyper-synchronous discharges from an aggregate of neurons in the brain and then recruits surrounding neurons to comprise one or both hemispheres of the brain (Kandel et al., 2000; Carey and Fuchs, 2001). In most patients presenting TLE, the development of seizures is preceded with an IPI such as the head trauma, stroke, SE, and infections like meningitis (Harvey et al., 1997; Fisher et al., 1998; Cendes, 2004). Nevertheless, it is difficult to predict the consequences of initial seizures in humans because of multiple contributing factors. These include differences in the etiology and age at the onset of seizures, the types, frequency and duration of seizures, interventions with AEDs and genetic components (Avoli et al., 2005; Guerrini et al., 2005). On the other hand, it is well known that epilepsy or seizures are linked with neurodegeneration in several areas of the brain (Wasterlain and Shirasaka, 1994; Jacobs et al., 2000; Armstrong, 2005). Furthermore, it is clear that both necrotic and apoptotic cell death contribute to neuronal damage in epileptogenic insults such as the SE, head injury or stroke (Fujikawa et al., 2000b; Fujikawa et al., 2000a; Henshall et al., 2000; Sahuquillo et al., 2004). It is supposed that multiple mechanisms and neurochemical modulators play roles in the initiation of epileptogenesis after an IPI. The term epileptogenesis refers to the transformation of the normal neuronal network into a long lasting chronically hyperexcitable state. Figure 1 illustrates major epileptogenic processes that emerge after an IPI and lead to the occurrence of SRMS.
Figure 1.
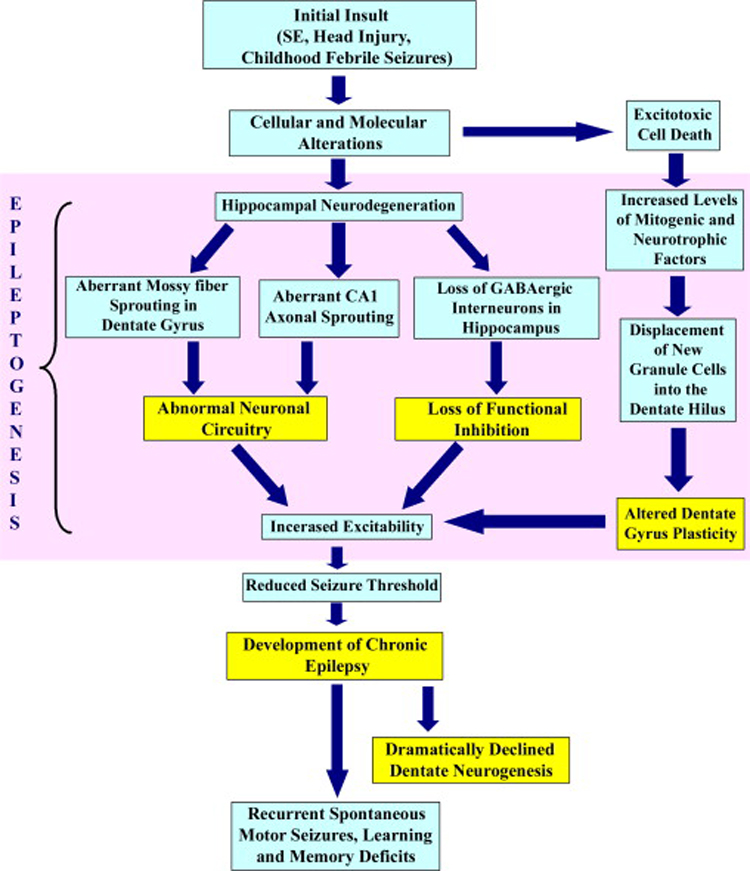
The various aspects of epileptogenesis after the initial brain insult and the evolution of the initial precipitating injury into chronic epilepsy and learning and memory deficits. An initial insult in the form of head injury or the status epilepticus (SE) typically leads to a number of cellular and molecular changes in the hippocampus. A transient surge in the proliferation of neural stem/progenitor cells also occurs in the dentate gyrus immediately after the SE resulting in abnormal neurogenesis. A multitude of alterations in the milieu of the dentate gyrus and the hippocampal CA1 and CA3 subfields lead to abnormal synaptic reorganization, the loss of functional inhibition by the GABAergic system and altered dentate gyrus plasticity, all of which augment the process of epileptogenesis. Collectively, these changes contribute to the occurrence of spontaneous recurrent motor seizures (SRMS) and learning and memory deficits during the chronic phase of epilepsy.
The brain injury resulting from seizures is a dynamic process that comprises multiple factors contributing to neuronal cell death. These include genetic factors, the extent of glutamate-mediated excitotoxicity leading to disturbances in the intracellular electrolyte metabolism, mitochondrial dysfunction, oxidative stress, growth factor withdrawal or depletion and increased concentration of cytokines (Ferriero, 2005). At cellular level, intense seizure activity typically initiates massive influx of calcium via voltage gated and N-methyl-D-aspartate (NMDA)-dependent ion channels (Van Den Pol et al., 1996). Elevated intracellular ions lead to biochemical cascades which trigger acute neuronal cell death after the SE (Fujikawa et al., 2000a). Additionally, high levels of intracellular calcium can induce generation of reactive oxygen species (ROS, also referred to as free radicles), uncoupling of mitochondria and activation of a wide range of catabolic enzymes that are capable of deteriorating cell function (Gupta and Dettbarn, 2003; Niquet and Wasterlain, 2004; Acharya and Katyare, 2005; Niquet et al., 2005).
3. Hippocampal neurodegeneration and synaptic reorganization after seizures
Acute seizures in the adult brain may lead to alterations in the synaptic plasticity, including the long term potentiation of synaptic responses. A wide range of neuropsychological deficits may follow the SE, which typically include learning and memory dysfunction and other cognitive deficits (Holmes et al., 2004; Holmes, 2006). Although multiple regions of the brain are affected with the SE induced through chemoconvulsants such as KA or pilocarpine, the hippocampal region has received the most attention because of its highly plastic nature and increased susceptibility to seizure-induced damage (Parent et al., 1997; Shetty and Turner, 1999a, b; Rao et al., 2006). After an IPI, characteristic pattern of hippocampal cell loss and shrinkage is seen later in life, when patients develop TLE (Sloviter, 1999). The injury inflicted by acute seizures in the hippocampus often includes considerable bilateral neurodegeneration in the DH, and CA1 and CA3 subfields, which eventually leads to a massive abnormal sprouting of mossy fibers into the dentate supragranular layer (Rao et al., 2006). Examples of degenerating neurons in both hippocampal and extrahippocampal regions, visualized through Fluoro-Jade B, Silver and TUNEL staining are shown in figure 2.
Figure 2.
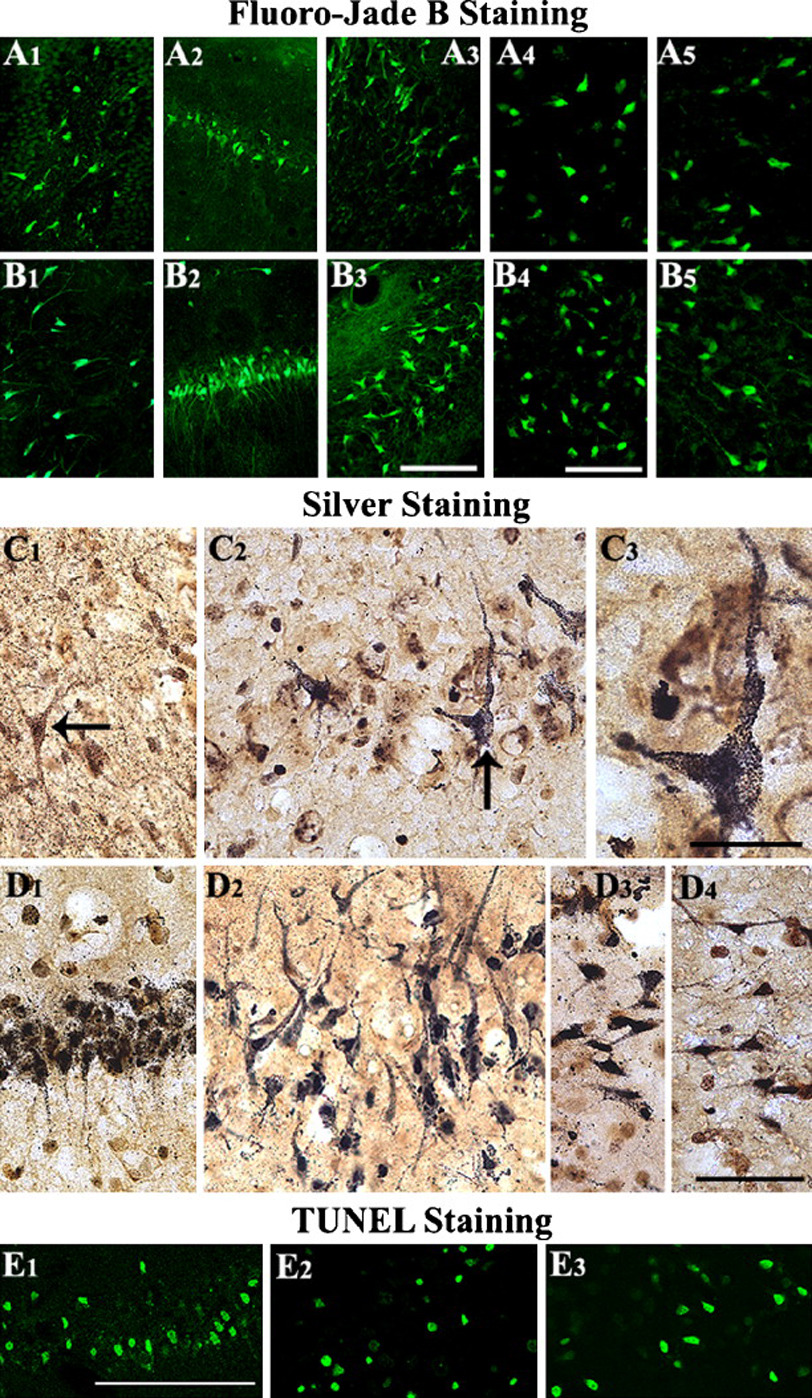
Neurodegeneration after the status epilepticus in hippocampal and extrahippocampal regions. The degenerating neurons were visualized at 24 hr after the status epilepticus through Fluoro-Jade B (A1-B5), silver (C1-D4), and TUNEL staining (E1-E3) in different regions of the hippocampus, amygdala, and the entorhinal cortex. Scale bars, A1-A3 and B1-B3 = 100µm; A4, A5, B4, B5 = 50µm; C1-C3 and D1-D4 = 20m; E = 200 µm. (Figure reproduced from: Rao et al., 2006; J Neurosci Res. 83(6):1088–1105).
3.1. Abnormal sprouting of mossy fibers in the dentate gyrus
The dentate gyrus (DG) has been the major focus of attention in TLE because of its higher threshold for seizures and its function as a “gate” to prevent the propagation of seizures into the hippocampus under normal conditions (Tauck and Nadler, 1985; Buckmaster and Dudek, 1999). However, in both epileptic human hippocampus and the KA treated rat hippocampus, axons of granule cells (mossy fibers) sprout aberrantly into the dentate supragranular layer (DSGL) as indicated in figure 3. The sprouted mossy fibers form new synapses on granule cell dendrites, which increase the overall excitatory connections between granule cells (Okazaki et al., 1995; Buckmaster et al., 2002; Scharfman, 2003). The aberrant axonal sprouting response of granule cells after hippocampal injury is likely a consequence of the degeneration of their post-synaptic target cells (CA3-pyramidal neurons) and/or afferent neurons (hilar mossy cells, Shetty and Turner, 1997b, 1999b; Shetty et al., 2005). Studies have shown that the extent of aberrant dentate mossy fiber sprouting positively correlates with both antidromically evoked burst firing and spontaneous seizures in KA models of TLE (Cronin and Dudek, 1988; Masukawa et al., 1989; Milgram et al., 1991; Masukawa et al., 1992; Mathern et al., 1993; Dudek et al., 1994; Okazaki et al., 1995; Buhl et al., 1996; Lynch and Sutula, 2000; Wuarin and Dudek, 2001; Cavazos et al., 2003), suggesting that the aberrant mossy fiber sprouting that ensues after the hippocampal injury contributes to the increased seizure susceptibility of the DG. Thus, prevention of aberrant mossy fiber sprouting may be important for blocking hyperexcitability and SRMS after the SE in both humans and animal models (Shetty et al., 2005).
Figure 3.
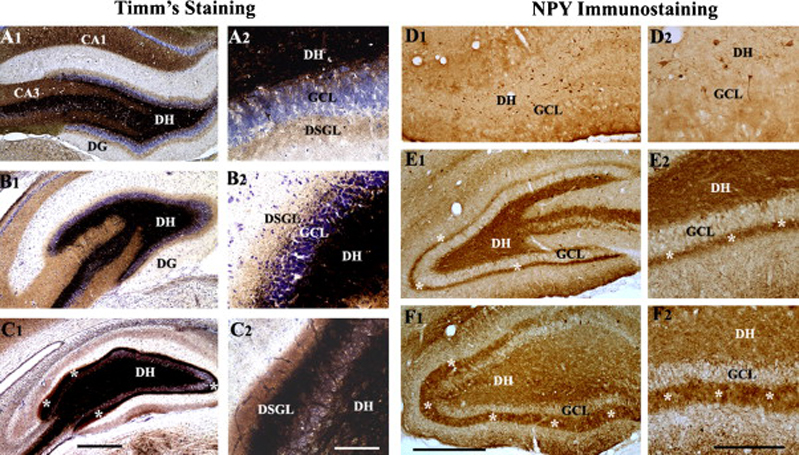
Aberrant mossy fiber sprouting after the status epilepticus in the dentate gyrus. The extent of the aberrant mossy fiber sprouting is illustrated for rats with moderate hippocampal injury (B1, B2 and E1, E2) and rats with severe hippocampal injury (C1,C2 and F1,F2), in comparison to age-matched intact rats (A1,2 and D1,D2) by Timm's histochemical staining (A1-C2) and neuropeptide Y (NPY) immunostaining (D1-F2). Note that, in comparison to rats exhibiting moderate hippocampal injury (B1, B2 and E1, E2), rats with severe hippocampal injury (C1, C2 and F1, F2) exhibit much robust aberrant sprouting of mossy fibers into the dentate supragranular layer (DSGL). DH, dentate hilus; GCL, granule cell layer. Scale bars, A, B1, C1 = 500 µm; A2, B2, C2 = 100 µm; D1, E1 and F1 = 500µm; D2, E2, F2 = 200 µm. (Figures reproduced from: Rao et al., 2006; J Neurosci Res. 83(6):1088–1105).
3.2. Abnormal sprouting of entorhinal axons in the CA1 subfield
Deafferentation of CA1 pyramidal neurons after KA-lesions (due to CA3 pyramidal cell loss) shows recovery of synaptic density over 2–3 months (Nadler et al., 1980a; Nadler et al., 1980b); but the source of axons participating in this reinnervation is mostly unknown. A study by Shetty (2002) investigated the contribution of the entorhinal cortex in this reinnervation at 3-months post-KA administration. The axons from the entorhinal cortex were visualized by anterograde axon tracing using injections of the biotinylated dextran-amine into the entorhinal cortex. In the CA1 region of the intact hippocampus, entorhinal axons were conspicuous in the alveus and the stratum lacunosum moleculare (SLM) but sparse in the stratum radiatum [SR, Fig. 4 (upper left panel)]. However, after KA-induced CA3-region injury, the density of entorhinal axons increased in the SR (375% of the intact hippocampus), as a large number of axons from the entorhinal fiber plexus in the SLM invaded the SR. The SR also exhibited wavy entorhinal axons filled with boutons and oriented parallel to the stratum pyramidale, suggesting some collateral sprouting from the entorhinal axons traversing the SR (Shetty, 2002). The sprouted fibers appear to come from both entorhinal fiber plexus in the SLM (translaminar sprouting) and entorhinal axons traversing the SR (intralaminar sprouting). The major contribution appears to be from the entorhinal plexus in the SLM (Fig 3). This aberrant sprouting may lead to altered afferent excitatory connectivity in the CA1 subfield and contribute to the persistent CA1 hyperexcitability that is typically observed after CA3-region neurodegeneration (Turner and Wheal, 1991). Indeed, a study using the pilocarpine model of TLE suggests that local application of convulsants to the medial entorhinal cortex leads to considerably enhanced epileptiform discharges in the CA1 subfield of pilocarpine treated rats in comparison to control rats (Wozny et al., 2005). Furthermore, single cell recordings of CA1 pyramidal neurons revealed that aberrant sprouting of entorhinal axons into the CA1 subfield leads to an altered cortical influence on CA1 neurons that eventually develops into hyperexcitability. The excitatory responses in the CA1 subfield were characterized by multiple after-discharges and strong paired-pulse facilitation in response to activation of the temporo-ammonic pathway. Thus, in epileptic rats, electrographic seizures may enter the hippocampus not only through the DG, but also via the temporo-ammonic pathway by shortcutting the trisynaptic hippocampal loop (Wozny et al., 2005).
Figure 4.
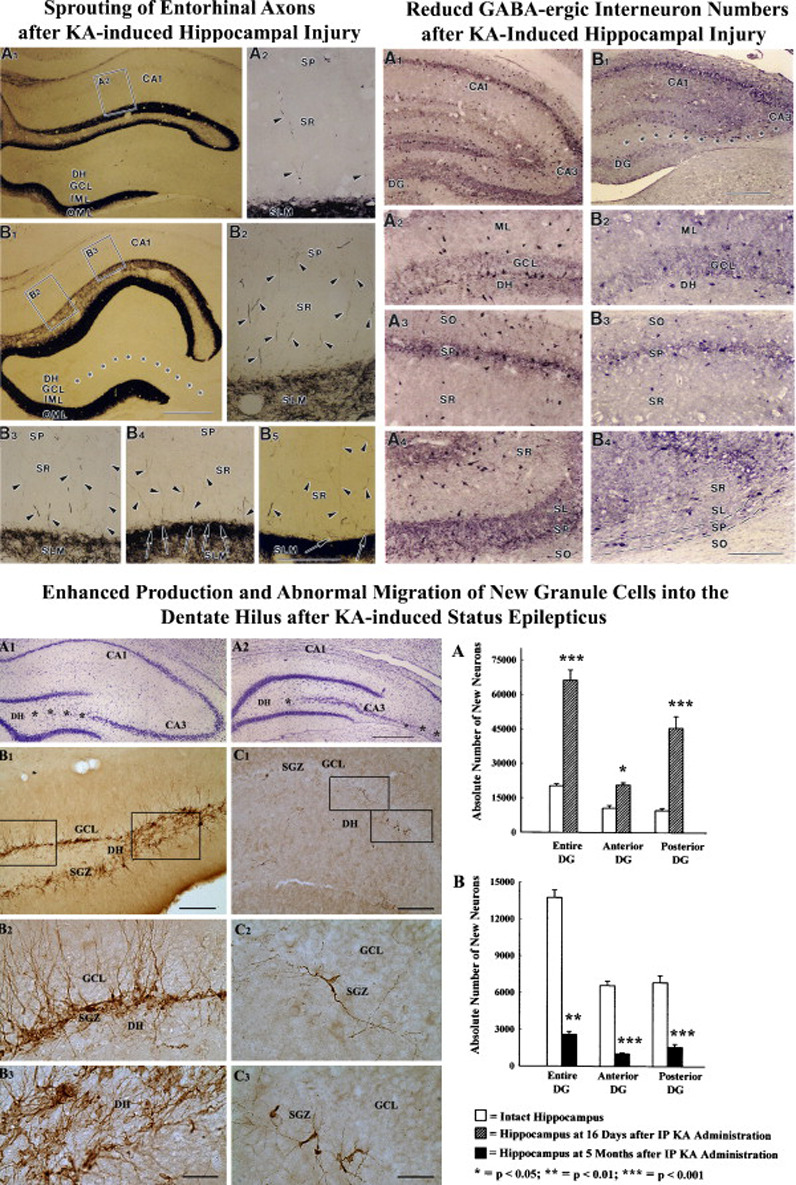
Top left panel: Morphology of the biotinylated dextran amine (BDA)-positive entorhinal axons in the CA3-lesioned hippocampus at 3 months after an intracerebroventricular administration of the kainic acid. A1, A2: Entorhinal axon of the alvear pathway traversing the CA1 stratum radiatum in an intact hippocampus showing wavy axons exhibiting a large number of en passant bouton (arrowheads). B1-B4: Region of the CA1 subfield from a CA3-lesioned hippocampus exhibiting a large number of horizontally oriented axons (arrows) filled with boutons (arrowheads), and branches (arrow in B3) and growth cone-like expansions at their termination in the outer thirds of the CA1 stratum radiatum. GCL, granule cell layer; IML, inner molecular layer; MML, middle molecular layer; OML, outer molecular layer; SLM, stratum lacunosum moleculare; SO, stratum oriens; SP, stratum pyramidale; SR, stratum radiatum. Scale bars, A1-A3, B2-B4 and C2 = 10 µm; B1 = 50 µm. (Figure reproduced from: Shetty, AK; 2002; Hippocampus; 12(4):534–542). Top right panel: Loss of GABA-ergic interneurons following kainic acid (KA) induced hippocampal injury. Note that KA induced injury reduces the density of GABA-ergic interneurons in the dentate gyrus, and CA1 and CA3 subfields (B1-B4), in comparison to the density typically observed in these regions of the naïve hippocampus (A1-A4). GCL, granule cell layer; DH, dentate hilus; SLM, stratum lacunosum moleculare; SO, stratum oriens; SP, stratum pyramidale; SR, stratum radiatum. (Figure reproduced from Shetty AK, Turner DA, J. Neurosci. 2006). Bottom panel: Hippocampal cytoarchitecture and distribution of newly formed doublecortin (DCX) immunopositive neurons in the dentate gyrus following intraperitoneal kainic acid (IPKA) injections. The photographs A1 and A2 show Nissl-stained sections from the septal and temporal regions of the hippocampus showing milder (A1) and severe (A2) neurodegeneration (Asterisks). The photographs B1, B2, B3 show the distribution of dramatically increased DCX immunopositive new neurons in the dentate gyrus at 16 days after IP KA injections. The photographs C1, C2, C3 show severely declined dentate neurogenesis (as revealed by only a few DCX immunopositive new neurons) at 5 months after KA injections. Scale bars, A1, A2 = 500 µm; B1, C1 = 200µm; B2, B3, C2 and C3 = 50µm. (Reproduced from: Hattiangady et al., 2004; Neurobiol Dis. 17(3):473–490).
3.3. Sprouting of CA3 axons
Siddiqui and Joseph (2005) using a KA model of TLE provide evidence for widespread CA3 structural reorganization in the form of sprouting of CA3 axons into multiple areas of the hippocampus and the entorhinal cortex. This includes an increase in the density of efferents to areas that normally receive CA3 afferent input such as the CA1 subfield and the subiculum. In addition, new efferents projected into the pre-and para- subicular regions and medial and lateral entorhinal cortices. Interestingly, a new CA3 Schaffer collateral projection to the entorhinal cortex was also observed. The widespread sprouting of CA3 axons to regions of the hippocampus and the entorhinal cortex likely explain how the epileptic hippocampus propagates the unconventional impulse excitation to cortical fields. Furthermore, because these cortical fields have a critical role in providing excitatory input into the hippocampus, this synaptic reorganization likely forms reverberating excitatory circuits (Siddiqui and Joseph, 2005). It is also likely that sprouting-related mechanisms explain the latency period (or quiescent phase) prior to the development of chronic TLE characterized by SRMS (Siddiqui and Joseph, 2005).
4. Changes in GABA-ergic interneurons after seizures
The prevailing hypothesis pertaining to links between GABA-ergic interneurons, epileptogenesis and TLE is that epileptogenesis results from a diminished GABA-mediated inhibition occurring through the degeneration of fractions of GABA-ergic interneurons. Inhibitory inputs from GABA-ergic hippocampal interneurons prevent the principal excitatory hippocampal neurons from becoming hyperexcitable under normal conditions (Freund and Buzsaki 1996). This is mediated by direct inhibitory inputs from these interneurons to both presynaptic axons, and soma and dendrites of principal excitatory neurons (Dvorak-Carbone and Schuman 1999; Yan et al. 2003). While the inhibitory inputs to presynaptic axons control the excitatory input coming from other regions (such as from the entorhinal cortex for dentate granule cells, dentate mossy fibers for CA3 pyramidal neurons and Schaeffer collaterals for CA1 pyramidal neurons), inhibitory input to soma and dendrites restrain the excitability of principal excitatory neurons (Karnup and Stelzer 1999). Thus, the overall inhibition mediated by GABA-ergic interneurons sets a threshold for the excitation of pyramidal cells, and the strength of interneuron input controls the level of discharges from principal hippocampal cells. This is evidenced by epileptic-like conditions in animal models following disinhibition of the principal excitatory neurons (Prince 1978; Dingledine and Gjerstad 1980). Therefore, it is plausible that reduced inhibitory input to principal hippocampal neurons due to loss of fractions of GABA-ergic interneurons underlies the persistent hyperexcitability in TLE (Franck et al. 1988; Cornish and Wheal 1989; Williams et al. 1993; Perez et al. 1996). In this section, we discuss the current understanding regarding the survival of GABA-ergic interneurons after acute seizures or the SE.
4.1. Alterations in hippocampal interneurons
Analyses of the hippocampus in animal models of TLE at early post-injury time-points suggested that GABA-ergic interneurons are least vulnerable to the SE or injury (Kohler et al., 1984; Franck et al., 1988; Davenport et al., 1990; Sloviter, 1991; Bekenstein and Lothman, 1993). However, analyses of subclasses of interneurons at later post-lesion time-points suggested a reduction in the number of interneurons (Sperk et al., 1986; Shetty and Turner, 1995a; Kobayashi and Buckmaster, 2003). Studies analyzing GABA-ergic [glutamate decarboxylase-67 (GAD-67) positive] interneurons at 1–6 months post-KA showed that reductions in GABA-ergic interneurons occur throughout the hippocampus after KA-induced hippocampal injury [Shetty and Turner, 2000, 2001; Figure 4 (upper right panel)]. Interestingly, parallel quantification of Nissl-stained interneurons mostly revealed no changes in interneuron density (Shetty and Turner, 2001), suggesting that reductions in GAD-67 positive interneuron density after KA-induced injury reflect down-regulation of GAD-67 protein expression in a major fraction of interneurons. Thus, the structural basis of the inhibitory system remains undisturbed following KA-induced hippocampal injury, especially the soma of interneurons and their efferent projections onto principal cells of the hippocampus. However, there is continued loss of functional inhibition in the hippocampus following KA-induced hippocampal injury (Cornish and Wheal, 1989; Perez et al., 1996). This suggests that persistent loss of GAD-67 within interneurons may be due to loss of both afferent connectivity and afferent neurotrophic support onto interneurons because of the loss of CA3 pyramidal and hilar neurons (Rocamora et al., 1992). Loss of both afferent connectivity and afferent neurotrophic support onto interneurons likely leads to less activation of these interneurons resulting in loss of functional inhibition.
Other factors, such as failure of release of GABA, activation of inhibitory autoreceptors, or down-regulation of GABA receptors could also be involved. On the contrary, studies of direct interneuron to principal cell inhibition in KA-lesioned hippocampus have indicated that these aspects of the inhibitory system remain mostly undisturbed (Bernard et al., 1998). Thus, persistently diminished number of GABA synthesizing interneurons may be the reason for continued decrease in functional inhibition. Changes in the function of GABA receptors may also contribute to reduced inhibitory function in epileptic conditions. Analysis of the DG for GABAA receptors in epileptic animals demonstrates that, while alpha-1 subunits of GABAA receptors exhibit reductions, the alpha-4 subunits of GABAA receptors increase in density (Brooks-Kayal et al., 1998). This is in contrast to increased alpha-1 subunit levels observed after early life SE (Raol et al., 2006a). Interestingly, enhanced expression of alpha-1 subunits of GABAA receptors in the DG after the SE through adeno-associated virus type 2 containing the alpha-4 subunit gene promoter induced a threefold increase in mean seizure free time after the SE and a 60% reduction in the number of rats exhibiting chronic epilepsy in the first four weeks after the SE (Raol et al., 2006b). These results underscore that alpha-1 subunit levels are important for maintaining adequate inhibitory function in the hippocampus. Thus, preservation of hippocampal GABA-ergic interneuron numbers or increased expression of GABA receptors via neuroprotective strategies may be critical for preventing chronic epilepsy development after SE.
4.2. Alterations in entorhinal cortex interneurons
Recently, there has been considerable interest in examining the role of entorhinal cortex in the induction of spontaneous seizures during chronic epilepsy (Kumar and Buckmaster, 2006). Studies from brains of TLE patients and animal models of TLE imply loss of layer III pyramidal neurons in the medial entorhinal cortex and hyperexcitability and hypersynchrony of less vulnerable layer II stellate neurons (Scharfman et al., 1998; Tolner et al., 2005; Kumar and Buckmaster, 2006, 2007; Kumar et al., 2007). It is hypothesized that hyperexcitability of stellate neurons leads to excessive, synchronous, excitatory synaptic input to the dentate granule cells (Buckmaster and Dudek, 1997a, b; Kobayashi and Buckmaster, 2003), which in turn contributes to dentate hyperexcitability and generation of SRMS. Investigation into the causes of hyperexcitability in stellate neurons did not detect any changes in their intrinsic electrophysiological properties and recurrent excitation but noted loss of inhibition (Buckmaster and Dudek, 1997a, b; Kobayashi and Buckmaster, 2003; Kumar et al., 2007). Loss of inhibition to stellate cells appeared to be due to loss of a fraction of GABA-ergic interneurons in layer III of the entorhinal cortex (Kumar and Buckmaster, 2006). Thus, changes in entorhinal cortex particularly the loss of GABA-ergic interneurons likely also contributes to the maintenance of chronic epilepsy. Indeed, higher seizure control after surgery when the resection of the hippocampus was combined with the resection of the entorhinal cortex observed in a recent study on human mesial TLE supports this possibility (Bonilha et al., 2007).
5. Dentate neurogenesis and temporal lobe epilepsy
Addition of new neurons to the dentate granule cell layer from proliferating neural stem/progenitor cells (NSCs) in the subgranular zone (SGZ) of the DG is maintained all through life in the mammalian CNS (Kaplan and Hinds, 1977; Kuhn et al., 1996; Cameron et al., 1998; Eriksson et al., 1998; Kornack and Rakic, 1999; Gage, 2002; Gould and Gross, 2002; Song et al., 2002; Emsley et al., 2005). Interestingly, hippocampal functions of learning and memory are closely linked to the extent of dentate neurogenesis (Gross, 2000; Feng et al., 2001; Shors et al., 2001; Hallbergson et al., 2003; Monje and Palmer, 2003). Moreover, changes in the milieu of NSCs in the SGZ can suppress or enhance dentate neurogenesis. For instance, cranial irradiation damages neurogenic niches in the SGZ, which leads to suppression of neurogenesis and impairments in learning and memory function (Monje et al., 2002; Monje and Palmer, 2003). On the other hand, several other types of acute brain/hippocampal injury such as ischemia, stroke and hypoxia considerably up-regulate dentate neurogenesis in the young adult brain (Choi et al., 2003; Felling and Levison, 2003). Additionally, continuous seizures such as SE induced via excitotoxins increases NSC proliferation and neurogenesis in the SGZ of the DG (Parent et al., 1997; Madsen et al., 2000; Nakagawa et al., 2000; Scott et al., 2000; Ekdahl et al., 2001; Hattiangady et al., 2004).
Hippocampal injury inflicted by excitotoxins such as KA also enhances the production of new neurons in the adult DG (Gray and Sundstrom, 1998). Typically, the SE or hippocampal injury induces an initial, transitory surge in the proliferation of NSCs, which leads to over production of new neurons during the first few weeks after injury (Parent et al., 1997; Nakagawa et al., 2000). This is likely due to the release of multiple mitogenic factors from dying neurons, deafferented granule cells and reactive glia, as earlier studies imply that multiple neurotrophic factors are up-regulated in the hippocampus following seizures or excitotoxic injury (Lowenstein et al., 1993; Shetty et al., 2003; Shetty et al., 2004). This may also be due to acute hyperexcitability in the DG that follows immediately after the SE or KA-induced injury (Sloviter et al., 2006). Nevertheless, dentate neurogenesis reaches baseline after 2–3 weeks of the insult with normalization of the rate of proliferation of NSCs (Parent et al., 1997; Nakagawa et al., 2000), which may parallel the normalization in the levels of neurotrophic factors (Shetty et al., 2003). Shortly after acute seizures or the SE, some of the newly born neurons migrate aberrantly into the DH (Parent and Lowenstein, 1997; Scharfman et al., 2000; Parent, 2002, 2003; Hattiangady et al., 2004; Figure 4 [Lower panel]). This is likely due to the hyperactivity induced overproduction of new neurons in the dentate SGZ, lack of space in the granule cell layer (GCL), and loss of expression of reelin, a secreted migration guidance cue that persists in the adult hippocampus (Gong et al., 2007). Despite their abnormal location, the intrinsic electrical properties of these ectopic neurons are mostly comparable to granule cells located in the GCL (Scharfman et al., 2000). However, their dendritic and synaptic structures are different from normal dentate granule cells (Shapiro et al., 2005; Shapiro and Ribak, 2005; Shapiro and Ribak, 2006). In addition, they frequently exhibit spontaneous epileptiform activity, which is never observed in normal granule cells (Scharfman et al., 2000). Recent studies further suggest that ectopic granule cells contribute to a lower seizure threshold in the epileptic hippocampus and are involved in supporting recurrent seizures in epileptic rats (Scharfman, 2002b; Scharfman et al., 2003).
Thus, following the SE, the DG exhibits abnormal circuitry in the DH, which is an additional epileptogenic change that likely contributes to the evolution of the IPI into chronic epilepsy. Furthermore, recent studies suggest that once the acute seizure-induced hippocampal injury evolves into chronic TLE characterized by SRMS, dentate neurogenesis declines dramatically in the chronically injured hippocampus (Hattiangady et al., 2004; Kralic et al., 2005), which is consistent with the observation in human TLE (Pirttila et al., 2005). Figure 4 (lower panels) demonstrates the extent of neurogenesis during the acute and chronic phases of TLE in a rat model. As fraction of newly-born neurons develop into GABA-ergic interneurons (Liu et al., 2003) and TLE is associated with decreased numbers of GABA-ergic interneurons, declined neurogenesis during chronic epilepsy may contribute to increased seizure-susceptibility of the DG. Likewise, hippocampal-dependent learning and memory deficits and depression observed in chronic TLE (Brown-Croyts et al., 2000; Oddo et al., 2003; Alessio et al., 2004) could be linked at least partially to the declined neurogenesis. Thus, neuroprotective approaches that block both aberrant neurogenesis occurring during the early period after the SE and dramatically decreased neurogenesis occurring at extended time-points after the SE may be useful for preventing chronic epilepsy as well as learning and memory deficits observed after SE.
6. Neuroprotective strategies for preventing chronic epilepsy
The onset of chronic epileptic seizures (also refereed to as SRMS) after brain insults such as SE, stroke or head trauma occurs after a delay. It is believed that multiple epileptogenic changes occur during this latent period. Thus, the latent period after an IPI provides an opportunity for applying effective intervention strategies that are capable of preventing the progression of initial seizure or injury induced neurodegeneration into chronic epilepsy, characterized by SRMS and learning and memory deficits. Moreover, early intervention after the initial insult may modify the progression of disease considerably. For example, shortening the duration of seizures or limiting their spread might prevent some of the neurodegeneration induced by acute seizures. Similarly, application of strategies that are efficacious for promoting the repair of disrupted circuitry may prevent the development of abnormal synaptic reorganization. Additionally, approaches that block abnormal DG neurogenesis may reduce seizure susceptibility of the DG (Parent, 2003; Jung et al., 2004).
From the beginning of medical history, continuous efforts have been made to treat seizures and in recent years, treatment modalities employed have been relatively successful in suppressing seizures in 50–65% of cases. Although pharmaceutical agents that suppress seizures (also called ‘antiepileptic drugs’), do not seem to have ‘anti-epileptogenic’ effects, neuroprotection may be possible with certain AEDs (Pitkanen, 2002). In addition, studies suggest that administration of gonadal steroids, neurotrophic factors and dietary interventions may be useful in this regard. However, to achieve maximum success via neuroprotective strategies, it is important to ascertain the efficacy of their administration at the right time (i.e. shortly after the SE or in the early part of the latent period) in sufficient dosage. This would allow determination of their ability for enhancing the function of endogenous repair systems without disturbing the delicate functioning of the CNS. Therefore, a combination of neuroprotective and antiepileptogenic strategies that are effective for combating the disease progression are needed, and not drugs that just suppress the symptoms of the disorder.
In animal models of TLE induced by KA or pilocarpine, neuronal damage mostly occurs in structures belonging to the circuit of initiation and maintenance of seizures (i.e. the DG and the hippocampus). However, some neurodegeneration also occurs in the propagation areas such as the entorhinal, perirhinal and piriform cortices, and thalamic and amygdalar nuclei. Ideal neuroprotection strategies after acute seizures should be capable of rescuing neurons in multiple brain regions that are vulnerable to seizures and be efficacious to prevent SRMS that usually arise weeks after the IPI (Sutula et al., 2003). Thus, interventions that not only protect neurons from dying after acute seizures but also forestall the evolution of initial seizures into SRMS are required. With increasing evidence for the progressive and cumulative adverse consequence of seizures in experimental models and TLE, an increased focus for developing neuroprotective interventions is vital for minimizing the incidence of chronic epilepsy. Recent advancements in animal/human studies enlighten the domain of antiepileptogenic and neuroprotective strategies for salvaging, protecting and repairing neurons in post-SE condition. Most of the studies on neuroprotection are based on animal experimental models of neurodegeneration (Pitkanen, 2002). Electrically and pharmacologically evolved seizures as well as different models of ischemia are frequently used. Preconditioning models have provided valuable insights into how repair systems work in the brain. Here we evaluate the current knowledge and recent developments concerning neuroprotection strategies for preventing or treating TLE via drug interventions, ketogenic diet therapy, administration of antioxidants and hormones, and neural cell transplantation.
7. Neuroprotection using antiepileptic drugs
The anticonvulsive mechanisms of conventional and newly introduced drugs vary considerably. The most common actions were shown on ion channels, GABA-ergic and glutamatergic metabolism, receptors or secondary messengers (Macdonald and Kelly, 1994, 1995). Extensive efforts have been made to achieve neuroprotection through effective seizure suppression with anticonvulsants and new compounds that may be neuroprotective through mechanisms other than anticonvulsant actions. A variety of AEDs have been tested in rat models of SE for their efficacy to prevent epilepsy, neurodegeneration and behavioral defects. Such initial insult modification by AEDs should be clearly differentiated from drugs that are capable of improving the long-term consequences of a brain insult when administered at delayed time-points after the insult. Numerous reports suggest that conventional as well as recently introduced anticonvulsants have some neuroprotective activity in models of ischemia (detailed in reviews by Pitkanen, 2002; Trojnar et al., 2002; Stepien et al., 2005). However, achieving considerable neuroprotection in models of seizures and epilepsy is difficult because of multiple alterations that concurrently ensue in the brain after an initial insult and contribute to the development of chronic epilepsy. It is plausible that ongoing seizures and AED treatment influence the biological processes in the brain and contribute to lasting impairments in cognitive function (Marsh et al., 2006b). The effects of AEDs on psychotropic behavior have been extensively discussed in earlier reviews (see Selai et al., 2005; Ettinger, 2006; Schmitz, 2006; Kalinin, 2007). In the following section, we confer neuroprotective effects of certain AEDs in animal models of seizures and epilepsy. Table 1 provides a brief outline of anti-epileptogenic effects of some of the conventional and new generation AEDs.
Table 1.
Antiepileptogenic effects of AEDs in experimental models of seizures
| Antiepileptic drug | Effect on Epileptogenesis |
|---|---|
| Benzodiazepines | +/− |
| Phenobarbital | + |
| Valproate | +/− |
| Topiramate | + |
| Gabapentin | +/− |
| Lamotrigine | + |
| Felbamate | + |
| Levetiracetam | +/− |
| Tiagabine | + |
| Vigabatrin | + |
(+) Neuroprotective action present; (+/−) variable data
7.1. Conventional Antiepileptic drugs
7.1.1. Benzodiazepines
The benzodiazepines produce a variety of effects by modulating the GABAA receptor (O'Brien, 2005; McNamara, 2006). First, they increase the frequency of opening of the associated chloride ion channels and hyperpolarize the membrane. These changes facilitate the inhibitory effects of the available GABA, and leads to sedatory and anxiolytic effects (Schwartz et al., 1995; O'Brien, 2005). Second, different benzodiazepines can have different affinities for GABAA receptors. Among these, diazepam, clobazam and clonazepam are well known anticonvulsants. Diazepam is widely used to limit the duration of seizure period in SE models of TLE, in order to reduce the SE-associated mortality and to avoid the variability in long-term consequences of SE arising from differences in the duration of SE among individual animals (Goodman, 1998). Marciani and colleagues (1993) reported neuroprotective effects of clonazepam with respect to inhibition of the epileptic activity and the prevention of CA1 pyramidal neuron loss at 10 minutes after bilateral carotid occlusion on Mongolian gerbils. Interestingly, very low doses of clonazepam (0.2 mg/Kg, i.p.) were effective for both attenuating tonic-clonic seizures and decreasing the mortality of rats from 15% to 3% following KA-induced seizures (MacGregor et al., 1997). When clonazepam was combined with a muscimol agonist chrolmethizol, the overall neuroprotective effect was enhanced (MacGregor et al., 1997). Likewise, diazepam treatment immediately after soman induced seizures considerably prevented pathophysiological alterations in the hippocampus as well as the piriform cortex (Bhagat et al., 2005).
Furthermore, Pitkanen and colleagues (2005) examined the effects of termination of seizures with diazepam (20 mg/Kg b.w.) at 2 or 3 hours after the commencement of SE in the amygdala electrical stimulation model of SE. This study demonstrated that treatment of the SE with diazepam at 2 hours after the onset of SE reduces the risk of epilepsy later in life. Interestingly, administration of diazepam as above reduced the percentage of epileptic animals to 42% compared to 94% in the vehicle group, and the animals that developed chronic epilepsy in the diazepam group displayed reduced frequency of SRMS and milder aberrant mossy fiber sprouting (Pitkanen et al., 2005). Additionally, a recent study demonstrates that, administration of diazepam (5 mg/Kg b.w) at one hour after the onset of KA-induced SE considerably reduces behavioral seizures, seizure-induced increases in net neurogenesis as well as the ectopic migration of newly born neurons into the DH (Dhanushkodi and Shetty, 2007). Thus, administration of benzodiazepines such as diazepam shortly after the induction of SE appears very useful for reducing seizures and minimizing seizure induced epileptogenic changes such as the aberrant mossy fiber sprouting and the ectopic migration of newly born granule cells. However, it remains to be determined whether rats treated with diazepam at one hour after the commencement of the SE would develop chronic epilepsy later in life. Moreover, it may be useful to explore a combination therapy of benzodiazepines with new generation AEDs for further enhancing neuroprotection and preventing chronic epilepsy development after the SE.
7.1.2. Phenobarbital
Phenobarbital (PHB) enhances the action of GABA through GABAA receptors and inhibits the action of glutamate by blocking sodium channels (Deckers et al., 2000; McNamara, 2006). Numerous neuroprotective as well as neurodegenerative properties have been suggested for PHB. In an elegant study by Sutula and co-workers (1992), it was demonstrated that co-treatment of PHB with KA administration and further 5 days of PHB treatment (at a dose of 60 mg/Kg b.w.) after the KA administration suppresses acute seizure activity, protects against excitotoxic damage in the DG, reduces aberrant mossy fiber sprouting, and abolishes the increased susceptibility of KA treated animals to kindling. Thus, early PHB administration after the onset of seizures appears useful for reducing the damage and abnormal synaptic reorganization in the DG. However, PHB treatment in this study did not protect against seizure-induced neurodegeneration in CA1 and CA3 subfields (Sutula et al., 1992). Another pre-treatment study with PHB (at a dose of 20 mg/Kg b.w.) initiated 45 minutes prior to the systemic KA injection demonstrated prevention of KA-induced deficits in spatial learning task (Brown-Croyts et al., 2000). Yet, no positive effects were observed when PHB treatment was initiated 2–3 hours after the administration of KA. The neuroprotective actions of PHB are believed to be due to free radical scavenging and cytochrome P450 induction properties of PHB. Thus, PHB is a useful neuroprotective agent when given either prior to or during the administration of chemoconvulsants. Contrarily, when administered after the onset of the SE, PHB does not appear to be beneficial for preventing the evolution of SE into chronic epilepsy.
7.1.3. Valproate
The valproic acid (VPA; 2-propylpentanoic acid) is the drug of choice for primary generalized epilepsies, and is used for the treatment of partial seizures (McNamara, 2006). Interestingly, the discovery of the VPA was accidental; its antiepileptic properties were recognized when it was used as a solvent for the experimental screening of new antiepileptic compounds (Krall et al., 1978a, b). The VPA inhibits the function of sodium and possibly T-type calcium channels, and enhances GABA-ergic transmission (Deckers et al., 2000; Czuczwar et al., 2001). Yet, short term infusions of VPA through the microdialysis probe does not prevent hippocampal neurodegeneration and accumulation of extracellular glutamate induced by potassium channel blocker 4-aminopyridine (Pena and Tapia, 2000). However, in a KA model of TLE, repetitive treatment with VPA for 40 days, commencing at 24 hours after the onset of the SE considerably protects against neurodegeneration in CA1 and CA3 subfields and the DH, and blocks the development of SRMS and deficits in emotional responses or spatial learning until 75 days after the SE (Bolanos et al., 1998). In contrast to these findings, a study by Brandt et al. (2006) using a rat model of SE induced by prolonged electrical stimulation of the basal amygdala demonstrates that VPA treatment beginning at 4 hours post-SE and ending at 4 weeks post-SE does not prevent the frequency or severity of SRMS but averts hippocampal neurodegeneration and behavioral impairments to some extent (Brandt et al., 2006). Furthermore, a recent study by Jessberger and associates (2007) shows that prolonged VPA administration commencing shortly after the SE is efficient for inhibiting the seizure-induced abnormal neurogenesis in the DG and hippocampal-dependent learning deficits.
Thus, based on currently available studies, it appears that the efficacy of prolonged administration of VPA after the onset of the SE for preventing chronic epilepsy development varies depending on the model employed. It is currently unknown whether beneficial effects of VPA for blocking chronic epilepsy observed in some studies persist once the VPA administration is terminated. To validate the usefulness of long-term administration of VPA for preventing the SE-induced chronic epilepsy will therefore need rigorous long-term studies in multiple SE models of chronic epilepsy in future. Nevertheless, there seems to be consensus regarding the beneficial effects of VPA administration for decreasing the SE induced hippocampal neurodegeneration, abnormal neurogenesis and learning impairments. Although the precise mechanisms underlying these beneficial effects are unknown, increased levels of multiple cell survival factors after VPA administration might be involved. These may comprise cAMP responsive element binding protein (CREB), brain-derived neurotrophic factor (BDNF), Bcl-2, mitogen-activated protein kinases (MAPK), the cell survival factor Akt, as VPA is known to regulate the levels of these factors involved in cell survival pathways (De Sarno et al., 2002; Loscher, 2002; Bachmann et al., 2005). Furthermore, VPA has been shown to suppress the seizure-induced expression of c-fos and c-jun. This is significant because these early genes are likely involved in the apoptosis and necrosis pathways (Szot et al., 2005).
7.2. New Antiepileptic drugs
The conventional AEDs were the mainstays of seizure treatment until the 1990s, when newer AEDs with equal or greater efficacy but with fewer toxic effects and better tolerability were developed (McNamara, 2006). In the following section, we discuss the available studies on neuroprotective properties of new generation AEDs.
7.2.1. Topiramate
The drug topiramate (TPM) is a potent anticonvulsant and is structurally different from other AEDs. Multiple mechanisms of action have been proposed for TPM. It exerts an inhibitory effect on sodium conductance leading to reduced duration of spontaneous bursts and decreased frequency of generated action potentials. Administration of TPM also enhances GABA action by unknown mechanisms, inhibits the AMPA subtype of glutamate receptors, and weakly inhibits carbonic anhydrase (Czuczwar and Przesmycki, 2001; McNamara, 2006). Studies concerning the neuroprotective properties suggest that administration of TPM results in a delayed occurrence and reduced severity of kindled seizures (Hanaya et al., 1998), and diminished hippocampal neuronal injury in the CA1 and CA3 subfields following seizures (Niebauer and Gruenthal, 1999). In the lithium-pilocarpine model of TLE, the administration of TPM treatment decreased seizure frequency and the development of SRMS with increasing dose (Kudin et al., 2004; Rigoulot et al., 2004; Suchomelova et al., 2006). Furthermore, it decreased neurodegeneration in CA1 and CA3 subfields and the DH (Rigoulot et al., 2004; Suchomelova et al., 2006) and increased the functioning of mitochondrial oxidative energy metabolism (Kudin et al., 2004). Administration of TPM has also been reported to be neuroprotective against ibotenate induced excitotoxic brain lesions in developing mouse (Sfaello et al., 2005). Studies on combination therapy of TPM with other AEDs indicated that TPM was useful for preventing neurodegeneration in hippocampal and ventral entorhinal cortices when combined with diazepam but not efficient for delaying the occurrence and frequency of SRMS (Francois et al., 2006). On the other hand, higher doses of TPM with budipine terminated SE and facilitated the survival of CA1 pyramidal neurons in a kindling model of SE (Fisher et al., 2004). A recent study suggests that administration of a lower dose of TPM (20mg/Kg b.w.) at 40 minutes after the pilocarpine induced SE followed with diazepam (4 mg/Kg b.w.) administration at 160 minutes after the SE prevents short-term memory deficits typically seen following the SE (Frisch et al., 2007). Collectively, the above studies suggest that administration of TPM as either monotherapy or as a combination therapy with other AEDs limits the extent of hippocampal neurodegeneration, leads to anti-seizure effects, and averts short-term memory deficits in animal models of SE. However, from the studies available hitherto, it does not appear that the administration of TPM after the onset of SE would be efficacious for preventing chronic epilepsy development.
7.2.2. Felbamate
The drug felbamate (FBM) is a broad spectrum AED introduced into the clinical practice for controlling seizures in patients affected by Lennox-Gastaut epilepsy, complex partial seizures or otherwise intractable epilepsies (Corradetti and Pugliese, 1998; Trojnar et al., 2002; McNamara, 2006). The anticonvulsive effects of FBM are mediated by several mechanisms, which include the blockade of sodium and voltage-dependent L-type calcium channels, potentiation of GABAergic neurotransmission, and reduced glutamate-mediated excitation by its action on NMDA receptors (Deckers et al., 2000; McNamara, 2006). The neuroprotective properties of this drug were first observed in the hippocampal slice model. It was observed that perfusion of slices with 1.2–1.6 mM concentrations of FBM reduces the incidence of the irreversible disappearance of the CA1 electrical responses induced by KA (Longo et al., 1995). Administration of FBM also decreases the amplitude of NMDA receptor mediated excitatory post-synaptic potentials (EPSPs) and propagation of epileptic discharges in electrically evoked synaptic potentiation of CA1 in rat hippocampal slices (Pugliese and Corradetti, 1996; Pugliese et al., 1996). In an experimental model of self-sustaining SE, FBM and fluorofelbamate, a FBM analogue, reduced the frequency and severity of SRMS (Mazarati et al., 2000, 2002). Thus, FBM appears to be a useful drug for controlling seizures. However, elucidation of its neuroprotective properties and its capability for thwarting chronic epilepsy after the SE require detailed long-term studies in animal models of TLE.
7.2.3. Levetiracetam
The antiepileptic activity for levetiracetam (LEV) is still obscure though it is known that it binds to 90 kDa integral membrane protein and likely modulates the calcium mediated neurotransmitter release (Janz et al., 1999; Bialer et al., 2004; Lynch et al., 2004). The available reports on neuroprotective actions of LEV indicate that administration of LEV treatment (at 54 mg/Kg b.w.) commencing at 30 minutes and ending on day 21 after the onset of seizures is not effective for reducing the SE-induced hippocampal damage (Pitkanen, 2002; Klitgaard and Pitkanen, 2003). A recent study, in a model of SE induced through electrical stimulation of the amygdala, demonstrates that prolonged (5–8 weeks) administration of LEV does not lead to antiepileptogenic or neuroprotective effects (Brandt et al., 2007). Furthermore, behavioral hyperactivity and learning deficits typically observed in epileptic rats were not affected with LEV treatment after the SE. Thus, the outlook for using LEV as antiepileptogenic and neuroprotective drug is bleak though it may be useful for mere control of seizures in the epileptic patients (Grosso et al., 2007; Heo et al., 2007; Schulze- Bonhage et al., 2007).
7.2.4. Gabapentin
Gabapentin (GBP), a structural analogue of GABA (Kelly, 1998), is known for increasing the synthesis of GABA and blocking the α2-δ1 subunits of voltage dependent calcium channels (Deckers et al., 2000; Field et al., 2006; McNamara, 2006). A study shows that young (postnatal day 35) animals treated with GBP for 40 days following the KA-induced SE exhibited reduced incidence of SRMS, a better pathology score, diminished aggressiveness, and no long-term adverse consequences on cognitive processes (Cilio et al., 2001). Thus, this drug has some promise but detailed investigations on its anti-epileptogenic neuroprotective properties are needed.
7.2.5. Lamotrigine
The AED lamotrigine (LTG) is a triazine compound capable of blocking voltage-dependent sodium-channel conductance. It inhibits depolarization of the glutamatergic presynaptic membrane leading to inhibition of glutamate release (Matsuo et al., 1996; Matsuo, 1999; Rogawski and Loscher, 2004). A study shows that pre-treatment of animals with LTG before KA administration prevents hippocampal cell loss, but does not prevent seizures at doses of 10 and 30 mg/Kg b.w. (Maj et al., 1998). On the other hand, in amygdala kindling model, LTG treatment at doses of 5, 10 and 20 mg/Kg reduces seizure severity (Maj et al., 1999). Likewise, pre-treatment of animals with LTG, prior to 3-nitropropionic acid induced neurotoxicity, prevents hippocampal and striatal lesions (Lee et al., 2000). Furthermore, Halonen and colleagues (2001a) demonstrate that administration of LTG (12.5 mg/Kg b.w.) twice a day for 2 weeks, starting at 60 minutes after an hour of perforant pathway stimulation reduces hippocampal damage in adult rats but fails to block the SE-induced spatial memory impairments. On the contrary, 15 mg/Kg b.w. dose of LTG prior to amygdala kindling model serves as an effective anticonvulsant but fails to alter kindling (Postma et al., 2000). Thus, the pre-treatment study and the study where treatment was commenced shortly after the SE suggest neuroprotective property for this drug. Nonetheless, it is difficult to predict its usefulness for blocking chronic epilepsy when administered after the induction of SE or acute seizures, as no such studies are currently available.
7.2.6. Tiagabine
The drug tiagabine (TGB), a novel GABAergic agonist, temporarily prolongs the presence of GABA in the synaptic cleft through delayed clearance (Czuczwar and Patsalos, 2001; Reijs et al., 2004). It increases synaptic GABA availability via inhibition of the GABA transporter GAT-1 on presynaptic neurons and glial cells (Czuczwar and Patsalos, 2001; Reijs et al., 2004). In the perforant pathway stimulation model of SE, Halonen et al. (1996) showed that sub-chronic administration of TGB at a dose of 50 mg/Kg b.w. per day completely prevented the occurrence of generalized clonic seizures during stimulation, reduced the loss of pyramidal cells in the CA3c and CA1 subfields of the hippocampus and diminished impairments in spatial memory associated with hippocampal damage (Halonen et al., 1996). Yet, the degeneration of somatostatin immunoreactive neurons in the DH could not be prevented with this treatment. Furthermore, there is no evidence so far to support the neuroprotective and antiepileptogenic properties of this drug when administered after the induction of the SE.
7.2.7. Vigabatrin
The drug vigabatrin (VGB), a close structural analog of GABA, binds irreversibly to the active site of GABA-transaminase. In vivo studies in human and animal subjects have shown that VGB significantly increases extracellular GABA concentrations in the brain (Sidhu et al., 1997; McNamara, 2006). A study shows that administration of VGB by osmotic minipumps (75 mg/Kg per day) for 2 months starting at 2 days after the induction of the SE with KA reduces hippocampal neurodegeneration (Jolkkonen et al., 1996). However, in the amygdala stimulation model of SE, neuroprotection was not observed with 10 weeks of VGB treatment (75 mg/Kg b.w. per day via subcutaneous minipumps) commencing at 2 days after the onset of SE (Halonen et al., 2001b). In the same vein, a study by Pitkanen et al. (1999) suggests that VGB treatment commencing at either 1 hour or 1 week after the onset of KA-induced SE does not lead to any neuroprotective effects. Moreover, in an electrically kindled model, 4 weeks of VGB treatment at a dose of 250 mg/Kg b.w. commencing at 48 hours after the stimulation did not lead to any neuroprotective effects on CA1 neurodegeneration (Lothman, 1996). On the other hand, Andre and colleagues (2001), report a clear neuroprotective effect of VGB when treatment was initiated at 10 minutes after the onset of the SE and continued for 45 days. The overall neuroprotection was almost complete in the CA3 subfield, considerable in the CA1 subfield and moderate in the DH (Andre et al., 2001). Thus, VGB works well when treatment is initiated very early after the onset of the SE. It does not seem efficacious however when the treatment is commenced after an hour of SE.
7.3. Conclusions
The extent of neuroprotective and antiepileptogenic effects of individual AEDs (conventional or new generation drugs) is still unclear because studies available so far demonstrate variable extent of neuroprotection with these drugs, depending on the timing of administration after the onset of SE and the animal model of SE employed in the study. Among the conventional drugs, early administration of diazepam or VPA after the SE appears promising for preventing chronic epilepsy. Therefore, well defined long-term studies in animal models as well as clinical trials with a combination of conventional AEDs (such as diazepam and VPA) are needed in future to fully gauze their neuroprotective and antiepileptogenic properties (Sankar, 2005). On the other hand, investigation on the neuroprotective properties of new generation drugs is still in infancy. The drugs such as TPM, LTG and VGB appear to have some anti-epileptogenic effects in animal models of epilepsy. Although many AEDs are effective in terms of their anticonvulsive effects, currently no individual AED can be viewed as a potential neuroprotective drug for preventing chronic epilepsy after an IPI (Loscher et al., 2006). Considering these, examining the effects of a combination of two or more drugs would be useful for ascertaining their efficacy for modifying the progression of epileptogenesis. However, it is plausible that the available AEDs are useful for only controlling seizures and deleterious for maintaining normal cognitive function, as also suggested by Sankar and Holmes (2004). Thus, while searching new drugs for treating epilepsy, it is important to find those that are useful not only for seizure suppression but also efficacious for preventing seizure-induced neurodegeneration and blocking multiple epileptogenic changes that ensue after an IPI.
8. Neuroprotection using the ketogenic diet
In spite of new developments in the AED research and availability of almost 20 AEDs, seizures remain unmanageable in many types of epileptic manifestations. Moreover, the AED therapy is associated with significant side-effects (Porter et al., 1997; Browne and Holmes, 2001; Wheless et al., 2001). From this perspective, since 1990s, the ketogenic diet (KD) is emerging as one of the effective therapies with relatively reduced side effects particularly in difficult-to-control epilepsies (Freeman et al., 2006, 2007). A number of studies suggest that KD is more effective for the management of refractory epilepsy in children than other currently available anticonvulsant medications (Freeman and Vining, 1998; Freeman et al., 1998; Vining et al., 1998; Kossoff, 2004; Kossoff et al., 2004; Kossoff and McGrogan, 2005). Moreover, the availability of several infant formulas of the KD makes this approach more convenient for treating children with epilepsy (Nordli et al., 2001; Klepper et al., 2002). In this section we describe neuroprotection potential of KD therapy for epilepsy.
8.1 Neuroprotective and disease modifying effects of the ketogenic diet
Several studies have implied that the KD has anticonvulsant and anti-epileptogenic roles in different rodent models of epilepsy. Freeman and colleagues (2002) reported that a calorie-restricted (CR) diet high in fats, with sufficient protein and limited carbohydrates could mimic the biochemical changes of starvation and could preserve its beneficial effects on seizures. A series of experiments carried out by Bough et al. (1999, 2000, 2003) assessed the effects of the KD on seizure severity and threshold. In one study, animals fed with KD for 5 weeks after pentylenetetrazole (PTZ) induced seizures exhibited longer latency period and elevated thresholds for seizure induction in comparison to controls (Bough and Eagles, 1999). When the caloric ratio of fat in the diet was increased from 80% to 90%, resistance to PTZ-induced seizures improved further (Bough et al., 2000). Moreover, electrophysiological studies suggest that Schaffer collateral stimulation in hippocampal slices of the KD-fed rats results in relatively fewer CA1 population spikes than in hippocampal slices of intact control animals (Stafstrom et al., 1999), which is likely mediated through augmentation of the inhibition (Bough et al., 2003). Rho et al. (1999) tested the age-dependent efficacy of the KD on flurothyl-induced seizures in mature (P51) and juvenile (P24) mice. Adult mice receiving the KD exhibited significantly longer latency periods to the onset of seizures than age-matched controls (Rho et al., 1999). Likewise, when mice were fed with the KD for 2 weeks, they displayed greater resistance to seizures induced by electroshock, which may be attributed to preservation of brain energy charge (Nakazawa et al., 1983). The ketotic rats that were treated with KA exhibited minimal hippocampal pyramidal cell damage, fewer and briefer SRMS, and diminished aberrant mossy fiber sprouting into the DSGL (Muller-Schwarze et al., 1999). Studies also suggest that the therapeutic effect of the KD depends on early intervention with the KD after the SE (Su et al., 2000). The precise mechanisms by which the KD works against development of epileptic seizures are still being worked out. However, indirect evidences suggest roles for ketone bodies such as the acetoacetate and acetone in the clinical effects of the KD therapy. For instance, pretreatment with acetoacetate blocks seizures in mice susceptible to audiogenic seizures (Rho et al., 2002). Likewise, seizures can be suppressed in a dose-dependent manner using intraperitoneal administration of acetone in animal models of epilepsy (Likhodii et al., 2003).
8.2. Metabolic effects of ketogenic diet therapy
The anticonvulsive effects of the KD are largely dependent on the maintenance of reduced blood glucose levels through hepatic metabolism and reduced body weight (Livingston, 1972; Greene et al., 2001, 2003). Figure 5 illustrates the effects of KD therapy on neurotransmitter metabolism. It appears that a reduction in the blood glucose level triggers a metabolic switch and causes the brain to burn ketones as the source of energy. Consequently, ketone metabolism gradually reduces neuronal excitability through effects on neurotransmitter levels and membrane potential. Numerous hypotheses have been proposed to explain the anticonvulsant activity of the KD. These include acidosis, which favors neuronal inhibition via proton-sensitive ion channels (Al-Mudallal et al., 1996), changes in electrolyte and water balance (Millichap and Jones, 1964; Millichap et al., 1964), direct inhibitory actions of fatty acids (Cunnane et al., 2002), alterations in neurotransmitters such as GABA and glutamate (Erecinska et al., 1996; Szot et al., 2001; Yudkoff et al., 2001a, b), changes in the energy metabolism, and functional alterations in mitochondria (Appleton and DeVivo, 1974; Pan et al., 1999). Moreover, the changes in neurotransmitter levels are important because of links between neurotransmitter levels and anticonvulsive and anti-epileptogenic effects. Cerebral acidosis induced by the KD reduces the activity of excitatory N-methyl-D-aspartate (NMDA) receptors (Swink et al., 1997). When seizures were induced by GABA receptor blockers such as picrotoxin, bicuculline and gamma-butyrolactone, the KD therapy was found to be more efficacious (Bough et al., 2003). Moreover, a study by Cheng et al. (2004) suggests that mild ketosis enhances the expression of both isoforms of GAD (GAD-65 and GAD-67) in the brain, which provides an indirect evidence for an increased GABA levels following the KD therapy. The above possibilities are supported by other reports, which include the observations that ketosis is associated with altered glutamate metabolism with diminished transamination of glutamate to aspartate and increased decarboxylation of glutamate to generate more GABA (Erecinska et al., 1996; Yudkoff et al., 1997). Taken together, it appears that the KD therapy leads to an increased GABA-ergic neurotransmission in the brain (Fig. 5).
Figure 5.
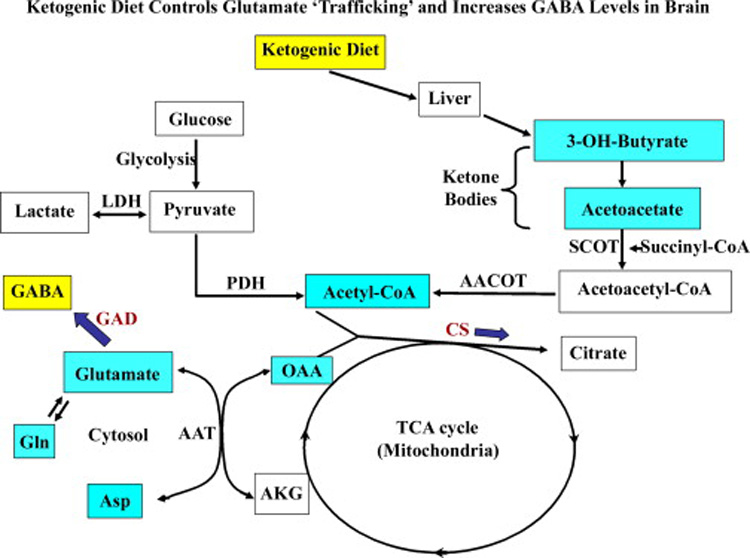
A schematic representation of the metabolism of glucose, ketone bodies and amino acids (excitatory neurotransmitters) in the brain. Approximately 90% of dietary calories derived from fats induce ketosis through fatty acid metabolism in the liver. The major ketone bodies produced by this route comprise 3-hydroxy-butyrate (3-OH-butyrate) and acetoacetate, which serve as fuel for high-energy demand of the brain in epileptic conditions. The ketone body acetoacetate is sequestered to acetoacetyl-CoA by succinyl-CoA transferase (SCOT) in the brain. In parallel, the pyruvate (through glycolysis) generates acetyl-CoA by pyruvate dehydrogenase complex (PDH) and a fraction of lactate through lactate dehydrogenase (LDH). The acetoacetyl-CoA routed via ketone bodies metabolism also generates excess pool of acetyl-CoA in the mitochondrial acetoacetyl-CoA thiolase (AACOT) reaction and enters the tricarboxylic acid (TCA) cycle. Hence, oxaloacetate (OAA) pool diminishes because of increased availability of substrates for key TCA cycle enzyme citrate synthetase (CS). As a result, less OAA is available for transamination reaction of aspartate aminotransferase (AAT) to produce aspartate, which in turn leads to increased glutamate pool for glutamic acid decarboxylase (GAD) and favors GABA synthesis. Alternatively, glutamine (Gln) production would likely reduce the glutamate load in neurons.
8.3. Effects of ketogenic diet on mitochondrial function
The seizure induced changes in mitochondria such as respiratory chain dysfunction, structural alterations, and increased free radical load through seepage of electrons from the electron transport chain may further enhance the susceptibility of the brain to other epileptic manifestations (Willmore et al., 1978; Kunz et al., 2000; Liang and Patel, 2004; Patel et al., 2004; Acharya and Katyare, 2005; Bonilha et al., 2006). During metabolic switch over, the fatty acids comprising 80 to 90% of total diet calories in the KD undergo beta-oxidation in mitochondria. This is presumed to be associated with altered balance of important substrates and neurotransmitters as discussed above. Sullivan et al. (2004a) demonstrated that KD therapy increases mitochondrial uncoupling protein (UCP2) activity and decreases reactive oxygen species (ROS) formation in the mouse hippocampus. The mitochondrial UCP dissipates mitochondrial oxidative energy metabolism in terms of ATP production and releases energy as heat. Furthermore, ketones have the ability to reduce ROS formation in isolated mitochondria, as suggested by the induction of glutathione peroxidase, an enzyme important for ROS metabolism and activity, in the rat hippocampus (Ziegler et al., 2003). Recently, a gene expression study by Bough and associates (2006) suggests that the anticonvulsant action of the KD therapy likely occurs through a coordinated upregulation of 19 proteasome-related transcripts that are important for enhanced oxidative phosphorylation. The role for mitochondrial biogenesis in neuronal survival in epilepsy is also supported by the observation that surviving dentate hilar neurons in humans with epilepsy contain more mitochondria than normal (Blumcke et al., 1999). Collectively, the occurrence of mitochondrial biogenesis, upregulation of multiple proteasome transcripts, increased production of the UCP2, reduced ROS generation, enhanced respiration rate of isolated mitochondria and enhanced alternative energy stores suggest activation of multiple neuroprotective changes following the KD therapy (Sullivan et al., 2004a, b; Andrews et al., 2005).
8.4. Effects of ketogenic diet on seizure induced apoptosis
The inhibitory effects of the KD on seizure-induced apoptosis or cell death have been reported in the past few years. It appears that the KD suppresses apoptosis via multiple mechanisms (Fig. 6). First, mice that were fed on the KD have an increased content of the calcium binding protein calbindin (McIntosh et al., 1998; Noh et al., 2005a). Typically, activation of the excitatory neurotransmitter receptors leads to increases in the free intracellular calcium via calcium influx into neurons (MacDermott et al., 1986), which eventually results in cell death in seizure conditions (Ure and Perassolo, 2000). Hence, increased calbindin that buffers increased intracellular calcium during hyperexcitability likely serves a neuroprotective role. Second, Noh et al. (2005b) report that the pro-apoptotic protein clusterin does not accumulate in the hippocampus of KD fed mice treated with KA, in contrast to mice treated with KA alone (Noh et al., 2005b). The KD also seems to block the KA induced cell death mediated by several other pro-apoptotic families of proteins, such as Bad, Bax and caspase-3 in the hippocampus (Noh et al., 2006). It is possible that decreased levels of ROS mediated by the KD will also regulate the Akt/Bad/14-3-3 cascade and thereby prevent further injury following seizures (Fig. 6).
Figure 6.
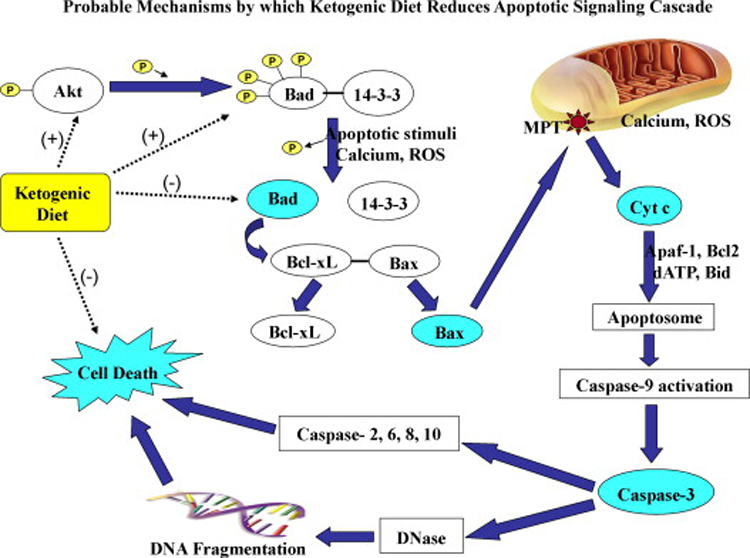
The potential role of ketogenic diet on mitochondrial dependent apoptotic signaling cascades. The ketogenic diet therapy is associated with increased activity of anti-apoptotic proteins like Akt and molecular chaperon 14-3-3 and reduced activity of pro-apoptotic proteins Bad, Bax and caspase-3. Akt phosphorylates Bad that forms a complex with 14-3-3. This prevents further activation of Bax that is involved in the mitochondrial permeability transition pore (MPT) formation and subsequent release of cytochrome c (cyt c) into the cytosol. Overall, the ketogenic diet helps to prevent the caspase-dependent apoptotic cell death.
8.5. Clinical relevance of ketogenic diet therapy
The efficiency of the KD is not just restricted to children afflicted with epilepsy but it is now shown to be beneficial across a wide variety of ages, seizure types and severities (Kossoff et al., 2002; Mady et al., 2003), as well as different etiologies (Kossoff et al., 2002; Kossoff and McGrogan, 2005). There is clear evidence supporting the view that KD also improves the long-term outcome in children with refractory epilepsy (Freeman, 2001; Hemingway et al., 2001; Marsh et al., 2006a). For example, one-year follow up study of the KD therapy in children with intractable epilepsy demonstrated ~90% reductions in the total number of seizures in ~50% of patients. Moreover, from the reports of the diet’s efficacy worldwide in recent years, it appears that approximately half of patients receiving the KD will have ~50% reduction in their seizures, and ~33% of patients receiving the KD will have 90% reductions in their seizures (Hassan et al., 1999; Kankirawatana et al., 2001; Coppola et al., 2002; Klepper et al., 2002; Francois et al., 2003; Kim et al., 2004; Vaisleib et al., 2004). Nevertheless, well structured studies in relation to composition of the KD, seizure type and severity and long-term efficacy are needed to fully understand the usefulness of this therapy for managing chronic epilepsy (Stafstrom et al., 2006). As most side effects of the diet are mild, the overall acceptability of this treatment in a majority of patients is better (Hemingway et al., 2001; Coppola et al., 2002). Early onset biochemical disturbances include hypoglycemia, hypertriglyceridemia (which may lead to pancreatitis), hypercholesterolemia, transient hyperuricemia, increased liver transaminases (especially in patients receiving VPA), hypoproteinemia, hypomagnesemia, hyponatremia and metabolic acidosis (Vining, 1999; Wheless, 2001; Coppola et al., 2002; Kang et al., 2004). Late-onset biochemical disturbances include secondary hypocarnitinemia, iron deficiency anemia, vitamin and mineral deficiencies, increased uric acid production, low serum bicarbonate levels, persistent hyponatremia, sustained metabolic acidosis, and dyslipidemias (Vining, 1999; Furth et al., 2000; Kossoff, 2004). However, in comparison to the ineffectiveness of the AED therapy for controlling seizures in refractory epilepsies, the KD therapy presumes importance for its long-term efficacy.
8.6. Conclusions
The available studies support the use of KD for treating chronic epilepsy, as it provides better seizure control than the AEDs. Furthermore, the anti-convulsive and anti-epileptogenic effects observed in animals treated with the KD supports the use of the KD as a neuroprotective agent against acute seizures or the SE. Nevertheless, the major limitation of the available studies is that the animals were pre-treated with the KD for prolonged periods before the induction of the SE. Therefore, it is still uncertain whether introduction of the KD after the SE or an IPI will be efficacious for preventing chronic epilepsy development and learning and memory impairments. Although improvements in GABA-ergic function and reduction in the apoptotic cascade observed in animals fed on the KD are suggestive of the potential neuroprotective role for the KD therapy (Fig. 5 and Fig. 6), it remains to be determined whether such improvements will also occur if the KD therapy is initiated after an IPI such as the SE. This necessitates detailed studies for validating the KD approach as a neuroprotective therapy for preventing the occurrence of chronic epilepsy after the IPI.
9. Neuroprotection via administration of neurotrophic factors
The neurotrophic factors appear to play key roles in pathophysiological conditions such as seizures (Jankowsky and Patterson, 2001). A variety of neurotrophic factors has potent effects on neuronal survival, differentiation, neurite outgrowth, neurotransmitter synthesis, synaptic plasticity and excitability (Weisenhorn et al., 1999). However, the epileptogenic or anti-epileptogenic effects of various neurotrophic factors following brain insults like seizures are still being studied. The major neurotrophic factors that are of interest in terms of epilepsy include the fibroblast growth factor-2 (FGF-2), BDNF, neurotrophin-3 (NT-3), nerve growth factor (NGF), glial cell line derived neurotrophic factor (GDNF), and the vascular endothelial growth factor (VEGF). While some studies imply that increased expression of many neurotrophic factors after brain injury or acute seizures contributes to the neuroprotection of the injured brain, other studies suggest that increases in some of the neurotrophic factors (such as BDNF and NGF) actually promote epileptogenic changes. In this segment, we discuss studies on different neurotrophic factors pertaining to epilepsy and try to identify the neurotrophic factors that are potentially useful as neuroprotective and anticonvulsive agents for administration after the SE for preventing chronic epilepsy development.
9.1. Potential of fibroblast growth factors
Fibroblast growth factors (FGFs), a family of polypeptide growth factors, play important roles in the development of embryo and adult tissue homeostasis (Yeoh and de Haan, 2007). Twenty two FGFs with molecular weights ranging from 17 to 34 kDa have been identified so far (Ornitz and Itoh, 2001; Yeoh and de Haan, 2007). The FGFs mediate their biological responses through high affinity binding to four trans-membrane proteins (FGFR1-FGFR4) with intrinsic tyrosine kinase activity (Coumoul and Deng, 2003; Zhao et al., 2007). It has been shown that the phosphorylation of FGFRs following binding of FGFs leads to activation of the downstream cytoplasmic signal transduction pathways (Itoh and Ornitz, 2004). The seizures have been shown to increase transcripts of FGF-2, FGF-5 as well as FGF receptors (Gomez-Pinilla et al., 1995). Furthermore, systemic administration of the recombinant human aFGF has been shown to have anticonvulsant properties in KA induced tonic-clonic epilepsy model where convulsions and mortality rate were decreased by 74–77% (Cuevas and Gimenez-Gallego, 1996). However, among the FGFs, the FGF-2 (or the basic FGF) has received the most attention as the promising candidate for neuroprotection against brain insults because studies have shown that the FGF-2 can protect neurons against glutamate-induced neurotoxicity (Mattson et al., 1993). Furthermore, FGF-2 mRNA and protein exhibit strong up-regulation after neuronal damage (Alzheimer and Werner, 2002). Moreover, in animal models of stroke, FGF-2 administration has proved effective against neuronal loss (Li and Stephenson, 2002).
The FGF-2 likely affords neuroprotection by interfering with a number of signaling pathways, including expression and gating of NMDA receptors, maintenance of calcium homeostasis, regulation of ROS-detoxifying enzymes, and strengthening of the anti-apoptotic pathways (Alzheimer and Werner, 2002). Thus, it appears that increased levels of FGF-2 during and after seizures can protect neurons from undergoing degeneration. Indeed, the administration of FGF-2 has neuroprotective effect against seizure-induced neuronal cell death and behavioral deficits (Liu et al., 1993; Liu and Holmes, 1997). In contrast to the above observations, a study by Zucchini et al. (2005) employing KA injections into FGF-2 knock-out and FGF-2 over-expressing mice shows that though epileptogenic seizures constitutively induce the expression of FGF-2 mRNA and synthesis of FGF-2 in astrocytes as well as in neurons of the hippocampus, the severity of seizures was not altered in the FGF-2 knockout mice but increased in the FGF-2 over-expressing mice. This suggests that considerably increased expression of FGF-2 increases the susceptibility to seizures. Thus, currently, it is not clear whether the neuroprotection afforded by the FGF-2 is efficacious for preventing the chronic epilepsy development after the SE or acute seizures. Moreover, it should be noted that the neuroprotection in the studies of Liu and colleagues (Liu et al., 1993; Liu and Holmes, 1997) depended on intracerebroventricular administration of the FGF-2 prior to the seizure onset. It is imperative that, for development of FGF-2 as a therapeutic agent for seizures, it is necessary to determine whether FGF-2 administration after the onset of seizures would offer neuroprotection, halt epileptogenesis, and prevent the progression into chronic epilepsy. In addition, administration of FGF-2 into the lateral ventricle after the onset of seizures is cumbersome. Hence, a clinically practicable approach of FGF-2 administration after seizures is necessary. A study shows that subcutaneous administration of FGF-2 can also reach brain regions, as FGF-2 can cross the BBB of the adult brain (Wagner et al., 1999). Therefore, in conditions such as seizures, because of the possible BBB disruption, the peripherally administered FGF-2 may readily enter brain regions undergoing seizures and provide neuroprotection. However, detailed studies are needed to examine whether peripheral FGF-2 treatment commencing after the onset of SE would provide significant neuroprotection, and prevent the progression of initial seizures into chronic epilepsy and learning and memory deficits.
9.2. Potential of neurotrophins as neuroprotective agents
The neurotrophins, one of the most well known families of neurotrophic factors, comprise the BDNF, the NT-3, the NGF, and the neurotrophin-4/5 (Leibrock et al., 1989; Ernfors et al., 1990; Hohn et al., 1990). These proteins mediate their action through binding to tyrosine kinase receptors (TrkA, TrkB, TrkC) and a low affinity neurotrophin receptor (p75NTR). The neurotrophins are important for neuronal survival, axon growth and path-finding, synaptic plasticity and neurotransmission in the CNS and PNS. They have been shown to exhibit neuroprotective properties in conditions such as brain injury and ischemia. Because of this and their ability to modulate the axonal reorganization after injury, there is considerable interest in understanding their role in epileptic conditions. In the following section, we discuss the available reports on neurotrophins BDNF, NT-3 and NGF pertaining to epilepsy, and evaluate whether any of these factors are potentially useful for neuroprotection after the SE or an IPI.
9.2.1. Usefulness of the brain-derived neurotrophic factor
Among the neurotrophins, the BDNF has received the most attention as a potential therapeutic target for treating TLE (see reviews by Koyama and Ikegaya, 2005; Simonato et al., 2006). It is well known that epileptogenic insults increase the synthesis of BDNF and activation of trkB receptors (Binder et al., 2001). A time-course study suggests that the concentration of the BDNF increases at an early post-seizure delay of 4 days in the hippocampus, in comparison to its level in the intact control hippocampus (Shetty et al., 2003). Figure 7 illustrates the concentration of BDNF at various time points after the induction of seizures. The increase in BDNF at early post-seizure delay agrees with mRNA studies in several animal models of epilepsy, which demonstrated that surviving hippocampal cells increase their levels of mRNA for BDNF after glutamate receptor activation (Ballarin et al., 1991; Ernfors et al., 1991a; Gall et al., 1991a; Isackson et al., 1991; Dugich-Djordjevic et al., 1992; Rocamora et al., 1992). Despite the well known positive effects of BDNF on neuron survival, many studies imply that increased BDNF at early time-points after the SE has proepileptogenic function. This is because of the following observations. First, the BDNF potentiates excitatory signals (Schinder and Poo, 2000) and reduces inhibitory synaptic transmission (Tanaka et al., 1997). Second, reduced BDNF signals such as that observed in BDNF+/− mutant mice, rats infused with trkB-receptor antibodies, and mice overexpressing truncated trkB are associated with diminished epileptogenesis (Kokaia et al., 1995; Binder et al., 2001). Third, absence of trkB receptors in synapsin-Cre conditional trkB−/− mice, where trkB is ablated in hippocampal granule cells and CA3 pyramidal neurons, prevents the development of kindling (He et al., 2004). Fourth, the transgenic mice that overexpress BDNF protein have more severe seizures in response to the excitotoxin KA (Croll et al., 1999). Fifth, intracerebroventricular administration of BDNF accelerates the development of kindling (Xu et al., 2004). However, the BDNF also seems to have some beneficial effects during the course of TLE. For instance, it has been shown that the BDNF amplifies GABA currents in oocytes expressing GABAA receptors transplanted from surgically removed specimens of human epileptic brains (Palma et al., 2005). Furthermore, a study shows that the BDNF favors survival and regeneration of hippocampal neurons damaged by the SE (Simonato et al., 2006).
Figure 7.
Concentration of Brain derived neurotrophic factor (BDNF) in hippocampi ipsi- and contralateral to unilateral kainic acid (KA) administration at different time-points after the KA administration. Note that the BDNF is significantly up-regulated at 4 days post-KA administration, reaches the level observed in the intact hippocampus at 45 days post-KA, and decreases below the control levels at 120 days post-KA. The hippocampus contralateral to KA administration retains baseline BDNF until 45 days post lesion but exhibits considerable decline at 120 day post-KA. (Figure reproduced from:Shetty et al., 2003; J Neurochem; 87:147–159).
From the above, it appears that the overall detrimental effects of increased BDNF are greater than the potential beneficial effects. This conclusion is also supported by the following observations. The aberrant sprouting of mossy fibers into the DSGL, a morphological change that occurs after injury or seizures in the hippocampus, is also linked to hyperexcitability in the DG of the injured hippocampus (Tauck and Nadler, 1985; Okazaki et al., 1995). Studies show that the degree of DSGL sprouting after hippocampal injury correlates with both antidromically evoked burst firing and development of spontaneous seizures, which normally do not arise from the DG of the intact hippocampus (Cronin and Dudek, 1988; Mathern et al., 1993; Mello et al., 1993; Okazaki et al., 1995). Although the progression of aberrant mossy fiber sprouting is slow and usually takes about 4 months to show the maximal level of sprouting, the initiation of this sprouting occurs soon after the hippocampal injury (Shetty and Turner, 1995a; Shetty et al., 2005). Therefore, it is possible that the pathological up-regulation of BDNF at early post-lesion time-points plays a role in the initiation of aberrant dentate mossy fiber sprouting after the hippocampal injury. Indeed, at early post-lesion, trkB immunostaining exhibits a clear increase in regions where mossy fibers are present (Katoh-Semba et al., 1999). From this, it appears that the BDNF released from mossy fibers immediately after KA induced degeneration of CA3 pyramidal neurons and dentate hilar cells induces morphological changes in granule cells by mediating signals to the cells by binding to trkB at cell surfaces. Thus, a newly synthesized, anterogradely transported, and secreted BDNF likely influences the initial sprouting of axons from granule cells (i.e. mossy fibers). Indeed, a study suggests that intrahippocampal infusion of BDNF into intact adult rats initiates mossy fiber sprouting and seizure activity (Zhu and Roper, 2001; Scharfman, 2002a). Another study suggests that excessive activation of L-type calcium channels causes granule cells to express BDNF, and the release of BDNF into the extracellular space stimulates TrkB receptors present on the hilar segment of the mossy fibers which leads to axonal branching in mossy fibers. This branching eventually results in development of hyperexcitable dentate circuits (Koyama et al., 2004). Thus, it appears that increased BDNF levels after the SE promote epileptogenesis. From this perspective, administration of the BDNF (or increasing BDNF levels in the brain through other strategies) shortly after the SE may enhance the speed of progression of the SE-induced injury into chronic epilepsy through increased epileptogenesis. Therefore, development of strategies that induce long-term suppression of BDNF after the SE appear beneficial for diminishing the mossy fiber sprouting as well as the incidence of chronic epilepsy after an IPI.
Interestingly, BDNF is naturally down-regulated under chronic epileptic conditions (Shetty et al., 2003). This down-regulation likely reflects an adaptive mechanism to minimize seizures that originate from the hippocampus, as the re-organized hippocampal circuitry is seizure-prone and likely contributes to the persistent hyperexcitability observed in the hippocampus at extended time-points after KA-induced seizures (Tauck and Nadler, 1985; Okazaki et al., 1995). Indeed, the massive down-regulation of the calcium binding protein calbindin observed in dentate granule cells supports the hyperexcitable state of the hippocampus at extended time-points after KA-induced seizures (Shetty and Turner, 1995b; Shetty and Hattiangady, 2007a). This is because an electrophysiological study has shown that granule cells that lack calbindin represent cells exhibiting hyperexcitability (Nagerl et al., 2000). A reduced concentration of BDNF in the chronically epileptic hippocampus appears advantageous, as BDNF up-regulation increases the vulnerability of certain brain areas to seizures or even causes seizures. For example, application of BDNF to hippocampal slices from intact control rats enhances the efficacy of excitatory mossy fiber synapse on to CA3 pyramidal cells (Scharfman, 1997). In hippocampal slices from pilocarpine treated rats, BDNF enhances responses to stimulation of mossy fiber collaterals in the DSGL (Scharfman et al., 1999). Moreover, BDNF exposure in these epileptic animals leads to seizure like events, suggesting that BDNF is likely more potent after seizure or injury induced mossy fiber sprouting. Therefore, increasing BDNF concentration during chronic epileptic conditions may be detrimental.
9.2.2. Efficacy of Nerve growth factor
Like the BDNF, increased NGF levels have been observed in the hippocampus shortly after an injury or acute seizures (Gall et al., 1991b; Lauterborn et al., 1994; Shetty et al., 2003). The extent of up-regulation of NGF seen at 4 days post-seizures however endures until 120 days after KA-induced seizures (Shetty et al., 2003). Similar to the BDNF, increased NGF levels after acute seizures are considered proepileptogenic. This is because intraventricular infusions of the anti-NGF IgGs retards the development of kindling and blocks the aberrant mossy fiber sprouting (Van der Zee et al., 1995). Likewise, intraventricular infusion of the NGF accelerates kindling development and increases mossy fiber sprouting (Adams et al., 1997). Furthermore, inhibition of the NGF binding to trkA receptors retards the development of kindling (Li et al., 2005). Additionally, a study in a KA model of epilepsy shows that the robust sprouting of mossy fibers into the DSGL occurs between 45 days and 120 days after KA-induced seizures (Shetty et al., 2003). As this period of robust aberrant mossy fiber sprouting parallels the period of up-regulation of NGF, it appears that the NGF plays a major role in the progression of the aberrant mossy fiber sprouting, an important epileptogenic change observed after acute seizures or an IPI. From the above, it is clear that increasing NGF levels after the SE or an IPI is not beneficial for thwarting the progression of the SE into chronic epilepsy. Rather, it appears that long-term suppression of NGF following the SE has promise for reducing the aberrant mossy fiber sprouting and spontaneous seizures.
9.2.3. Effectiveness of neurotrophin-3
The NT-3 and its receptor trkC are found in the hippocampal formation and the amygdala, the regions associated with epilepsy (Lamballe et al., 1991; Ernfors et al., 1992; Merlio et al., 1992; Bengzon et al., 1993; Xu et al., 2002). A quantitative study on NT-3 protein demonstrates that the concentration of NT-3 remains comparable to that of the intact hippocampus at 4 days post-seizures but shows moderate up-regulation at 45 days post-seizures and reaches the baseline level at 120 post-seizures (Shetty et al., 2003). Based on these observations, it is unlikely that NT-3 is involved in the initiation of epileptogenic processes such as the aberrant mossy fiber sprouting. However, because of moderately increased levels during the period of robust mossy fiber sprouting (i.e. between 45 and 120 days post-seizures), its involvement in the progression of the mossy fiber sprouting cannot be ruled out. The ability of NT-3 to induce mossy fiber sprouting following continuous infusion in the absence of electrical activation supports this possibility (Xu et al., 2002). Additionally, NT-3 knock out mice exhibit delayed kindling development (Elmer et al., 1997). On the other hand, continuous NT-3 infusion in kindled animals retarded behavioral seizures and inhibited kindling-induced mossy fiber sprouting into the DSGL (Xu et al., 2002). When taken together, it appears that increased levels of NT-3 in epileptic conditions such as after kindling actually inhibits mossy fiber sprouting, even though it has the propensity to promote mossy fiber sprouting under normal conditions. The inhibitory effect of NT-3 under epileptic conditions likely involves down-regulation of the high affinity trkA and trkC receptors and attenuation of trk phosphorylation, which in turn leads to a loss of responsiveness to NGF and NT-3 (Xu et al., 2002). Therefore, it is tempting to speculate that greatly increasing NT-3 levels following the SE will be beneficial for suppressing the aberrant sprouting of mossy fibers in the DG. As this aberrant sprouting contributes to the development of chronic epilepsy, such a strategy may also decrease the incidence of spontaneous seizures during the chronic phase. Certainly, appropriate strategies to increase NT-3 levels after the SE need to be developed and long term effects of such increase in NT-3 levels ought to be analyzed in future to fully comprehend the link between NT-3 and epileptogenesis.
9.3. Efficiency of glial cell line derived neurotrophic factor
The GDNF, a member of a subclass of the TGF-β family of neurotrophic factors, is well known to promote the survival and differentiation of dopaminergic neurons (Lin et al., 1993). The GDNF also enhances high-affinity uptake of dopamine in these cells. Pertaining to epilepsy, it has been observed that the GDNF plays a neuroprotective role in kindling and kindling-induced structural changes through mechanisms that employ Ret and p140NCAM receptor system (Airaksinen and Saarma, 2002; Paratcha et al., 2003). The GDNF also binds to a family of low-affinity co-receptors known as GDNF family receptor alpha’s (Airaksinen and Saarma, 2002). Pre-treatment with rhGDNF prevents the KA induced seizure activity and neuronal cell loss in hippocampal, thalamic and amygdaloid regions (Martin et al., 1995). Acute increase in the GDNF transcripts in dentate granule cells and induction of Ret receptor expression in the hilus and GFRα1 receptor throughout the hippocampus has been observed after kindling (Kokaia et al., 1999). Similarly, suppression of the development and persistence of hippocampal kindling was observed in mice lacking the GFRα2 receptors (Nanobashvili et al., 2000). Moreover, Li et al. (2002) demonstrated that intraventricular administration of GDNF prevents kindling-induced increase in hilar area and blocks mossy fiber sprouting in the CA3 region of the hippocampus. A study by Yoo et al (2006) showed that pre-treatment of animals with adenoviral-vector-derived GDNF (Ad-GDNF) leads to the suppression of the KA-induced tonic-clonic convulsions and significant reductions in apoptotic cells in the CA3 and DH regions. In the same vein, a recent study by Kanter-Schlifke et al (2007) shows that introduction of Ad-GDNF either prior or after KA treatment results in decreased frequency, duration, and induction-threshold to generalized seizures in rats. Collectively, these results suggest that GDNF is a useful neuroprotective agent against SE-induced epileptogenesis. However, long-term studies in multiple SE models of epilepsy are needed to validate GDNF as a potent neuroprotective agent capable of either preventing or considerably reducing the evolution of SE into chronic epilepsy.
9.4. Usefulness of the vascular endothelial growth factor
The VEGF, one of the main factors in induction of angiogenesis in the brain (Krum et al., 2002), is implicated in enhancing the blood–brain barrier (BBB) permeability (Yancopoulos et al., 2000) and inflammation (Proescholdt et al., 1999). The VEGF is also expressed in neurons and glial cells, and the concentration of VEGF increases after seizures (Croll et al., 2004). The actions of the VEGF are mediated through VEGF receptors 1 and 2, which are primarily localized on endothelial cells. However, these receptors are also found in neurons and glia, and in conditions such as injury, the VEGF receptors on neurons and glia exhibit upregulation (Croll et al., 2004). It has been proposed that seizure-produced VEGF acts on both neural and epithelial cells, which in turn promotes neurotrophic effects and increases the BBB permeability and inflammatory reactions (Croll et al., 2004; Simonato et al., 2005). From this point of view, increasing the levels of VEGF shortly after the SE may not be beneficial. On the other hand, when recombinant human VEGF was bath applied to adult rat hippocampal slices taken from chronically epileptic rats, it significantly reduced the amplitude of responses elicited by Schaffer collateral stimulation and spontaneous discharges in areas CA1–CA3 and the dentate gyrus (McCloskey et al., 2005). Thus, VEGF could be a ‘double-edged sword’ in epilepsy, as promptly suggested by Croll et al (2004). It seems that increasing VEGF levels in chronic epileptic conditions is useful for reducing spontaneous seizures. Long-term studies on the efficacy of increased levels of VEGF on spontaneous seizures in animal models of chronic epilepsy are needed in future to validate this possibility.
9.5. Conclusions
A differential expression of neurotrophic factors after insults such as seizures indicates that different stages of physiological alterations contributing to epileptogenesis may be modulated by these factors. The epileptogenic or anti-epileptogenic effects of these factors in susceptible brain regions assume importance with respect to devising novel neuroprotective strategies. Based on these, neurotrophic factors and their receptors seem to be important targets for anticonvulsant drug development. Investigations performed so far on the levels and potential functions of various neurotrophic factors in epileptic conditions suggest that increasing the levels of BDNF and NGF are not useful for suppressing epileptogenesis and preventing chronic epilepsy development after the SE or an IPI. On the contrary, long-term suppression of the activity of these neurotrophic factors appears beneficial. Furthermore, increasing the levels of the GDNF and NT-3 may also be advantageous for suppressing chronic epilepsy development because of their ability to suppress epileptogenic changes such as the aberrant mossy fiber sprouting. Regarding FGF-2 and VEGF, it is still unresolved whether it is proepileptogenic or antiepileptogenic, as both scenarios have been presented in different studies. Overall, rigorous studies testing whether long-term administration of distinct neurotrophic factors (or the strategies that maintain higher levels of distinct neurotrophic factors in the brain) after the onset of the SE is effective for preventing epilepsy are needed for further advances in this field.
10. Efficacy of antioxidants as neuroprotective compounds against epilepsy
Oxidative injury, resulting from excessive release of free radicals, likely contributes to the initiation and progression of epilepsy after brain injury. Therefore, antioxidant therapies aimed at reducing oxidative stress have received considerable attention in the treatment of epilepsy. These approaches may also restrain tissue damage and favorably alter the clinical course of the disease (Costello and Delanty, 2004). In this section, we discuss the efficacy of two distinct antioxidants (resveratrol and curcumin) for reducing excitotoxic injury and epilepsy in animal models of TLE.
10.1. Resveratrol
Resveratrol (3, 5, 4’-tri-hydroxy stilbene), a naturally occurring phytoalexin present in high concentrations in the skin of red grapes, belongs to the polyphenol group of plant compounds. After the awareness of reduced cardiac risk in the consumers of red wine, popularly referred as “French paradox” (i.e. low incidence of cardiovascular events in spite of diet relatively high in saturated fat), great interest has emerged in resveratrol, which is the active constituent of red wine (Tredici et al., 1999; Sun et al., 2002; Ikeda et al., 2003). Resveratrol exists in cis and trans isomeric forms; however, the trans isomer is the major form which contributes to its biological activity (Fremont, 2000). Resveratrol mediates a wide range of biological activities with multisystem benefits. It has been shown that resveratrol can protect the heart from ischemia using an isolated rat heart model (Sato et al., 2000a, b); inhibit the peroxidation of low density lipoproteins (LDL) via free radical scavenging mechanisms (Belguendouz et al., 1997). It can also act as an anti-inflammatory agent (Jang and Pezzuto, 1999; Manna et al., 2000).
Studies on the effects of resveratrol on neurons in cell culture and animal models suggest that resveratrol is a potent neuroprotective compound that mediates its effects mainly via inhibition of the oxidative stress. The examples include the following. First, in the presence of resveratrol, the neuronal cell death induced by ethanol is reduced (Sun et al., 2001; Sun and Sun, 2001), and the apoptosis of cells induced by oxidized lipoproteins is suppressed (Sun et al., 1997; Draczynska-Lusiak et al., 1998a, b). Second, both co- and post-treatments with resveratrol in culture attenuated hippocampal cell death and intracellular ROS accumulation produced by sodium nitroprusside (Bastianetto et al., 2000). Studies suggest that the anti-apoptotic effects of resveratrol are mediated by inhibition of mitochondrial cell death pathway involving activation of caspase (Nicolini et al., 2001). Third, besides inhibition of the oxidative stress, there is also evidence for other effects of resveratrol in protecting cells from injury. These include anti-inflammatory effects of resveratrol in astrocytes and the ability of resveratrol to inhibit nitric oxide (NO) production induced by cytokines (Li and Sun, 1998; Li et al., 1999). Fourth, resveratrol has been shown to protect cultured neurons against amyloid beta-peptide (Jang and Surh, 2003; Han et al., 2004), a neurotoxic peptide that likely plays a critical role in the neuropathology of Alzheimer’s disease. Fifth, resveratrol pre-treatment was found to be neuroprotective against oxidative stress in rat models of stroke (Sinha et al., 2002; Wang et al., 2002). Analyses of resveratrol in the brain after systemic administration suggested that resveratrol crosses the BBB and its activity in the brain lasts for up to 4 hrs (Wang et al., 2002). Thus, resveratrol is a potent neuroprotective compound. However, the relevance of the above findings to clinical situations remains to be demonstrated.
The protective effects of resveratrol pertaining to TLE include the following. A study on chronic administration of resveratrol suggests that resveratrol pre-treatment partially protects rat hippocampal neurons against KA-induced damage in vivo (Virgili and Contestabile, 2000). Acute resveratrol pre-treatment was also found to be associated with reductions in the severity of KA-induced SE (Gupta et al., 2002). Another study demonstrates that administration of resveratrol 30 minutes prior to KA diminishes KA-induced neuronal cell death in the hippocampus (Wang et al., 2004). The resveratrol mediated attenuation of neuronal cell death was associated with suppression of activated astrocytes and microglia. As increased oxidative stress is a key factor in the mechanisms of KA-induced neurotoxicity, the neuroprotective effect afforded by resveratrol in this study supports the purported ability of the resveratrol to act as free radical scavenger to protect against neuronal damage caused by excitotoxic insults. How does resveratrol protect neurons from excitotoxic damage? Perfusion with trans-resveratrol (10–100 µM) significantly suppresses glutamate-induced currents in postsynaptic CA1 pyramidal neurons demonstrating that trans-resveratrol inhibits the postsynaptic glutamate receptors, which probably explains its anti-excitotoxic feature (Gao et al., 2006). Further, its inhibitory action on voltage-activated potassium currents implicated in neuronal apoptosis may contribute to its neuroprotective effects (Gao and Hu, 2005). When organotypic hippocampal cultures exposed to oxygen-glucose deprivation (OGD) were treated with 10, 25 and 50 µM concentrations of resveratrol, cell death was reduced to 22, 20 and 13% respectively in comparison to 46% cell death in vehicle treated group (Gao and Hu, 2005; Zamin et al., 2006). A recent study, in addition, demonstrates that just 24 hours of exposure of astrocytes to resveratrol modulates their basal glutamate uptake (Vieira de Almeida et al., 2007). Thus, the available reports clearly indicate neuroprotective function for resveratrol. However, long term studies on the effects of resveratrol administration in SE models are needed to validate this compound as a useful drug for preventing epilepsy after brain injury or SE.
10.2. Curcumin
Curcumin, a phenolic pigment, is a natural antioxidant isolated from the medicinal plant Curcuma Longa Linn. When KA was injected 18 hours after the curcumin treatment and tissues were analyzed after 48 hours, there was marked reduction in the neuronal cell death and caspase-3 immunoreactivity, and inhibition of reactive astrocyte expression (Shin et al., 2007). Curcumin inhibits NO production in the lipopolysaccharide (LPS) stimulated microglial cells (Jung et al., 2006) and thereby prevents microglial cell mediated neurodegeneration in neurons. In fact, KA-induced increase in NO levels was shown to be reversed by administration of manganese complexes of curcumin and diacetylcurcumin, likely by their NO scavenging activity (Sumanont et al., 2004, 2006). In oxidative stress conditions, curcumin administration has shown to decrease lipid peroxidation, mitochondrial dysfunction, and apoptotic indices (Wang et al., 2005). Furthermore, a recent study using traumatic brain injury model demonstrates that supplementation of curcumin in the diet dramatically reduces oxidative damage, normalizes the levels of BDNF, synapsin I and CREB, and counteracts the cognitive impairments caused by traumatic brain injury (Wu et al., 2006). Thus, because of its anti-apoptotic, anti-inflammatory and anti-oxidant properties, curcumin therapy via diet may be useful for preventing chronic epilepsy after an IPI.
10.3. Conclusions
Both resveratrol and curcumin would likely be considered as powerful neuroprotective drugs against acute seizures or the status epilepticus in future because of their capability to provide neuroprotection in excitotoxic conditions. However, one caveat is that the positive effects in studies conducted so far were observed when these antioxidant compounds were administered prior to the onset of seizures. For promotion of these compounds as neuroprotective agents against SE, it will be essential to determine whether their administration after the onset of SE would also offer neuroprotection. Additionally, it is critical to determine in future studies whether the antioxidant-mediated neuroprotection is adequate for preventing the SE-induced epileptogenesis, the development of SRMS, and learning and memory impairments. Careful scrutiny in chronic seizure prototypes is crucial in future studies for endorsing these compounds as reliable neuroprotective agents against SE-induced TLE.
11. Hormones and neuroprotection
The effects of hormones, either peripheral or endogenous on the nervous system have been well established. Especially, gonadal steroids like estrogen, progesterone and their precursors are proven to have direct effects on neurotransmitter receptors (Hoffman et al., 2006). The cyclical changes in these steroids are believed to be important in the pathogenesis of catamenial epilepsy because susceptibility to seizures during menstrual cycle are linked to serum hormone levels (Reddy, 2004). Secondly, endogenous neurosteroid metabolism plays important roles in the growth and maturation of the brain (Stoffel-Wagner, 2001). Apart from steroids, erythropoietin is proposed to have neuroprotective role in several neurological insults (Siren and Ehrenreich, 2001). The antioxidant properties of pineal gland hormone melatonin are suggested to have anti-epileptogenic role as well (Martin et al., 2002; Chung and Han, 2003). In this section, we discuss the neuroprotective role of these hormones.
11.1. Potential of estrogens
Estrogen, the sex hormone belonging to the steroid hormone super family, is involved in various biological actions (Veliskova, 2007). Estrogens mostly mediate their actions by activation of specific estrogen receptors (ER) that are widely distributed in the brain and expressed on both neurons and glia (Mhyre and Dorsa, 2006). Because of their effects on neuronal excitability, it is believed that estrogens play a role in epilepsy. Additionally, an association has been observed between seizures and fluctuations of the sex hormone levels during the ovarian cycles in some women with epilepsy, a condition termed catamenial epilepsy (Logothetis et al., 1959; Backstrom, 1976; Herzog et al., 1997). Administration of low doses of estradiol have been shown to prevent KA induced somatostatin-immunoreactive hilar neuronal loss in ovariectomized rat hippocampus (Azcoitia et al., 1998). Similarly, a single estradiol pre-treatment before KA insult delays the onset of SE and reduces the overall neurodegeneration in the CA3 subfield and the DG (Veliskova et al., 2000). A study by Reibel et al., (2000) also demonstrates reduced pyramidal cell loss after chronic supplementation of estradiol in SE-induced neurodegenerative consequences in ovariectomized rats. As ovariectomy is also associated with changes in GABA levels, GABA synthesizing enzymes, and the KCC2 transporter that contributes to chloride gradients and critical for GABAA receptor-mediated actions (Nakamura et al., 2004), the exact mechanisms involved in the estrogen mediated neuroprotective effects against SE-induced hippocampal neurodegeneration are unknown. Recently, the interaction between estrogen and NPY has been proposed as a potential mechanism for neuroprotective effects of estrogen (Veliskova, 2007). This is because estrogen administration increases NPY expression in the dentate hilus, and NPY has inhibitory function as well as neuroprotective properties (Silva et al., 2003).
However, there are concerns about using estradiol as a neuroprotective compound in epileptic conditions, as it increases KA receptor subunit KA2, and inhibits glutamate uptake, both of which would likely facilitate excitotoxic injury (Scharfman and MacLusky, 2006). Moreover, estradiol can enhance actions of NMDA receptors (Zamani et al., 2004) and inhibit GABAergic transmission by decreasing the effects of GABA at GABAA receptors in CA1 pyramidal cells (Rudick and Woolley, 2001). Interestingly, estrogen is shown to influence BDNF gene, and BDNF potentiates several of the glutamatergic pathways in hippocampus and other brain regions (see the review by Scharfman and Maclusky, 2005). Thus, both anticonvulsant and proconvulsant actions have been reported for estrogens. A simple rule for the effects of estrogens on seizures cannot be applied due to the complexity of action of these hormones on neuronal excitability (see the review by Veliskova, 2007 for details). Additionally, estrogen, being an anabolic steroid, may not be suitable as an exogenous neuroprotective compound due to its multiple physiological effects.
11.2. Efficacy of progesterone
Unlike the confusion regarding the link between estrogen and seizures, the studies of progesterone on seizures suggest that progesterone administration leads to anticonvulsant effects (Scharfman and MacLusky, 2006). The ability of progesterone (PROG) to suppress seizures has been shown to be dose-dependent, as low but not high physiological levels of PROG suppressed seizures and reduced hippocampal damage (Hoffman et al., 2003). In laboratory animals, injection of PROG results in increased seizure threshold, or delayed onset of seizures following the administration of convulsants (Belelli et al., 1989; Frye and Bayon, 1998; Rogawski, 2003). In the same vein, clinical studies have shown that PROG decreases seizure frequency in women with epilepsy (Herzog, 1999). The effects of PROG on neuronal excitability are associated with endogenous neurosteroid metabolism. For instance, administration of very low doses of PROG leads to increased metabolism and formation allopregnanolone that modulates GABAA receptor activity (Baulieu et al., 1996). Allopregnanolone binds to specific neurosteroid binding sites on GABAA receptors (Kokate et al., 1994), and levels of allopregnanolone and GABAA receptors are positively correlated with PROG levels (Baulieu et al., 1989). On the contrary, administration of a higher dose of PROG does not induce anti-convulsive effects (Hoffman et al., 2003).
11.3. Efficiency of other neurosteroids
The other neurosteroids such as pregnenolone sulfate (PREGS) and DHEA are shown to potentiate the effects of NMDA in increasing intracellular calcium levels. The PREGS, an endogenous neurosteroid synthesized by glia, mostly acts as a potent convulsant when injected intracerebroventricularly and intraperitoneally (Williamson et al., 2004). Furthermore, it is known that the PREGS is a negative allosteric modulator of GABAA receptors and a positive modulator of the NMDA receptors. Interestingly, a recent study by evaluating the effects of PREGS in a genetic animal model of absence epilepsy suggests that PREGS reduces the number and duration of epileptic spike-wave discharges when microinjected into the peri-oral region of the primary somatosensory cortex (Citraro et al., 2006). However, it was found that the effects of PREGS were complex and depended upon both the dose and the site of administration.
11.4. Usefulness of erythropoietin
Erythropoietin (EPO), a chief hormone in erythropoiesis, is induced by hypoxia, hypoglycemia, strong neuronal depolarization and excessive oxygen radicals (Marti et al., 1997; Chikuma et al., 2000). Exogenous EPO administration may lessen seizure severity, elongate seizure latencies and reduce neuronal damage in rats subjected to KA-induced seizures (Brines et al., 2000). EPO has also been suggested to be protective against seizures, via regulating neurotransmitter/neuropeptide release. It has been shown that EPO increases glutamate uptake and desensitizes glutamate receptors (Morishita et al., 1997). Recent evidence indicates that systemic administration of recombinant human EPO protects BBB and reduces the severity, latency, and duration of PTZ induced convulsions (Uzum et al., 2006). The manner in which EPO provides neuroprotection is currently unclear. Diverse cell types have been demonstrated to produce EPO and many cells besides erythroid progenitors express the EPO receptors, including the brain. The discovery that astrocytes produce EPO in response to hypoxia and that the EPO could protect nearby neuronal cells from ischemic injury in vivo (Sakanaka et al., 1998) added further support for the pleiotropic nature of this cytokine. One of the suggested mechanisms of action is through prevention of apoptosis and immune-modulation; however, further studies with respect to epileptic seizures are needed to assess its overall efficacy for preventing epilepsy after SE.
11.5. Melatonin as a neuroprotectant
In addition to its well known effects on circadian rhythms, melatonin has been shown to be useful for enhancing immunity, preventing cancer, slowing down the aging process, and preventing oxidative stress related tissue damage (Brzezinski, 1997; Czeisler, 1997). Pertaining to epilepsy, studies in several animal models suggest that melatonin is capable of restraining acute seizures and diminishing chronic epileptic manifestations in humans. For instance, following excitotoxic seizures, melatonin deficiency (via pinealectomy) causes greater levels of neurodegeneration (Manev et al., 1996), suggesting that endogenous melatonin likely plays a neuroprotective role against seizures. Likewise, chronic treatment with melatonin reduces the PTZ-induced seizure incidence and mortality rate in male gerbils (Champney et al., 1996). Sedative as well as non-sedative doses of melatonin are efficacious for suppressing generalized seizures and seizure scores in animal models of kindling, PTZ-induced seizures (Albertson et al., 1981; Mevissen and Ebert, 1998) and elevating the electroconvulsive threshold in a maximum electroshock mice model (Borowicz et al., 1999). Furthermore, in patients with drug resistant TLE, though baseline melatonin levels decrease, seizures induce 3-fold increase in melatonin (Bazil et al., 2000), which likely suggest endogenous anticonvulsant activity in response to seizures. It has been suggested that increased melatonin levels after seizures represent a protective mechanism against repetitive seizures (Bazil et al., 2000).
Indeed, considerable evidence indicates that melatonin has anticonvulsant and neuroprotective properties. First, melatonin administration prior to KA attenuates seizures and death of pyramidal neurons in the CA3 region when compared to KA administration group without melatonin (Giusti et al., 1996a, b; Mohanan and Yamamoto, 2002; Chung and Han, 2003; de Lima et al., 2005; Lee et al., 2006). This neuroprotective action of melatonin against KA-induced excitotoxicity is thought to be exerted either through the activation of neuronal Akt by direct action on hippocampal neurons or through the increased expression of astroglial GDNF, which subsequently activates neuronal PI3K/Akt pathway (Lee et al., 2006). Second, melatonin administration protects against mitochondrial damage and increased lipid peroxidation, DNA breakages, microglial activation in the hippocampal neurons induced by KA (Mohanan and Yamamoto, 2002; Chung and Han, 2003; Lee et al., 2006). Third, melatonin administration is associated with enhanced survival of glial cells, which may promote neuronal survival after brain injury (Borlongan et al., 2000). Fourth, melatonin modulates the electrical activity of the neurons by acting on plasma membrane receptors (Acuna-Castroviejo et al., 1995) and facilitating the GABA-ergic function (Wan et al., 1999), both of which likely contribute to anticonvulsive effects (Dawson and Encel, 1993; Golombek et al., 1996). Fifth, melatonin has been shown to inhibit brain glutamate receptors and NO production (Bikjdaouene et al., 2003; Yahyavi-Firouz-Abadi et al., 2006). The GABA-ergic effect of melatonin possibly occurs through changes in membrane ion permeability with increased chloride ion influx through GABAA-dependent chloride channels (Rosenstein et al., 1990). Melatonin likely attenuates neuronal excitability through inhibition of the glutamate-mediated response of the striatum to motor cortex stimulation, where inhibitory effects of melatonin was suggested to be associated with decreased calcium influx (Escames et al., 2001). Such inhibition could be mediated by either NMDA or L-arginine receptors (Escames et al., 2004). The dose dependant anticonvulsant property of melatonin was shown to be associated with decreased aspartate, glutamate, nitrite and elevated GABA and taurin levels in various brain regions (Bikjdaouene et al., 2003). In untreated epileptic patients, melatonin production is shown to be increased (Schapel et al., 1995) and its diurnal variation in epileptic children is altered (Molina-Carballo et al., 1994). Moreover, it is administered as adjunctive anticonvulsant therapy in severe infantile myoclonic epilepsy (Molina-Carballo et al., 1997). Thus, melatonin seems to be very effective in preventing seizures particularly when administered prior to excitotoxic challenge, and for suppressing seizures during chronic epilepsy. However, it is unknown whether melatonin administration after the onset of SE would prevent the development of chronic epilepsy or simply delay the process (Ananth et al., 2003).
11.6. Conclusions
The neuroprotective effects of steroids chiefly depend on their metabolism. The major cytochrome P450 type enzymes and aromatase are shown to play active roles in endogenous metabolism of neurosteroids in the brain. The SE-induced epileptogenic alterations are not prevented by estrogen in post-SE treatment strategies. Moreover, suppressing GABA-ergic function and enhancing glutamatergic action indicates a pro-convulsive role for estrogen. On the contrary, low doses of PROG and subsequent in vivo conversion of PROG to allopregnanolone appears beneficial through its effects on GABAergic system. The functions of endogenous neurosteroids with respect to epileptogenesis are still being studied. Dose dependent antiepileptogenic effects of melatonin are promising. Altered BBB permeability in epileptic seizures could allow accumulation of peripheral hormones like EPO that could serve as a potent neuroprotectant. Additionally, role of astrocytes that can produce EPO in stress conditions assumes importance in chronic epileptic seizures. In conclusion, PROG, endogenous neurosteroids, melatonin and EPO appear to be important hormone candidates for future studies in neuroprotective strategies pertaining to epilepsy.
12. Neuroprotective Effects of Neural Cell Transplants
Although multipotent and self-renewing neural stem cells (NSCs) persist in both neurogenic (neuron producing) and non-neurogenic regions of the adult CNS, the capacity of the adult mammalian CNS for self-repair is limited (Turner and Shetty, 2003; Shetty and Hattiangady, 2007b). This is because, in conditions such as injury or disease, the endogenous NSCs fail to form adequate new neurons to replace the lost neurons. Moreover, the plasticity of residual neurons in response to injury and connectivity of new neurons derived from NSCs after the injury or the SE are abnormal and detrimental to brain function in many instances (Shetty and Turner, 1996; Parent et al., 1997; Scharfman et al., 2000; Hattiangady et al., 2004; McCloskey et al., 2006). Therefore, the possibility of replacing lost neurons through grafting of fresh fetal post-mitotic neurons or NSCs expanded in culture has received great attention (Turner and Shetty, 2003; Shetty and Hattiangady, 2007b). Additionally, cell transplantation may be more favorable than single neurotrophic factor delivery for treating epilepsy because grafted neurons that are specific to the injured area may secrete multitude of beneficial trophic factors, in addition to providing additional synapses and facilitating the repair of disrupted circuits (Shetty et al., 2000, 2005;Hattiangady et al., 2006).
Studies demonstrate that grafting of specific fetal cells is useful for replacing the lost neurons and exerting a significant functional recovery in Parkinson’s disease. This approach has the potential to treat other neurodegenerative disorders including the TLE and stroke (Shetty and Turner, 1996; Freed et al., 2001; Isacson and Sladek, 1999; Kordower et al., 1998; Sanberg et al., 1997; Turner and Shetty, 2003). Studies in adult animal models of neurodegenerative diseases suggest that a more complete functional recovery with grafts requires integration of grafts with the host involving at least partial reconstitution of the damaged circuitry (Dunnett et al., 1997). Appropriate reconstitution of the disrupted host circuitry by specific neural grafts may also prevent, inhibit, or even reverse the formation of aberrant circuitry after lesions. Hence, studies on the efficacy of grafts of fetal hippocampal cells in different hippocampal lesion models are of interest for developing graft-mediated therapy for TLE, stroke, and head injury (Mudrick and Baimbridge, 1991; Holmes et al., 1992; Freeman et al., 1995; Soares et al., 1995; Shetty and Turner, 1995a, 1996, 1997a, b, 2000; Shetty et al., 2000; Zaman et al., 2000, 2001). Significant reductions in GABA-ergic interneuron number, functional disinhibition, aberrant sprouting of axons, appear to lead to hippocampal hyperexcitability in both TLE and KA-lesion models. Therefore, treatment strategies aimed at preventing hyperexcitability and seizures through grafting will need to develop ways to: (i) facilitate appropriate reinnervation of deafferented neurons; (ii) increase the efficacy of inhibition by activating the deafferented host GABA-ergic interneurons through appropriate re-innervation; (iii) suppress the aberrant axonal sprouting; and (iv) prevent the evolution of the IPI into chronic epilepsy characterized by SRMS. In this section, we discuss the available studies addressing the above issues using cell grafts in models of TLE.
12.1. Efficacy of fetal cell grafting for hippocampal repair after injury
The most extensive neural cell grafting studies pertaining to the TLE were performed in an intracerebroventricular KA (ICV KA) model. The primary aims of these studies are to test the efficacy of fetal hippocampal cell grafts for mediating appropriate reconstruction of the disrupted hippocampal circuitry and for suppressing epileptogenic changes following a relatively milder hippocampal injury induced by ICV KA administration (Shetty and Turner, 1996; Turner and Shetty, 2003; Shetty et al., 2005). In addition, preliminary grafting studies have been performed in an intraperitoneal KA (IP KA) model of TLE with the aim of testing the potential of fetal hippocampal cells placed at early time points after SE for averting chronic epilepsy development (Rao and Shetty, 2004).
12.1.1. Graft cell survival and graft-host connectivity
In the KA-lesioned CA3 region, the cell survival of grafts placed at 4 days post-lesion and analyzed at one-month post-grafting was greater for hippocampal cell grafts (CA3 grafts, 69% of injected cells, CA1 grafts, 42%) than non-hippocampal cell grafts (striatal cell grafts, 12% survival; Zaman et al., 2000). Thus, adequate survival of grafted cells in the lesioned adult hippocampus depends on the specificity of donor cells to the region of grafting. Yet, specific grafts exhibit reduced cell survival (31%) if they are placed at a prolonged post-lesion delay, suggesting decreased receptivity of the lesioned host with time after lesion (Zaman et al., 2001). Furthermore, specific CA3 cell grafts placed at 45 days post-lesion and analyzed at one-year post-grafting exhibit survival of 36% of grafted cells, which is equivalent to the graft cell survival seen at one-month post-grafting for these grafts (Fig 8, upper panel; Zaman and Shetty, 2001). Thus, grafted cells that survive the first month of grafting exhibit prolonged survival in this epilepsy model. Additionally, graft cell survival in this TLE model could be improved significantly via pre-treatment and grafting of donor hippocampal cells with FGF-2 (Zaman and Shetty, 2003). Analyses of axonal projections from CA3 grafts placed at 4-days post-lesion using cross-species grafting and mouse specific M6 staining showed robust axon projections into the denervated areas of CA1 and dentate regions (Shetty et al., 2005). CA3 grafts located close to the lesioned CA3 also showed a high propensity for establishing projections into the contralateral hippocampus (Shetty et al., 2000). Septal projections were robust from CA3 cell grafts, regardless of their intrahippocampal location (Shetty et al., 2000). Further, hippocampal grafts containing CA3 pyramidal neurons acquire robust mossy fiber inputs from the host. Thus, robust and specific interchange of axons occurs between the host and hippocampal cell grafts containing CA3 cells, suggesting that specific hippocampal cell grafting can restore the disrupted circuitry in the lesioned adult hippocampus.
Figure 8.
Top panel: Survival and morphology of fetal CA3 cell grafts placed close to the lesioned CA3 region of the adult hippocampus (marked by asterisks) at 45 days post-lesion and analyzed at 1 year postgrafting. A1 and A2 show a Nissl-stained section of the rat hippocampus containing fetal CA3 transplant (outlined by asterisks) located just below the degenerated CA3 cell layer (indicated by interrupted lines). B1, B2 show NeuN positive neurons within the graft in a neighboring section. DH, dentate hilus. Scale bar: A1, B1 = 400µm; A2, B2 = 50µm. (Reproduced from: Zaman, V and Shetty, AK; 2001; Neurobiol Dis.; 8:942–95). Bottom panel: Effects of CA3 or CA1 cell grafts on aberrant mossy fiber sprouting analyzed at one year post- grafting. Note dense mossy fiber sprouting in the dentate supragranular layer in the lesion only group (A1, D2) and a marked reduction in mossy fiber sprouting (B1, D3) following grafting of fetal CA3 cells but not with CA1 cells (C1, D4). B2 shows dense innervation of fetal CA3 cells grafted into the degenerated host CA3 region by host mossy fibers. C2 shows lack of host mossy fiber growth into the fetal CA1 graft. DH, dentate hilus. Scale bar: Scale bar: A1, B1, C1=500µm; B2, C2, D1–4 = 100µm.
12.1.2. Effect of fetal hippocampal cell grafting on the number of host GABA-ergic interneurons
The density of interneurons positive for GAD-67 (a synthesizing enzyme of GABA) has been measured in the adult hippocampus at 6-months after KA-induced hippocampal injury with grafting of either fetal mixed-hippocampal, CA3, or CA1 cells into the injured CA3-region at early post-lesion, in comparison to the “lesion-only” and intact hippocampi (Shetty and Turner, 2000). In dentate and CA1 regions of the lesioned hippocampus receiving grafts containing either mixed hippocampal or CA3 cells, the GAD-67 interneuron density was significantly greater than the “lesion-only” hippocampus but was comparable to the intact hippocampus. Nevertheless, in the lesioned hippocampus receiving CA1 cell grafts, it remained comparable to the “lesion-only” hippocampus. Thus, grafts containing CA3 cells restore lesion-induced depletions in host GABA-ergic interneurons, likely by reinnervation of denervated and GAD-67 deficient interneurons. This specific graft-mediated effect is beneficial because re-activation of interneurons after hippocampal injury could ameliorate both loss of functional inhibition and hyperexcitability.
12.1.3. Effect of fetal hippocampal cell grafting on aberrant sprouting of host mossy fibers
The long-term effects of CA3 and CA1 cell grafts placed into the lesioned CA3 region at 4 days post-lesion on aberrant sprouting of host mossy fibers into the DSGL have been measured using Timm’s staining and quantitative analyses (Fig 8, lower panel; Shetty et al., 2005). Analysis of mossy fiber sprouting at 1–12 months post-lesion in “lesion-only” hippocampus showed that the maximal extent of DSGL sprouting occurs by 4 months post-lesion and persists at the same level at 12 months post-lesion (Shetty et al., 2003). However, in hippocampus receiving CA3 grafts, there was a clear reduction in both width and the density of mossy fiber sprouting into the DSGL compared to “lesion-only” hippocampus. Reductions were significant in all regions of the DG (73–80%, Fig 8, lower panel). The density of DSGL sprouting was comparable to that of the control hippocampus. In hippocampus receiving CA3 grafts, sprouts of host mossy fibers from the principal mossy fiber bundle invaded grafts and densely innervated larger CA3 pyramidal neurons. In contrast, in hippocampus receiving CA1 grafts, the extent of DSGL mossy fiber sprouting remained greater than the intact hippocampus and grafts did not receive host mossy fibers (Fig. 8, lower panel). The CA3 graft-mediated suppression of sprouting is long-term because, the hippocampus receiving CA3 grafts analyzed at 12 months post-lesion exhibited similar minimal level of sprouting as the hippocampus receiving CA3 grafts analyzed at 4-months post-lesion. Prolonged suppression of aberrant sprouting in rats receiving CA3 grafts but not in rats receiving CA1 cell grafts suggests that the development of post-KA aberrant mossy fiber circuitry can be prevented by placement of grafts containing CA3 cells at early post-lesion. Presumably, this suppression is dependent on a numerically dense innervation of a more appropriate postsynaptic target within the graft (CA3 neurons) than is present within the DSGL. Suppression of aberrant sprouting is likely useful because the sprouted mossy fibers in DSGL are believed to contribute to the generation and propagation of seizures in the DG.
12.1.4. Effect of fetal hippocampal cell grafting on calbindin expression
The loss of CA3-pyramidal neurons in hippocampus after ICV KA leads to a permanent loss of calbindin in a major fraction of dentate granule cells and CA1 pyramidal cells (, 2007a). It is possible that enduring loss of calbindin in the DG and the CA1 subfield after CA3- lesion is due to disruption of the hippocampal circuitry. If this is true, then, fetal CA3 grafts placed into the lesioned-CA3 should restore calbindin by restitution of the disrupted circuitry. Recently, the effects of fetal CA3 and CA1 cell grafting into the lesioned CA3 region of adult rats at early post-lesion time-point on hippocampal calbindin has been examined. Calbindin in dentate granule and CA1 pyramidal cells of grafted animals was evaluated at 6-months post-lesion using immunostaining. Calbindin in the “lesion-only” hippocampus was dramatically reduced at 6-months post-lesion, compared to the intact hippocampus (Shetty and Hattiangady, 2007a). However, calbindin in the lesioned hippocampus receiving CA3 cell grafts was restored to the level of the intact hippocampus (Shetty and Hattiangady, 2007a). In contrast, calbindin in the lesioned hippocampus receiving CA1 grafts remained less than the intact hippocampus. Thus, specific cell grafting restores the injury-induced loss of calbindin in the adult hippocampus, likely via restitution of the disrupted circuitry. Since the loss of calbindin after hippocampal injury is linked to hyperexcitability (Nagerl et al., 2000), re-expression of calbindin in both dentate gyrus and CA1 subfield following CA3 cell grafting may suggest that specific cell grafting is efficacious for ameliorating injury-induced hyperexcitability in the adult hippocampus. Electrophysiological studies of KA-lesioned hippocampus receiving CA3 cell grafts are required in future to validate this possibility.
12.1.5. Efficacy of neural cell grafting for preventing SE-induced development of chronic seizures
Preliminary grafting studies have been performed in an intraperitoneal KA (IP KA) model of TLE with the aim of testing the potential of fetal hippocampal cells placed at early time points after SE for exhibiting adequate survival, suppressing epileptogenic changes, and averting chronic epilepsy development (Rao and Shetty, 2004). The results obtained so far are very promising. Because of partial lesions in both CA3 and CA1 regions in this model, mixed fetal hippocampal cells were used as donor cell for grafting. To enhance hippocampal recovery, cells were grafted bilaterally into the hippocampus (3 sites on each side). The results suggest that hippocampal cell grafting after SE-induced hippocampal injury considerably suppresses the development of chronic epilepsy. In ~50% of IP KA treated rats receiving grafts, no behavioral seizures were observed until 20 weeks after grafting. In the remaining half of transplanted rats, seizures were less pronounced at all time-points measured in this study (Rao and Shetty, 2004). In contrast, in the IP KA-treated group receiving no grafts, all rats exhibited robust SRMS. Thus, hippocampal cell grafting at 4 days after an IPI appears efficacious to suppress the development of chronic epilepsy. However, to validate the efficacy of this therapeutic approach, rigorous long-term studies are needed in future using both behavioral and video-EEG measurements.
12.2. Additional cell grafting studies that are relevant to preventing TLE after an IPI
The ability to restrain seizures through grafting of different cell types into different regions of the brain has also been tested in several other studies. In this section, some of the significant studies are discussed.
12.2.1. Efficacy of grafting cells that are engineered to produce GABA
Thompson and Suchomelova (2004) performed bilateral grafting of conditionally immortalized neurons engineered to produce GABA into the substantia nigra of rats at 45–65 days after the SE induced by lithium-pilocarpine and measured the effects of these grafts on SRMS at 1–10 days post-grafting. Interestingly, animals that received GABA-producing cells exhibited fewer SRMS than animals that received only control cells but the survival of grafted GABA-producing cells appeared to be limited. These results imply that cells genetically engineered to secrete GABA have the ability to diminish SRMS when transplanted into seizure-modulating nuclei. However, the long-term efficacy of this approach is currently unknown because of poor survival of grafted cells observed in the host brain in this model.
Studies on the effects of GABA-producing cells on the development of seizures after grafting into the dentate gyrus (Thompson, 2005) or the substantia nigra (Castillo et al., 2006) demonstrated raised GABA levels, increased electrical seizure threshold and delayed development of behavioral seizures in the kindling model of epilepsy (Thompson, 2005) and decreased seizures in the KA model (Castillo et al., 2006). However, as grafting was done prior to the induction of seizures or an IPI in these studies, this approach is not clinically practicable. Additionally, the long-term survival of cells engineered to produce GABA in the epileptic brain is unknown. In this context, grafting of fetal GABA-ergic cells may be beneficial for long-term functional recovery, as the survival of these cells can be enhanced through grafting of these cells with a cocktail of neurotrophic factors. Indeed, a recent study shows that fetal GABA-ergic cells from the E15 lateral ganglionic eminance pre-treated and grafted with neurotrophic factors at 4 days after the SE exhibit long-term survival in the hippocampus (Hattiangady et al., 2007). In addition, a vast majority of these cells differentiated into GABA-ergic interneurons, and subclasses of interneurons derived from grafts expressed markers such as parvalbumin, calbindin, calretinin and NPY. More importantly, rats that received these grafts after the SE exhibited ~80% reduction in the frequency of SRMS on a long-term basis, in comparison to rats that received only sham grafting surgery (Hattiangady et al., 2007).
12.2.2. Usefulness of grafting cells that are engineered to produce adenosine
A series of elegant experiments by Boison and associates tested the effects of grafts of encapsulated fibroblasts engineered to release the brain’s endogenous anticonvulsant adenosine on seizures (Boison et al., 1999; Boison et al., 2002; Huber et al., 2001; and Boison, 2005). Transplantation of these cells into the ventricles of rats kindled in the hippocampus resulted in reduced behavioral seizures and afterdischarges. However, this positive effect was transient and did not last beyond 2 weeks after grafting because of poor survival of the fibroblasts in the host brain. Assessment of the efficiency of transplants of mouse myoblasts engineered to release adenosine demonstrated reduced seizures for ~3 weeks in 50% of the animals (Fedele et al., 2004; Guttinger et al., 2005). Yet, the long-term efficacy of these transplants for reducing chronic seizures is currently unknown. The transient nature of positive effects in these studies suggests that either the grafted cells fail to exhibit enduring survival in an ectopic location or the amount of adenosine released by these cells becomes inadequate over time.
Recently, Li and colleagues (2007) analyzed the effects of embryonic stem (ES) cell derived neural precursors that are engineered to release adenosine through biallelic genetic disruption of the adenosine kinase gene (Adk−/−) to suppress seizures in a kindling model of TLE. The donor ES cells were differentiated into neural precursor cells in vitro following the genetic modification and then grafted into the hippocampus of rats prior to kindling. Animals that received the adenosine releasing grafts displayed sustained protection from developing generalized seizures. The therapeutic effect mediated by Adk(−/−) neural precursors appeared to be due to graft-mediated adenosine release, as seizures could transiently be elicited through blocking of adenosine A1 receptors and histological analysis at 26 days revealed the presence of surviving grafted cells in the hippocampus expressing mature neuronal markers. These results are promising for mediating antiepileptogenic effects through stem cell-mediated adenosine delivery in epileptic conditions. However, for further advances, it is important to rigorously test the usefulness of this approach with such grafts placed after the induction of SE or kindling.
12.3. Conclusions
In both TLE and animal models of TLE, the SE or head injury induced hippocampal neurodegeneration and the subsequent epileptogenic changes are believed to be responsible for the occurrence of SRMS. As the development of chronic TLE after the SE or head injury-induced neurodegeneration occurs progressively, finding apt interventions that both block the basic processes influencing the development of chronic TLE and promote compensatory repair processes after brain injury are of great importance. In this context, neural grafting studies are very relevant. The grafting studies performed early after a unilateral ICV KA administration (a model of mild hippocampal injury and TLE) demonstrate the following: replacement of the degenerated neurons in the injured hippocampus with grafting of specific fetal cells facilitates apt structural recovery of the lesioned adult hippocampus. This comprises restoration of the disrupted circuitry, suppression of the aberrant mossy fiber sprouting, and restoration of the GABA-ergic interneuron numbers and calbindin re-expression (Shetty and Turner, 1996, 1997b, a, 2000; Shetty et al., 2005; Shetty and Hattiangady, 2007a). However, the efficacy of this grafting intervention for functional recovery (particularly suppression of chronic SRMS) could not be tested, as rats treated unilaterally with ICV KA under anesthesia do not exhibit SRMS until 3 months post-KA and exhibit only occasional SRMS at 4–5 months post-ICV KA (Hattiangady et al., 2004). The preliminary studies using IP KA model suggest that fetal hippocampal cell grafting at 4 days after SE has the potential to suppress the progression of the initial seizure-induced hippocampal injury into chronic TLE (Rao and Shetty, 2004). To fully ascertain the efficacy of this intervention for preventing the development of chronic TLE after the SE-induced hippocampal injury, rigorous long-term studies are needed.
Pertaining to other cell types, grafts of fetal GABA-ergic cells from the lateral ganglionic eminance (Hattiangady et al., 2007), and grafts of embryonic stem (ES) cell derived neural precursors engineered to release adenosine (Li et al., 2007) show considerable promise for mediating antiepileptogenic effects in epilepsy models. However, for validating these approaches, it is important to rigorously test the usefulness of this approach in multiple SE models of epilepsy. In addition, a combination therapy along with cell transplantation may be more useful for inducing functional recovery in epileptic conditions. This may include a combination of: (i) fetal hippocampal cell transplants with delivery of neurotrophic factors (such as GDNF) via gene therapy; (ii) fetal hippocampal cell or GABA-ergic cell transplants with exposure of animals to enriched environment; (iii) fetal hippocampal cell transplants with neural precursors or NSCs engineered to release adenosine; (iv) fetal hippocampal cell transplants with neural precursors or NSCs engineered to release GABA; and (v) fetal hippocampal cell or GABA-ergic cell transplants with most promising AEDs. Such studies are clearly needed in future to make further advances in cell therapy for TLE.
Despite the promise of several cell transplantation strategies in animal models discussed above, none of these approaches is ready for clinical application. This is because there is no conclusive evidence hitherto to support that neural grafts placed after an IPI are capable of effectively preventing the occurrence of spontaneous seizures. Furthermore, none of the studies performed so far has demonstrated antiepileptogenic properties of grafts on a long-term basis using extensive video-EEG analyses. These criteria need to be met before considering any clinical application of cell therapy for TLE. In addition, the use of fetal cell grafting strategy for widespread clinical application is complicated because of the difficulty in obtaining large amounts of fetal tissue and ethical concerns regarding the use of fetal cells. Identifying alternative sources of cells are critical in this regard. Although NSCs from the hippocampus and other regions provide a rich source of donor cells for grafting following their expansion in culture (Shetty and Turner, 1998; Shetty, 2004), studies on the efficacy of such cells for suppressing seizures in animal models of TLE are still in infancy (see the review by Shetty and Hattiangady, 2007b for details). Detailed studies on the efficacy of NSC grafts in animal models of epilepsy are therefore additional requirements for further progress in this field.
13. Overall conclusions
The epileptogenesis is a dynamic process with major modifications taking place at multiple levels, which include synaptic plasticity, aberrant reorganization of the neuronal circuitry, alterations in interneuron number and function, and changes in dentate neurogenesis. The latency period found between the initial precipitating insult and the development of chronic epilepsy offers a useful window for application of promising neuroprotective strategies. Ideally, the initial insult modification strategy should be efficacious for inducing anti-seizure, antiepileptogenic and disease-modifying effects. If these criteria are met, such an intervention would likely prevent or at least minimize the progression of an IPI or the SE into chronic epilepsy characterized by SRMS and cognitive impairments. Thus, the window between the initial precipitating insult and the development of chronic epilepsy is important for applying promising neuroprotection strategies against epileptogenesis. After going through the various intervention strategies examined so far in epilepsy models, the following conclusions emerge.
First, the standard treatment with appropriate AEDs after an IPI or the SE appears useful as an anti-seizure strategy. Yet, it should be noted that this treatment is more beneficial if given early after the onset of acute seizures or brain injury. When given early after the SE, some of the AEDs were able to minimize neuronal cell death, and curb the aberrant reorganization of neuronal circuitry and other epileptogenic changes to some extent. Despite this, currently no individual AED can be projected as potential neuroprotectant capable of thwarting epileptogenesis, as adequate data from animal and human studies are lacking. Among the conventional drugs, early administration of diazepam or VPA after the SE appears promising for preventing chronic epilepsy. Among the new generation AEDs, only TPM, LTG and VGB seem to have some anti-epileptogenic effects. Considering these, examining the effects of a combination of two or more AEDs may be useful for ascertaining their ability to modify the progression of epileptogenesis.
Second, the high fat KD is gaining considerable importance for treating or preventing epilepsy because of relatively fewer side-effects and usefulness in managing refractory epilepsies. Despite some biochemical complications, neuroprotective effects of KD are quite evident. Nonetheless, it is still uncertain whether introduction of the KD after the SE or an IPI will be efficacious for preventing chronic epilepsy development and learning and memory impairments. Therefore, detailed studies are needed to validate the KD approach as a potent neuroprotective therapy for preventing epileptogenesis and the occurrence of chronic epilepsy after an IPI or the SE.
Third, the neurotrophic factors and their receptors appear to be important targets for anticonvulsant drug development. From the studies so far, it emerges that blocking the activity of BDNF and NGF has promise for suppressing epileptogenesis and preventing chronic epilepsy development after the SE or an IPI. Additionally, in view of their ability to suppress epileptogenic changes, strategies that increase the levels of GDNF and NT-3 may be advantageous for suppressing chronic epilepsy development after the SE. Clearly, additional long-term studies are needed to fully ascertain the utility of these approaches.
Fourth, because of their ability to provide neuroprotection in excitotoxic conditions, the antioxidants resveratrol and curcumin are promising as powerful neuroprotective drugs against acute seizures or the SE in future, as these compounds have almost nil side-effects, considerable anti-apoptotic properties and ability to protect neurons against seizures. Nonetheless, it will be essential to determine in future whether the antioxidant-mediated neuroprotection is adequate for preventing the SE-induced epileptogenesis, the development of SRMS, and learning and memory impairments. Among the major gonadal steroids, low-dose PROG administration appears useful for suppressing seizures as well as enhancing GABA-ergic function. In contrast, because of both anticonvulsant and proconvulsant actions, estrogen administration may not be useful for restraining chronic epilepsy development after the SE. On the other hand, some of the steroid metabolites, endogenous neurosteroids and EPO have some promise.
Fifth, the cell transplantation therapy (using either fetal hippocampal cells or NSCs as donor cells) is emerging as an alternative therapeutic strategy for preventing and treating chronic epilepsy. Appropriate fetal cell grafting shortly after the SE induced injury appears efficacious for blocking multiple processes of epileptogenesis and facilitating useful compensatory repair mechanisms for restoring the abnormal neural circuitry after an IPI. Effective structural recovery, suppression of excitatory inputs, restoration of GABAergic function and calbindin re-expression in the lesioned adult hippocampus is some of the fruitful indications foretelling neuroprotection efficacy of neural cell transplants. However, detailed studies in animal models of seizures are needed before this approach can be applied clinically. Additionally, availability of fetal cells is severely limited, which necessitates the development of alternative sources of donor cells such as NSCs or embryonic stem cells for grafting in future (SanMartin and Borlongan, 2006; Yasuhara et al., 2006a, b; Shetty and Hattiangady, 2007b). Additionally, rigorous characterization of these donor cell types following grafting in animal models of epilepsy are needed to determine their efficacy for thwarting chronic epilepsy after an IPI. Thus, the prospect of cell transplantation therapy for TLE looks promising but is not ready for clinical application.
Taken together, it is apparent that promising neuroprotective strategies need to be employed immediately after the initial precipitating injuries for forestalling the development of abnormal excitatory circuitry in the injured brain. Potential neuroprotection and treatment strategies for preventing chronic epilepsy are illustrated in Figure 9. The interventions such as the administration of drugs, the KD, the neurotrophic factors, the antioxidants and neural cell transplantation would be more efficacious if applied immediately after an IPI, as this early intervention may considerably prevent hippocampal neurodegeneration and abnormal reorganization of the hippocampal circuitry. The application of these interventions during the latent period (i.e. before the commencement of SRMS) may also be useful for preventing chronic epilepsy and learning and memory impairments. On the other hand, the application of interventions during the early phase of chronic epilepsy may be useful for reducing chronic seizures and improving cognitive functions. Additionally, pre-conditioning strategies such as regular dietary intake of antioxidants like resveratrol and curcumin may prove beneficial for both minimizing neurodegeneration in susceptible neuronal targets of the SE and for preserving normal brain function during and after initial insults.
Figure 9.
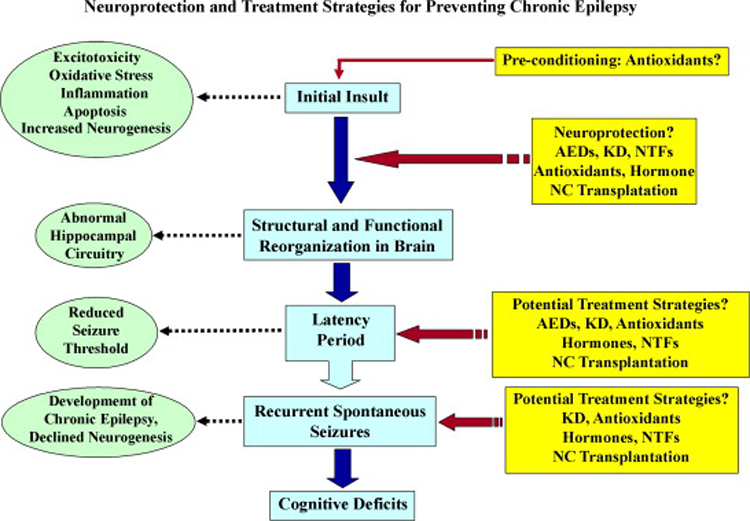
Potential neuroprotection and treatment strategies for preventing the chronic epilepsy development after an initial precipitating injury. An IPI leads to abnormal structural and functional reorganization of the brain circuitry, which gradually develops into chronic epilepsy characterized by spontaneous recurrent motor seizures (SRMS). A number of molecular as well as organizational level alterations occur during this gradual process. The neuroprotective interventions such as administration of the anti-epileptic drugs (AEDs), the ketogenic diet therapy (KD), the neurotrophic factors such as GDNF and NT-3, the antioxidants such as resveratrol and Curcumin and transplants of cells containing fetal hippocampal cells or neural stem cells may be beneficial immediately after an IPI for preventing subsequent epileptogenesis. The potential treatment strategies either during or after latency periods may also help in controlling the SRMS and cognitive deficits. Additionally, pre-conditioning with natural anti-oxidants may be effective in reducing IPI or seizure induced brain damage and the consequent epileptogenesis.
Acknowledgements
We gratefully acknowledge the support from National Institute of Neurological Disorders and Stroke (R01 NS54780, R01 NS43507 and R01 NS29482 to A.K.S), the National Institute for Aging (R01 AG20924 to A.K.S.) and the Department of Veterans Affairs (VA Merit Awards to A.K.S.) for our research group's contributions to the area of research discussed in this review.
List of Abbreviations
- AAT
Aspartate aminotransferase
- AACOT
Acetoacetyl-CoA thiolase
- Ad-GDNF
Adenoviral vector derived glial cell line derived neurotrophic factor
- AED
Anti-epileptic drug
- AKG
Alpha keto glutarate
- aFGF
Acidic fibroblast growth factor
- BBB
Blood brain barrier
- BDA
Biotinylated dextran amine
- BDNF
Brain derived neurotrophic factor
- bFGF (FGF-2)
Basic fibroblast growth factor or Fibroblast growth factor-2
- BHB
Beta hydroxy butyrate
- cAMP
Cyclic adenosine mono phosphate
- CNS
Central nervous system
- CR
Calorie restricted
- CS
Citrate synthetase
- CREB
cAMP responsive element binding protein
- Cyt c
Cytochrome c
- DCX
Doublecortin
- DG
Dentate gyrus
- DH
Dentate hilus
- DHEA
Dehydroepiandrosterone
- DSGL
Dentate supragranular layer
- EEG
Electroencephalogram
- EPO
Erythropoietin
- EPSP
Excitatory post-synaptic potential
- ERK
Extracellular-signal-regulated kinase
- FBM
Felbamate
- GABA
Gamma amino butyric acid
- GABAA
GABA receptor A subtype
- GAD
Glutamic acid decarboxylase
- GAT-1
GABA transporter type 1
- GBP
Gabapentin
- GCL
Granule cell layer
- GDNF
Glial cell derived neurotrophic factor
- Gln
Glutamine
- ICV KA
Intracerebroventricular kainic acid
- IPI
Initial precipitating injury
- IP KA
Intraperitoneal kainic acid
- IPSP
Inhibitory post-synaptic potential
- KA
Kainic acid
- KD
Ketogenic diet
- LDH
Lactate dehydrogenase
- LEV
Levetiracetam
- LDL
Low density lipoprotein
- LPS
Lipopolysaccharide
- LTP
Long-term potentiation
- MAPK
Mitogen-activated protein kinase
- MPT
Mitochondrial permeability transition pore
- NGF
Nerve growth factor
- NMDA
N-methyl-D-aspartate
- NO
Nitric oxide
- NPY
Neuropeptide Y
- NSC
Neural stem/progenitor cells
- NTF
Neurotrophic factor
- NT-3
Neurotrophin-3
- OAA
Oxaloacetate
- OGD
Oxygen-glucose deprivation
- PHB
Phenobarbital
- PI3K
Phosohatidylinositol-3-kinase
- PNS
Peripheral nervous system
- PROG
Progesterone
- PREG
Pregnenolone
- PREGS
Pregnenolone sulfate
- PTZ
Pentelenetetrazole
- PDH
Pyruvate dehydrogenase
- rhGDNF
Recombinant human GDNF
- ROS
Reactive oxygen species
- SCOT
Succinyl-CoA transferase
- SE
Status epilepticus
- SGZ
Subgranular zone
- SLM
Stratum lacunosum moleculare
- SO
Stratum oriens
- SP
Stratum pyramidale
- SR
Stratum radiatum
- SRMS
Spontaneous recurrent motor seizures
- TCA
Tricarboxylic acid
- TGB
Tiagabine
- TGF-β
Transforming growth factor beta
- TLE
Temporal lobe epilepsy
- TPM
Topiramate
- UCP
Uncoupling protein
- VEGF
Vascular endothelial growth factor
- VGB
Vigabatrin
- VPA
Valproic acid
- 3-OH-butyrate
3-hydroxy-butyrate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acharya MM, Katyare SS. Structural and functional alterations in mitochondrial membrane in picrotoxin-induced epileptic rat brain. Exp Neurol. 2005;192:79–88. doi: 10.1016/j.expneurol.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Acuna-Castroviejo D, Escames G, Macias M, Munoz Hoyos A, Molina Carballo A, Arauzo M, Montes R. Cell protective role of melatonin in the brain. J Pineal Res. 1995;19:57–63. doi: 10.1111/j.1600-079x.1995.tb00171.x. [DOI] [PubMed] [Google Scholar]
- Adams B, Sazgar M, Osehobo P, Van der Zee CE, Diamond J, Fahnestock M, Racine RJ. Nerve growth factor accelerates seizure development, enhances mossy fiber sprouting, and attenuates seizure-induced decreases in neuronal density in the kindling model of epilepsy. J Neurosci. 1997;17:5288–5296. doi: 10.1523/JNEUROSCI.17-14-05288.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Airaksinen MS, Saarma M. The GDNF family: signalling, biological functions and therapeutic value. Nature Rev. 2002;3:383–394. doi: 10.1038/nrn812. [DOI] [PubMed] [Google Scholar]
- Ajmone-Cat MA, Iosif RE, Ekdahl CT, Kokaia Z, Minghetti L, Lindvall O. Prostaglandin E2 and BDNF levels in rat hippocampus are negatively correlated with status epilepticus severity: no impact on survival of seizure-generated neurons. Neurobiol Dis. 2006;23:23–35. doi: 10.1016/j.nbd.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Al-Mudallal AS, LaManna JC, Lust WD, Harik SI. Diet-induced ketosis does not cause cerebral acidosis. Epilepsia. 1996;37:258–261. doi: 10.1111/j.1528-1157.1996.tb00022.x. [DOI] [PubMed] [Google Scholar]
- Albertson TE, Peterson SL, Stark LG, Lakin ML, Winters WD. The anticonvulsant properties of melatonin on kindled seizures in rats. Neuropharmacol. 1981;20:61–66. doi: 10.1016/0028-3908(81)90043-5. [DOI] [PubMed] [Google Scholar]
- Alessio A, Damasceno BP, Camargo CH, Kobayashi E, Guerreiro CA, Cendes F. Differences in memory performance and other clinical characteristics in patients with mesial temporal lobe epilepsy with and without hippocampal atrophy. Epilepsy Behav. 2004;5:22–27. doi: 10.1016/j.yebeh.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Alzheimer C, Werner S. Fibroblast growth factors and neuroprotection. Adv Exp MedBiol. 2002;513:335–351. doi: 10.1007/978-1-4615-0123-7_12. [DOI] [PubMed] [Google Scholar]
- Ananth C, Gopalakrishnakone P, Kaur C. Protective role of melatonin in domoic acid-induced neuronal damage in the hippocampus of adult rats. Hippocampus. 2003;13:375–387. doi: 10.1002/hipo.10090. [DOI] [PubMed] [Google Scholar]
- Andre V, Ferrandon A, Marescaux C, Nehlig A. Vigabatrin protects against hippocampal damage but is not antiepileptogenic in the lithium-pilocarpine model of temporal lobe epilepsy. Epilepsy Res. 2001;47:99–117. doi: 10.1016/s0920-1211(01)00299-6. [DOI] [PubMed] [Google Scholar]
- Andrews ZB, Horvath B, Barnstable CJ, Elsworth J, Yang L, Beal MF, Roth RH, Matthews RT, Horvath TL. Uncoupling protein-2 is critical for nigral dopamine cell survival in a mouse model of Parkinson's disease. J Neurosci. 2005;25:184–191. doi: 10.1523/JNEUROSCI.4269-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleton DB, DeVivo DC. An animal model for the ketogenic diet. Epilepsia. 1974;15:211–227. doi: 10.1111/j.1528-1157.1974.tb04943.x. [DOI] [PubMed] [Google Scholar]
- Armstrong DD. Epilepsy-induced microarchitectural changes in the brain. Pediatr Dev Pathol. 2005;8:607–614. doi: 10.1007/s10024-005-0054-3. [DOI] [PubMed] [Google Scholar]
- Avoli M, Louvel J, Pumain R, Kohling R. Cellular and molecular mechanisms of epilepsy in the human brain. Prog Neurobiol. 2005;77:166–200. doi: 10.1016/j.pneurobio.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Azcoitia I, Sierra A, Garcia-Segura LM. Estradiol prevents kainic acid-induced neuronal loss in the rat dentate gyrus. NeuroReport. 1998;9:3075–3079. doi: 10.1097/00001756-199809140-00029. [DOI] [PubMed] [Google Scholar]
- Bachmann RF, Schloesser RJ, Gould TD, Manji HK. Mood stabilizers target cellular plasticity and resilience cascades: implications for the development of novel therapeutics. Mol Neurobiol. 2005;32:173–202. doi: 10.1385/MN:32:2:173. [DOI] [PubMed] [Google Scholar]
- Backstrom T. Epileptic seizures in women related to plasma estrogen and progesterone during the menstrual cycle. Acta neurologica Scandinavica. 1976;54:321–347. doi: 10.1111/j.1600-0404.1976.tb04363.x. [DOI] [PubMed] [Google Scholar]
- Ballarin M, Ernfors P, Lindefors N, Persson H. Hippocampal damage and kainic acid injection induce a rapid increase in mRNA for BDNF and NGF in the rat brain. Exp Neurol. 1991;114:35–43. doi: 10.1016/0014-4886(91)90082-n. [DOI] [PubMed] [Google Scholar]
- Barde YA. What, if anything, is a neurotrophic factor? TrendsNeurosci. 1988;11:343–346. doi: 10.1016/0166-2236(88)90055-0. [DOI] [PubMed] [Google Scholar]
- Bastianetto S, Zheng WH, Quirion R. Neuroprotective abilities of resveratrol and other red wine constituents against nitric oxide-related toxicity in cultured hippocampal neurons. Brit J Pharmacol. 2000;131:711–720. doi: 10.1038/sj.bjp.0703626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baulieu EE, Schumacher M, Koenig H, Jung-Testas I, Akwa Y. Progesterone as a neurosteroid: actions within the nervous system. Cellular and molecular Neurobiol. 1996;16:143–154. doi: 10.1007/BF02088173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baulieu EE. Contragestion and other clinical applications of RU 486, an antiprogesterone at the receptor. Science. 1989;245:1351–1357. doi: 10.1126/science.2781282. [DOI] [PubMed] [Google Scholar]
- Bazil CW, Short D, Crispin D, Zheng W. Patients with intractable epilepsy have low melatonin, which increases following seizures. Neurology. 2000;55:1746–1748. doi: 10.1212/wnl.55.11.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekenstein JW, Lothman EW. Dormancy of inhibitory interneurons in a model of temporal lobe epilepsy. Science. 1993;259:97–100. doi: 10.1126/science.8093417. [DOI] [PubMed] [Google Scholar]
- Belelli D, Bolger MB, Gee KW. Anticonvulsant profile of the progesterone metabolite 5 alpha-pregnan-3 alpha-ol-20-one. Eur J Pharmacol. 1989;166:325–329. doi: 10.1016/0014-2999(89)90077-0. [DOI] [PubMed] [Google Scholar]
- Belguendouz L, Fremont L, Linard A. Resveratrol inhibits metal ion-dependent and independent peroxidation of porcine low-density lipoproteins. Biochem Pharmacol. 1997;53:1347–1355. doi: 10.1016/s0006-2952(96)00820-9. [DOI] [PubMed] [Google Scholar]
- Bengzon J, Kokaia Z, Ernfors P, Kokaia M, Leanza G, Nilsson OG, Persson H, Lindvall O. Regulation of neurotrophin and trkA, trkB and trkC tyrosine kinase receptor messenger RNA expression in kindling. Neuroscience. 1993;53:433–446. doi: 10.1016/0306-4522(93)90207-v. [DOI] [PubMed] [Google Scholar]
- Bernard C, Esclapez M, Hirsch JC, Ben-Ari Y. Interneurones are not so dormant in temporal lobe epilepsy: a critical reappraisal of the dormant basket cell hypothesis. Epilepsy Res. 1998;32:93–103. doi: 10.1016/s0920-1211(98)00043-6. [DOI] [PubMed] [Google Scholar]
- Bethmann K, Brandt C, Loscher W. Resistance to Phenobarbital Extends to Phenytoin in a Rat Model of Temporal Lobe Epilepsy. Epilepsia. 2007;48:816–826. doi: 10.1111/j.1528-1167.2007.00980.x. [DOI] [PubMed] [Google Scholar]
- Bhagat YA, Obenaus A, Hamilton MG, Mikler J, Kendall EJ. Neuroprotection from soman-induced seizures in the rodent: evaluation with diffusion- and T2-weighted magnetic resonance imaging. Neurotoxicol. 2005;26:1001–1013. doi: 10.1016/j.neuro.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Bialer M, Johannessen SI, Kupferberg HJ, Levy RH, Perucca E, Tomson T. Progress report on new antiepileptic drugs: a summary of the Seventh Eilat Conference (EILAT VII); Epilepsy Res; 2004. pp. 1–48. [DOI] [PubMed] [Google Scholar]
- Bikjdaouene L, Escames G, Leon J, Ferrer JM, Khaldy H, Vives F, Acuna-Castroviejo D. Changes in brain amino acids and nitric oxide after melatonin administration in rats with pentylenetetrazole-induced seizures. J Pineal Res. 2003;35:54–60. doi: 10.1034/j.1600-079x.2003.00055.x. [DOI] [PubMed] [Google Scholar]
- Binder DK, Croll SD, Gall CM, Scharfman HE. BDNF and epilepsy: too much of a good thing? Trends Neurosci. 2001;24:47–53. doi: 10.1016/s0166-2236(00)01682-9. [DOI] [PubMed] [Google Scholar]
- Binder DK, Routbort MJ, McNamara JO. Immunohistochemical evidence of seizure-induced activation of trk receptors in the mossy fiber pathway of adult rat hippocampus. J Neurosci. 1999;19:4616–4626. doi: 10.1523/JNEUROSCI.19-11-04616.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumcke I, Zuschratter W, Schewe JC, Suter B, Lie AA, Riederer BM, Meyer B, Schramm J, Elger CE, Wiestler OD. Cellular pathology of hilar neurons in Ammon's horn sclerosis. J Comp Neurol. 1999;414:437–453. doi: 10.1002/(sici)1096-9861(19991129)414:4<437::aid-cne2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Boison D, Scheurer L, Tseng JL, Aebischer P, Mohler H. Seizure suppression in kindled rats by intraventricular grafting of an adenosine releasing synthetic polymer. Exp Neurol. 1999;160:164–174. doi: 10.1006/exnr.1999.7209. [DOI] [PubMed] [Google Scholar]
- Boison D, Huber A, Padrun V, Deglon N, Aebischer P, Mohler H. Seizure suppression by adenosine-releasing cells is independent of seizure frequency. Epilepsia. 2002;43:788–796. doi: 10.1046/j.1528-1157.2002.33001.x. [DOI] [PubMed] [Google Scholar]
- Boison D. Adenosine and epilepsy: from therapeutic rationale to new therapeutic strategies. Neuroscientist. 2005;11:25–36. doi: 10.1177/1073858404269112. [DOI] [PubMed] [Google Scholar]
- Bolanos AR, Sarkisian M, Yang Y, Hori A, Helmers SL, Mikati M, Tandon P, Stafstrom CE, Holmes GL. Comparison of valproate and phenobarbital treatment after status epilepticus in rats. Neurology. 1998;51:41–48. doi: 10.1212/wnl.51.1.41. [DOI] [PubMed] [Google Scholar]
- Bonilha L, Yasuda CL, Rorden C, Li LM, Tedeschi H, de Oliveira E, Cendes F. Does resection of the medial temporal lobe improve the outcome of temporal lobe epilepsy surgery? Epilepsia. 2007;48:571–578. doi: 10.1111/j.1528-1167.2006.00958.x. [DOI] [PubMed] [Google Scholar]
- Borlongan CV, Yamamoto M, Takei N, Kumazaki M, Ungsuparkorn C, Hida H, Sanberg PR, Nishino H. Glial cell survival is enhanced during melatonin-induced neuroprotection against cerebral ischemia. Faseb J. 2000;14:1307–1317. doi: 10.1096/fj.14.10.1307. [DOI] [PubMed] [Google Scholar]
- Borowicz KK, Kaminski R, Gasior M, Kleinrok Z, Czuczwar SJ. Influence of melatonin upon the protective action of conventional anti-epileptic drugs against maximal electroshock in mice. Eur Neuropsychopharmacol. 1999;9:185–190. doi: 10.1016/s0924-977x(98)00022-4. [DOI] [PubMed] [Google Scholar]
- Bough KJ, Eagles DA. A ketogenic diet increases the resistance to pentylenetetrazole-induced seizures in the rat. Epilepsia. 1999;40:138–143. doi: 10.1111/j.1528-1157.1999.tb02066.x. [DOI] [PubMed] [Google Scholar]
- Bough KJ, Schwartzkroin PA, Rho JM. Calorie restriction and ketogenic diet diminish neuronal excitability in rat dentate gyrus in vivo. Epilepsia. 2003;44:752–760. doi: 10.1046/j.1528-1157.2003.55502.x. [DOI] [PubMed] [Google Scholar]
- Bough KJ, Wetherington J, Hassel B, Pare JF, Gawryluk JW, Greene JG, Shaw R, Smith Y, Geiger JD, Dingledine RJ. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann Neurol. 2006;60:223–235. doi: 10.1002/ana.20899. [DOI] [PubMed] [Google Scholar]
- Bough KJ, Yao SG, Eagles DA. Higher ketogenic diet ratios confer protection from seizures without neurotoxicity. Epilepsy Res. 2000;38:15–25. doi: 10.1016/s0920-1211(99)00077-7. [DOI] [PubMed] [Google Scholar]
- Bradshaw RA. Nerve growth factor. Ann Rev Biochem. 1978;47:191–216. doi: 10.1146/annurev.bi.47.070178.001203. [DOI] [PubMed] [Google Scholar]
- Brandt C, Gastens AM, Sun M, Hausknecht M, Loscher W. Treatment with valproate after status epilepticus: effect on neuronal damage, epileptogenesis, and behavioral alterations in rats. Neuropharmacol. 2006;51:789–804. doi: 10.1016/j.neuropharm.2006.05.021. [DOI] [PubMed] [Google Scholar]
- Brandt C, Glien M, Gastens AM, Fedrowitz M, Bethmann K, Volk HA, Potschka H, Loscher W. Prophylactic treatment with levetiracetam after status epilepticus: Lack of effect on epileptogenesis, neuronal damage, and behavioral alterations in rats. Neuropharmacol. 2007;53:207–221. doi: 10.1016/j.neuropharm.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Brines ML, Ghezzi P, Keenan S, Agnello D, de Lanerolle NC, Cerami C, Itri LM, Cerami A. Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. Proc Nat Acad Sci. 2000;97:10526–10531. doi: 10.1073/pnas.97.19.10526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks-Kayal AR, Shumate MD, Jin H, Rikhter TY, Coulter DA. Selective changes in single cell GABA (A) receptor subunit expression and function in temporal lobe epilepsy. Nat Med. 1998;4(10):1166–1172. doi: 10.1038/2661. [DOI] [PubMed] [Google Scholar]
- Brown-Croyts LM, Caton PW, Radecki DT, McPherson SL. Phenobarbital pre-treatment prevents kainic acid-induced impairments in acquisition learning. Life Sci. 2000;67:643–650. doi: 10.1016/s0024-3205(00)00658-5. [DOI] [PubMed] [Google Scholar]
- Browne TR, Holmes GL. Epilepsy. New England J Med. 2001;344:1145–1151. doi: 10.1056/NEJM200104123441507. [DOI] [PubMed] [Google Scholar]
- Brzezinski A. Melatonin in humans. New England J Med. 1997;336:186–195. doi: 10.1056/NEJM199701163360306. [DOI] [PubMed] [Google Scholar]
- Buckmaster PS, Dudek FE. Network properties of the dentate gyrus in epileptic rats with hilar neuron loss and granule cell axon reorganization. J Neurophysiol. 1997a;77:2685–2696. doi: 10.1152/jn.1997.77.5.2685. [DOI] [PubMed] [Google Scholar]
- Buckmaster PS, Dudek FE. Neuron loss, granule cell axon reorganization, and functional changes in the dentate gyrus of epileptic kainate-treated rats. J Comp Neurol. 1997b;385:385–404. [PubMed] [Google Scholar]
- Buckmaster PS, Dudek FE. In vivo intracellular analysis of granule cell axon reorganization in epileptic rats. J Neurophysiol. 1999;81:712–721. doi: 10.1152/jn.1999.81.2.712. [DOI] [PubMed] [Google Scholar]
- Buckmaster PS, Zhang GF, Yamawaki R. Axon sprouting in a model of temporal lobe epilepsy creates a predominantly excitatory feedback circuit. J Neurosci. 2002;22:6650–6658. doi: 10.1523/JNEUROSCI.22-15-06650.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhl EH, Otis TS, Mody I. Zinc-induced collapse of augmented inhibition by GABA in a temporal lobe epilepsy model. Science. 1996;271:369–373. doi: 10.1126/science.271.5247.369. [DOI] [PubMed] [Google Scholar]
- Cameron HA, Hazel TG, McKay RD. Regulation of neurogenesis by growth factors and neurotransmitters. J Neurobiol. 1998;36:287–306. [PubMed] [Google Scholar]
- Carey K, Fuchs S. Pediatric pharmacology. What you need to know for the next pediatric call. Emergency Medical Services. 2001;30:27–34. 37-28, 40. [PubMed] [Google Scholar]
- Castillo CG, Mendoza S, Freed WJ, Giordano M. Intranigral transplants of immortalized GABAergic cells decrease the expression of kainic acid-induced seizures in the rat. Behav Brain Res. 2006;171:109–115. doi: 10.1016/j.bbr.2006.03.025. [DOI] [PubMed] [Google Scholar]
- Cavazos JE, Zhang P, Qazi R, Sutula TP. Ultrastructural features of sprouted mossy fiber synapses in kindled and kainic acid-treated rats. J Comp Neurol. 2003;458:272–292. doi: 10.1002/cne.10581. [DOI] [PubMed] [Google Scholar]
- Cendes F. Febrile seizures and mesial temporal sclerosis. Curr Opin Neurol. 2002;17:161–164. doi: 10.1097/00019052-200404000-00013. [DOI] [PubMed] [Google Scholar]
- Champney TH, Hanneman WH, Legare ME, Appel K. Acute and chronic effects of melatonin as an anticonvulsant in male gerbils. J PinealRes. 1996;20:79–83. doi: 10.1111/j.1600-079x.1996.tb00243.x. [DOI] [PubMed] [Google Scholar]
- Cheng CM, Hicks K, Wang J, Eagles DA, Bondy CA. Caloric restriction augments brain glutamic acid decarboxylase-65 and -67 expression. J Neurosci Res. 2004;77:270–276. doi: 10.1002/jnr.20144. [DOI] [PubMed] [Google Scholar]
- Chikuma M, Masuda S, Kobayashi T, Nagao M, Sasaki R. Tissue-specific regulation of erythropoietin production in the murine kidney, brain, and uterus. Am J Physiol. 2000;279:E1242–E1248. doi: 10.1152/ajpendo.2000.279.6.E1242. [DOI] [PubMed] [Google Scholar]
- Choi JS, Kim SY, Park HJ, Cha JH, Choi YS, Kang JE, Chung JW, Chun MH, Lee MY. Upregulation of gp130 and differential activation of STAT and p42/44 MAPK in the rat hippocampus following kainic acid-induced seizures. Mol Brain Res. 2003;119:10–18. doi: 10.1016/j.molbrainres.2003.08.010. [DOI] [PubMed] [Google Scholar]
- Chung SY, Han SH. Melatonin attenuates kainic acid-induced hippocampal neurodegeneration and oxidative stress through microglial inhibition. J Pineal Res. 2003;34:95–102. doi: 10.1034/j.1600-079x.2003.00010.x. [DOI] [PubMed] [Google Scholar]
- Cilio MR, Bolanos AR, Liu Z, Schmid R, Yang Y, Stafstrom CE, Mikati MA, Holmes GL. Anticonvulsant action and long-term effects of gabapentin in the immature brain. Neuropharmacol. 2001;40:139–147. doi: 10.1016/s0028-3908(00)00103-9. [DOI] [PubMed] [Google Scholar]
- Citraro R, Russo E, Di Paola ED, Ibbadu GF, Gratteri S, Marra R, De Sarro G. Effects of some neurosteroids injected into some brain areas of WAG/Rij rats, an animal model of generalized absence epilepsy. Neuropharmacol. 2006;50:1059–1071. doi: 10.1016/j.neuropharm.2006.02.011. [DOI] [PubMed] [Google Scholar]
- Clough R, Statnick M, Maring-Smith M, Wang C, Eells J, Browning R, Dailey J, Jobe P. Fetal raphe transplants reduce seizure severity in serotonin-depleted GEPRs. NeuroReport. 1996;8:341–346. doi: 10.1097/00001756-199612200-00067. [DOI] [PubMed] [Google Scholar]
- Coppola G, Veggiotti P, Cusmai R, Bertoli S, Cardinali S, Dionisi-Vici C, Elia M, Lispi ML, Sarnelli C, Tagliabue A, Toraldo C, Pascotto A. The ketogenic diet in children, adolescents and young adults with refractory epilepsy: an Italian multicentric experience. Epilepsy Res. 2002;48:221–227. doi: 10.1016/s0920-1211(01)00315-1. [DOI] [PubMed] [Google Scholar]
- Cornish SM, Wheal HV. Long-term loss of paired pulse inhibition in the kainic acid-lesioned hippocampus of the rat. Neuroscience. 1989;28:563–571. doi: 10.1016/0306-4522(89)90005-5. [DOI] [PubMed] [Google Scholar]
- Corradetti R, Pugliese AM. Electrophysiological effects of felbamate. Life Sci. 1998;63:1075–1088. doi: 10.1016/s0024-3205(98)00230-6. [DOI] [PubMed] [Google Scholar]
- Costello DJ, Delanty N. Oxidative injury in epilepsy: potential for antioxidant therapy? Expert Rev Neurotherapeutics. 2004;4:541–553. doi: 10.1586/14737175.4.3.541. [DOI] [PubMed] [Google Scholar]
- Coumoul X, Deng CX. Roles of FGF receptors in mammalian development and congenital diseases. Birth Defects Res C Embryo Today. 2003;69:286–304. doi: 10.1002/bdrc.10025. [DOI] [PubMed] [Google Scholar]
- Croll SD, Goodman JH, Scharfman HE. Vascular endothelial growth factor (VEGF) in seizures: a double-edged sword. Adv Exp Med Biol. 2004;548:57–68. doi: 10.1007/978-1-4757-6376-8_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croll SD, Suri C, Compton DL, Simmons MV, Yancopoulos GD, Lindsay RM, Wiegand SJ, Rudge JS, Scharfman HE. Brain-derived neurotrophic factor transgenic mice exhibit passive avoidance deficits, increased seizure severity and in vitro hyperexcitability in the hippocampus and entorhinal cortex. Neuroscience. 1999;93:1491–1506. doi: 10.1016/s0306-4522(99)00296-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronin J, Dudek FE. Chronic seizures and collateral sprouting of dentate mossy fibers after kainic acid treatment in rats. Brain Res. 1988;474:181–184. doi: 10.1016/0006-8993(88)90681-6. [DOI] [PubMed] [Google Scholar]
- Cuevas P, Gimenez-Gallego G. Antiepileptic effects of acidic fibroblast growth factor examined in kainic acid-mediated seizures in the rat. Neurosci Lett. 1996;203:66–68. doi: 10.1016/0304-3940(95)12254-0. [DOI] [PubMed] [Google Scholar]
- Cunnane SC, Musa K, Ryan MA, Whiting S, Fraser DD. Potential role of polyunsaturates in seizure protection achieved with the ketogenic diet. Prostaglandins, leukotrienes, and essential fatty acids. 2002;67:131–135. doi: 10.1054/plef.2002.0409. [DOI] [PubMed] [Google Scholar]
- Czeisler CA. Commentary: evidence for melatonin as a circadian phase-shifting agent. J Biol Rhythms. 1997;12:618–623. doi: 10.1177/074873049701200617. [DOI] [PubMed] [Google Scholar]
- Czuczwar M, Kis J, Swiader M, Turski WA, Przesmycki K. Evaluation of interaction between valproate and baclofen in the formalin test in mice. Pol J Pharmacol. 2001;53:51–54. [PubMed] [Google Scholar]
- Czuczwar SJ, Patsalos PN. The new generation of GABA enhancers. Potential in the treatment of epilepsy. CNS drugs. 2001;15:339–350. doi: 10.2165/00023210-200115050-00001. [DOI] [PubMed] [Google Scholar]
- Czuczwar SJ, Przesmycki K. Felbamate, gabapentin and topiramate as adjuvant antiepileptic drugs in experimental models of epilepsy. Pol J Pharmacol. 2001;53:65–68. [PubMed] [Google Scholar]
- Dantz D, Bewersdorf J, Fruehwald-Schultes B, Kern W, Jelkmann W, Born J, Fehm HL, Peters A. Vascular endothelial growth factor: a novel endocrine defensive response to hypoglycemia. J Clin Endocrinol Metab. 2002;87:835–840. doi: 10.1210/jcem.87.2.8215. [DOI] [PubMed] [Google Scholar]
- Davenport CJ, Brown WJ, Babb TL. Sprouting of GABAergic and mossy fiber axons in dentate gyrus following intrahippocampal kainate in the rat. Exp Neurol. 1990;109:180–190. doi: 10.1016/0014-4886(90)90072-z. [DOI] [PubMed] [Google Scholar]
- Dawson D, Encel N. Melatonin and sleep in humans. J Pineal Res. 1993;15:1–12. doi: 10.1111/j.1600-079x.1993.tb00503.x. [DOI] [PubMed] [Google Scholar]
- de Lima E, Soares JM, Jr., del Carmen Sanabria Garrido Y, Gomes Valente S, Priel MR, Chada Baracat E, Abrao Cavalheiro E, da Graca Naffah-Mazzacoratti M, Amado D. Effects of pinealectomy and the treatment with melatonin on the temporal lobe epilepsy in rats. Brain Res. 2005;1043:24–31. doi: 10.1016/j.brainres.2005.02.027. [DOI] [PubMed] [Google Scholar]
- De Sarno P, Li X, Jope RS. Regulation of Akt and glycogen synthase kinase-3 beta phosphorylation by sodium valproate and lithium. Neuropharmacol. 2002;43:1158–1164. doi: 10.1016/s0028-3908(02)00215-0. [DOI] [PubMed] [Google Scholar]
- Deckers CL, Czuczwar SJ, Hekster YA, Keyser A, Kubova H, Meinardi H, Patsalos PN, Renier WO, Van Rijn CM. Selection of antiepileptic drug polytherapy based on mechanisms of action: the evidence reviewed. Epilepsia. 2000;41:1364–1374. doi: 10.1111/j.1528-1157.2000.tb00111.x. [DOI] [PubMed] [Google Scholar]
- Detour J, Schroeder H, Desor D, Nehlig A. A 5-month period of epilepsy impairs spatial memory, decreases anxiety, but spares object recognition in the lithium-pilocarpine model in adult rats. Epilepsia. 2005;46:499–508. doi: 10.1111/j.0013-9580.2005.38704.x. [DOI] [PubMed] [Google Scholar]
- Devinsky O. Diagnosis and treatment of temporal lobe epilepsy. Rev Neurol Dis. 2004;1:2–9. [PubMed] [Google Scholar]
- Dhanushkodi A, Shetty AK. Effects of diazepam administration at one-hour after the induction of status epilepticus on production and abnormal migration of newly born neurons in the adult dentate gyrus. Soc. Neurosci. Abstr. 2007:457.2. [Google Scholar]
- Dingledine R, Gjerstad L. Reduced inhibition during epileptiform activity in the in vitro hippocampal slice. J. Physiol. 1980;305:297–313. doi: 10.1113/jphysiol.1980.sp013364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draczynska-Lusiak B, Chen YM, Sun AY. Oxidized lipoproteins activate NF-kappaB binding activity and apoptosis in PC12 cells. NeuroReport. 1998a;9:527–532. doi: 10.1097/00001756-199802160-00028. [DOI] [PubMed] [Google Scholar]
- Draczynska-Lusiak B, Doung A, Sun AY. Oxidized lipoproteins may play a role in neuronal cell death in Alzheimer disease. Molecular and chemical neuropathology / sponsored by the International Society for Neurochemistry and the World Federation of Neurology and research groups on neurochemistry and cerebrospinal fluid. 1998b;33:139–148. doi: 10.1007/BF02870187. [DOI] [PubMed] [Google Scholar]
- Dudek FE, Obenaus A, Schweitzer JS, Wuarin JP. Functional significance of hippocampal plasticity in epileptic brain: electrophysiological changes of the dentate granule cells associated with mossy fiber sprouting. Hippocampus. 1994;4:259–265. doi: 10.1002/hipo.450040306. [DOI] [PubMed] [Google Scholar]
- Dugich-Djordjevic MM, Tocco G, Willoughby DA, Najm I, Pasinetti G, Thompson RF, Baudry M, Lapchak PA, Hefti F. BDNF mRNA expression in the developing rat brain following kainic acid-induced seizure activity. Neuron. 1992;8:1127–1138. doi: 10.1016/0896-6273(92)90133-x. [DOI] [PubMed] [Google Scholar]
- Dunnett SB, Kendall AL, Watts C, Torres EM. Neuronal cell transplantation for Parkinson's and Huntington's diseases. British Med Bull. 1997;53:757–776. doi: 10.1093/oxfordjournals.bmb.a011646. [DOI] [PubMed] [Google Scholar]
- Dvorak-Carbone H, Schuman EM. Experience-dependent modifications of hippocampal place cell firing. Hippocampus. 1999;1:193–206. doi: 10.1002/hipo.450010207. [DOI] [PubMed] [Google Scholar]
- Ekdahl CT, Mohapel P, Elmer E, Lindvall O. Caspase inhibitors increase short-term survival of progenitor-cell progeny in the adult rat dentate gyrus following status epilepticus. Eur J Neurosci. 2001;14:937–945. doi: 10.1046/j.0953-816x.2001.01713.x. [DOI] [PubMed] [Google Scholar]
- Emsley JG, Mitchell BD, Kempermann G, Macklis JD. Adult neurogenesis and repair of the adult CNS with neural progenitors, precursors, and stem cells. Prog Neurobiol. 2005;75:321–341. doi: 10.1016/j.pneurobio.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Engel J., Jr. Excitation and inhibition in epilepsy. Can J Neurological Sci. 1996;23:167–174. doi: 10.1017/s0317167100038464. [DOI] [PubMed] [Google Scholar]
- Engel J., Jr. Intractable epilepsy: definition and neurobiology. Epilepsia. 2001;42 Suppl. 6:3. doi: 10.1046/j.1528-1157.2001.0420s6003.x. [DOI] [PubMed] [Google Scholar]
- Elmer E, Kokaia M, Ernfors P, Ferencz I, Kokaia Z, Lindvall O. Suppressed kindling epileptogenesis and perturbed BDNF and TrkB gene regulation in NT-3 mutant mice. Exp Neurol. 1997;145:93–103. doi: 10.1006/exnr.1997.6478. [DOI] [PubMed] [Google Scholar]
- Erecinska M, Nelson D, Daikhin Y, Yudkoff M. Regulation of GABA level in rat brain synaptosomes: fluxes through enzymes of the GABA shunt and effects of glutamate, calcium, and ketone bodies. J Neurochem. 1996;67:2325–2334. doi: 10.1046/j.1471-4159.1996.67062325.x. [DOI] [PubMed] [Google Scholar]
- Eriksson PS, Perfilieva E, Bjork-Eriksson T, Alborn AM, Nordborg C, Peterson DA, Gage FH. Neurogenesis in the adult human hippocampus. Nature Med. 1998;4:1313–1317. doi: 10.1038/3305. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Bengzon J, Kokaia Z, Persson H, Lindvall O. Increased levels of messenger RNAs for neurotrophic factors in the brain during kindling epileptogenesis. Neuron. 1991a;7:165–176. doi: 10.1016/0896-6273(91)90084-d. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Ibanez CF, Ebendal T, Olson L, Persson H. Molecular cloning and neurotrophic activities of a protein with structural similarities to nerve growth factor: developmental and topographical expression in the brain. Proc Nat Acad Sci. 1990;87:5454–5458. doi: 10.1073/pnas.87.14.5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernfors P, Merlio JP, Persson H. Cells expressing mRNA for neurotrophins and their receptors during embryonic rat development. Eur J Neurosci. 1992;4:1140–1158. doi: 10.1111/j.1460-9568.1992.tb00141.x. [DOI] [PubMed] [Google Scholar]
- Escames G, Leon J, Lopez LC, Acuna-Castroviejo D. Mechanisms of N-methyl-D-aspartate receptor inhibition by melatonin in the rat striatum. J Neuroendocrinol. 2004;16:929–935. doi: 10.1111/j.1365-2826.2004.01250.x. [DOI] [PubMed] [Google Scholar]
- Escames G, Macias M, Leon J, Garcia J, Khaldy H, Martin M, Vives F, Acuna-Castroviejo D. Calcium-dependent effects of melatonin inhibition of glutamatergic response in rat striatum. J Neuroendocrinol. 2001;13:459–466. doi: 10.1046/j.1365-2826.2001.00656.x. [DOI] [PubMed] [Google Scholar]
- Ettinger AB. Psychotropic effects of antiepileptic drugs. Neurology. 2006;67:1916–1925. doi: 10.1212/01.wnl.0000247045.85646.c0. [DOI] [PubMed] [Google Scholar]
- Felling RJ, Levison SW. Enhanced neurogenesis following stroke. J Neurosci Res. 2003;73:277–283. doi: 10.1002/jnr.10670. [DOI] [PubMed] [Google Scholar]
- Feng R, Rampon C, Tang YP, Shrom D, Jin J, Kyin M, Sopher B, Miller MW, Ware CB, Martin GM, Kim SH, Langdon RB, Sisodia SS, Tsien JZ. Deficient neurogenesis in forebrain-specific presenilin-1 knockout mice is associated with reduced clearance of hippocampal memory traces. Neuron. 2001;32:911–926. doi: 10.1016/s0896-6273(01)00523-2. [DOI] [PubMed] [Google Scholar]
- Fedele DE, Koch P, Scheurer L, Simpson EM, Mohler H, Brustle O, Boison D. Engineering embryonic stem cell derived glia for adenosine delivery. Neurosci Lett. 2004;370:160–165. doi: 10.1016/j.neulet.2004.08.031. [DOI] [PubMed] [Google Scholar]
- Ferencz I, Kokaia M, Elmer E, Keep M, Kokaia Z, Lindvall O. Suppression of kindling epileptogenesis in rats by intrahippocampal cholinergic grafts. Eur. J. Neurosci. 1998;10:213–220. doi: 10.1046/j.1460-9568.1998.00033.x. [DOI] [PubMed] [Google Scholar]
- Ferriero DM. Protecting neurons. Epilepsia. 2005;46 Suppl 7:45–51. doi: 10.1111/j.1528-1167.2005.00302.x. [DOI] [PubMed] [Google Scholar]
- Field MJ, Cox PJ, Stott E, Melrose H, Offord J, Su TZ, Bramwell S, Corradini L, England S, Winks J, Kinloch RA, Hendrich J, Dolphin AC, Webb T, Williams D. Identification of the alpha2-delta-1 subunit of voltage-dependent calcium channels as a molecular target for pain mediating the analgesic actions of pregabalin. Proc Nat Acad Sci. 2006;103:17537–17542. doi: 10.1073/pnas.0409066103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher PD, Sperber EF, Moshe SL. Hippocampal sclerosis revisited. Brain Dev. 1998;20:563–573. doi: 10.1016/s0387-7604(98)00069-2. [DOI] [PubMed] [Google Scholar]
- Fisher A, Wang X, Cock HR, Thom M, Patsalos PN, Walker MC. Synergism between topiramate and budipine in refractory status epilepticus in the rat. Epilepsia. 2004;45:1300–1307. doi: 10.1111/j.0013-9580.2004.26404.x. [DOI] [PubMed] [Google Scholar]
- Foldvary-Schaefer N, Harden C, Herzog A, Falcone T. Hormones and seizures. Cleveland Clinic journal of medicine. 2004;71 Suppl 2:S11–S18. doi: 10.3949/ccjm.71.suppl_2.s11. [DOI] [PubMed] [Google Scholar]
- Franck JE, Kunkel DD, Baskin DG, Schwartzkroin PA. Inhibition in kainate-lesioned hyperexcitable hippocampi: physiologic, autoradiographic, and immunocytochemical observations. J Neurosci. 1988;8:1991–2002. doi: 10.1523/JNEUROSCI.08-06-01991.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francois J, Koning E, Ferrandon A, Nehlig A. The combination of topiramate and diazepam is partially neuroprotective in the hippocampus but not antiepileptogenic in the lithium-pilocarpine model of temporal lobe epilepsy. Epilepsy Res. 2006;72:147–163. doi: 10.1016/j.eplepsyres.2006.07.014. [DOI] [PubMed] [Google Scholar]
- Francois LL, Manel V, Rousselle C, David M. Ketogenic regime as anti-epileptic treatment: its use in 29 epileptic children. Arch Pediatr. 2003;10:300–306. doi: 10.1016/s0929-693x(03)00030-7. [DOI] [PubMed] [Google Scholar]
- Freed CR, Greene PE, Breeze RE, Tsai WY, DuMouchel W, Kao R, Dillon S, Winfield H, Culver S, Trojanowski JQ, Eidelberg D, Fahn S. Transplantation of embryonic dopamine neurons for severe Parkinson's disease. New England J Med. 2001;344:710–719. doi: 10.1056/NEJM200103083441002. [DOI] [PubMed] [Google Scholar]
- Freeman J, Kelly M, Freeman J. The Ketogenic Diet: A Treatment for Epilepsy. 3rd ed. NY: Demos Medical Publishers; 2002. [Google Scholar]
- Freeman TB, Olanow CW, Hauser RA, Nauert GM, Smith DA, Borlongan CV, Sanberg PR, Holt DA, Kordower JH, Vingerhoets FJ, et al. Bilateral fetal nigral transplantation into the postcommissural putamen in Parkinson's disease. Ann Neurol. 1995;38:379–388. doi: 10.1002/ana.410380307. [DOI] [PubMed] [Google Scholar]
- Freeman J, Veggiotti P, Lanzi G, Tagliabue A, Perucca E. The ketogenic diet: from molecular mechanisms to clinical effects. Epilepsy Res. 2006;68:145–180. doi: 10.1016/j.eplepsyres.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Freeman JM. The ketogenic diet and epilepsy. Nestle Nutrition workshop series. 2001;5:307–318. doi: 10.1159/000061835. discussion 318–321. [DOI] [PubMed] [Google Scholar]
- Freeman JM, Kossoff EH, Hartman AL. The ketogenic diet: one decade later. Pediatrics. 2007;119:535–543. doi: 10.1542/peds.2006-2447. [DOI] [PubMed] [Google Scholar]
- Freeman JM, Vining EP. Ketogenic diet: a time-tested, effective, and safe method for treatment of intractable childhood epilepsy. Epilepsia. 1998;39:450–451. doi: 10.1111/j.1528-1157.1998.tb01400.x. [DOI] [PubMed] [Google Scholar]
- Freeman JM, Vining EP, Pillas DJ, Pyzik PL, Casey JC, Kelly LM. The efficacy of the ketogenic diet-1998: a prospective evaluation of intervention in 150 children. Pediatrics. 1998;102:1358–1363. doi: 10.1542/peds.102.6.1358. [DOI] [PubMed] [Google Scholar]
- Freund TF, Buzsaki G. Interneurons of the hippocampus. Hippocampus. 1996;6:347–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Fremont L. Biological effects of resveratrol. Life sciences. 2000;66:663–673. doi: 10.1016/s0024-3205(99)00410-5. [DOI] [PubMed] [Google Scholar]
- Frisch C, Kudin AP, Elger CE, Kunz WS, Helmstaedter C. Amelioration of water maze performance deficits by topiramate applied during pilocarpine-induced status epilepticus is negatively dose-dependent. Epilepsy Res. 2007;73:173–180. doi: 10.1016/j.eplepsyres.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Frye CA, Bayon LE. Seizure activity is increased in endocrine states characterized by decline in endogenous levels of the neurosteroid 3 alpha,5 alpha-THP. Neuroendocrinol. 1998;68:272–280. doi: 10.1159/000054375. [DOI] [PubMed] [Google Scholar]
- Fujikawa DG, Itabashi HH, Wu A, Shinmei SS. Status epilepticus-induced neuronal loss in humans without systemic complications or epilepsy. Epilepsia. 2000a;41:981–991. doi: 10.1111/j.1528-1157.2000.tb00283.x. [DOI] [PubMed] [Google Scholar]
- Fujikawa DG, Shinmei SS, Cai B. Seizure-induced neuronal necrosis: implications for programmed cell death mechanisms. Epilepsia. 2000b;41 Suppl 6:S9–S13. doi: 10.1111/j.1528-1157.2000.tb01549.x. [DOI] [PubMed] [Google Scholar]
- Furth SL, Casey JC, Pyzik PL, Neu AM, Docimo SG, Vining EP, Freeman JM, Fivush BA. Risk factors for urolithiasis in children on the ketogenic diet. PediatricNnephrol (Berlin, Germany) 2000;15:125–128. doi: 10.1007/s004670000443. [DOI] [PubMed] [Google Scholar]
- Gage FH. Neurogenesis in the adult brain. J Neurosci. 2002;22:612–613. doi: 10.1523/JNEUROSCI.22-03-00612.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gall C, Lauterborn J, Bundman M, Murray K, Isackson P. Seizures and the regulation of neurotrophic factor and neuropeptide gene expression in brain. Epilepsy Res Suppl. 1991a;4:225–245. [PubMed] [Google Scholar]
- Gall C, Murray K, Isackson PJ. Kainic acid-induced seizures stimulate increased expression of nerve growth factor mRNA in rat hippocampus. Brain Res Mol Brain Res. 1991b;9:113–123. doi: 10.1016/0169-328x(91)90136-l. [DOI] [PubMed] [Google Scholar]
- Gao ZB, Chen XQ, Hu GY. Inhibition of excitatory synaptic transmission by trans-resveratrol in rat hippocampus. Brain Res. 2006;1111:41–47. doi: 10.1016/j.brainres.2006.06.096. [DOI] [PubMed] [Google Scholar]
- Gao ZB, Hu GY. Trans-resveratrol, a red wine ingredient, inhibits voltage-activated potassium currents in rat hippocampal neurons. Brain Res. 2005;1056:68–75. doi: 10.1016/j.brainres.2005.07.013. [DOI] [PubMed] [Google Scholar]
- Giusti P, Franceschini D, Petrone M, Manev H, Floreani M. In vitro and in vivo protection against kainate-induced excitotoxicity by melatonin. J PinealRes. 1996a;20:226–231. doi: 10.1111/j.1600-079x.1996.tb00263.x. [DOI] [PubMed] [Google Scholar]
- Giusti P, Lipartiti M, Franceschini D, Schiavo N, Floreani M, Manev H. Neuroprotection by melatonin from kainate-induced excitotoxicity in rats. Faseb J. 1996b;10:891–896. doi: 10.1096/fasebj.10.8.8666166. [DOI] [PubMed] [Google Scholar]
- Golombek DA, Pevet P, Cardinali DP. Melatonin effects on behavior: possible mediation by the central GABAergic system. Neurosci Biobehavioral Rev. 1996;20:403–412. doi: 10.1016/0149-7634(95)00052-6. [DOI] [PubMed] [Google Scholar]
- Gomez-Pinilla F, van der Wal EA, Cotman CW. Possible coordinated gene expressions for FGF receptor, FGF-5, and FGF-2 following seizures. Exp Neurol. 1995;133:164–174. doi: 10.1006/exnr.1995.1019. [DOI] [PubMed] [Google Scholar]
- Gong C, Wang TW, Huang HS, Parent JM. Reelin regulates neuronal progenitor migration in intact and epileptic hippocampus. J Neurosci. 2007;27:1803–1811. doi: 10.1523/JNEUROSCI.3111-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman JH. Experimental models of status epilepticus. In: Peterson SL, Albertson TE, editors. Neuropharmacology Methods in Epilepsy Research. Boca Raton: CRC Press; 1998. pp. 95–125. [Google Scholar]
- Gould E, Gross CG. Neurogenesis in adult mammals: some progress and problems. J Neurosci. 2002;22:619–623. doi: 10.1523/JNEUROSCI.22-03-00619.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray WP, Sundstrom LE. Kainic acid increases the proliferation of granule cell progenitors in the dentate gyrus of the adult rat. Brain Res. 1998;790:52–59. doi: 10.1016/s0006-8993(98)00030-4. [DOI] [PubMed] [Google Scholar]
- Greene AE, Todorova MT, McGowan R, Seyfried TN. Caloric restriction inhibits seizure susceptibility in epileptic EL mice by reducing blood glucose. Epilepsia. 2001;42:1371–1378. doi: 10.1046/j.1528-1157.2001.17601.x. [DOI] [PubMed] [Google Scholar]
- Greene AE, Todorova MT, Seyfried TN. Perspectives on the metabolic management of epilepsy through dietary reduction of glucose and elevation of ketone bodies. J Neurochem. 2003;86:529–537. doi: 10.1046/j.1471-4159.2003.01862.x. [DOI] [PubMed] [Google Scholar]
- Gross CG. Neurogenesis in the adult brain: death of a dogma. Nature Rev. 2000;1:67–73. doi: 10.1038/35036235. [DOI] [PubMed] [Google Scholar]
- Grosso S, Cordelli DM, Franzoni E, Coppola G, Capovilla G, Zamponi N, Verrotti A, Morgese G, Balestri P. Efficacy and safety of levetiracetam in infants and young children with refractory epilepsy. Seizure. 2007 doi: 10.1016/j.seizure.2007.02.004. PMID: 17368928. [DOI] [PubMed] [Google Scholar]
- Guerrini R, Bonanni P, Parmeggiani L, Hallett M, Oguni H. Pathophysiology of myoclonic epilepsies. Adv Neurol. 2005;95:23–46. [PubMed] [Google Scholar]
- Gupta RC, Dettbarn WD. Prevention of kainic acid seizures-induced changes in levels of nitric oxide and high-energy phosphates by 7-nitroindazole in rat brain regions. BrainRes. 2003;981:184–192. doi: 10.1016/s0006-8993(03)03034-8. [DOI] [PubMed] [Google Scholar]
- Gupta YK, Briyal S, Chaudhary G. Protective effect of trans-resveratrol against kainic acid-induced seizures and oxidative stress in rats. Pharmacol Biochem Behav. 2002;71:245–249. doi: 10.1016/s0091-3057(01)00663-3. [DOI] [PubMed] [Google Scholar]
- Guttinger M, Padrun V, Pralong WF, Boison D. Seizure suppression and lack of adenosine A1 receptor desensitization after focal long-term delivery of adenosine by encapsulated myoblasts. Exp Neurol. 2005;193:53–64. doi: 10.1016/j.expneurol.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Gwinn RP, Kondratyev A, Gale K. Time-dependent increase in basic fibroblast growth factor protein in limbic regions following electroshock seizures. Neuroscience. 2002;114(2):403–409. doi: 10.1016/s0306-4522(02)00265-8. [DOI] [PubMed] [Google Scholar]
- Hallbergson AF, Gnatenco C, Peterson DA. Neurogenesis and brain injury: managing a renewable resource for repair. J Clin Invest. 2003;112:1128–1133. doi: 10.1172/JCI20098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halonen T, Nissinen J, Jansen JA, Pitkanen A. Tiagabine prevents seizures, neuronal damage and memory impairment in experimental status epilepticus. Eur J Pharmacol. 1996;299:69–81. doi: 10.1016/0014-2999(95)00835-7. [DOI] [PubMed] [Google Scholar]
- Halonen T, Nissinen J, Pitkanen A. Chronic elevation of brain GABA levels beginning two days after status epilepticus does not prevent epileptogenesis in rats. Neuropharmacoly. 2001a;40:536–550. doi: 10.1016/s0028-3908(00)00183-0. [DOI] [PubMed] [Google Scholar]
- Halonen T, Nissinen J, Pitkanen A. Effect of lamotrigine treatment on status epilepticus-induced neuronal damage and memory impairment in rat. Epilepsy Res. 2001b;46:205–223. doi: 10.1016/s0920-1211(01)00278-9. [DOI] [PubMed] [Google Scholar]
- Han YS, Zheng WH, Bastianetto S, Chabot JG, Quirion R. Neuroprotective effects of resveratrol against beta-amyloid-induced neurotoxicity in rat hippocampal neurons: involvement of protein kinase C. Brit J Pharmacol. 2004;141:997–1005. doi: 10.1038/sj.bjp.0705688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanaya R, Sasa M, Ujihara H, Ishihara K, Serikawa T, Iida K, Akimitsu T, Arita K, Kurisu K. Suppression by topiramate of epileptiform burst discharges in hippocampal CA3 neurons of spontaneously epileptic rat in vitro. Brain Res. 1998;789:274–282. doi: 10.1016/s0006-8993(98)00116-4. [DOI] [PubMed] [Google Scholar]
- Harvey AS, Berkovic SF, Wrennall JA, Hopkins IJ. Temporal lobe epilepsy in childhood: clinical, EEG, and neuroimaging findings and syndrome classification in a cohort with new-onset seizures. Neurology. 1997;49:960–968. doi: 10.1212/wnl.49.4.960. [DOI] [PubMed] [Google Scholar]
- Hassan AM, Keene DL, Whiting SE, Jacob PJ, Champagne JR, Humphreys P. Ketogenic diet in the treatment of refractory epilepsy in childhood. PediatricNeurol. 1999;21:548–552. doi: 10.1016/s0887-8994(99)00045-4. [DOI] [PubMed] [Google Scholar]
- Hattiangady B, Rao MS, Shetty AK. Chronic temporal lobe epilepsy is associated with severely declined dentate neurogenesis in the adult hippocampus. Neurobiol Dis. 2004;17:473–490. doi: 10.1016/j.nbd.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Hattiangady B, Rao MS, Zaman V, Shetty AK. Incorporation of embryonic CA3 cell grafts into the adult hippocampus at 4-months after injury: effects of combined neurotrophic supplementation and caspase inhibition. Neuroscience. 2006;139:1369–1383. doi: 10.1016/j.neuroscience.2006.01.058. [DOI] [PubMed] [Google Scholar]
- Hattiangady B, Rao MS, Rai KS, Shetty AK. Efficacy of striatal precursor cell grafting into the hippocampus for suppressing the progression of status epilepticus into chronic epilepsy. Cell Transplant. 2007;16:325. [Google Scholar]
- Hemingway C, Freeman JM, Pillas DJ, Pyzik PL. The ketogenic diet: a 3- to 6-year follow-up of 150 children enrolled prospectively. Pediatrics. 2001;108:898–905. doi: 10.1542/peds.108.4.898. [DOI] [PubMed] [Google Scholar]
- Henshall DC, Chen J, Simon RP. Involvement of caspase-3-like protease in the mechanism of cell death following focally evoked limbic seizures. J Neurochem. 2000;74:1215–1223. doi: 10.1046/j.1471-4159.2000.741215.x. [DOI] [PubMed] [Google Scholar]
- He XP, Kotloski R, Nef S, Luikart BW, Parada LF, McNamara JO. Conditional deletion of TrkB but not BDNF prevents epileptogenesis in the kindling model. Neuron. 2004;43:31–42. doi: 10.1016/j.neuron.2004.06.019. [DOI] [PubMed] [Google Scholar]
- Heo K, Lee BI, Yi SD, Huh K, Kim JM, Lee SA, Shin DJ, Song HK, Lee SK, Kim JY, Lu S, Dubois C, Tonner F. Efficacy and safety of levetiracetam as adjunctive treatment of refractory partial seizures in a multicentre open-label single-arm trial in Korean patients. Seizure. 2007;16:402–409. doi: 10.1016/j.seizure.2007.02.011. [DOI] [PubMed] [Google Scholar]
- Herzog AG, Klein P, Ransil BJ. Three patterns of catamenial epilepsy. Epilepsia. 1997;38:1082–1088. doi: 10.1111/j.1528-1157.1997.tb01197.x. [DOI] [PubMed] [Google Scholar]
- Herzog AG. Progesterone therapy in women with epilepsy: a 3-year follow-up. Neurology. 1999;52:1917–1918. doi: 10.1212/wnl.52.9.1917-a. [DOI] [PubMed] [Google Scholar]
- Hicks AU, Hewlett K, Windle V, Chernenko G, Ploughman M, Jolkkonen J, Weiss S, Corbett D. Enriched environment enhances transplanted subventricular zone stem cell migration and functional recovery after stroke. Neuroscience. 2007;146:31–40. doi: 10.1016/j.neuroscience.2007.01.020. [DOI] [PubMed] [Google Scholar]
- Hoffman GE, Merchenthaler I, Zup SL. Neuroprotection by ovarian hormones in animal models of neurological disease. Endocrine. 2006;29:217–231. doi: 10.1385/ENDO:29:2:217. [DOI] [PubMed] [Google Scholar]
- Hoffman GE, Moore N, Fiskum G, Murphy AZ. Ovarian steroid modulation of seizure severity and hippocampal cell death after kainic acid treatment. Exp Neurol. 2003;182:124–134. doi: 10.1016/s0014-4886(03)00104-3. [DOI] [PubMed] [Google Scholar]
- Hohn A, Leibrock J, Bailey K, Barde YA. Identification and characterization of a novel member of the nerve growth factor/brain-derived neurotrophic factor family. Nature. 1990;344:339–341. doi: 10.1038/344339a0. [DOI] [PubMed] [Google Scholar]
- Holmes GL. Clinical evidence that epilepsy is a progressive disorder with special emphasis on epilepsy syndromes that do progress. Adv Neurol. 2006;97:323–331. [PubMed] [Google Scholar]
- Holmes GL, Thompson JL, Huh K, Stuart JD, Carl GF. Effects of neural transplantation on seizures in the immature genetically epilepsy-prone rat. Exp Neurol. 1992;116:52–63. doi: 10.1016/0014-4886(92)90175-p. [DOI] [PubMed] [Google Scholar]
- Holmes MD, Silbergeld DL, Drouhard D, Wilensky AJ, Ojemann LM. Effect of vagus nerve stimulation on adults with pharmacoresistant generalized epilepsy syndromes. Seizure. 2004;13:340–345. doi: 10.1016/j.seizure.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Huber A, Padrun V, Deglon N, Aebischer P, Mohler H, Boison D. Grafts of adenosine-releasing cells suppress seizures in kindling epilepsy. Proc Nat Acad Sci. 2001;98:7611–7616. doi: 10.1073/pnas.131102898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda K, Negishi H, Yamori Y. Antioxidant nutrients and hypoxia/ischemia brain injury in rodents. Toxicol. 2003;189:55–61. doi: 10.1016/s0300-483x(03)00152-5. [DOI] [PubMed] [Google Scholar]
- Isacson O, Sladek JR., Jr. Cellular and molecular treatments of neurological diseases. Exp Neurol. 1999;159:1–3. doi: 10.1006/exnr.1999.7155. [DOI] [PubMed] [Google Scholar]
- Isackson PJ, Huntsman MM, Murray KD, Gall CM. BDNF mRNA expression is increased in adult rat forebrain after limbic seizures: temporal patterns of induction distinct from NGF. Neuron. 1991;6:937–948. doi: 10.1016/0896-6273(91)90234-q. [DOI] [PubMed] [Google Scholar]
- Itoh N, Ornitz DM. Evolution of the Fgf and Fgfr gene families. Trends Genet. 2004;20:563–569. doi: 10.1016/j.tig.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Jacobs KM, Graber KD, Kharazia VN, Parada I, Prince DA. Postlesional epilepsy: the ultimate brain plasticity. Epilepsia. 2000;41 Suppl 6:S153–S161. doi: 10.1111/j.1528-1157.2000.tb01574.x. [DOI] [PubMed] [Google Scholar]
- Jang JH, Surh YJ. Protective effect of resveratrol on beta-amyloid-induced oxidative PC12 cell death. Free Rad Biol Med. 2003;34:1100–1110. doi: 10.1016/s0891-5849(03)00062-5. [DOI] [PubMed] [Google Scholar]
- Jang M, Pezzuto JM. Cancer chemopreventive activity of resveratrol. Drugs under experimental and clinical research. 1999;25:65–77. [PubMed] [Google Scholar]
- Jankowsky JL, Patterson PH. The role of cytokines and growth factors in seizures and their sequelae. Prog Neurobiol. 2001;63:125–149. doi: 10.1016/s0301-0082(00)00022-8. [DOI] [PubMed] [Google Scholar]
- Janz R, Goda Y, Geppert M, Missler M, Sudhof TC. SV2A and SV2B function as redundant Ca2+ regulators in neurotransmitter release. Neuron. 1999;24:1003–1016. doi: 10.1016/s0896-6273(00)81046-6. [DOI] [PubMed] [Google Scholar]
- Jessberger S, Nakashima K, Clemenson GD, Jr., Mejia E, Mathews E, Ure K, Ogawa S, Sinton CM, Gage FH, Hsieh J. Epigenetic modulation of seizure-induced neurogenesis and cognitive decline. J Neurosci. 2007;27:5967–5975. doi: 10.1523/JNEUROSCI.0110-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolkkonen J, Halonen T, Jolkkonen E, Nissinen J, Pitkanen A. Seizure-induced damage to the hippocampus is prevented by modulation of the GABAergic system. Neuroreport. 1996;7:2031–2035. doi: 10.1097/00001756-199608120-00036. [DOI] [PubMed] [Google Scholar]
- Jung KH, Chu K, Kim M, Jeong SW, Song YM, Lee ST, Kim JY, Lee SK, Roh JK. Continuous cytosine-b-D-arabinofuranoside infusion reduces ectopic granule cells in adult rat hippocampus with attenuation of spontaneous recurrent seizures following pilocarpine-induced status epilepticus. Eur J Neurosci. 2004;19:3219–3226. doi: 10.1111/j.0953-816X.2004.03412.x. [DOI] [PubMed] [Google Scholar]
- Jung KK, Lee HS, Cho JY, Shin WC, Rhee MH, Kim TG, Kang JH, Kim SH, Hong S, Kang SY. Inhibitory effect of curcumin on nitric oxide production from lipopolysaccharide-activated primary microglia. Life sciences. 2006;79:2022–2031. doi: 10.1016/j.lfs.2006.06.048. [DOI] [PubMed] [Google Scholar]
- Kalinin VV. Suicidality and antiepileptic drugs: is there a link? Drug Saf. 2007;30:123–142. doi: 10.2165/00002018-200730020-00003. [DOI] [PubMed] [Google Scholar]
- Kandel ER, Schwartz JH, Jessell TM. New York: McGraw-Hill, Health Professions Division; Principles of neural science. 2000
- Kang HC, Chung DE, Kim DW, Kim HD. Early- and late-onset complications of the ketogenic diet for intractable epilepsy. Epilepsia. 2004;45:1116–1123. doi: 10.1111/j.0013-9580.2004.10004.x. [DOI] [PubMed] [Google Scholar]
- Kang HJ, Schuman EM. Neurotrophin-induced modulation of synaptic transmission in the adult hippocampus. J Physiol, Paris. 1995b;89:11–22. doi: 10.1016/0928-4257(96)80547-x. [DOI] [PubMed] [Google Scholar]
- Kankirawatana P, Jirapinyo P, Kankirawatana S, Wongarn R, Thamanasiri N. Ketogenic diet: an alternative treatment for refractory epilepsy in children. Journal of the Medical Association of Thailand. 2001;84:1027–1032. [PubMed] [Google Scholar]
- Kanter-Schlifke I, Georgievska B, Kirik D, Kokaia M. Seizure suppression by GDNF gene therapy in animal models of epilepsy. Mol Ther. 2007;15:1106–1113. doi: 10.1038/sj.mt.6300148. [DOI] [PubMed] [Google Scholar]
- Kaplan MS, Hinds JW. Neurogenesis in the adult rat: electron microscopic analysis of light radioautographs. Science. 1977;197:1092–1094. doi: 10.1126/science.887941. [DOI] [PubMed] [Google Scholar]
- Karnup S, Stelzer A. Temporal overlap of excitatory and inhibitory afferent input in guinea-pig CA1 pyramidal cells. J. Physiol. 1999;516:485–504. doi: 10.1111/j.1469-7793.1999.0485v.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kralic JE, Ledergerber DA, Fritschy JM. Disruption of the neurogenic potential of the dentate gyrus in a mouse model of temporal lobe epilepsy with focal seizures. Eur J Neurosci. 2005;22:1916–1927. doi: 10.1111/j.1460-9568.2005.04386.x. [DOI] [PubMed] [Google Scholar]
- Katoh-Semba R, Takeuchi IK, Inaguma Y, Ito H, Kato K. Brain-derived neurotrophic factor, nerve growth and neurotrophin-3 selected regions of the rat brain following kainic acid-induced seizure activity. Neurosci Res. 1999;35:19–29. doi: 10.1016/s0168-0102(99)00059-0. [DOI] [PubMed] [Google Scholar]
- Kelly KM. Gabapentin. Antiepileptic mechanism of action. Neuropsychobiol. 1998;38:139–144. doi: 10.1159/000026529. [DOI] [PubMed] [Google Scholar]
- Khaibullina AA, Rosenstein JM, Krum JM. Vascular endothelial growth factor promotes neurite maturation in primary CNS neuronal cultures. Brain Res Dev Brain Res. 2004;148:59–68. doi: 10.1016/j.devbrainres.2003.09.022. [DOI] [PubMed] [Google Scholar]
- Kim DW, Kang HC, Park JC, Kim HD. Benefits of the nonfasting ketogenic diet compared with the initial fasting ketogenic diet. Pediatrics. 2004;114:1627–1630. doi: 10.1542/peds.2004-1001. [DOI] [PubMed] [Google Scholar]
- Klepper J, Leiendecker B, Bredahl R, Athanassopoulos S, Heinen F, Gertsen E, Florcken A, Metz A, Voit T. Introduction of a ketogenic diet in young infants. J Inherited Metab Dis. 2002;25:449–460. doi: 10.1023/a:1021238900470. [DOI] [PubMed] [Google Scholar]
- Klitgaard H, Pitkanen A. Antiepileptogenesis, neuroprotection, and disease modification in the treatment of epilepsy: focus on levetiracetam. Epileptic Disord. 2003;5 Suppl 1:S9–S16. [PubMed] [Google Scholar]
- Kobayashi M, Buckmaster PS. Reduced inhibition of dentate granule cells in a model of temporal lobe epilepsy. J Neurosci. 2003;23:2440–2452. doi: 10.1523/JNEUROSCI.23-06-02440.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler C, Chan-Palay V, Wu JY. Septal neurons containing glutamic acid decarboxylase immunoreactivity project to the hippocampal region in the rat brain. Anatomy and embryology. 1984;169:41–44. doi: 10.1007/BF00300585. [DOI] [PubMed] [Google Scholar]
- Kokaia M, et al. Seizure suppression in kindling epilepsy by intracerebral implants of GABA - but not by noradrenalin-releasing polymer matrices. Exp. Brain Res. 1994;100:385–394. doi: 10.1007/BF02738399. [DOI] [PubMed] [Google Scholar]
- Kokaia M, Ernfors P, Kokaia Z, Elmer E, Jaenisch R, Lindvall O. Suppressed epileptogenesis in BDNF mutant mice. Exp Neurol. 1995;133:215–224. doi: 10.1006/exnr.1995.1024. [DOI] [PubMed] [Google Scholar]
- Kokaia Z, Airaksinen MS, Nanobashvili A, Larsson E, Kujamaki E, Lindvall O, Saarma M. GDNF family ligands and receptors are differentially regulated after brain insults in the rat. Eur J Neurosci. 1999;11:1202–1216. doi: 10.1046/j.1460-9568.1999.00513.x. [DOI] [PubMed] [Google Scholar]
- Kokaia Z, Kelly ME, Elmer E, Kokaia M, McIntyre DC, Lindvall O. Seizure-induced differential expression of messenger RNAs for neurotrophins and their receptors in genetically fast and slow kindling rats. Neuroscience. 1996;75:197–207. doi: 10.1016/0306-4522(96)00257-6. [DOI] [PubMed] [Google Scholar]
- Kokate TG, Svensson BE, Rogawski MA. Anticonvulsant activity of neurosteroids: correlation with gamma-aminobutyric acid-evoked chloride current potentiation. J Pharmacol Exp Therap. 1994;270:1223–1229. [PubMed] [Google Scholar]
- Kolb B, Morshead C, Gonzalez C, Kim M, Gregg C, Shingo T, Weiss S. Growth factor-stimulated generation of new cortical tissue and functional recovery after stroke damage to the motor cortex of rats. J Cereb Blood Flow Metab. 2007;27:983–997. doi: 10.1038/sj.jcbfm.9600402. [DOI] [PubMed] [Google Scholar]
- Kordower JH, Freeman TB, Chen EY, Mufson EJ, Sanberg PR, Hauser RA, Snow B, Olanow CW. Fetal nigral grafts survive and mediate clinical benefit in a patient with Parkinson's disease. Mov Disord. 1998;13:383–393. doi: 10.1002/mds.870130303. [DOI] [PubMed] [Google Scholar]
- Kornack DR, Rakic P. Continuation of neurogenesis in the hippocampus of the adult macaque monkey. Proc Nat Acad Sci. 1999;96:5768–5773. doi: 10.1073/pnas.96.10.5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kossoff EH. More fat and fewer seizures: dietary therapies for epilepsy. Lancet Neurol. 2004;3:415–420. doi: 10.1016/S1474-4422(04)00807-5. [DOI] [PubMed] [Google Scholar]
- Kossoff EH, McGrogan JR. Worldwide use of the ketogenic diet. Epilepsia. 2005;46:280–289. doi: 10.1111/j.0013-9580.2005.42704.x. [DOI] [PubMed] [Google Scholar]
- Kossoff EH, McGrogan JR, Freeman JM. Benefits of an all-liquid ketogenic diet. Epilepsia. 2004;45:1163. doi: 10.1111/j.0013-9580.2004.18504.x. [DOI] [PubMed] [Google Scholar]
- Kossoff EH, Pyzik PL, McGrogan JR, Vining EP, Freeman JM. Efficacy of the ketogenic diet for infantile spasms. Pediatrics. 2002;109:780–783. doi: 10.1542/peds.109.5.780. [DOI] [PubMed] [Google Scholar]
- Koyama R, Yamada MK, Fujisawa S, Katoh-Semba R, Matsuki N, Ikegaya Y. Brainderived neurotrophic factor induces hyperexcitable reentrant circuits in the dentate gyrus. J Neurosci. 2004;24:7215–7224. doi: 10.1523/JNEUROSCI.2045-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama R, Ikegaya Y. To BDNF or not to BDNF: that is the epileptic hippocampus. Neuroscientist. 2005;11:282–287. doi: 10.1177/1073858405278266. [DOI] [PubMed] [Google Scholar]
- Kralic JE, Ledergerber DA, Fritschy JM. Disruption of the neurogenic potential of the dentate gyrus in a mouse model of temporal lobe epilepsy with focal seizures. Eur J Neurosci. 2005;22:1916–1927. doi: 10.1111/j.1460-9568.2005.04386.x. [DOI] [PubMed] [Google Scholar]
- Krall RL, Penry JK, Kupferberg HJ, Swinyard EA. Antiepileptic drug development: I. History and a program for progress. Epilepsia. 1978a;19:393–408. doi: 10.1111/j.1528-1157.1978.tb04506.x. [DOI] [PubMed] [Google Scholar]
- Krall RL, Penry JK, White BG, Kupferberg HJ, Swinyard EA. Antiepileptic drug development: II. Anticonvulsant drug screening. Epilepsia. 1978b;19:409–428. doi: 10.1111/j.1528-1157.1978.tb04507.x. [DOI] [PubMed] [Google Scholar]
- Krum JM, Mani N, Rosenstein JM. Angiogenic and astroglial responses to vascular endothelial growth factor administration in adult rat brain. Neuroscience. 2002;110:589–604. doi: 10.1016/s0306-4522(01)00615-7. [DOI] [PubMed] [Google Scholar]
- Kudin AP, Debska-Vielhaber G, Vielhaber S, Elger CE, Kunz WS. The mechanism of neuroprotection by topiramate in an animal model of epilepsy. Epilepsia. 2004;45:1478–1487. doi: 10.1111/j.0013-9580.2004.13504.x. [DOI] [PubMed] [Google Scholar]
- Kuhn HG, Dickinson-Anson H, Gage FH. Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J Neurosci. 1996;16:2027–2033. doi: 10.1523/JNEUROSCI.16-06-02027.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar SS, Buckmaster PS. Hyperexcitability, interneurons, and loss of GABAergic synapses in entorhinal cortex in a model of temporal lobe epilepsy. J Neurosci. 2006;26:4613–4623. doi: 10.1523/JNEUROSCI.0064-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar SS, Buckmaster PS. Neuron-specific nuclear antigen NeuN is not detectable in gerbil subtantia nigra pars reticulata. Brain Res. 2007 doi: 10.1016/j.brainres.2007.01.027. PMID: 17291468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar SS, Jin X, Buckmaster PS, Huguenard JR. Recurrent circuits in layer II of medial entorhinal cortex in a model of temporal lobe epilepsy. J Neurosci. 2007;27:1239–1246. doi: 10.1523/JNEUROSCI.3182-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz WS, Kudin AP, Vielhaber S, Blumcke I, Zuschratter W, Schramm J, Beck H, Elger CE. Mitochondrial complex I deficiency in the epileptic focus of patients with temporal lobe epilepsy. Ann Neurol. 2000;48:766–773. [PubMed] [Google Scholar]
- Lamballe F, Klein R, Barbacid M. The trk family of oncogenes and neurotrophin receptors. Princess Takamatsu symposia. 1991;22:153–170. [PubMed] [Google Scholar]
- Lauterborn JC, Isackson PJ, Gall CM. Seizure-induced increases in NGF mRNA exhibit different time courses across forebrain regions and are biphasic in hippocampus. Exp Neurol. 1994;125:22–40. doi: 10.1006/exnr.1994.1003. [DOI] [PubMed] [Google Scholar]
- Lee SH, Chun W, Kong PJ, Han JA, Cho BP, Kwon OY, Lee HJ, Kim SS. Sustained activation of Akt by melatonin contributes to the protection against kainic acid-induced neuronal death in hippocampus. J Pineal Res. 2006;40:79–85. doi: 10.1111/j.1600-079X.2005.00283.x. [DOI] [PubMed] [Google Scholar]
- Lee WT, Shen YZ, Chang C. Neuroprotective effect of lamotrigine and MK-801 on rat brain lesions induced by 3-nitropropionic acid: evaluation by magnetic resonance imaging and in vivo proton magnetic resonance spectroscopy. Neuroscience. 2000;95:89–95. doi: 10.1016/s0306-4522(99)00410-8. [DOI] [PubMed] [Google Scholar]
- Leibrock J, Lottspeich F, Hohn A, Hofer M, Hengerer B, Masiakowski P, Thoenen H, Barde YA. Molecular cloning and expression of brain-derived neurotrophic factor. Nature. 1989;341:149–152. doi: 10.1038/341149a0. [DOI] [PubMed] [Google Scholar]
- Li T, Steinbeck JA, Lusardi T, Koch P, Lan JQ, Wilz A, Segschneider M, Simon RP, Brustle O, Boison D. Suppression of kindling epileptogenesis by adenosine releasing stem cell-derived brain implants. Brain. 2007;130:1276–1288. doi: 10.1093/brain/awm057. [DOI] [PubMed] [Google Scholar]
- Li Q, Stephenson D. Postischemic administration of basic fibroblast growth factor improves sensorimotor function and reduces infarct size following permanent focal cerebral ischemia in the rat. Exp Neurol. 2002;177:531–537. doi: 10.1006/exnr.2002.7994. [DOI] [PubMed] [Google Scholar]
- Li S, Xu B, Martin D, Racine RJ, Fahnestock M. Glial cell line-derived neurotrophic factor modulates kindling and activation-induced sprouting in hippocampus of adult rats. Exp Neurol. 2002;178:49–58. doi: 10.1006/exnr.2002.8036. [DOI] [PubMed] [Google Scholar]
- Li W, Sun G. Polyphenolic antioxidants on cytokine-induced iNOS and sPLA2 in an importalized astrocyte cell line (DITNC) In: Parker L, Ong A, editors. Biological Oxidation & Antioxidants, Molecular Mechanisms & Health Effects. Champaign: AOCS Press; 1998. pp. 90–103. [Google Scholar]
- Li W, Xia J, Sun G. Cytokine induction of iNOS and sPLA2 in immortalized astrocytes (DITNC): Response to genistein and pyrrolidine dithiocarbamate. J Interfon Cytokine Res. 1999;19:121–127. doi: 10.1089/107999099314261. [DOI] [PubMed] [Google Scholar]
- Liang LP, Patel M. Mitochondrial oxidative stress and increased seizure susceptibility in Sod2(−/+) mice. Free Rad Biol Med. 2004;36:542–554. doi: 10.1016/j.freeradbiomed.2003.11.029. [DOI] [PubMed] [Google Scholar]
- Likhodii SS, Burnham WM. Calorie restriction may not elevate the threshold of PTZ-induced seizures. Epilepsy Res. 2003;57:77–78. doi: 10.1016/j.eplepsyres.2003.10.006. author reply 79–80. [DOI] [PubMed] [Google Scholar]
- Lin LF, Doherty DH, Lile JD, Bektesh S, Collins F. GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science. 1993;260:1130–1132. doi: 10.1126/science.8493557. [DOI] [PubMed] [Google Scholar]
- Lindvall O, Bengzon J, Elmér E, Kokaia M, Kokaia Z. In: Functional Neural Transplantation. Dunnett SB, Björklund A, editors. New York: Raven; 1994. pp. 387–413. [Google Scholar]
- Litt B, Esteller R, Echauz J, D'Alessandro M, Shor R, Henry T, Pennell P, Epstein C, Bakay R, Dichter M, Vachtsevanos G. Epileptic seizures may begin hours in advance of clinical onset: a report of five patients. Neuron. 2001;30:51–64. doi: 10.1016/s0896-6273(01)00262-8. [DOI] [PubMed] [Google Scholar]
- Li C, Watanabe G, Weng Q, Jin W, Furuta C, Suzuki AK, Kawaguchi M, Taya K. Expression of nerve growth factor (NGF), and its receptors TrkA and p75 in the reproductive organs of the adult male rats. Zool Sci. 2005;22:933–937. doi: 10.2108/zsj.22.933. [DOI] [PubMed] [Google Scholar]
- Liu H, Kaur J, Dashtipour K, Kinyamu R, Ribak CE, Friedman LK. Suppression of hippocampal neurogenesis is associated with developmental stage, number of perinatal seizure episodes, and glucocorticosteroid level. Exp Neurol. 2003;184:196–213. doi: 10.1016/s0014-4886(03)00207-3. [DOI] [PubMed] [Google Scholar]
- Liu Z, D'Amore PA, Mikati M, Gatt A, Holmes GL. Neuroprotective effect of chronic infusion of basic fibroblast growth factor on seizure-associated hippocampal damage. Brain Res. 1993;626:335–338. doi: 10.1016/0006-8993(93)90598-h. [DOI] [PubMed] [Google Scholar]
- Liu Z, Holmes GL. Basic fibroblast growth factor is highly neuroprotective against seizure-induced long-term behavioural deficits. Neuroscience. 1997;76:1129–1138. doi: 10.1016/s0306-4522(96)00412-5. [DOI] [PubMed] [Google Scholar]
- Livingston S. Dietary treatment of epilepsy. In: Charles LS, Thomas C, editors. Comprehensive Management of Epilepsy in Infancy, Childhood and Adolescence. Springfield, IL: 1972. pp. 378–405. [Google Scholar]
- Logothetis J, Harner R, Morrell F, Torres F. The role of estrogens in catamenial exacerbation of epilepsy. Neurology. 1959;9:352–360. doi: 10.1212/wnl.9.5.352. [DOI] [PubMed] [Google Scholar]
- Longo R, Domenici MR, Scotti de Carolis A, Sagratella S. Felbamate selectively blocks in vitro hippocampal kainate-induced irreversible electrical changes. Life sciences. 1995;56:PL409–PL414. doi: 10.1016/0024-3205(95)00158-3. [DOI] [PubMed] [Google Scholar]
- Loscher W. Basic pharmacology of valproate: a review after 35 years of clinical use for the treatment of epilepsy. CNS drugs. 2002;16:669–694. doi: 10.2165/00023210-200216100-00003. [DOI] [PubMed] [Google Scholar]
- Loscher W, Poulter MO, Padjen AL. Major targets and mechanisms of antiepileptic drugs and major reasons for failure. Adv Neurol. 2006;97:417–427. [PubMed] [Google Scholar]
- Lothman EW. Neurobiology as a basis for rational polypharmacy. Section overview for rational polypharmacy conference. Epilepsy Res Suppl. 1996;11:3–7. [PubMed] [Google Scholar]
- Lowenstein DH, Seren MS, Longo FM. Prolonged increases in neurotrophic activity associated with kainate-induced hippocampal synaptic reorganization. Neuroscience. 1993;56:597–604. doi: 10.1016/0306-4522(93)90359-n. [DOI] [PubMed] [Google Scholar]
- Lynch BA, Lambeng N, Nocka K, Kensel-Hammes P, Bajjalieh SM, Matagne A, Fuks B. The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc Nat Acad Sci. 2004;101:9861–9866. doi: 10.1073/pnas.0308208101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Sutula T. Recurrent excitatory connectivity in the dentate gyrus of kindled and kainic acid-treated rats. J Neurophusiol. 2000;83:693–704. doi: 10.1152/jn.2000.83.2.693. [DOI] [PubMed] [Google Scholar]
- Macdonald RL, Kelly KM. Mechanisms of action of currently prescribed and newly developed antiepileptic drugs. Epilepsia. 1994;35 Suppl 4:S41–S50. doi: 10.1111/j.1528-1157.1994.tb05955.x. [DOI] [PubMed] [Google Scholar]
- Macdonald RL, Kelly KM. Antiepileptic drug mechanisms of action. Epilepsia. 1995;36 Suppl 2:S2–S12. doi: 10.1111/j.1528-1157.1995.tb05996.x. [DOI] [PubMed] [Google Scholar]
- MacGregor DG, Graham DI, Stone TW. The attenuation of kainate-induced neurotoxicity by chlormethiazole and its enhancement by dizocilpine, muscimol, and adenosine receptor agonists. Exp Neurol. 1997;148:110–123. doi: 10.1006/exnr.1997.6625. [DOI] [PubMed] [Google Scholar]
- Madsen TM, Treschow A, Bengzon J, Bolwig TG, Lindvall O, Tingstrom A. Increased neurogenesis in a model of electroconvulsive therapy. Biol Psych. 2000;47:1043–1049. doi: 10.1016/s0006-3223(00)00228-6. [DOI] [PubMed] [Google Scholar]
- Mady MA, Kossoff EH, McGregor AL, Wheless JW, Pyzik PL, Freeman JM. The ketogenic diet: adolescents can do it, too. Epilepsia. 2003;44:847–851. doi: 10.1046/j.1528-1157.2003.57002.x. [DOI] [PubMed] [Google Scholar]
- Maj R, Fariello RG, Pevarello P, Varasi M, McArthur RA, Salvati P. Anticonvulsant activity of PNU-151774E in the amygdala kindled model of complex partial seizures. Epilepsia. 1999;40:1523–1528. doi: 10.1111/j.1528-1157.1999.tb02035.x. [DOI] [PubMed] [Google Scholar]
- Maj R, Fariello RG, Ukmar G, Varasi M, Pevarello P, McArthur RA, Salvati P. PNU-151774E protects against kainate-induced status epilepticus and hippocampal lesions in the rat. Eur J Pharmacol. 1998;359:27–32. doi: 10.1016/s0014-2999(98)00554-8. [DOI] [PubMed] [Google Scholar]
- Manev H, Uz T, Kharlamov A, Joo JY. Increased brain damage after stroke or excitotoxic seizures in melatonin-deficient rats. Faseb J. 1996;10:1546–1551. doi: 10.1096/fasebj.10.13.8940301. [DOI] [PubMed] [Google Scholar]
- Manford M, Hart YM, Sander JW, Shorvon SD. National General Practice Study of Epilepsy (NGPSE): partial seizure patterns in a general population. Neurology. 1992;42:1911–1917. doi: 10.1212/wnl.42.10.1911. [DOI] [PubMed] [Google Scholar]
- Manna SK, Mukhopadhyay A, Aggarwal BB. Resveratrol suppresses TNF-induced activation of nuclear transcription factors NF-kappa B, activator protein-1, and apoptosis: potential role of reactive oxygen intermediates and lipid peroxidation. J Immunol. 2000;164:6509–6519. doi: 10.4049/jimmunol.164.12.6509. [DOI] [PubMed] [Google Scholar]
- Marciani MG, Santone G, Sancesario G, Massa R, Stanzione P, Bernardi G. Protective effect of clonazepam on ischemic brain damage induced by 10-minute bilateral carotid occlusion in Mongolian gerbils. Functional Neurol. 1993;8:115–120. [PubMed] [Google Scholar]
- Marsh EB, Freeman JM, Kossoff EH, Vining EP, Rubenstein JE, Pyzik PL, Hemingway C. The outcome of children with intractable seizures: a 3- to 6-year follow-up of 67 children who remained on the ketogenic diet less than one year. Epilepsia. 2006a;47:425–430. doi: 10.1111/j.1528-1167.2006.00439.x. [DOI] [PubMed] [Google Scholar]
- Marsh ED, Brooks-Kayal AR, Porter BE. Seizures and antiepileptic drugs: does exposure alter normal brain development? Epilepsia. 2006b;47:1999–2010. doi: 10.1111/j.1528-1167.2006.00894.x. [DOI] [PubMed] [Google Scholar]
- Marti HH, Gassmann M, Wenger RH, Kvietikova I, Morganti-Kossmann MC, Kossmann T, Trentz O, Bauer C. Detection of erythropoietin in human liquor: intrinsic erythropoietin production in the brain. Kidney Int. 1997;51:416–418. doi: 10.1038/ki.1997.55. [DOI] [PubMed] [Google Scholar]
- Martin D, Miller G, Rosendahl M, Russell DA. Potent inhibitory effects of glial derived neurotrophic factor against kainic acid mediated seizures in the rat. Brain Res. 1995;683:172–178. doi: 10.1016/0006-8993(95)00369-2. [DOI] [PubMed] [Google Scholar]
- Martin V, Sainz RM, Antolin I, Mayo JC, Herrera F, Rodriguez C. Several antioxidant pathways are involved in astrocyte protection by melatonin. J Pineal Res. 2002;33:204–212. doi: 10.1034/j.1600-079x.2002.02113.x. [DOI] [PubMed] [Google Scholar]
- Masukawa LM, Higashima M, Kim JH, Spencer DD. Epileptiform discharges evoked in hippocampal brain slices from epileptic patients. Brain Res. 1989;493:168–174. doi: 10.1016/0006-8993(89)91012-3. [DOI] [PubMed] [Google Scholar]
- Masukawa LM, Uruno K, Sperling M, O'Connor MJ, Burdette LJ. The functional relationship between antidromically evoked field responses of the dentate gyrus and mossy fiber reorganization in temporal lobe epileptic patients. Brain Res. 1992;579:119–127. doi: 10.1016/0006-8993(92)90750-4. [DOI] [PubMed] [Google Scholar]
- Mathern GW, Cifuentes F, Leite JP, Pretorius JK, Babb TL. Hippocampal EEG excitability and chronic spontaneous seizures are associated with aberrant synaptic reorganization in the rat intrahippocampal kainate model. Electroencephal and Clin Neurophysiol. 1993;87:326–339. doi: 10.1016/0013-4694(93)90186-y. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Kumar KN, Wang H, Cheng B, Michaelis EK. Basic FGF regulates the expression of a functional 71 kDa NMDA receptor protein that mediates calcium influx and neurotoxicity in hippocampal neurons. J Neurosci. 1993;13:4575–4588. doi: 10.1523/JNEUROSCI.13-11-04575.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo F. Lamotrigine. Epilepsia. 1999;40 Suppl 5:S30–S36. doi: 10.1111/j.1528-1157.1999.tb00917.x. [DOI] [PubMed] [Google Scholar]
- Matsuo F, Gay P, Madsen J, Tolman KG, Rollins DE, Risner ME, Lai AA. Lamotrigine high-dose tolerability and safety in patients with epilepsy: a double-blind, placebo-controlled, eleven-week study. Epilepsia. 1996;37:857–862. doi: 10.1111/j.1528-1157.1996.tb00038.x. [DOI] [PubMed] [Google Scholar]
- Matsuzaki H, Tamatani M, Yamaguchi A, Namikawa K, Kiyama H, Vitek MP, Mitsuda N, Tohyama M. Vascular endothelial growth factor rescues hippocampal neurons from glutamate-induced toxicity: signal transduction cascades. Faseb J. 2001;15:1218–1220. [PubMed] [Google Scholar]
- Maurice T, Phan VL, Urani A, Kamei H, Noda Y, Nabeshima T. Neuroactive neurosteroids as endogenous effectors for the sigma1 (sigma1) receptor: pharmacological evidence and therapeutic opportunities. Jap J Pharmacol. 1999;81:125–155. doi: 10.1254/jjp.81.125. [DOI] [PubMed] [Google Scholar]
- Mazarati AM, Baldwin RA, Sofia RD, Wasterain CG. Felbamate in experimental model of status epilepticus. Epilepsia. 2000;41:123–127. doi: 10.1111/j.1528-1157.2000.tb00130.x. [DOI] [PubMed] [Google Scholar]
- Mazarati AM, Sofia RD, Wasterlain CG. Anticonvulsant and antiepileptogenic effects of fluorofelbamate in experimental status epilepticus. Seizure. 2002;11:423–430. doi: 10.1053/seiz.2002.0677. [DOI] [PubMed] [Google Scholar]
- McCloskey DP, Hintz TM, Pierce JP, Scharfman HE. Stereological methods reveal the robust size and stability of ectopic hilar granule cells after pilocarpine-induced status epilepticus in the adult rat. Eur J Neurosci. 2006;24:2203–2210. doi: 10.1111/j.1460-9568.2006.05101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCloskey DP, Croll SD, Scharfman HE. Depression of synaptic transmission by vascular endothelial growth factor in adult rat hippocampus and evidence for increased efficacy after chronic seizures. J Neurosci. 2005;25:8889–8897. doi: 10.1523/JNEUROSCI.2577-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh TK, Saatman KE, Raghupathi R, Graham DI, Smith DH, Lee VM, Trojanowski JQ. The Dorothy Russell Memorial Lecture. The molecular and cellular sequelae of experimental traumatic brain injury: pathogenetic mechanisms. Neuropathol Aapplied Neurobiol. 1998;24:251–267. doi: 10.1046/j.1365-2990.1998.00121.x. [DOI] [PubMed] [Google Scholar]
- McKeown MJ, McNamara JO. When do epileptic seizures really begin? Neuron. 2001;30:1–3. doi: 10.1016/s0896-6273(01)00253-7. [DOI] [PubMed] [Google Scholar]
- McNamara JO. Pharmacotherapy of the Epilepsies. In: Brunton L, Laszo J, Parker K, editors. Goodman and Gilman’s The Pharmacological Basis of Therapeutics. NY: The McGraw-Hill Medical Publishing Division; 2006. pp. 501–525. [Google Scholar]
- Mello LE, Cavalheiro EA, Tan AM, Kupfer WR, Pretorius JK, Babb TL, Finch DM. Circuit mechanisms of seizures in the pilocarpine model of chronic epilepsy: cell loss and mossy fiber sprouting. Epilepsia. 1993;34:985–995. doi: 10.1111/j.1528-1157.1993.tb02123.x. [DOI] [PubMed] [Google Scholar]
- Mellon SH, Griffin LD, Compagnone NA. Biosynthesis and action of neurosteroids. Brain Res Brain Res Rev. 2001;37:3–12. doi: 10.1016/s0165-0173(01)00109-6. [DOI] [PubMed] [Google Scholar]
- Merlio JP, Ernfors P, Jaber M, Persson H. Molecular cloning of rat trkC and distribution of cells expressing messenger RNAs for members of the trk family in the rat central nervous system. Neuroscience. 1992;51:513–532. doi: 10.1016/0306-4522(92)90292-a. [DOI] [PubMed] [Google Scholar]
- Mevissen M, Ebert U. Anticonvulsant effects of melatonin in amygdala-kindled rats. Neurosci Lett. 1998;257:13–16. doi: 10.1016/s0304-3940(98)00790-3. [DOI] [PubMed] [Google Scholar]
- Mhyre AJ, Dorsa DM. Estrogen activates rapid signaling in the brain: role of estrogen receptor alpha and estrogen receptor beta in neurons and glia. Neuroscience. 2006;138:851–858. doi: 10.1016/j.neuroscience.2005.10.019. [DOI] [PubMed] [Google Scholar]
- Milgram NW, Yearwood T, Khurgel M, Ivy GO, Racine R. Changes in inhibitory processes in the hippocampus following recurrent seizures induced by systemic administration of kainic acid. Brain Res. 1991;551:236–246. doi: 10.1016/0006-8993(91)90938-r. [DOI] [PubMed] [Google Scholar]
- Millichap JG, Jones JD. Acid-Base, Electrolyte, and Amino-Acid Metabolism in Children with Petit Mal. Etiologic Significance and Modification by Anticonvulsant Drugs and the Ketogenic Diet. Epilepsia. 1964;90:239–255. doi: 10.1111/j.1528-1157.1964.tb03331.x. [DOI] [PubMed] [Google Scholar]
- Millichap JG, Jones JD, Rudis BP. Mechanism of Anticonvulsant Action of Ketogenic Diet. Studies in Animals with Experimental Seizures and in Children with Petit Mal Epilepsy. American journal of diseases of children. 1964;107:593–604. [PubMed] [Google Scholar]
- Mohanan PV, Yamamoto HA. Preventive effect of melatonin against brain mitochondria DNA damage, lipid peroxidation and seizures induced by kainic acid. Toxicol Lett. 2002;129:99–105. doi: 10.1016/s0378-4274(01)00475-1. [DOI] [PubMed] [Google Scholar]
- Molina-Carballo A, Acuna-Castroviejo D, Rodriguez-Cabezas T, Munoz-Hoyos A. Effects of febrile and epileptic convulsions on daily variations in plasma melatonin concentration in children. J Pineal Res. 1994;16:1–9. doi: 10.1111/j.1600-079x.1994.tb00075.x. [DOI] [PubMed] [Google Scholar]
- Molina-Carballo A, Munoz-Hoyos A, Reiter RJ, Sanchez-Forte M, Moreno-Madrid F, Rufo-Campos M, Molina-Font JA, Acuna-Castroviejo D. Utility of high doses of melatonin as adjunctive anticonvulsant therapy in a child with severe myoclonic epilepsy: two years' experience. J Pineal Res. 1997;23:97–105. doi: 10.1111/j.1600-079x.1997.tb00341.x. [DOI] [PubMed] [Google Scholar]
- Monje ML, Mizumatsu S, Fike JR, Palmer TD. Irradiation induces neural precursor-cell dysfunction. Nature Med. 2002;8:955–962. doi: 10.1038/nm749. [DOI] [PubMed] [Google Scholar]
- Monje ML, Palmer T. Radiation injury and neurogenesis. Curr Opinion Neurol. 2003;16:129–134. doi: 10.1097/01.wco.0000063772.81810.b7. [DOI] [PubMed] [Google Scholar]
- Monnet FP, Mahe V, Robel P, Baulieu EE. Neurosteroids, via sigma receptors, modulate the [3H]norepinephrine release evoked by N-methyl-D-aspartate in the rat hippocampus. Proc Nat Acad Sci. 1995;92:3774–3778. doi: 10.1073/pnas.92.9.3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita E, Masuda S, Nagao M, Yasuda Y, Sasaki R. Erythropoietin receptor is expressed in rat hippocampal and cerebral cortical neurons, and erythropoietin prevents in vitro glutamate-induced neuronal death. Neuroscience. 1997;76:105–116. doi: 10.1016/s0306-4522(96)00306-5. [DOI] [PubMed] [Google Scholar]
- Mudrick LA, Baimbridge KG. Hippocampal neurons transplanted into ischemically lesioned hippocampus: anatomical assessment of survival, maturation and integration. Experimental Brain Res. 1991;86:233–247. doi: 10.1007/BF00228948. [DOI] [PubMed] [Google Scholar]
- Muller-Schwarze AB, Tandon P, Liu Z, Yang Y, Holmes GL, Stafstrom CE. Ketogenic diet reduces spontaneous seizures and mossy fiber sprouting in the kainic acid model. NeuroReport. 1999;10:1517–1522. doi: 10.1097/00001756-199905140-00023. [DOI] [PubMed] [Google Scholar]
- Nadler JV, Perry BW, Cotman CW. Selective reinnervation of hippocampal area CA1 and the fascia dentata after destruction of CA3-CA4 afferents with kainic acid. Brain research. 1980a;182:1–9. doi: 10.1016/0006-8993(80)90825-2. [DOI] [PubMed] [Google Scholar]
- Nadler JV, Perry BW, Gentry C, Cotman CW. Degeneration of hippocampal CA3 pyramidal cells induced by intraventricular kainic acid. J Comp Neurol. 1980b;192:333–359. doi: 10.1002/cne.901920209. [DOI] [PubMed] [Google Scholar]
- Nagerl UV, Mody I, Jeub M, Lie AA, Elger CE, Beck H. Surviving granule cells of the sclerotic human hippocampus have reduced Ca(2+) influx because of a loss of calbindin-D(28k) in temporal lobe epilepsy. J Neurosci. 2000;20:1831–1836. doi: 10.1523/JNEUROSCI.20-05-01831.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa E, Aimi Y, Yasuhara O, Tooyama I, Shimada M, McGeer PL, Kimura H. Enhancement of progenitor cell division in the dentate gyrus triggered by initial limbic seizures in rat models of epilepsy. Epilepsia. 2000;41:10–18. doi: 10.1111/j.1528-1157.2000.tb01498.x. [DOI] [PubMed] [Google Scholar]
- Nakamura NH, Rosell DR, Akama KT, McEwen BS. Estrogen and ovariectomy regulate mRNA and protein of glutamic acid decarboxylases and cation-chloride cotransporters in the adult rat hippocampus. Neuroendocrinol. 2004;80:308–323. doi: 10.1159/000083657. [DOI] [PubMed] [Google Scholar]
- Nakazawa M, Kodama S, Matsuo T. Effects of ketogenic diet on electroconvulsive threshold and brain contents of adenosine nucleotides. Brain Dev. 1983;5:375–380. doi: 10.1016/s0387-7604(83)80042-4. [DOI] [PubMed] [Google Scholar]
- Nanobashvili A, Airaksinen MS, Kokaia M, Rossi J, Asztely F, Olofsdotter K, Mohapel P, Saarma M, Lindvall O, Kokaia Z. Development and persistence of kindling epilepsy are impaired in mice lacking glial cell line-derived neurotrophic factor family receptor alpha 2. Proc Natl Acad Sci. 2000;97:12312–12317. doi: 10.1073/pnas.97.22.12312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolini G, Rigolio R, Miloso M, Bertelli AA, Tredici G. Anti-apoptotic effect of trans-resveratrol on paclitaxel-induced apoptosis in the human neuroblastoma SH-SY5Y cell line. Neurosc Lett. 2001;302:41–44. doi: 10.1016/s0304-3940(01)01654-8. [DOI] [PubMed] [Google Scholar]
- Niebauer M, Gruenthal M. Topiramate reduces neuronal injury after experimental status epilepticus. Brain Res. 1999;837:263–269. doi: 10.1016/s0006-8993(99)01615-7. [DOI] [PubMed] [Google Scholar]
- Niquet J, Liu H, Wasterlain CG. Programmed neuronal necrosis and status epilepticus. Epilepsia. 2005;46 Suppl 5:43–48. doi: 10.1111/j.1528-1167.2005.01025.x. [DOI] [PubMed] [Google Scholar]
- Niquet J, Wasterlain CG. Bim, Bad, and Bax: a deadly combination in epileptic seizures. The J Clinical Invest. 2004;113:960–962. doi: 10.1172/JCI21478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh HS, Kang SS, Kim DW, Kim YH, Park CH, Han JY, Cho GJ, Choi WS. Ketogenic diet increases calbindin-D28k in the hippocampi of male ICR mice with kainic acid seizures. Epilepsy Res. 2005a;65:153–159. doi: 10.1016/j.eplepsyres.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Noh HS, Kim DW, Kang SS, Cho GJ, Choi WS. Ketogenic diet prevents clusterin accumulation induced by kainic acid in the hippocampus of male ICR mice. Brain Res. 2005b;1042:114–118. doi: 10.1016/j.brainres.2005.01.097. [DOI] [PubMed] [Google Scholar]
- Noh HS, Kim YS, Kim YH, Han JY, Park CH, Kang AK, Shin HS, Kang SS, Cho GJ, Choi WS. Ketogenic diet protects the hippocampus from kainic acid toxicity by inhibiting the dissociation of bad from 14-3-3. J Neurosci Res. 2006;84:1829–1836. doi: 10.1002/jnr.21057. [DOI] [PubMed] [Google Scholar]
- Nordli DR, Jr., Kuroda MM, Carroll J, Koenigsberger DY, Hirsch LJ, Bruner HJ, Seidel WT, De Vivo DC. Experience with the ketogenic diet in infants. Pediatrics. 2001;108:129–133. doi: 10.1542/peds.108.1.129. [DOI] [PubMed] [Google Scholar]
- O'Brien CP. Benzodiazepine use, abuse, and dependence. J Clin Psych. 2005;66 Suppl 2:28–33. [PubMed] [Google Scholar]
- Oddo S, Solis P, Consalvo D, Giagante B, Silva W, D'Alessio L, Centurion E, Saidon P, Kochen S. Mesial temporal lobe epilepsy and hippocampal sclerosis: cognitive function assessment in Hispanic patients. Epilepsy Behav. 2003;4:717–722. doi: 10.1016/j.yebeh.2003.09.008. [DOI] [PubMed] [Google Scholar]
- Okazaki MM, Evenson DA, Nadler JV. Hippocampal mossy fiber sprouting and synapse formation after status epilepticus in rats: visualization after retrograde transport of biocytin. The J Comp Neurol. 1995;352:515–534. doi: 10.1002/cne.903520404. [DOI] [PubMed] [Google Scholar]
- Ornitz DM, Itoh N. Fibroblast growth factors. Genome biology. 2001;2:S3005. doi: 10.1186/gb-2001-2-3-reviews3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palma E, Torchia G, Limatola C, Trettel F, Arcella A, Cantore G, Di Gennaro G, Manfredi M, Esposito V, Quarato PP, Miledi R, Eusebi F. BDNF modulates GABAA receptors microtransplanted from the human epileptic brain to Xenopus oocytes. Proc Nat Acad Sci. 2005;102:1667–1672. doi: 10.1073/pnas.0409442102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan JW, Bebin EM, Chu WJ, Hetherington HP. Ketosis and epilepsy: 31P spectroscopic imaging at 4.1 T. Epilepsia. 1999;40:703–707. doi: 10.1111/j.1528-1157.1999.tb00766.x. [DOI] [PubMed] [Google Scholar]
- Paratcha G, Ledda F, Ibanez CF. The neural cell adhesion molecule NCAM is an alternative signaling receptor for GDNF family ligands. Cell. 2003;113:867–879. doi: 10.1016/s0092-8674(03)00435-5. [DOI] [PubMed] [Google Scholar]
- Parent JM. The role of seizure-induced neurogenesis in epileptogenesis and brain repair. Epilepsy Res. 2002;50:179–189. doi: 10.1016/s0920-1211(02)00078-5. [DOI] [PubMed] [Google Scholar]
- Parent JM. Injury-induced neurogenesis in the adult mammalian brain. Neuroscientist. 2003;9:261–272. doi: 10.1177/1073858403252680. [DOI] [PubMed] [Google Scholar]
- Parent JM, Lowenstein DH. Mossy fiber reorganization in the epileptic hippocampus. Curr Opinion Neurol. 1997;10:103–109. doi: 10.1097/00019052-199704000-00006. [DOI] [PubMed] [Google Scholar]
- Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, Lowenstein DH. Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci. 1997;17:3727–3738. doi: 10.1523/JNEUROSCI.17-10-03727.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AB, de Graaf RA, Mason GF, Kanamatsu T, Rothman DL, Shulman RG, Behar KL. Glutamatergic neurotransmission and neuronal glucose oxidation are coupled during intense neuronal activation. J Cereb Blood Flow Metab. 2004;24:972–985. doi: 10.1097/01.WCB.0000126234.16188.71. [DOI] [PubMed] [Google Scholar]
- Pena F, Tapia R. Seizures and neurodegeneration induced by 4-aminopyridine in rat hippocampus in vivo: role of glutamate- and GABA-mediated neurotransmission and of ion channels. Neuroscience. 2000;101:547–561. doi: 10.1016/s0306-4522(00)00400-0. [DOI] [PubMed] [Google Scholar]
- Perez Y, Morin F, Beaulieu C, Lacaille JC. Axonal sprouting of CA1 pyramidal cells in hyperexcitable hippocampal slices of kainate-treated rats. Eur J Neurosci. 1996;8:736–748. doi: 10.1111/j.1460-9568.1996.tb01259.x. [DOI] [PubMed] [Google Scholar]
- Pirttila TJ, Manninen A, Jutila L, Nissinen J, Kalviainen R, Vapalahti M, Immonen A, Paljarvi L, Karkola K, Alafuzoff I, Mervaala E, Pitkanen A. Cystatin C expression is associated with granule cell dispersion in epilepsy. Ann Neurol. 2005;58:211–223. doi: 10.1002/ana.20545. [DOI] [PubMed] [Google Scholar]
- Pitkanen A. Drug-mediated neuroprotection and antiepileptogenesis: animal data. Neurology. 2002;59:S27–S33. doi: 10.1212/wnl.59.9_suppl_5.s27. [DOI] [PubMed] [Google Scholar]
- Pitkanen A, Nissinen J, Jolkkonen E, Tuunanen J, Halonen T. Effects of vigabatrin treatment on status epilepticus-induced neuronal damage and mossy fiber sprouting in the rat hippocampus. Epilepsy Res. 1999;33:67–85. doi: 10.1016/s0920-1211(98)00074-6. [DOI] [PubMed] [Google Scholar]
- Pitkanen A, Kharatishvili I, Narkilahti S, Lukasiuk K, Nissinen J. Administration of diazepam during status epilepticus reduces development and severity of epilepsy in rat. Epilepsy Res. 2005;63:27–42. doi: 10.1016/j.eplepsyres.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Plassart-Schiess E, Baulieu EE. Neurosteroids: recent findings. Brain Res Brain Res Rev. 2001;37:133–140. doi: 10.1016/s0165-0173(01)00113-8. [DOI] [PubMed] [Google Scholar]
- Porter R, Meldrum B, Macdonald R, Dicter M, Dam M, Treiman D, Chadwick D, Gram L, Pedley T. Antiepileptic drugs. In: Engel J, Pedley T, editors. Epilepsy: A Comprehensive Textbook. NY: Lippincott-Raven; 1997. pp. 1379–1670. [Google Scholar]
- Postma T, Krupp E, Li XL, Post RM, Weiss SR. Lamotrigine treatment during amygdala-kindled seizure development fails to inhibit seizures and diminishes subsequent anticonvulsant efficacy. Epilepsia. 2000;41:1514–1521. doi: 10.1111/j.1499-1654.2000.001514.x. [DOI] [PubMed] [Google Scholar]
- Prince D. Neurophysiology of epilepsy. Annu. Rev. Neurosci. 1978;1:395–415. doi: 10.1146/annurev.ne.01.030178.002143. [DOI] [PubMed] [Google Scholar]
- Proescholdt MA, Heiss JD, Walbridge S, Muhlhauser J, Capogrossi MC, Oldfield EH, Merrill MJ. Vascular endothelial growth factor (VEGF) modulates vascular permeability and inflammation in rat brain. J Neuropathol Exp Neurol. 1999;58:613–627. doi: 10.1097/00005072-199906000-00006. [DOI] [PubMed] [Google Scholar]
- Pugliese AM, Corradetti R. Effects of the antiepileptic drug felbamate on long-term potentiation in the CA1 region of rat hippocampal slices. Neuroscience letters. 1996;215:21–24. doi: 10.1016/s0304-3940(96)12948-7. [DOI] [PubMed] [Google Scholar]
- Pugliese AM, Passani MB, Pepeu G, Corradetti R. Felbamate decreases synaptic transmission in the CA1 region of rat hippocampal slices. J Pharmacol Exp Ther. 1996;279:1100–1108. [PubMed] [Google Scholar]
- Rao M, Shetty A. Fetal hippocampal cell transplantation after status epilepticus suppress the development of spontaneous motor seizures in a rat model of temporal lobe epilepsy. Soc Neurosci Abstr. 2004:454.16. [Google Scholar]
- Rao MS, Hattiangady B, Reddy DS, Shetty AK. Hippocampal neurodegeneration, spontaneous seizures, and mossy fiber sprouting in the F344 rat model of temporal lobe epilepsy. J Neurosci Res. 2006;83:1088–1105. doi: 10.1002/jnr.20802. [DOI] [PubMed] [Google Scholar]
- Raol YH, Zhang G, Lund IV, Porter BE, Maronski MA, Brooks-Kayal AR. Increased GABA(A)-receptor alpha1-subunit expression in hippocampal dentate gyrus after early-life status epilepticus. Epilepsia. 2006a;47:1665–1673. doi: 10.1111/j.1528-1167.2006.00640.x. [DOI] [PubMed] [Google Scholar]
- Raol YH, Lund IV, Bandyopadhyay S, Zhang G, Roberts DS, Wolfe JH, Russek SJ, Brooks-Kayal AR. Enhancing GABA(A) receptor alpha 1 subunit levels in hippocampal dentate gyrus inhibits epilepsy development in an animal model of temporal lobe epilepsy. J Neurosci. 2006b;26:11342–11346. doi: 10.1523/JNEUROSCI.3329-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy DS. Role of neurosteroids in catamenial epilepsy. Epilepsy Res. 2004;62:99–118. doi: 10.1016/j.eplepsyres.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Reibel S, Andre V, Chassagnon S, Andre G, Marescaux C, Nehlig A, Depaulis A. Neuroprotective effects of chronic estradiol benzoate treatment on hippocampal cell loss induced by status epilepticus in the female rat. Neurosci Lett. 2000;281:79–82. doi: 10.1016/s0304-3940(00)00784-9. [DOI] [PubMed] [Google Scholar]
- Reijs R, Aldenkamp AP, De Krom M. Mood effects of antiepileptic drugs. Epilepsy Behav. 2004;5 Suppl 1:S66–S76. doi: 10.1016/j.yebeh.2003.11.009. [DOI] [PubMed] [Google Scholar]
- Rho JM, Anderson GD, Donevan SD, White HS. Acetoacetate, acetone, and dibenzylamine (a contaminant in l-(+)-beta-hydroxybutyrate) exhibit direct anticonvulsant actions in vivo. Epilepsia. 2002;43:358–361. doi: 10.1046/j.1528-1157.2002.47901.x. [DOI] [PubMed] [Google Scholar]
- Rho JM, Kim DW, Robbins CA, Anderson GD, Schwartzkroin PA. Age-dependent differences in flurothyl seizure sensitivity in mice treated with a ketogenic diet. Epilepsy Res. 1999;37:233–240. doi: 10.1016/s0920-1211(99)00068-6. [DOI] [PubMed] [Google Scholar]
- Rigoulot MA, Koning E, Ferrandon A, Nehlig A. Neuroprotective properties of topiramate in the lithium-pilocarpine model of epilepsy. J Pharmacol Exp Ther. 2004;308:787–795. doi: 10.1124/jpet.103.057091. [DOI] [PubMed] [Google Scholar]
- Rocamora N, Palacios JM, Mengod G. Limbic seizures induce a differential regulation of the expression of nerve growth factor, brain-derived neurotrophic factor and neurotrophin-3, in the rat hippocampus. Mol Brain Res. 1992;13:27–33. doi: 10.1016/0169-328x(92)90041-9. [DOI] [PubMed] [Google Scholar]
- Rogawski MA. Progesterone, neurosteroids, and the hormonal basis of catamenial epilepsy. Annals of Neurology. 2003;53:288–291. doi: 10.1002/ana.10534. [DOI] [PubMed] [Google Scholar]
- Rogawski MA, Loscher W. The neurobiology of antiepileptic drugs. Nature Rev. 2004;5:553–564. doi: 10.1038/nrn1430. [DOI] [PubMed] [Google Scholar]
- Rosenstein JM, Mani N, Khaibullina A, Krum JM. Neurotrophic effects of vascular endothelial growth factor on organotypic cortical explants and primary cortical neurons. J Neurosci. 2003;23:11036–11044. doi: 10.1523/JNEUROSCI.23-35-11036.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenstein RE, Chuluyan HE, Diaz MC, Cardinali DP. GABA as a presumptive paracrine signal in the pineal gland. Evidence on an intrapineal GABAergic system. Brain Res Bull. 1990;25:339–344. doi: 10.1016/0361-9230(90)90080-j. [DOI] [PubMed] [Google Scholar]
- Rudick CN, Woolley CS. Estrogen regulates functional inhibition of hippocampal CA1 pyramidal cells in the adult female rat. J Neurosci. 2001;21:6532–6543. doi: 10.1523/JNEUROSCI.21-17-06532.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahuquillo J, Mena MP, Vilalta A, Poca MA. Moderate hypothermia in the management of severe traumatic brain injury: a good idea proved ineffective? Curr Pharmaceutical design. 2004;10:2193–2004. doi: 10.2174/1381612043384051. [DOI] [PubMed] [Google Scholar]
- Sakanaka M, Wen TC, Matsuda S, Masuda S, Morishita E, Nagao M, Sasaki R. In vivo evidence that erythropoietin protects neurons from ischemic damage. Proc Nat Acad Sci. 1998;95:4635–4640. doi: 10.1073/pnas.95.8.4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanberg PR, Borlongan CV, Koutouzis TK, Norgren RB, Jr., Cahill DW, Freeman TB. Human fetal striatal transplantation in an excitotoxic lesioned model of Huntington's disease. Ann NY Acad Sci. 1997;831:452–460. doi: 10.1111/j.1749-6632.1997.tb52217.x. [DOI] [PubMed] [Google Scholar]
- Sankar R. Neuroprotection in epilepsy: the Holy Grail of antiepileptogenic therapy. Epilepsy Behav. 2005;7 Suppl 3:S1–S2. doi: 10.1016/j.yebeh.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Sankar R, Holmes GL. Mechanisms of action for the commonly used antiepileptic drugs: relevance to antiepileptic drug-associated neurobehavioral adverse effects. Journal of child Neurol. 2004;19 Suppl 1:S6–S14. doi: 10.1177/088307380401900102. [DOI] [PubMed] [Google Scholar]
- SanMartin A, Borlongan CV. Cell transplantation: toward cell therapy. Cell Transplant. 2006;15:665–673. doi: 10.3727/000000006783981666. [DOI] [PubMed] [Google Scholar]
- Sato M, Maulik G, Bagchi D, Das DK. Myocardial protection by protykin, a novel extract of trans-resveratrol and emodin. Free Rad Res. 2000a;32:135–144. doi: 10.1080/10715760000300141. [DOI] [PubMed] [Google Scholar]
- Sato M, Ray PS, Maulik G, Maulik N, Engelman RM, Bertelli AA, Bertelli A, Das DK. Myocardial protection with red wine extract. J Cardiovasc Pharmacol. 2000b;35:263–268. doi: 10.1097/00005344-200002000-00013. [DOI] [PubMed] [Google Scholar]
- Schapel GJ, Beran RG, Kennaway DL, McLoughney J, Matthews CD. Melatonin response in active epilepsy. Epilepsia. 1995;36:75–78. doi: 10.1111/j.1528-1157.1995.tb01669.x. [DOI] [PubMed] [Google Scholar]
- Scharfman H. Does BDNF Contribute to Temporal Lobe Epilepsy? Epilepsy Curr. 2002a;2:92–94. doi: 10.1046/j.1535-7597.2002.t01-1-00033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE. Hyperexcitability in combined entorhinal/hippocampal slices of adult rat after exposure to brain-derived neurotrophic factor. J Neurophysiol. 1997;78:1082–1095. doi: 10.1152/jn.1997.78.2.1082. [DOI] [PubMed] [Google Scholar]
- Scharfman HE. Does the Development of a GABAergic Phenotype by Hippocampal Dentate Gyrus Granule Cells Contribute to Epileptogenesis. Epilepsy Curr. 2002b;2:63. doi: 10.1046/j.1535-7597.2002.00023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE. Insight into Molecular Mechanisms of Catamenial Epilepsy. Epilepsy Curr. 2003;3:86–88. doi: 10.1046/j.1535-7597.2003.03305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, Goodman JH, Du F, Schwarcz R. Chronic changes in synaptic responses of entorhinal and hippocampal neurons after amino-oxyacetic acid (AOAA)-induced entorhinal cortical neuron loss. J Neurophysiol. 1998;80:3031–3046. doi: 10.1152/jn.1998.80.6.3031. [DOI] [PubMed] [Google Scholar]
- Scharfman HE, Goodman JH, Sollas AL. Actions of brain-derived neurotrophic factor in slices from rats with spontaneous seizures and mossy fiber sprouting in the dentate gyrus. J Neurosci. 1999;19:5619–5631. doi: 10.1523/JNEUROSCI.19-13-05619.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, Goodman JH, Sollas AL. Granule-like neurons at the hilar/CA3 border after status epilepticus and their synchrony with area CA3 pyramidal cells: functional implications of seizure-induced neurogenesis. J Neurosci. 2000;20:6144–6158. doi: 10.1523/JNEUROSCI.20-16-06144.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, Maclusky NJ. Similarities between actions of estrogen and BDNF in the hippocampus: coincidence or clue? Trends Neurosci. 2005;28:79–85. doi: 10.1016/j.tins.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Scharfman HE, MacLusky NJ. The influence of gonadal hormones on neuronal excitability, seizures, and epilepsy in the female. Epilepsia. 2006;47:1423–1440. doi: 10.1111/j.1528-1167.2006.00672.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, Sollas AE, Berger RE, Goodman JH, Pierce JP. Perforant path activation of ectopic granule cells that are born after pilocarpine-induced seizures. Neuroscience. 2003;121:1107–1029. doi: 10.1016/s0306-4522(03)00481-0. [DOI] [PubMed] [Google Scholar]
- Schinder AF, Berninger B, Poo M. Postsynaptic target specificity of neurotrophin-induced presynaptic potentiation. Neuron. 2000;25:151–163. doi: 10.1016/s0896-6273(00)80879-x. [DOI] [PubMed] [Google Scholar]
- Schmitz B. Effects of antiepileptic drugs on mood and behavior. Epilepsia. 2006;47 Suppl 2:28–33. doi: 10.1111/j.1528-1167.2006.00684.x. [DOI] [PubMed] [Google Scholar]
- Schulze-Bonhage A, Hefft S, Oehl B. Termination of complex partial status epilepticus by intravenous levetiracetam - a case report. J Neurol Neurosurg Psych. 2007 doi: 10.1136/jnnp.2006.113951. PMID: 17353254. [DOI] [PubMed] [Google Scholar]
- Schwartz RD, Yu X, Katzman MR, Hayden-Hixson DM, Perry JM. Diazepam, given postischemia, protects selectively vulnerable neurons in the rat hippocampus and striatum. J Neurosci. 1995;15:529–539. doi: 10.1523/JNEUROSCI.15-01-00529.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott BW, Wojtowicz JM, Burnham WM. Neurogenesis in the dentate gyrus of the rat following electroconvulsive shock seizures. Exp Neurol. 2000;165:231–236. doi: 10.1006/exnr.2000.7458. [DOI] [PubMed] [Google Scholar]
- Selai C, Bannister D, Trimble M. Antiepileptic drugs and the regulation of mood and quality of life (QOL): the evidence from epilepsy. Epilepsia. 2005;46 Suppl 4:50–57. doi: 10.1111/j.1528-1167.2005.463010.x. [DOI] [PubMed] [Google Scholar]
- Sfaello I, Baud O, Arzimanoglou A, Gressens P. Topiramate prevents excitotoxic damage in the newborn rodent brain. Neurobiol Dis. 2005;20:837–848. doi: 10.1016/j.nbd.2005.05.019. [DOI] [PubMed] [Google Scholar]
- Shapiro LA, Korn MJ, Ribak CE. Newly generated dentate granule cells from epileptic rats exhibit elongated hilar basal dendrites that align along GFAP-immunolabeled processes. Neuroscience. 2005;136:823–831. doi: 10.1016/j.neuroscience.2005.03.059. [DOI] [PubMed] [Google Scholar]
- Shapiro LA, Ribak CE. Integration of newly born dentate granule cells into adult brains: hypotheses based on normal and epileptic rodents. Brain Res Rev. 2005;48:43–56. doi: 10.1016/j.brainresrev.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Shapiro LA, Ribak CE. Newly born dentate granule neurons after pilocarpine-induced epilepsy have hilar basal dendrites with immature synapses. Epilepsy Res. 2006;69:53–66. doi: 10.1016/j.eplepsyres.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Shetty AK. Entorhinal axons exhibit sprouting in CA1 subfield of the adult hippocampus in a rat model of temporal lobe epilepsy. Hippocampus. 2002;12:534–542. doi: 10.1002/hipo.10031. [DOI] [PubMed] [Google Scholar]
- Shetty AK. Progenitor cells from the CA3 region of the embryonic day 19 rat hippocampus generate region-specific neuronal phenotypes in vitro. Hippocampus. 2004;14:595–614. doi: 10.1002/hipo.10206. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Hattiangady B. Restoration of calbindin after fetal hippocampal CA3 cell grafting into the injured hippocampus in a rat model of temporal lobe epilepsy. Hippocampus. 2007a doi: 10.1002/hipo.20311. [Epub ahead of print] PMID: 17604349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty AK, Hattiangady B. Prospects of Stem Cell Therapy for Temporal Lobe Epilepsy. Stem Cells. 2007b doi: 10.1634/stemcells.2007-0313. [Epub ahead of print] PMID: 17600108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty AK, Rao MS, Hattiangady B, Zaman V, Shetty GA. Hippocampal neurotrophin levels after injury: Relationship to the age of the hippocampus at the time of injury. J Neurosci Res. 2004;78:520–532. doi: 10.1002/jnr.20302. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Turner DA. Enhanced cell survival in fetal hippocampal suspension transplants grafted to adult rat hippocampus following kainate lesions: a three-dimensional graft reconstruction study. Neuroscience. 1995a;67:561–582. doi: 10.1016/0306-4522(95)00025-e. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Turner DA. Intracerebroventricular kainic acid administration in adult rat alters hippocampal calbindin and non-phosphorylated neurofilament expression. J Comp Neurol. 1995b;363:581–599. doi: 10.1002/cne.903630406. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Turner DA. Development of fetal hippocampal grafts in intact and lesioned hippocampus. Prog Neurobiol. 1996;50:597–653. doi: 10.1016/s0301-0082(96)00048-2. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Turner DA. Development of long-distance efferent projections from fetal hippocampal grafts depends upon pathway specificity and graft location in kainate-lesioned adult hippocampus. Neuroscience. 1997a;76:1205–1219. doi: 10.1016/s0306-4522(96)00413-7. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Turner DA. Fetal hippocampal cells grafted to kainate-lesioned CA3 region of adult hippocampus suppress aberrant supragranular sprouting of host mossy fibers. Exp Neurol. 1997b;143:231–245. doi: 10.1006/exnr.1996.6363. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Turner DA. In vitro survival and differentiation of neurons derived from epidermal growth factor-responsive postnatal hippocampal stem cells: inducing effects of brain-derived neurotrophic factor. J Neurobiol. 1998;35:395–425. doi: 10.1002/(sici)1097-4695(19980615)35:4<395::aid-neu7>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Turner DA. Fetal hippocampal grafts containing CA3 cells restore host hippocampal glutamate decarboxylase-positive interneuron numbers in a rat model of temporal lobe epilepsy. J Neurosci. 2000;20:8788–8801. doi: 10.1523/JNEUROSCI.20-23-08788.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty AK, Turner DA. Glutamic acid decarboxylase-67-positive hippocampal interneurons undergo a permanent reduction in number following kainic acid-induced degeneration of ca3 pyramidal neurons. Exp Neurol. 2001;169:276–297. doi: 10.1006/exnr.2001.7668. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Zaman V, Hattiangady B. Repair of the injured adult hippocampus through graft-mediated modulation of the plasticity of the dentate gyrus in a rat model of temporal lobe epilepsy. J Neurosci. 2005;25:8391–8401. doi: 10.1523/JNEUROSCI.1538-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty AK, Zaman V, Shetty GA. Hippocampal neurotrophin levels in a kainate model of temporal lobe epilepsy: a lack of correlation between brain-derived neurotrophic factor content and progression of aberrant dentate mossy fiber sprouting. J Neurochem. 2003;87:147–159. doi: 10.1046/j.1471-4159.2003.01979.x. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Zaman V, Turner DA. Pattern of long-distance projections from fetal hippocampal field CA3 and CA1 cell grafts in lesioned CA3 of adult hippocampus follows intrinsic character of respective donor cells. Neuroscience. 2000;99:243–255. doi: 10.1016/s0306-4522(00)00178-0. [DOI] [PubMed] [Google Scholar]
- Shin HJ, Lee JY, Son E, Lee DH, Kim HJ, Kang SS, Cho GJ, Choi WS, Roh GS. Curcumin attenuates the kainic acid-induced hippocampal cell death in the mice. Neurosci Lett. 2007 doi: 10.1016/j.neulet.2007.01.060. PMID: 17300872. [DOI] [PubMed] [Google Scholar]
- Shors TJ, Miesegaes G, Beylin A, Zhao M, Rydel T, Gould E. Neurogenesis in the adult is involved in the formation of trace memories. Nature. 2001;410:372–376. doi: 10.1038/35066584. [DOI] [PubMed] [Google Scholar]
- Siddiqui AH, Joseph SA. CA3 axonal sprouting in kainate-induced chronic epilepsy. Brain Res. 2005;1066:129–146. doi: 10.1016/j.brainres.2005.10.066. [DOI] [PubMed] [Google Scholar]
- Sidhu RS, Del Bigio MR, Tuor UI, Seshia SS. Low-dose vigabatrin (gamma-vinyl GABA)-induced damage in the immature rat brain. Exp Neurol. 1997;144:400–405. doi: 10.1006/exnr.1997.6412. [DOI] [PubMed] [Google Scholar]
- Silva AP, Pinheiro PS, Carvalho AP, Carvalho CM, Jakobsen B, Zimmer J, Malva JO. Activation of neuropeptide Y receptors is neuroprotective against excitotoxicity in organotypic hippocampal slice cultures. Faseb J. 2003;17:1118–1120. doi: 10.1096/fj.02-0885fje. [DOI] [PubMed] [Google Scholar]
- Simonato M, Tongiorgi E, Kokaia M. Angels and demons: neurotrophic factors and epilepsy. Trends Pharamcol Sci. 2006;27:631–638. doi: 10.1016/j.tips.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Sinha K, Chaudhary G, Gupta YK. Protective effect of resveratrol against oxidative stress in middle cerebral artery occlusion model of stroke in rats. Life sciences. 2002;71:655–665. doi: 10.1016/s0024-3205(02)01691-0. [DOI] [PubMed] [Google Scholar]
- Siren AL, Ehrenreich H. Erythropoietin--a novel concept for neuroprotection. Eur Arch Psych ClinNeurosci. 2001;251:179–184. doi: 10.1007/s004060170038. [DOI] [PubMed] [Google Scholar]
- Sloviter RS. Permanently altered hippocampal structure, excitability, and inhibition after experimental status epilepticus in the rat: the "dormant basket cell" hypothesis and its possible relevance to temporal lobe epilepsy. Hippocampus. 1991;1:41–66. doi: 10.1002/hipo.450010106. [DOI] [PubMed] [Google Scholar]
- Sloviter RS. Status epilepticus-induced neuronal injury and network reorganization. Epilepsia. 1999;40 Suppl 1:S34–S39. doi: 10.1111/j.1528-1157.1999.tb00876.x. discussion S40-31. [DOI] [PubMed] [Google Scholar]
- Sloviter RS. The neurobiology of temporal lobe epilepsy: too much information, not enough knowledge. Comptes Rendus Biologies. 2005;328:143–153. doi: 10.1016/j.crvi.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Sloviter RS, Zappone CA, Harvey BD, Frotscher M. Kainic acid-induced recurrent mossy fiber innervation of dentate gyrus inhibitory interneurons: possible anatomical substrate of granule cell hyper-inhibition in chronically epileptic rats. J Comp Neurol. 2006;494:944–960. doi: 10.1002/cne.20850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares HD, Sinson GP, McIntosh TK. Fetal hippocampal transplants attenuate CA3 pyramidal cell death resulting from fluid percussion brain injury in the rat. J Neurotrauma. 1995;12:1059–1067. doi: 10.1089/neu.1995.12.1059. [DOI] [PubMed] [Google Scholar]
- Song HJ, Stevens CF, Gage FH. Neural stem cells from adult hippocampus develop essential properties of functional CNS neurons. Nature Neurosci. 2002;5:438–445. doi: 10.1038/nn844. [DOI] [PubMed] [Google Scholar]
- Sperk G, Wieser R, Widmann R, Singer EA. Kainic acid induced seizures: changes in somatostatin, substance P and neurotensin. Neuroscience. 1986;17:1117–1126. doi: 10.1016/0306-4522(86)90081-3. [DOI] [PubMed] [Google Scholar]
- Stafstrom CE, Moshe SL, Swann JW, Nehlig A, Jacobs MP, Schwartzkroin PA. Models of pediatric epilepsies: strategies and opportunities. Epilepsia. 2006;47:1407–1414. doi: 10.1111/j.1528-1167.2006.00674_1.x. [DOI] [PubMed] [Google Scholar]
- Stafstrom CE, Wang C, Jensen FE. Electrophysiological observations in hippocampal slices from rats treated with the ketogenic diet. Dev Neurosci. 1999;21:393–399. doi: 10.1159/000017389. [DOI] [PubMed] [Google Scholar]
- Stepien K, Tomaszewski M, Czuczwar SJ. Profile of anticonvulsant activity and neuroprotective effects of novel and potential antiepileptic drugs--an update. Pharmacol Rep. 2005;57:719–733. [PubMed] [Google Scholar]
- Stoffel-Wagner B. Neurosteroid metabolism in the human brain. Eur J Endocrinol. 2001;145:669–679. doi: 10.1530/eje.0.1450669. [DOI] [PubMed] [Google Scholar]
- Strine TW, Kobau R, Chapman DP, Thurman DJ, Price P, Balluz LS. Psychological distress, comorbidities, and health behaviors among U.S. adults with seizures: results from the 2002 National Health Interview Survey. Epilepsia. 2005;46:1133–1139. doi: 10.1111/j.1528-1167.2005.01605.x. [DOI] [PubMed] [Google Scholar]
- Su SW, Cilio MR, Sogawa Y, Silveira DC, Holmes GL, Stafstrom CE. Timing of ketogenic diet initiation in an experimental epilepsy model. Dev Brain Res. 2000;125:131–138. doi: 10.1016/s0165-3806(00)00130-9. [DOI] [PubMed] [Google Scholar]
- Suchomelova L, Baldwin RA, Kubova H, Thompson KW, Sankar R, Wasterlain CG. Treatment of experimental status epilepticus in immature rats: dissociation between anticonvulsant and antiepileptogenic effects. Pediatric Res. 2006;59:237–243. doi: 10.1203/01.pdr.0000196333.16608.30. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Rippy NA, Dorenbos K, Concepcion RC, Agarwal AK, Rho JM. The ketogenic diet increases mitochondrial uncoupling protein levels and activity. Ann Neurol. 2004a;55:576–580. doi: 10.1002/ana.20062. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Dragicevic NB, Deng JH, Bai Y, Dimayuga E, Ding Q, Chen Q, Bruce-Keller AJ, Keller JN. Proteasome inhibition alters neural mitochondrial homeostasis and mitochondria turnover. J Biol Chem. 2004b;279:20699–20707. doi: 10.1074/jbc.M313579200. [DOI] [PubMed] [Google Scholar]
- Sumanont Y, Murakami Y, Tohda M, Vajragupta O, Matsumoto K, Watanabe H. Evaluation of the nitric oxide radical scavenging activity of manganese complexes of curcumin and its derivative. Biol Pharmaceutical Bull. 2004;27:170–173. doi: 10.1248/bpb.27.170. [DOI] [PubMed] [Google Scholar]
- Sumanont Y, Murakami Y, Tohda M, Vajragupta O, Watanabe H, Matsumoto K. Prevention of kainic acid-induced changes in nitric oxide level and neuronal cell damage in the rat hippocampus by manganese complexes of curcumin and diacetylcurcumin. Life sciences. 2006;78:1884–1891. doi: 10.1016/j.lfs.2005.08.028. [DOI] [PubMed] [Google Scholar]
- Sun AY, Chen YM, James-Kracke M, Wixom P, Cheng Y. Ethanol-induced cell death by lipid peroxidation in PC12 cells. Neurochem Res. 1997;22:1187–1192. doi: 10.1023/a:1021968526696. [DOI] [PubMed] [Google Scholar]
- Sun AY, Ingelman-Sundberg M, Neve E, Matsumoto H, Nishitani Y, Minowa Y, Fukui Y, Bailey SM, Patel VB, Cunningham CC, Zima T, Fialova L, Mikulikova L, Popov P, Malbohan I, Janebova M, Nespor K, Sun GY. Ethanol and oxidative stress. Alcohol Clin Exp Res. 2001;25:237S–243S. doi: 10.1097/00000374-200105051-00038. [DOI] [PubMed] [Google Scholar]
- Sun AY, Simonyi A, Sun GY. The "French Paradox" and beyond: neuroprotective effects of polyphenols. Free Rad Biol Med. 2002;32:314–318. doi: 10.1016/s0891-5849(01)00803-6. [DOI] [PubMed] [Google Scholar]
- Sun AY, Sun GY. Ethanol and oxidative mechanisms in the brain. J Biomed Sci. 2001;8:37–43. doi: 10.1007/BF02255969. [DOI] [PubMed] [Google Scholar]
- Sutula T, Cavazos J, Golarai G. Alteration of long-lasting structural and functional effects of kainic acid in the hippocampus by brief treatment with phenobarbital. J Neurosci. 1992;12:4173–4187. doi: 10.1523/JNEUROSCI.12-11-04173.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutula TP, Hagen J, Pitkanen A. Do epileptic seizures damage the brain? Curr Opinion Neurol. 2003;16:189–195. doi: 10.1097/01.wco.0000063770.15877.bc. [DOI] [PubMed] [Google Scholar]
- Svensson B, Peters M, Konig HG, Poppe M, Levkau B, Rothermundt M, Arolt V, Kogel D, Prehn JH. Vascular endothelial growth factor protects cultured rat hippocampal neurons against hypoxic injury via an antiexcitotoxic, caspase-independent mechanism. J Cereb Blood Flow Metab. 2002;22:1170–1175. doi: 10.1097/01.wcb.0000037988.07114.98. [DOI] [PubMed] [Google Scholar]
- Swink TD, Vining EP, Freeman JM. The ketogenic diet: 1997. Adv Pediatrics. 1997;44:297-29. [PubMed] [Google Scholar]
- Szot P, Weinshenker D, Rho JM, Storey TW, Schwartzkroin PA. Norepinephrine is required for the anticonvulsant effect of the ketogenic diet. Brain Res Dev Brain Res. 2001;129:211–214. doi: 10.1016/s0165-3806(01)00213-9. [DOI] [PubMed] [Google Scholar]
- Szot P, White SS, Shen DD, Anderson GD. Valproic acid, but not lamotrigine, suppresses seizure-induced c-fos and c-Jun mRNA expression. Mol Brain Res. 2005;135:285–289. doi: 10.1016/j.molbrainres.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Saito H, Matsuki N. Inhibition of GABAA synaptic responses by brain-derived neurotrophic factor (BDNF) in rat hippocampus. J Neurosci. 1997;17:2959–2966. doi: 10.1523/JNEUROSCI.17-09-02959.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauck DL, Nadler JV. Evidence of functional mossy fiber sprouting in hippocampal formation of kainic acid-treated rats. J Neurosci. 1985;5:1016–1022. doi: 10.1523/JNEUROSCI.05-04-01016.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson KW, Suchomelova LM. Transplants of cells engineered to produce GABA suppress spontaneous seizures. Epilepsia. 2004;45:4–12. doi: 10.1111/j.0013-9580.2004.29503.x. [DOI] [PubMed] [Google Scholar]
- Thompson KW. Genetically engineered cells with regulatable GABA production can affect afterdischarges and behavioral seizures after transplantation into the dentate gyrus. Neuroscience. 2005;133:1029–1037. doi: 10.1016/j.neuroscience.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Tolner EA, Kloosterman F, Kalitzin SN, da Silva FH, Gorter JA. Physiological changes in chronic epileptic rats are prominent in superficial layers of the medial entorhinal area. Epilepsia. 2005;46 Suppl 5:72–81. doi: 10.1111/j.1528-1167.2005.01012.x. [DOI] [PubMed] [Google Scholar]
- Tredici G, Miloso M, Nicolini G, Galbiati S, Cavaletti G, Bertelli A. Resveratrol, map kinases and neuronal cells: might wine be a neuroprotectant? Drugs Under Experimental and Clinical Research. 1999;25:99–103. [PubMed] [Google Scholar]
- Trojnar MK, Malek R, Chroscinska M, Nowak S, Blaszczyk B, Czuczwar SJ. Neuroprotective effects of antiepileptic drugs. Pol J Pharmacol. 2002;54:557–566. [PubMed] [Google Scholar]
- Turner DA, Wheal HV. Excitatory synaptic potentials in kainic acid-denervated rat CA1 pyramidal neurons. J Neurosci. 1991;11:2786–2794. doi: 10.1523/JNEUROSCI.11-09-02786.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner DA, Shetty AK. Clinical prospects for neural grafting therapy for hippocampal lesions and epilepsy. Neurosurgery. 2003;52:632–644. doi: 10.1227/01.neu.0000047825.91205.e6. [DOI] [PubMed] [Google Scholar]
- Ure JA, Perassolo M. Update on the pathophysiology of the epilepsies. J Neurol Sci. 2000;177:1–17. doi: 10.1016/s0022-510x(00)00356-7. [DOI] [PubMed] [Google Scholar]
- Uzum G, Sarper Diler A, Bahcekapili N, Ziya Ziylan Y. Erythropoietin prevents the increase in blood-brain barrier permeability during pentylentetrazol induced seizures. Life sciences. 2006;78:2571–2576. doi: 10.1016/j.lfs.2005.10.027. [DOI] [PubMed] [Google Scholar]
- Vaisleib II, Buchhalter JR, Zupanc ML. Ketogenic diet: outpatient initiation, without fluid, or caloric restrictions. Pediatric Neurol. 2004;31:198–202. doi: 10.1016/j.pediatrneurol.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Van Den Pol AN, Obrietan K, Belousov A. Glutamate hyperexcitability and seizure-like activity throughout the brain and spinal cord upon relief from chronic glutamate receptor blockade in culture. Neuroscience. 1996;74:653–674. doi: 10.1016/0306-4522(96)00153-4. [DOI] [PubMed] [Google Scholar]
- Van der Zee CE, Rashid K, Le K, Moore KA, Stanisz J, Diamond J, Racine RJ, Fahnestock M. Intraventricular administration of antibodies to nerve growth factor retards kindling and blocks mossy fiber sprouting in adult rats. J Neurosci. 1995;15:5316–5323. doi: 10.1523/JNEUROSCI.15-07-05316.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veliskova J. Estrogens and epilepsy: why are we so excited? Neuroscientist. 2007;13:77–88. doi: 10.1177/1073858406295827. [DOI] [PubMed] [Google Scholar]
- Veliskova J, Velisek L, Galanopoulou AS, Sperber EF. Neuroprotective effects of estrogens on hippocampal cells in adult female rats after status epilepticus. Epilepsia. 2000;41 Suppl 6:S30–S35. doi: 10.1111/j.1528-1157.2000.tb01553.x. [DOI] [PubMed] [Google Scholar]
- Vieira de Almeida LM, Pineiro CC, Leite MC, Brolese G, Tramontina F, Feoli AM, Gottfried C, Goncalves CA. Resveratrol Increases Glutamate Uptake, Glutathione Content, and S100B Secretion in Cortical Astrocyte Cultures. Cell Mol Neurobiol. 2007 doi: 10.1007/s10571-007-9152-2. [Epub ahead of print] PMID: 17554623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vining EP. Clinical efficacy of the ketogenic diet. Epilepsy research. 1999;37:181–190. doi: 10.1016/s0920-1211(99)00070-4. [DOI] [PubMed] [Google Scholar]
- Vining EP, Freeman JM, Ballaban-Gil K, Camfield CS, Camfield PR, Holmes GL, Shinnar S, Shuman R, Trevathan E, Wheless JW. A multicenter study of the efficacy of the ketogenic diet. Arch Neurol. 1998;55:1433–1437. doi: 10.1001/archneur.55.11.1433. [DOI] [PubMed] [Google Scholar]
- Virgili M, Contestabile A. Partial neuroprotection of in vivo excitotoxic brain damage by chronic administration of the red wine antioxidant agent, trans-resveratrol in rats. Neuroscience letters. 2000;281:123–126. doi: 10.1016/s0304-3940(00)00820-x. [DOI] [PubMed] [Google Scholar]
- Wagner JP, Black IB, DiCicco-Bloom E. Stimulation of neonatal and adult brain neurogenesis by subcutaneous injection of basic fibroblast growth factor. J Neurosci. 1999;19:6006–6016. doi: 10.1523/JNEUROSCI.19-14-06006.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Q, Man HY, Liu F, Braunton J, Niznik HB, Pang SF, Brown GM, Wang YT. Differential modulation of GABAA receptor function by Mel1a and Mel1b receptors. Nature Neurosci. 1999;2:401–403. doi: 10.1038/8062. [DOI] [PubMed] [Google Scholar]
- Wang Q, Sun AY, Simonyi A, Jensen MD, Shelat PB, Rottinghaus GE, MacDonald RS, Miller DK, Lubahn DE, Weisman GA, Sun GY. Neuroprotective mechanisms of curcumin against cerebral ischemia-induced neuronal apoptosis and behavioral deficits. J Neurosci Res. 2005;82:138–148. doi: 10.1002/jnr.20610. [DOI] [PubMed] [Google Scholar]
- Wang Q, Xu J, Rottinghaus GE, Simonyi A, Lubahn D, Sun GY, Sun AY. Resveratrol protects against global cerebral ischemic injury in gerbils. Brain Res. 2002;958:439–447. doi: 10.1016/s0006-8993(02)03543-6. [DOI] [PubMed] [Google Scholar]
- Wang Q, Yu S, Simonyi A, Rottinghaus G, Sun GY, Sun AY. Resveratrol protects against neurotoxicity induced by kainic acid. Neurochem Res. 2004;29:2105–2112. doi: 10.1007/s11064-004-6883-z. [DOI] [PubMed] [Google Scholar]
- Wasterlain CG, Shirasaka Y. Seizures, brain damage and brain development. Brain Dev. 1994;16:279–295. doi: 10.1016/0387-7604(94)90025-6. [DOI] [PubMed] [Google Scholar]
- Weisenhorn DM, Roback J, Young AN, Wainer BH. Cellular aspects of trophic actions in the nervous system. Int Rev Cytol. 1999;189:177–265. doi: 10.1016/s0074-7696(08)61388-1. [DOI] [PubMed] [Google Scholar]
- Wheless JW. The ketogenic diet: an effective medical therapy with side effects. J Child Neurol. 2001;16:633–635. doi: 10.1177/088307380101600901. [DOI] [PubMed] [Google Scholar]
- Wheless JW, Baumgartner J, Ghanbari C. Vagus nerve stimulation and the ketogenic diet. Neurologic clinics. 2001;19:371–407. doi: 10.1016/s0733-8619(05)70023-2. [DOI] [PubMed] [Google Scholar]
- Williams S, Vachon P, Lacaille J-C. Monosynaptic GABA-mediated inhibitory postsynaptic potentials in CA1 pyramidal cells of hyperexcitable hippocampal slices from kainic acid treated rats. Neuroscience. 1993;52:541–554. doi: 10.1016/0306-4522(93)90404-4. [DOI] [PubMed] [Google Scholar]
- Williamson J, Mtchedlishvili Z, Kapur J. Characterization of the convulsant action of pregnenolone sulfate. Neuropharmacol. 2004;46:856–864. doi: 10.1016/j.neuropharm.2003.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willmore LJ, Hurd RW, Sypert GW. Epileptiform activity initiated by pial iontophoresis of ferrous and ferric chloride on rat cerebral cortex. Brain Res. 1978;152:406–410. doi: 10.1016/0006-8993(78)90273-1. [DOI] [PubMed] [Google Scholar]
- Wozny C, Gabriel S, Jandova K, Schulze K, Heinemann U, Behr J. Entorhinal cortex entrains epileptiform activity in CA1 in pilocarpine-treated rats. Neurobiol Dis. 2005;19:451–460. doi: 10.1016/j.nbd.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Wu A, Ying Z, Gomez-Pinilla F. Dietary curcumin counteracts the outcome of traumatic brain injury on oxidative stress, synaptic plasticity, and cognition. Exp Neurol. 2006;197:309–317. doi: 10.1016/j.expneurol.2005.09.004. [DOI] [PubMed] [Google Scholar]
- Wuarin JP, Dudek FE. Excitatory synaptic input to granule cells increases with time after kainate treatment. J Neurophysiol. 2001;85:1067–1077. doi: 10.1152/jn.2001.85.3.1067. [DOI] [PubMed] [Google Scholar]
- Xu B, Michalski B, Racine RJ, Fahnestock M. Continuous infusion of neurotrophin-3 triggers sprouting, decreases the levels of TrkA and TrkC, and inhibits epileptogenesis and activity-dependent axonal growth in adult rats. Neuroscience. 2002;115:1295–1308. doi: 10.1016/s0306-4522(02)00384-6. [DOI] [PubMed] [Google Scholar]
- Xu B, Michalski B, Racine RJ, Fahnestock M. The effects of brain-derived neurotrophic factor (BDNF) administration on kindling induction, Trk expression and seizure-related morphological changes. Neuroscience. 2004;126:521–531. doi: 10.1016/j.neuroscience.2004.03.044. [DOI] [PubMed] [Google Scholar]
- Yahyavi-Firouz-Abadi N, Tahsili-Fahadan P, Riazi K, Ghahremani MH, Dehpour AR. Involvement of nitric oxide pathway in the acute anticonvulsant effect of melatonin in mice. Epilepsy Res. 2006;68:103–113. doi: 10.1016/j.eplepsyres.2005.09.057. [DOI] [PubMed] [Google Scholar]
- Yan J, Zhang Y, Roder J, McDonald RJ. Aging effects on spatial tuning of hippocampal place cells. Exp. Brain Res. 2003;150:184–193. doi: 10.1007/s00221-003-1396-6. [DOI] [PubMed] [Google Scholar]
- Yancopoulos GD, Davis S, Gale NW, Rudge JS, Wiegand SJ, Holash J. Vascular-specific growth factors and blood vessel formation. Nature. 2000;407:242–248. doi: 10.1038/35025215. [DOI] [PubMed] [Google Scholar]
- Yasuhara T, Matsukawa N, Hara K, Yu G, Xu L, Maki M, Kim SU, Borlongan CV. Transplantation of human neural stem cells exerts neuroprotection in a rat model of Parkinson's disease. J Neurosci. 2006a:12497–12511. doi: 10.1523/JNEUROSCI.3719-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuhara T, Matsukawa N, Yu G, Xu L, Mays RW, Kovach J, Deans RJ, Hess DC, Carroll JE, Borlongan CV. Behavioral and histological characterization of intrahippocampal grafts of human bone marrow-derived multipotent progenitor cells in neonatal rats with hypoxic-ischemic injury. Cell Transplant. 2006b;15:231–238. doi: 10.3727/000000006783982034. [DOI] [PubMed] [Google Scholar]
- Yeoh JS, de Haan G. Fibroblast growth factors as regulators of stem cell self-renewal and aging. Mech. Ageing and Dev. 2007;128:17–24. doi: 10.1016/j.mad.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Yoo YM, Lee CJ, Lee U, Kim YJ. Neuroprotection of adenoviral-vector-mediated GDNF expression against kainic-acid-induced excitotoxicity in the rat hippocampus. Exp Neurol. 2006;200:407–417. doi: 10.1016/j.expneurol.2006.02.132. [DOI] [PubMed] [Google Scholar]
- Yudkoff M, Daikhin Y, Nissim I, Grunstein R, Nissim I. Effects of ketone bodies on astrocyte amino acid metabolism. J Neurochem. 1997;69:682–692. doi: 10.1046/j.1471-4159.1997.69020682.x. [DOI] [PubMed] [Google Scholar]
- Yudkoff M, Daikhin Y, Nissim I, Lazarow A, Nissim I. Brain amino acid metabolism and ketosis. J Neurosci Res. 2001a;66:272–281. doi: 10.1002/jnr.1221. [DOI] [PubMed] [Google Scholar]
- Yudkoff M, Daikhin Y, Nissim I, Lazarow A, Nissim I. Ketogenic diet, amino acid metabolism, and seizure control. J Neurosci Res. 2001b;66:931–940. doi: 10.1002/jnr.10083. [DOI] [PubMed] [Google Scholar]
- Zaman V, Shetty AK. Fetal hippocampal CA3 cell grafts transplanted to lesioned CA3 region of the adult hippocampus exhibit long-term survival in a rat model of temporal lobe epilepsy. Neurobiol Dis. 2001;8:942–952. doi: 10.1006/nbdi.2001.0440. [DOI] [PubMed] [Google Scholar]
- Zaman V, Shetty AK. Pretreatment of donor cells with FGF-2 enhances survival of fetal hippocampal CA3 cell transplants in the chronically lesioned young adult hippocampus. Exp Neurol. 2003;183:11–24. doi: 10.1016/s0014-4886(03)00167-5. [DOI] [PubMed] [Google Scholar]
- Zaman V, Turner DA, Shetty AK. Survival of grafted fetal neural cells in kainic acid lesioned CA3 region of adult hippocampus depends upon cell specificity. Exp Neurol. 2000;161:535–561. doi: 10.1006/exnr.1999.7304. [DOI] [PubMed] [Google Scholar]
- Zaman V, Turner DA, Shetty AK. Prolonged postlesion transplantation delay adversely influences survival of both homotopic and heterotopic fetal hippocampal cell grafts in Kainate-lesioned CA3 region of adult hippocampus. Cell transplant. 2001;10:41–52. [PubMed] [Google Scholar]
- Zamani MR, Levy WB, Desmond NL. Estradiol increases delayed, N-methyl-D-aspartate receptor-mediated excitation in the hippocampal CA1 region. Neuroscience. 2004;129:243–254. doi: 10.1016/j.neuroscience.2004.06.082. [DOI] [PubMed] [Google Scholar]
- Zamin LL, Dillenburg-Pilla P, Argenta-Comiran R, Horn AP, Simao F, Nassif M, Gerhardt D, Frozza RL, Salbego C. Protective effect of resveratrol against oxygen-glucose deprivation in organotypic hippocampal slice cultures: Involvement of PI3-K pathway. Neurobiol Dis. 2006;24:170–182. doi: 10.1016/j.nbd.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Zhao M, Li D, Shimazu K, Zhou YX, Lu B, Deng CX. Fibroblast Growth Factor Receptor-1 is Required for Long-Term Potentiation, Memory Consolidation, and Neurogenesis. Biol Psych. 2007;62:381–390. doi: 10.1016/j.biopsych.2006.10.019. [DOI] [PubMed] [Google Scholar]
- Zhu WJ, Roper SN. Brain-derived neurotrophic factor enhances fast excitatory synaptic transmission in human epileptic dentate gyrus. Ann Neurol. 2001;50:188–194. doi: 10.1002/ana.1074. [DOI] [PubMed] [Google Scholar]
- Ziegler DR, Ribeiro LC, Hagenn M, Siqueira IR, Araujo E, Torres IL, Gottfried C, Netto CA, Goncalves CA. Ketogenic diet increases glutathione peroxidase activity in rat hippocampus. Neurochem Res. 2003;28:1793–1797. doi: 10.1023/a:1026107405399. [DOI] [PubMed] [Google Scholar]
- Zucchini S, Barbieri M, Simonato M. Alterations in seizure susceptibility and in seizure-induced plasticity after pharmacologic and genetic manipulation of the fibroblast growth factor-2 system. Epilepsia. 2005;46 Suppl 5:52–58. doi: 10.1111/j.1528-1167.2005.01009.x. [DOI] [PubMed] [Google Scholar]