

Abstract
Age-related macular degeneration (AMD) represents a leading cause of blindness worldwide. While the clinical and histopathological aspects of AMD are well characterized, its etiology and pathogenesis remain unclear. Recent findings suggest a role for immunologic processes in AMD pathogenesis, including the age-related generation of extracellular deposits inside the Brusch membrane and beneath the retinal pigment epithelium, recruitment of macrophages for clearance of these deposits, complement activation, recruitment of tissue-destructive macrophages, microglial activation and accumulation, and proinflammatory effects of chronic inflammation by Chlamydia pneumoniae. This review discusses the evidence for the role of inflammation in human AMD and in animal models of AMD.
Keywords: Age-related macular degeneration (AMD), Inflammation, Complement, Macrophage, Microglia
Introduction
Immune-mediated processes have long been associated with a variety of ocular diseases—most characteristically, uveitis. While age-related macular degeneration (AMD) is not a classical inflammatory disease per se, a growing body of evidence suggests that inflammatory and immunologic events might play a role in AMD. In this review, we discuss AMD and the current evidence for the immunological basis of its pathogenesis.
Worldwide, AMD is a leading cause of blindness for individuals aged 55 and over [1]. In the US alone, at least 1.75 million individuals suffer from AMD, and this number promises to climb with the overall aging of the population. According to some estimates, in fact, by the year 2020, AMD will affect up to 2.95 million people in the US [2].
AMD is characterized by degenerative changes in the macula, the central region of the retina bearing the highest concentration of photoreceptors (primarily, cone cells) and therefore facilitating central vision and visual acuity. Accordingly, this degeneration of the macula leads to central vision loss, as well as impairment in the ability to read fine print or recognize faces. The earliest clinical manifestation and pathological feature of AMD is the development of drusen, extracellular deposits of glycoproteins, lipids, and cellular debris located inside Bruch's membrane and beneath the retinal pigment epithelium (RPE). A few small drusen can be found in healthy individuals over age 50, but the presence of large or numerous drusen confers significant risk for AMD [3, 4].
Clinically and histologically, AMD is generally classified into two major subtypes: dry AMD and wet AMD. Dry AMD progresses more slowly and manifests with drusen, geographic atrophy of RPE, and photoreceptor dysfunction and degeneration. Wet AMD, on the other hand, is more debilitating and often presents after dry AMD. The key feature of wet AMD is choroidal neovascularization (CNV), the growth of new blood vessels from the choroid into the region underlying the RPE or extending past the RPE into the subretinal space and retina. This choroidal neovascularization can lead to leakage of blood into the subretinal space, which, along with RPE atrophy and photoreceptor degeneration, leads to vision loss.
Risk factors for AMD include smoking, body mass index, diet, and light exposure and studies have linked genetics, oxidative stress, RPE senescence, hypoxia, and many other factors to AMD. Still, the exact etiology and pathogenesis of the disease remain largely unclear [5–9]. An ever-growing collection of evidence suggests that immunologic events play a key role in the pathogenesis of AMD.
Unlike uveitis, which manifests mainly with infiltrates of inflammatory cells, edema from inflammatory vascular damage, and inflammatory tissue damage, the inflammatory cells in the AMD lesions are much less prominent and often neglected, despite the fact that macrophages including giant cells and activated microglia have been described in both dry and wet AMD (Fig. 1) [10–13]. Here, we discuss the inflammatory aspects of AMD in humans—including the intersections between human AMD and the complement system, macrophages, microglia, and other related immune components—as well as the animal models of AMD that have arisen from and contributed to our understanding of the immunopathology of human AMD.
Fig. 1.
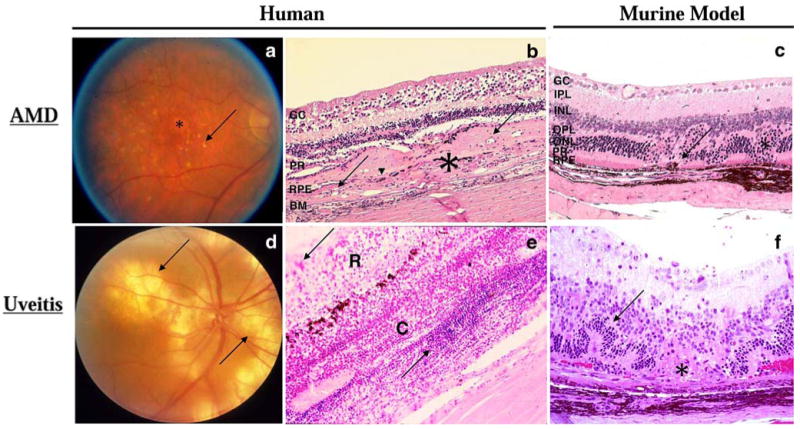
Fundoscopic and histologic representations of AMD and uveitis. a Fundoscopy of a human eye with AMD. Many drusen (arrow) and regions of RPE atrophy (asterisk) are visible by fundoscopy in a patient with dry AMD. b Photomicrograph of a human retina with wet AMD. Photoreceptor cells and most RPE (arrowhead) are replaced by a thick layer of fibrovascular tissue (asterisk) including small neovascular lumens (arrows). Ganglion cell layer, GC; photoreceptor layer, PR; RPE layer, RPE; and Bruch membrane, BM. (H & E, original magnification ×100). c Photomicrograph of a Cx3cr1−/−/Ccl2−/− mouse retina, a model for AMD. Choroidal neovascularization (arrows) and photoreceptor lesions (asterisk) are observed. Ganglion cell layer, GC; inner plexiform layer, IPL; inner nuclear layer, INL; outer plexiform layer, OPL; outer nuclear layer, ONL; photoreceptor layer, PR; and RPE layer, RPE. (H & E, original magnification ×200). d Fundoscopy of a human uveitic eye. Multiple large subretinal infiltrates (arrows) are visible by fundoscopy in a patient with Vogt–Koyanagi–Harada syndrome. e Photomicrograph of a human retina with uveitis. Massive inflammatory cellular infiltration (arrows) is seen in the edematous retina (R) and a thickened choroid (C) of a patient with sympathetic ophthalmia. (H & E, original magnification ×100). f Photomicrograph of a B10A mouse retina with experimental autoimmune uveitis. Focal retinal outer layer destruction (asterisk), retinal folds (arrow), and retinal and choroidal inflammatory cells are visible. (H & E, original magnification ×200)
Complement system in AMD
The complement system is a component of the innate immune system comprising over 30 soluble and membrane-bound proteins [14, 15]. The complement system is divided into three main pathways: classical, lecithin, and alternative. Briefly, the classical pathway is activated largely by immune complexes (antibody bound to antigen); the mannose-binding lectin pathway is activated primarily by mannose and N-acetyl glucosamine residues that are particularly abundant on bacterial cell surfaces, and the alternative pathway is initiated by a variety of activating substances including microbial surfaces and polysaccharides. Overall, activation of these pathways results in a proinflammatory response including generation of membrane attack complexes (MAC) which mediate cell lysis, release of chemokines to attract inflammatory cells to the site of damage, and enhancement of capillary permeability [14, 15].
The complement system is continuously activated at low levels in the normal eye and intraocular complement regulatory proteins tightly regulate this spontaneous complement activation to maintain complement activity at a level that promotes elimination of potential pathogens without damaging healthy tissue. Complement dysregulation leading to overactive complement activity can therefore cause immune-mediated ocular damage. Complement dysregulation has in fact been well characterized in autoimmune anterior uveitis, in which ocular specimens contain deposits of immune complexes such as immunoglobulin G (IgG) and complement component 3 (C3), as well as tumor necrosis factor (TNF)-α and interleukin (IL)-1 [16]. Recent studies suggest that the dysregulation of complement activity might be similarly implicated in AMD pathogenesis [17].
Complement components and complement regulatory proteins in human AMD
Complement components in human AMD
Studies in human eyes with AMD have suggested a role for the complement system in AMD. Mullins et al. [18] identified the presence of the complement component 5 (C5) and the MAC consisting of complement components 5b–9 (C5b–9) in drusen from human eyes, including AMD eyes. Similarly, C3a and C5a have been localized to drusen, RPE cells, and Bruch membrane in human AMD [19]. A variety of other studies have similarly demonstrated the presence of immune complexes, complement, and/or complement regulatory proteins in drusen and in nearby RPE [20–23]. More recently, genetic analyses for single-nucleotide polymorphisms (SNP) spanning the complement genes C3 and C5 has linked the arginine to guanine at amino acid 80 (R80G) SNP in the C3 gene to AMD (OR=1.7 for individuals bearing one copy of the R80G SNP and OR=2.6 for those bearing two copies of the SNP) [24].
Complement factor H in human AMD
Complement factor H (CFH) is a negative regulator of the complement system. More specifically, it impairs activation of the alternative pathway by inhibiting several steps of this pathway and by promoting degradation of activated complement components [25]. CFH binds heparin on cell surfaces to prevent complement-mediated damage to heparin and the cells bearing them. It also binds C-reactive protein (CRP) to inhibit CRP-mediated activation of the alternate pathway in response to damaged tissue [26, 27]. CFH is expressed in a variety of human ocular tissues, including sclera, RPE, retina, and choroids and studies in mice demonstrate that the ocular expression of CFH increases with age [28]. Thus, CFH might accumulate with age to inhibit alternate pathway activation and, accordingly, impairments in CFH function might create a procomplement, i.e., proinflammatory, environment leading to AMD.
Indeed, a host of recent genetic analyses in human AMD patients have generated significant interest in the role of CFH in AMD. The chromosome 1q32 region has long been considered to possess a susceptibility locus for AMD [29]. Edwards, Haines, Klein, Hageman and their colleagues [30–33] recently reported in four independent studies that the tyrosine to histidine at position 402 (Y402H) SNP in the CFH gene in the 1q32 region increases the risk of AMD. A subsequent meta-analysis by Thakkinstian et al. [34] supported the role of the Y402H CFH in AMD, suggesting that up to 50% of all AMD is associated with this polymorphism. We and others have also confirmed the association between CFH and AMD in different populations of the world [35–37]. Histological studies have demonstrated that CFH Y402H and its alternative splice product, factor-H-like protein 1 (FHL-1), which includes residue 402, are present in drusen-like deposits in human eyes [38, 39].
Recent studies by Skerka and Laine and their colleagues have demonstrated the functional significance of the AMD-associated Y402H CFH variant. Skerka et al. [38] demonstrated that the risk variants of CFH (from YH402 and HH402 patients) and of FHL-1 (recombinant H402 FHL-1) exhibit reduced affinity for heparin and CRP. Laine et al., in contrast, found no difference in binding to heparin but confirmed impaired binding to CRP by the risk-conferring variants of CFH. Similar results were obtained using recombinant abbreviated fragments of CFH containing the Y402 or H402 residues [39], further supporting the critical role of position 402 in mediating CRP binding. Overall, these findings suggest that the risk of AMD in patients bearing the Y402H CFH SNP might be attributed to decreased inhibition of CRP-mediated complement activation and possibly, decreased protection of heparin-bearing self-cells from complement effects. CFH and FHL-1 also possess decreased binding affinity for RPE and endothelial cells, and this reduction in binding correlates to reduced complement regulation [38]. Therefore, the studies above highlight the potential role of impaired CFH-mediated inhibition of the alternative complement pathway in AMD pathogenesis.
The link between CFH and AMD has been further strengthened by a variety of in vitro studies. For example, oxidative stress has long been associated with AMD, and long-term treatment of RPE cells with oxidized photoreceptors, but not nonoxidized photoreceptors, impairs CFH secretion. Presence of proinflammatory cytokines such as IL-6 also decreases CFH secretion by RPE cells [40]. Thus, it is possible that not only genetic variation in CFH but also AMD-associated processes such as oxidative stress that impair CFH synthesis and/or secretion might lead to overactivation of the alternative pathway and, ultimately, AMD pathology. Interestingly, deficiency or impaired function of complement factor H is also associated with membranoproliferative glomerulonephritis type II, a renal condition which is associated with dense deposits that might result from a similar inflammatory process as that which produces drusen seen in AMD [41].
Other important complement regulatory proteins in human AMD
Other complement regulatory components have also been linked to AMD, albeit with a smaller arsenal of evidence than that available for CFH. Vitronectin and clusterin, for example, are two negative regulators of the complement cascade that are present in drusen, including drusen in patients with AMD [21, 22]. Moreover, RPE cells near drusen show increased levels of cytoplasmic vitronectin, perhaps a compensatory response to complement attack [22]. Another complement inhibitor, cluster of differentiation 46 (CD46; also known as membrane cofactor protein), is similarly present in drusen and expressed at high levels in RPE cells adjacent to drusen [22, 38]. RPE cells in the Cx3cr1−/−/Ccl2−/− double-knockout murine model of AMD (see “Microglia in AMD”) also exhibit increased CD46 staining by immunohistochemistry [42].
Complement activating substances have also been associated with AMD. Recent epidemiological studies have demonstrated an association between complement activating molecules factor B (BF) in human AMD. Gold et al. screened BF and complement component 2 (C2) genes in two independent cohorts of patients with or without AMD and found that two haplotypes—L9H BF/E318D C2 and R32Q BF/a variant in intron 10 of C2—were protective for AMD. Given that most evidence regarding complement in AMD involves the alternative, rather than classical complement pathway, Gold and his colleagues hypothesized that the functional significance of these haplotypes arises from the BF variants and that the association with the C2 variants was due simply to the strong linkage disequilibrium between the two. These findings, coupled with prior studies documenting that at least one of these variants, R32Q BF, leads to impaired complement activation activity, suggest that these protective variants might exert their beneficial effect by modulating complement activation [43].
Analysis of CRP, an alternative complement pathway activator, in participants in the Age Related Eye Diseases Study demonstrated that the multivariate-adjusted odds ratio for AMD was 2.03 in patients with CRP levels more than 2 standard deviations above the mean [44]. Similarly, participants of the Rotterdam Study with elevated serum levels of CRP possessed increased risk of developing both early and late AMD [45].
Various constituents of the AMD lesion have also been linked to complement pathway activation. Accumulation of lipofuscin and A2E, the bis-retinoid component of lipofuscin, are early pathologic features of AMD, and in vitro photooxidation of A2E in RPE cells leads to complement activation [46]. Amyloid beta, an aggregate found in drusen, is also found in the brains of patients with Alzheimer's disease, where it has been demonstrated in vitro to activate complement [47]. Colocalization of amyloid beta with C3 activation products in human AMD eyes suggests that amyloid beta in AMD lesions might similarly activate complement [48].
Complement components and complement regulatory proteins in animal models of AMD
Complement components in animal models of CNV
Though animal models of AMD generated by directly altering complement components are lacking, several investigators have studied the effect of complement component deficiency in murine models of laser-induced CNV. Based on prior findings in humans that phototherapy for ocular conditions increases the risk of neovascularization, laser injury has commonly served as a model of experimental CNV. Like human CNV, animal models of laser-induced CNV are associated with leakage of fluorescein dye on fluorescein angiography. Histologically, this is marked by disruption of Brusch membrane, ingrowth of neovasculature into the subretinal space, and degeneration of photoreceptors near the lesion [49]. Given these similarities to CNV lesions in wet AMD, the laser-induced model has been used widely in studies evaluating the role of complement in these lesions.
Studies in animal models of laser-induced CNV demonstrate that CNV complexes accumulate C3 and the C5b–9 MAC and that these components are capable of upregulating the proangiogenic cytokine vascular endothelial growth factor (VEGF) [19, 50]. Murine models of genetic C3 deficiency, genetic C3a receptor deficiency, genetic C5 deficiency, genetic C5a receptor deficiency, or complement 6 (C6) deficiency by anti-C6 antibodies exhibit decreased complement and MAC levels at CNV sites, as well as decreased severity of laser-induced CNV [19, 50, 51]. C3a inhibition by neutralizing antibodies, inhibition of C3a receptors, C5a inhibition by neutralizing antibodies, and inhibition of C5a receptor similarly reduces laser-induced CNV in mice, suggesting a role for complement in laser-induced CNV and, thus, possibly AMD-associated CNV [19].
The targets described thus far—C3, C5, etc—are implicated in alternative complement pathway, as well as in the terminal complement pathway shared by all three complement pathways. This raises the important question of whether the classical or lectin pathways are also important in CNV. Bora et al. addressed this question by studying the impact of deficiency or inhibition of C4 and C1q, mediators of the classical and lectin complement pathways, respectively, in laser-induced CNV. The group found that inhibition of C4 and C1q had no effect on the murine model of laser-induced CNV, further supporting the role of the alternate pathway laser-induced CNV and AMD [51]. Moreover, the case for the alternative pathway is further strengthened by human and animal model studies of other alternative pathway components described in this review, including CFH and BF [30–33, 43].
Complement factor H in animal models of AMD
Based upon the epidemiological findings suggesting a role for impaired CFH function in human AMD, Coffey et al. developed complement factor H (cfh) knockout (cfh−/−) murine model of AMD. Compared to age-matched wild-type mice, cfh−/− exhibit an age-related decrease in visual acuity and impaired photoreceptor function. Moreover, retina of cfh−/− mice exhibit C3 deposition and signs of stress injury, as well as ultrastructural changes including disorganization of photoreceptor outer segments. Interestingly, cfh−/− mice had less basal membrane thickening and less sub-RPE electron dense drusen-like material than age-matched wild-type controls. These findings suggest that impaired or deficient complement factor H might mediate vision loss and retinal damage by overactivation of complement but that this same complement-mediated inflammatory cell recruitment might also serve to more effectively clear deposits. Alternatively, Coffey et al. proposed that cfh might itself be a major component or activator of drusen biogenesis and, thus, its absence might impair drusen biogenesis [52].
Other important complement regulatory proteins in animal models of CNV
Although animal models of AMD involving manipulation of complement regulatory proteins other than complement factor H are presently lacking, two studies have looked at the impact of these regulatory proteins in laser-induced CNV.
As discussed above, SNPs in the complement activating protein, BF, have been shown to be protective against human AMD [43]. At least one of these SNPs exhibits decreased complement activation ability, and thus it is possible that decreased BF-mediated complement activation may be protective against AMD in these individuals. Indeed, murine ablation of BF using small interfering RNA against BF followed by laser injury leads to reduced assembly of the C5b-9 MAC, decreased retinal expression of VEGF, and markedly reduced laser-induced CNV (8% incidence, versus 82–92% incidence in controls) [51]. Conversely, mice deficient in CD59 (cd59−/−), a negative regulator of complement, exhibit increased deposition of the C5a-9 MAC complex in CNV lesions, as well as more severe laser-induced CNV. Intraperitoneal or intravitreal administration of recombinant CD59 protein reverses this affect, leading to decreased MAC deposition, reduced expression of angiogenic factors, and dramatically decreased CNV development in the cd59−/− mice (13% incidence of CNV versus 94% in controls) [53]. These data in murine models of laser-induced CNV support studies in human AMD that overactivity of the complement system, at least in part, mediates AMD pathogenesis, including CNV.
Macrophages in AMD
Although classical ocular inflammatory diseases such as uveitis are mediated largely by T cells, macrophages also play an important role. Macrophages are the main components in granulomatous uveitis such as that associated with sarcoidosis, sympathetic ophthalmia, and Vogt–Koyanagi–Harada disease [54]. Macrophages are also one of the predominant inflammatory cells in anterior uveitis and experimental anterior uveitis [55, 56]. Moreover, many proinflammatory cytokines (IL-1, IL-6, TNF-α) and chemokines (chemokine (C-C motif) ligand 2 (CCL2), CCL3, CCL4, CCL5, IL-8, CX3CL1) that are produced by macrophages have been detected in various types of uveitic eyes [57].
Macrophages and the related giant cells have similarly been demonstrated in histologic specimens from patients with AMD, especially in regions of RPE atrophy, breakdown of Bruch's membrane, and choroidal neovascularization [10, 11, 13, 58]. Moreover, as with uveitis, chemokines that mediate macrophage recruitment to the retina have also been associated with AMD. Here, we discuss the role of the CCL2 chemokine and macrophages in human AMD and in animal models of AMD.
CCL2–CCR2 macrophage chemokinesis in human AMD
Chemokines expressed in the eye bind to chemokine receptors on inflammatory cells such as macrophages to promote the mobilization of these cells out of nearby vasculature and into ocular tissue, as well as between various ocular regions. It is thought that alterations or deficiencies in these chemokines or their receptors lead to impaired chemokine-mediated migration of inflammatory cells and, ultimately, AMD [59]. One chemokine that has generated particular interest is CCL2 and its receptor CCR2.
CCR2 is a chemokine receptor found on macrophages which binds the CCL2 (also known as monocyte chemoattractant protein-1) chemokine. Deficiencies in CCR2 lead to decreased leukocyte adhesion to microvasculature, as well as decreased extravasation of monocyte from the circulatory system into surrounding tissues. Moreover, deficiencies in either CCL2 or CCR2 impair macrophage accumulation induced by proinflammatory stimuli without affecting baseline levels of macrophage recruitment to the same tissues [60, 61].
To date, there are no direct studies of CCL2 or CCR2 in human AMD. However, altered expression of CCR2 has been implicated in studies of apolipoprotein E (ApoE) polymorphism in human AMD. Through an independent study and a meta-analysis of previously published studies, Bojanowski et al. demonstrated that the ApoE-112R variant (also known as E4) is associated with decreased risk of AMD and that the ApoE4 variant correlates with decreased expression of CCL2 in RPE cells. Based upon this finding, the group surmised that the ApoE4 variant lowers AMD risk by decreasing CCL2-mediated macrophage recruitment to the retina [62]. This suggests that macrophages play a pathologic, rather than restorative or adaptive, role in AMD.
CCL2–CCR2 macrophage chemokinesis in animal models of AMD
A study of Ccl2 and Ccr2 knockout in mice, however, suggests the opposite—that macrophages serve a beneficial role in the retina. Ambati et al. found that Ccl2- and Ccr2-deficient mice (Ccl2−/− and Ccr2−/−, respectively) develop funduscopically visible AMD-like lesions starting around 9 months of age. Ccl2−/− and Ccr2−/− mice also exhibit histological signs of AMD including lipofuscin granules and Brusch membrane thickening and ultrastructural signs of AMD such as increased melanosomes and lipofuscin granules. By 16 and 18 months of age, respectively, these mice also develop geographic RPE atrophy and choroidal neovascularization, respectively. In addition, retina from Ccl2−/− and Ccr2−/− mice have increased A2E accumulation, a biochemical sign of AMD [63].
Ambati et al. also demonstrated that drusen components such as C5, IgG, and advanced glycosylation products which accumulate in the eyes of Ccl2−/− and Ccr2−/− mice upregulate Ccr2 expression by RPE cells and choroidal endothelial cells. The group surmised that increased Ccr2 expression due to these deposits promotes macrophage infiltration to clear the deposits. Indeed, they demonstrated, in vitro, that macrophages adhere to IgG and C5 and facilitate their clearance. This was supported by findings of an age-dependent increase in Ccl2 expression in RPE of wild-type mice, as well as a similar age-related increase in macrophage infiltration in wild-type mice. Ccl2- and Ccr2-deficient mice showed less age-related macrophage accumulation. As such, the group surmised that impaired Ccl2 and Ccr2-mediated macrophage accumulation in the Ccl2−/−- and Ccr2−/− mice leads to impaired clearance of accumulated deposits of IgG, C5, advanced glycosylation products, and other drusen components and, ultimately, AMD pathology [63].
Thus, it appears that while it is generally accepted that macrophages accumulate in AMD lesions, it is still quite unclear whether these macrophages serve a maladaptive pro-AMD role, or whether they accumulate as an adaptive response to pathological deposits and tissue injury associated with AMD. The study of Ambati et al. in Ccr2- and Ccl2-deficient mice might suggest the latter role, at least during the early stage of AMD development, though correlative findings in human AMD are still necessary. In fact, the limited data we presently have regarding CCL2 in human AMD suggest an opposite function for macrophages than that which arises from the Ccr2- and Ccl2-deficient murine models. Indeed, the case of CCL2 and CCR2 is a harbinger of the perplexing, seemingly contradictory nature of other studies on the role of macrophages in human CNV and in animal models of CNV described below.
Macrophages are associated with CNV in human AMD
Macrophages have long been localized to sites of CNV in patients with human AMD [10, 13]. Examination of subfoveal neovascular membranes from patients with AMD by Lopez et al. revealed the presence of chronic inflammatory cells including macrophages [58]. Similarly, a prominent inflammatory component involving CD68+ macrophages was detected in rapidly progressive fibrovascular AMD membranes [64].
Grossniklaus et al. evaluated 20 specimens of human CNV, 14 of which were from AMD patients, and derived a possible mechanism by which macrophages might promote CNV development. In the study, Grossniklaus showed that macrophages express the proangiogenic factor VEGF and that they might therefore directly promote CNV. They also demonstrated that macrophages in these CNV samples express tissue factor, which may serve as a scaffold for vascularization [11].
Kamei et al. analyzed CNV membranes from patients with AMD and suggested another plausible function of the macrophages that accumulate at CNV lesions. They demonstrated that CNV membranes contain oxidized lipoproteins and that these same membranes stain positive for macrophages bearing scavenger receptors for oxidized proteins, including lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) and scavenger receptor that binds phosphatidylserine and oxidized lipoprotein. This study, as well as prior studies showing increased oxidized lipoprotein levels with age in normal eyes and AMD eyes, suggests that the macrophages seen in AMD and in CNV lesions might serve to ingest oxidized low-density lipoprotein that accumulates with age [65, 66]. The study did not, itself, demonstrate whether these macrophages serve maladaptive functions leading to CNV development or adaptive responses to the accumulation of oxidized protein and/or other pathological processes associated with age and AMD. However, Kamei et al. emphasized similarities between AMD and atherosclerosis, including their new finding that AMD choroidal neovascularization lesions, like those in atherosclerosis, house macrophages which uptake oxidized lipoproteins. In light of these similarities and the well-accepted pathologic role of macrophages in producing atherosclerotic lesions, they surmised that macrophage accumulation might also serve a pathological role in AMD [65].
Overall, though these studies bring to light several plausible mechanisms by which macrophages may have a pathogenic role for CNV in human AMD, none are able to clearly demonstrate whether macrophages accumulate near CNV because they play a causative role in CNV or because they serve as an adaptive response against CNV-associated pathology. This is, of course, due to the largely observational nature of human studies: we can analyze human AMD samples, but we cannot manipulate macrophage levels in living humans to determine whether they increase or decrease the risk of CNV. Consequently, researchers have turned largely to animal models to manipulate macrophage levels and assess the impact of macrophage alterations on experimentally induced CNV models.
Manipulating macrophages in animal models of CNV
Two groups—Espinosa-Heidmann and Sakurai and their colleagues—simultaneously reported independent studies demonstrating a harmful role of macrophages in CNV. Both groups investigated the role of macrophage depletion by dichloromethylene diphosphonate (Cl2MD)-containing liposomes on laser-induced CNV. When these liposomes are ingested by macrophages, the Cl2MD enzyme is released and its accumulation within lysosomes presumably induces apoptosis. Espinosa-Heidmann et al. administered Cl2MD liposomes to mice and documented its ability to deplete circulating monocytes and choroidal macrophages. Macrophage depletion by Cl2MD-liposome treatment ameliorated laser-induced CNV, as determined histologically and by fluorescein angiography [67].
Sakurai et al. used a similar strategy of macrophage depletion with Cl2MD liposomes. Their study showed that treatment of mice with Cl2MD liposomes prior to or immediately after laser injury led to decreased macrophage recruitment and decreased CNV volume, as well as decreased CNV as visualized by fluorescein angiography. Moreover, macrophage depletion was also correlated to decreased VEGF protein at the laser lesion [68].
In stark contrast to these findings by Espinosa-Heidman and Sakurai, Apte and his colleagues reported that macrophages have the opposite effect on laser-induced CNV: they inhibit laser-induced CNV. In their study, Apte and his associates induced laser injury in IL-10-deficient mice (IL-10−/−). IL-10−/− mice exhibit increased macrophage recruitment and reduced severity of laser-induced CNV. Moreover, inhibition of macrophage entry by anti-CD11b or anti-F4/80 treatment or by IL-10 treatment reverses this IL-10−/−-related amelioration of laser-induced CNV. Conversely, IL-10-overexpressing transgenic mice exhibit decreased macrophage infiltration into the retina and, in turn, a substantial increase in susceptibility to laser-induced CNV. Based on prior studies that laser-induced CNV is mediated through interactions of CD95L with CD95 on choroidal blood vessels, the group the showed that macrophages are capable of killing CD95+ cells on necrotic retina and that that macrophages from mice deficient in CD95L lost their ability to inhibit laser-induced CNV. Thus, Apte et al. hypothesized that RPE injury from laser treatment upregulates CD95L expression in macrophages and that these CD95L-bearing macrophages decrease CNV by killing CD95-positive neovascular endothelium cells [69].
Studies of macrophage depletion have therefore produced differing findings: Espinosa-Heidmann and Sakurai and their associates demonstrate that Cl2MD-liposome-induced macrophage depletion decreases laser-induced CNV, presumably by decreased macrophage-mediated VEGF secretion. In contrast, Apte et al. suggest that choroidal neovascular cell killing by macrophages decreases risk of laser-induced CNV. In response to the discordance between their findings and those previously reported by Espinosa-Heidmann and Sukarai, Apte et al. demonstrated that liposomes enter budding CNV endothelial cells and, thus, release of the Cl2MD enzyme in these endothelial cells might kill the CNV endothelium. They therefore hypothesized that the improvement after Cl2MD-liposome treatment might be due not from macrophage depletion but from direct injury to CNV endothelium [69].
The chemokine CCL2 and its receptors CCR2 described above have also been employed in the study of CNV in mice. Tsutsumi et al. utilized Ccr2 knockout mice (Ccr2−/−) to suggest that macrophages play an angiogenic, pro-CNV role in CNV. Ccr2−/− mice exhibit decreased infiltration of macrophages into the retina after laser injury, and this decrease in macrophages is associated with a reduction in laser-induced CNV. Moreover, macrophages isolated from eyes after laser injury express angiogenic factors and exhibit angiogenic activity in vitro [70]. Similarly, Yamada et al. [71] showed that treatment of mice with atorvastatin decreases Ccl2 levels, thereby reducing ocular infiltration of macrophages and decreasing laser-induced CNV volume. Both of these studies showing decreased laser-induced CNV in the absence of Ccr2 or Ccr2 are in stark contrast with the previously described study by Ambati et al. in which deficiencies of these chemokines led to AMD features, including CNV, in senescent mice [63, 70, 71]. Indeed, these contradictory findings in Ccr2- or Ccl2-deficient mice with laser-induced CNV versus CNV associated with senescence highlights the potentially dissimilar mechanisms underlying laser-induced CNV and CNV associated with age-related maculopathy. It is possible, for example, that laser-induced CNV, as a response to acute injury, is mediated by an overactive inflammatory response. As such, quelling inflammatory cell recruitment by Ccr2 or Ccl2 deficiency might decrease CNV induced by laser injury. In contrast, in the absence of laser injury, Ccr2 or Ccl2 deficiency might lead to impaired macrophage-mediated clearance of drusen deposits, creating a nidus for low-grade inflammation. This inflammation, over time, might recruit other inflammatory cells, including macrophages and nonmacrophages, which potentiate tissue damage and promote CNV development. For example, complement activation has, as described above, been shown to promote VEGF secretion by retinal cells and, thus, worsened CNV in senescent Ccr2−/− or Ccrl−/− mice might be due, in part, to increased complement-mediated VEGF secretion due to impaired drusen clearance.
Macrophages in AMD—friend or foe?
Overall, these studies of human AMD and animal models of AMD present conflicting evidence regarding the role of macrophages. Some studies suggest a beneficial role of macrophages, whereas others suggest that macrophage accumulation in AMD lesions is a pathological event. One possible explanation for these conflicting findings is a dual role of macrophages: anti-inflammatory macrophages which are recruited to clear drusen and other deposits seen with age, as well as proinflammatory macrophages which result from the low-grade inflammation of AMD lesions and which directly induce tissue damage [59, 72]. If so, a deficiency of the former result and/or an excess of the latter result might lead to AMD pathogenesis. In other words, before AMD develops, macrophages function as housekeepers in maintaining balance of waste deposit. After AMD develops, especially wet AMD, macrophages function as inflammatory stimulators thereby exacerbating the lesion.
Indeed, macrophages are a diverse group of cells inhabiting a broad spectrum of phenotypes and activities. At polar ends of this spectrum are M1 and M2 macrophages, two distinct subsets of macrophages [73]. A recent review by Mantovani et al. describes how these polarized M1 and M2 macrophages differ with regard to receptor expression, cytokine expression, and effector function. M1 macrophages possess considerable proinflammatory abilities, including tissue-damage activities, release of proinflammatory cytokines, and production of reactive oxygen and nitrogen species. In contrast, M2 macrophages are less inflammatory, serving to promote scavenging, immunoregulation, and/or tissue remodeling, including angiogenesis [73]. It is possible, then, that M2 macrophages serve a beneficial purpose by scavenging deposits such as drusen and that M1 macrophages might be harmful through their proinflammatory actions and ability to induce tissue damage. In addition, a subset of M2 macrophages at a later state of AMD pathogenesis or through acute injury via laser treatment might promote angiogenesis. If so, these diverse subsets of macrophages might explain the seemingly conflicting data regarding the role of macrophages in AMD.
As an example, consider how this theory might intersect with the role of CCR2–CCL2-mediated recruitment of macrophages in AMD. M2 macrophages which express scavenger receptors also express CCR2 receptor [73]. Thus, inhibition of CCR2, as in the Ccr2−/− murine model of AMD by Ambati et al, might impair recruitment and/or activity of M2 macrophages essential for scavenging debris. Inefficient scavenging of debris by M2 macrophages might trigger a cascade of events which eventually leads to AMD pathology, including CNV [63]. In contrast, these same M2 macrophages that express CCR2 also exhibit proangiogenic activity [73]. Thus, as Tsutsumi et al. describe, Ccr2−/− mice might actually exhibit decreased laser-induced CNV, which as we discussed above, presumably occurs via a different mechanism that CNV following development of AMD-like lesions [70]. Overall, our growing understanding of macrophage heterogeneity raises an important question regarding the role of macrophages in AMD, especially in light of conflicting theories regarding the beneficial and harmful roles of macrophages in AMD pathogenesis. Further studies in the M1 and M2 subsets of macrophages as well as the differential role of these macrophages in AMD are necessary to parse out the various functions of macrophages in preventing and/or inducing AMD pathology.
Microglia in AMD
The retina possesses two populations of myeloid-derived cells which have been given various terms such as perivascular macrophages and parenchymal microglia [74]. The perivascular macrophages of the retinal microglia are resident immune cells that entered the inner retina alongside blood vessels during embryological development [75]. Quiescent stellate-shaped retina microglia can be activated by retinal injury, leading to their conversion to enlarged ameboid microglia that migrate from the inner retina to photoreceptor-bearing outer retina. There, they facilitate phagocytosis of cellular debris and secrete cytokines, chemokines, and neurotoxins.
Microglia have been implicated in classical ocular inflammatory disease such as uveitis. In early experimental autoimmune uveitis (EAU), microglial migration to and activation at photoreceptor leads to the generation of TNF-α and peroxynitrite prior to infiltration of macrophages, suggesting that microglia might initiate EAU pathogenesis [76]. Whether microglia initiate the recruitment of cells and cause amplification of inflammation is unclear. Another notion is that migration to sites of injury represents regulation and containment without evoking inflammatory damage.
Microglia in human AMD
Recent studies in humans and animal models suggest a role for microglia in AMD. Indeed, chronic microglia activation is associated with various neurodegenerative diseases. Endogenous triggers alert microglia cells rapidly in the degenerating retina, leading to local proliferation, migration, enhanced phagocytosis, and secretion of cytokines, chemokines, and neurotoxins. This amplified immunological cascade and the loss of limiting control mechanisms may contribute significantly to retinal tissue damage and proapoptotic events [77]. Activated microglia have been identified in areas the outer retina and subretinal regions of human AMD eyes [12]. In vitro studies involving treatment of healthy photoreceptors with activated microglia suggest that activated microglia can induce injury to normal photoreceptor cells [78]. Thus, it is possible that microglia found in outer retina of AMD eyes might be pathogenic.
Given the documented presence of activated microglia in outer retina of human AMD eyes and the potential of these activated microglia to cause photoreceptor injury, a variety of investigators have studied the microglia chemokine receptor CX3CR1 in human AMD and in animal models of AMD. CX3CR1 is a G-protein coupled receptor found on inflammatory cells, including microglia, macrophages, astrocytes, and T cells. Its natural ligand, CX3CL1, also known as fractalkine–neurotactin, is a chemokine that binds to CX3CR1 receptors on leukocytes to promote the mobilization of these leukocytes to sites of inflammation as well as their subsequent activation [79]. CX3CR1 is expressed constitutively in the retina and other ocular tissues, where it may mediate influx of microglia and macrophages to clear accumulated deposits [80].
Two common SNPs of the CX3CR1 gene, V249I and T280M, have been implicated in a variety of diseases, including age-related diseases [81, 85]. We have demonstrated previously that these V249I and T280M CX3CR1 SNPs are associated with an increased risk of AMD [81]. The T280M CX3CR1 SNP is associated with decreased expression of CX3CR1 and lower CX3CR1 expression in the macula compared to perimacular retinal regions. In contrast, there were no significant differences in CX3CR1 expression between the macula and periphery in normal eyes [81, 82].
Functional studies suggest that inflammatory cells bearing the risk-conferring CX3CR1 variants might exhibit altered chemotaxis. McDermott et al. [83], for example, demonstrated that the T280M CX3CR1 SNP results in decreased CX3CL1-induced chemotaxis of leukocytes and Moatti et al. [84] showed that peripheral blood mononuclear cells expressing the V249I CX3CR1 SNP exhibited decreased binding sites for CX3CL1 ligand. Cells expressing both SNPs exhibit decreased CCL2-induced chemotaxis in the presence of CX3CL1 [85]. It is therefore hypothesized that decreased CX3CR1-mediated microglial migration, either by decreased expression as noted by Tuo et al. or by impaired chemotaxis due to these risk-conferring CX3CR1 SNPs, might mediate AMD pathogenesis.
Microglia in animal models of AMD
In light of these studies suggesting a link between impaired CX3CR1 function and human AMD and the previously described Ccl2−/− murine model of AMD, Tuo et al. determined whether a combined approach of Ccl2 deficiency and Cx3cr1 deficiency might produce a more consistent, more early-onset murine model of AMD [42]. The Ccl2−/−/Cx3cr1−/− double-knockout murine model of AMD showed fundoscopic findings of AMD at 6–9 weeks of age, as well as classical histological signs of AMD including focal thickening of the Bruch membrane, drusen, local RPE hypopigmentation and vacuolation, photoreceptor outer segment disorganization, and photoreceptor atrophy. The double-knockout mice also developed ultrastructural signs of AMD, as well as biochemical signs such as A2E accumulation. Interestingly, Tuo and his colleagues [42] also noted that retinal lesions in the Ccl2−/−/Cx3cr1−/− mice showed signs of microglial infiltration. This study by Tuo et al. [42] suggests that Ccl2 and/or Cx3cr1 depletion might play a role in microglial accumulation and, thus, AMD pathogenesis [12].
Indeed, Combadiere et al. recently demonstrated a direct link between CX3CR1 and microglial accumulation in AMD. Combadiere and his associates demonstrated that CX3CR1-positive cells in the outer retina and subretina space of human AMD eyes, as well in drusen and near choroidal neovascularization, are microglial cells. They demonstrated that Cx3cr1−/− mice exhibit signs of retinal degeneration. In Cx3cr1−/− mice, Cx3cr1 deficiency did not affect homing of microglia to the inner retina but increased accumulation of the microglia in the subretinal space near RPE cells [85]. These bloated accumulated microglia formed drusen-like deposits between the outer segment and RPE, and similar CX3CR1-positive bloated subretinal microglia were identified in human AMD eyes [85]. More recently, Xu et al. reported that aging C57BL/6 mice show increasing fundus autofluorescence by confocal scanning laser ophthalmoloscopy, a technique commonly employed to diagnose and monitor retinal lesions. By confocal microscopy, Xu and colleagues [86] showed that these fundus autofluorescence signals consisted of perivascular and subretinal microglia which increased with age and contained lipofuscin granules. These findings further support the notion that AMD lesions such as drusen are derived, at least in part, through accumulated subretinal microglia.
In summary, this interplay between human and animal studies of microglia in AMD suggests that photoreceptor injury in AMD is associated with migration of microglia from inner retina to outer retina where it is possible that they induce damage to nearby healthy photoreceptors. Moreover, altered chemotaxis of microglia due to decreased CX3CR1 expression or impaired CX3CR1 function is associated with human AMD. An animal model of Cx3cr1 deficiency suggests that impaired Cx3cr1-mediated microglial chemotaxis may prevent egress of microglia from the outer retina, facilitating their accumulation there and leading to drusen-like deposits and, possibly, further photoreceptor injury.
Chlamydia pneumoniae infection and AMD
Some evidence for the role of inflammation in AMD also arises from links between past infections and AMD. Specifically, several investigators have correlated prior Chlamydia pneumoniae, an intracellular pathogen that has been associated with inflammatory diseases such as atherosclerosis, with AMD. Two studies by Kalayoglu and Robman and their colleagues [87, 88] demonstrated that serum antibodies for C. pneumoniae proteins are associated with increased risk of AMD development and progression. A later study by Kalayoglu et al. reported that CNV specimens from AMD patients are significantly more likely to show evidence of C. pneumoniae by immunohistochemistry, when compared to non-AMD CNV and non-AMD eyes. Through in vitro studies, Kalayoglu and his associates demonstrated that macrophages produce VEGF after infection with C. pneumoniae [89]. In contrast to these findings, two recent studies by Robman and Kessler and their colleagues [90, 91] failed to show an association between C. pneumoniae and AMD or AMD CNV, respectively. Indeed, these negative findings, as well as the fact that many of the earlier positive studies possessed relatively small sample sizes, highlight the need for further studies to confirm or reject the association between C. pneumoniae and AMD.
C. pneumoniae is an obligate intracellular pathogen, which can exist as a persistent asymptomatic infection, resulting in chronic inflammation [92]. As described above, inflammation is a key facet of AMD pathogenesis and thus, with regard to studies demonstrating the presence of C. pneumoniae in retina of AMD CNV, the question arises whether these C. pneumoniae play a role in pathogenesis or whether they are coincidentally carried to the lesions by macrophages responding to or mediating other pathologic events. If, indeed, there is a relationship between AMD lesions and C. pneumoniae, the important question of the role of the pathogen, if any, in AMD pathogenesis.
C. pneumoniae exhibits a variety of proinflammatory behaviors, some of which are also implicated in AMD. As a microbe, Chlamydia can also activate the alternative pathway of complement, which as discussed above, is strongly implicated in AMD pathogenesis. Indeed, chronic inflammation from C. pneumoniae might be the trigger that activates the alternative complement pathway and ultimately leads to complement overactivity in patients with the Y402H risk variant of CFH, which as we have discussed above leads to impaired complement inhibitory function by CFH. Macrophage accumulation has also been associated with AMD, and prior studies have shown that C. pneumoniae infection promotes differentiation of monocytes to macrophages, as well as migration of monocytes to target tissues [93, 94]. In fact, monocytes infected by C. pneumoniae upregulate various adhesion molecules and exhibit enhanced migration through the blood–brain barrier [94]. This raises the question of whether Chlamydia can similarly enhance monocyte entry through the blood retinal barrier, as well as subsequent macrophage accumulation associated with AMD. Moreover, C.-pneumoniae-infected macrophages secrete proinflammatory and proangiogenic cytokines, perhaps increasing the risk of AMD and CNV, respectively. Oxidized lipids and proteins have long been associated with AMD lesions and, as described above, studies in human AMD eyes have demonstrated that macrophages, which accumulate in AMD eyes, express oxidized protein receptors such as LOX-1. C. pneumoniae has been shown in vitro to enhance LOX-1 expression in cells and, by this mechanism, might similarly increase macrophage accumulation in AMD eyes [95].
Overall, however, despite these potential links between C. pneumoniae and AMD, further studies are necessary to strengthen the evidence of a role of Chlamydial infection in AMD. Firstly, additional studies on human AMD—either seroepidemiological investigations or studies of C. pneumoniae presence in AMD eyes—are necessary, especially given the conflicting evidence available to date. Secondly, further studies are necessary to elucidate the potential pathophysiological impact of C. pneumoniae infection on AMD development. Lastly, animal models of C. pneumoniae, which already exist for the study of other diseases such as atherosclerosis, should be employed toward the study of AMD to determine whether there is a relationship and to further elucidate the possible underlying mechanisms.
Summary
Though there is considerable evidence from studies in both humans and animals demonstrating a role for inflammatory and immunological events in AMD pathogenesis, the time course and specific molecular mechanisms by which these processes lead to AMD are unclear. Thus far, the evidence suggests that the inflammatory underpinnings of AMD pathology are numerous and that an abnormality at any one of many processes might lead to AMD pathogenesis.
The current evidence suggests that the cumulative effect of insults to the retina, including oxidative stress, endoplasmic reticulum stress, and accumulation of RPE byproducts might lead to deposition of glycoproteins, lipids, and cellular debris inside Bruch's membrane and beneath the RPE. In a healthy eye, these small drusen deposits activate macrophage chemotactic ligands, leading to deposit clearance by M2 macrophages. Indeed, macrophages have been demonstrated to clear at least some drusen components and to express scavenger receptors [63, 65]. An overwhelming burden of deposits or impaired macrophage-mediated clearance of deposits, perhaps by Ccl2 deficiency [63], might lead to accumulation of these deposits which are proinflammatory, leading to recruitment of tissue-destructive M1 macrophages, as well as complement activation. Moreover, these accumulated deposits might interfere with the transport of macromolecules, including oxygen, between the choroidal vessels and the retina, thereby injuring RPE and photoreceptor cells and creating yet another nidus for inflammation.
Complement activity is tightly regulated and, thus, any genetic polymorphism or environmental stressor which activates complement (for example, increased C-reactive protein levels or A2E photooxidation) or impaired complement regulation (for example, the Y402H CFH polymorphism) leads to overactivity of the complement system, resulting in inflammatory damage to the retina [30–33, 38, 39, 45, 46]. Moreover, complement activation also leads to VEGF upregulation, promoting CNV [19, 50]. Influx of tissue-destructive M1 macrophages also potentiates ocular inflammation, leading to further tissue damage. At this point, M2 macrophages involved in tissue remodeling might also serve proangiogenic functions [73].
Photoreceptor injury from these processes or from age-related retinal insults such as oxidative injury promotes migration of microglia from the inner retina to the outer retina, where the microglia secrete cytokines, chemokines, and neurotoxins [12, 75, 77]. As such, microglia in the outer retina might potentiate inflammation and, as has been demonstrated in vitro, these activated microglia might directly cause photoreceptor injury [78]. Genetic variants or deficiencies in the CX3CR1 gene might prevent egress of microglia from the outer retina, allowing them to accumulate there, where they become components of drusen-like deposits and cause further retinal damage [12, 42, 81–85].
Though further studies are needed, recent evidence suggests that prior C. pneumoniae infection might also be implicated in AMD pathogenesis [87–91]. Though further studies are warranted, prior studies suggest showing that C. pneumoniae infection activates the alternative complement pathway and promotes macrophage activation and recruitment suggest that the role of C. pneumoniae, if any, in AMD might intersect with one or more of the immunologic processes described here [94, 95].
In sum, the evidence for the role of immunologic processes in the pathogenesis of AMD is compelling both in humans and in animal models. Further studies are necessary to clarify the precise mechanisms and time course underlying AMD development and, in turn, develop novel therapies for the prevention and/or treatment of AMD.
Contributor Information
Mrinali Patel, Section of Immunopathology, Laboratory of Immunology, National Eye Institute, National Institutes of Health, 10 Center Drive, 10/10N103, NIH/NEI, Bethesda, MD 20892-1857, USA; Howard Hughes Medical Institute, Chevy Chase, MD, USA.
Chi-Chao Chan, Section of Immunopathology, Laboratory of Immunology, National Eye Institute, National Institutes of Health, 10 Center Drive, 10/10N103, NIH/NEI, Bethesda, MD 20892-1857, USA, e-mail: chanc@nei.nih.gov.
References
- 1.Klein R, Peto T, Bird A, Vannewkirk MR. The epidemiology of age-related macular degeneration. Am J Ophthalmol. 2004;137:486–495. doi: 10.1016/j.ajo.2003.11.069. [DOI] [PubMed] [Google Scholar]
- 2.Friedman DS, O'Colmain BJ, Munoz B, Tomany SC, McCarty C, de Jong PT, Nemesure B, Mitchell P, Kempen J. Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol. 2004;122:564–572. doi: 10.1001/archopht.122.4.564. [DOI] [PubMed] [Google Scholar]
- 3.Klein R, Klein BE, Linton KL. Prevalence of age-related maculopathy. The Beaver Dam Eye Study. Ophthalmology. 1992;99:933–943. doi: 10.1016/s0161-6420(92)31871-8. [DOI] [PubMed] [Google Scholar]
- 4.Klein R, Klein BE, Jensen SC, Meuer SM. The five-year incidence and progression of age-related maculopathy: the Beaver Dam Eye Study. Ophthalmology. 1997;104:7–21. doi: 10.1016/s0161-6420(97)30368-6. [DOI] [PubMed] [Google Scholar]
- 5.Ambati J, Ambati BK, Yoo SH, Ianchulev S, Adamis AP. Age-related macular degeneration: etiology, pathogenesis, and therapeutic strategies. Surv Ophthalmol. 2003;48:257–293. doi: 10.1016/s0039-6257(03)00030-4. [DOI] [PubMed] [Google Scholar]
- 6.Beatty S, Koh H, Phil M, Henson D, Boulton M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2000;45:115–134. doi: 10.1016/s0039-6257(00)00140-5. [DOI] [PubMed] [Google Scholar]
- 7.Clemons TE, Milton RC, Klein R, Seddon JM, Ferris FL., III Risk factors for the incidence of advanced age-related macular degeneration in the age-related eye disease study (AREDS) AREDS report no. 19. Ophthalmology. 2005;112:533–539. doi: 10.1016/j.ophtha.2004.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scholl HP, Fleckenstein M, Charbel IP, Keilhauer C, Holz FG, Weber BH. An update on the genetics of age-related macular degeneration. Mol Vis. 2007;13:196–205. [PMC free article] [PubMed] [Google Scholar]
- 9.Taylor HR, Munoz B, West S, Bressler NM, Bressler SB, Rosenthal FS. Visible light and risk of age-related macular degeneration. Trans Am Ophthalmol Soc. 1990;88:163–173. [PMC free article] [PubMed] [Google Scholar]
- 10.Dastgheib K, Green WR. Granulomatous reaction to Bruch's membrane in age-related macular degeneration. Arch Ophthalmol. 1994;112:813–818. doi: 10.1001/archopht.1994.01090180111045. [DOI] [PubMed] [Google Scholar]
- 11.Grossniklaus HE, Ling JX, Wallace TM, Dithmar S, Lawson DH, Cohen C, Elner VM, Elner SG, Sternberg P., Jr Macrophage and retinal pigment epithelium expression of angiogenic cytokines in choroidal neovascularization. Mol Vis. 2002;8:119–126. [PubMed] [Google Scholar]
- 12.Gupta N, Brown KE, Milam AH. Activated microglia in human retinitis pigmentosa, late-onset retinal degeneration, and age-related macular degeneration. Exp Eye Res. 2003;76:463–471. doi: 10.1016/s0014-4835(02)00332-9. [DOI] [PubMed] [Google Scholar]
- 13.Penfold PL, Madigan MC, Gillies MC, Provis JM. Immunological and aetiological aspects of macular degeneration. Prog Retin Eye Res. 2001;20:385–414. doi: 10.1016/s1350-9462(00)00025-2. [DOI] [PubMed] [Google Scholar]
- 14.Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344:1058–1066. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 15.Walport MJ. Complement. Second of two parts. N Engl J Med. 2001;344:1140–1144. doi: 10.1056/NEJM200104123441506. [DOI] [PubMed] [Google Scholar]
- 16.Brito BE, O'Rourke LM, Pan Y, Anglin J, Planck SR, Rosenbaum JT. IL-1 and TNF receptor-deficient mice show decreased inflammation in an immune complex model of uveitis. Invest Ophthalmol Vis Sci. 1999;40:2583–2589. [PubMed] [Google Scholar]
- 17.Zipfel PF, Heinen S, Jozsi M, Skerka C. Complement and diseases: defective alternative pathway control results in kidney and eye diseases. Mol Immunol. 2006;43:97–106. doi: 10.1016/j.molimm.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 18.Mullins RF, Russell SR, Anderson DH, Hageman GS. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 2000;14:835–846. [PubMed] [Google Scholar]
- 19.Nozaki M, Raisler BJ, Sakurai E, Sarma JV, Barnum SR, Lambris JD, Chen Y, Zhang K, Ambati BK, Baffi JZ, Ambati J. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc Natl Acad Sci USA. 2006;103:2328–2333. doi: 10.1073/pnas.0408835103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson LV, Ozaki S, Staples MK, Erickson PA, Anderson DH. A potential role for immune complex pathogenesis in drusen formation. Exp Eye Res. 2000;70:441–449. doi: 10.1006/exer.1999.0798. [DOI] [PubMed] [Google Scholar]
- 21.Hageman GS, Mullins RF, Russell SR, Johnson LV, Anderson DH. Vitronectin is a constituent of ocular drusen and the vitronectin gene is expressed in human retinal pigmented epithelial cells. FASEB J. 1999;13:477–484. doi: 10.1096/fasebj.13.3.477. [DOI] [PubMed] [Google Scholar]
- 22.Johnson LV, Leitner WP, Staples MK, Anderson DH. Complement activation and inflammatory processes in Drusen formation and age related macular degeneration. Exp Eye Res. 2001;73:887–896. doi: 10.1006/exer.2001.1094. [DOI] [PubMed] [Google Scholar]
- 23.Crabb JW, Miyagi M, Gu X, Shadrach K, West KA, Sakaguchi H, Kamei M, Hasan A, Yan L, Rayborn ME, Salomon RG, Hollyfield JG. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci USA. 2002;99:14682–14687. doi: 10.1073/pnas.222551899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yates JR, Sepp T, Matharu BK, Khan JC, Thurlby DA, Shahid H, Clayton DG, Hayward C, Morgan J, Wright AF, Armbrecht AM, Dhillon B, Deary IJ, Redmond E, Bird AC, Moore AT. Complement C3 variant and the risk of age-related macular degeneration. N Engl J Med. 2007;357:553–561. doi: 10.1056/NEJMoa072618. [DOI] [PubMed] [Google Scholar]
- 25.Alsenz J, Schulz TF, Lambris JD, Sim RB, Dierich MP. Structural and functional analysis of the complement component factor H with the use of different enzymes and monoclonal antibodies to factor H. Biochem J. 1985;232:841–850. doi: 10.1042/bj2320841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodriguez de CS, Esparza-Gordillo J, Goicoechea de JE, Lopez-Trascasa M, Sanchez-Corral P. The human complement factor H: functional roles, genetic variations and disease associations. Mol Immunol. 2004;41:355–367. doi: 10.1016/j.molimm.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 27.Prosser BE, Johnson S, Roversi P, Herbert AP, Blaum BS, Tyrrell J, Jowitt TA, Clark SJ, Tarelli E, Uhrin D, Barlow PN, Sim RB, Day AJ, Lea SM. Structural basis for complement factor H linked age-related macular degeneration. J Exp Med. 2007;204:2277–2283. doi: 10.1084/jem.20071069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mandal MN, Ayyagari R. Complement factor H: spatial and temporal expression and localization in the eye. Invest Ophthalmol Vis Sci. 2006;47:4091–4097. doi: 10.1167/iovs.05-1655. [DOI] [PubMed] [Google Scholar]
- 29.Fisher SA, Abecasis GR, Yashar BM, Zareparsi S, Swaroop A, Iyengar SK, Klein BE, Klein R, Lee KE, Majewski J, Schultz DW, Klein ML, Seddon JM, Santangelo SL, Weeks DE, Conley YP, Mah TS, Schmidt S, Haines JL, Pericak-Vance MA, Gorin MB, Schulz HL, Pardi F, Lewis CM, Weber BH. Meta-analysis of genome scans of age-related macular degeneration. Hum Mol Genet. 2005;14:2257–2264. doi: 10.1093/hmg/ddi230. [DOI] [PubMed] [Google Scholar]
- 30.Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, Henning AK, SanGiovanni JP, Mane SM, Mayne ST, Bracken MB, Ferris FL, Ott J, Barnstable C, Hoh J. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, Hageman JL, Stockman HA, Borchardt JD, Gehrs KM, Smith RJ, Silvestri G, Russell SR, Klaver CC, Barbazetto I, Chang S, Yannuzzi LA, Barile GR, Merriam JC, Smith RT, Olsh AK, Bergeron J, Zernant J, Merriam JE, Gold B, Dean M, Allikmets R. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci USA. 2005;102:7227–7232. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P, Spencer KL, Kwan SY, Noureddine M, Gilbert JR, Schnetz-Boutaud N, Agarwal A, Postel EA, Pericak-Vance MA. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–421. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 33.Edwards AO, Ritter R, III, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 34.Thakkinstian A, Han P, McEvoy M, Smith W, Hoh J, Magnusson K, Zhang K, Attia J. Systematic review and meta-analysis of the association between complement factor H Y402H polymorphisms and age-related macular degeneration. Hum Mol Genet. 2006;15:2784–2790. doi: 10.1093/hmg/ddl220. [DOI] [PubMed] [Google Scholar]
- 35.Tuo J, Ning B, Bojanowski CM, Lin ZN, Ross RJ, Reed GF, Shen D, Jiao X, Zhou M, Chew EY, Kadlubar FF, Chan CC. Synergic effect of polymorphisms in ERCC6 5¢¢ flanking region and complement factor H on age-related macular degeneration predisposition. Proc Natl Acad Sci USA. 2006;103:9256–9261. doi: 10.1073/pnas.0603485103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ross RJ, Bojanowski CM, Wang JJ, Chew EY, Rochtchina E, Ferris FL, III, Mitchell P, Chan CC, Tuo J. The LOC387715 polymorphism and age-related macular degeneration: replication in three case-control samples. Invest Ophthalmol Vis Sci. 2007;48:1128–1132. doi: 10.1167/iovs.06-0999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haddad S, Chen CA, Santangelo SL, Seddon JM. The genetics of age-related macular degeneration: a review of progress to date. Surv Ophthalmol. 2006;51:316–363. doi: 10.1016/j.survophthal.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 38.Skerka C, Lauer N, Weinberger AA, Keilhauer CN, Suhnel J, Smith R, Schlotzer-Schrehardt U, Fritsche L, Heinen S, Hartmann A, Weber BH, Zipfel PF. Defective complement control of factor H (Y402H) and FHL-1 in age-related macular degeneration. Mol Immunol. 2007;44:3398–3406. doi: 10.1016/j.molimm.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 39.Laine M, Jarva H, Seitsonen S, Haapasalo K, Lehtinen MJ, Lindeman N, Anderson DH, Johnson PT, Jarvela I, Jokiranta TS, Hageman GS, Immonen I, Meri S. Y402H polymorphism of complement factor H affects binding affinity to C-reactive protein. J Immunol. 2007;178:3831–3836. doi: 10.4049/jimmunol.178.6.3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen M, Forrester JV, Xu H. Synthesis of complement factor H by retinal pigment epithelial cells is down-regulated by oxidized photoreceptor outer segments. Exp Eye Res. 2007;84:635–645. doi: 10.1016/j.exer.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 41.Zipfel PF, Hellwage J, Friese MA, Hegasy G, Jokiranta ST, Meri S. Factor H and disease: a complement regulator affects vital body functions. Mol Immunol. 1999;36:241–248. doi: 10.1016/s0161-5890(99)00038-3. [DOI] [PubMed] [Google Scholar]
- 42.Tuo J, Bojanowski CM, Zhou M, Shen D, Ross RJ, Rosenberg KI, Cameron DJ, Yin C, Kowalak JA, Zhuang Z, Zhang K, Chan CC. Murine ccl2/cx3cr1 deficiency results in retinal lesions mimicking human age-related macular degeneration. Invest Ophthalmol Vis Sci. 2007;48:3827–3836. doi: 10.1167/iovs.07-0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gold B, Merriam JE, Zernant J, Hancox LS, Taiber AJ, Gehrs K, Cramer K, Neel J, Bergeron J, Barile GR, Smith RT, Hageman GS, Dean M, Allikmets R. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006;38:458–462. doi: 10.1038/ng1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seddon JM, Gensler G, Milton RC, Klein ML, Rifai N. Association between C-reactive protein and age-related macular degeneration. JAMA. 2004;291:704–710. doi: 10.1001/jama.291.6.704. [DOI] [PubMed] [Google Scholar]
- 45.Boekhoorn SS, Vingerling JR, Witteman JC, Hofman A, de Jong PT. C-reactive protein level and risk of aging macula disorder: the Rotterdam study. Arch Ophthalmol. 2007;125:1396–1401. doi: 10.1001/archopht.125.10.1396. [DOI] [PubMed] [Google Scholar]
- 46.Zhou J, Jang YP, Kim SR, Sparrow JR. Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epithelium. Proc Natl Acad Sci USA. 2006;103:16182–16187. doi: 10.1073/pnas.0604255103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rogers J, Cooper NR, Webster S, Schultz J, McGeer PL, Styren SD, Civin WH, Brachova L, Bradt B, Ward P. Complement activation by beta-amyloid in Alzheimer disease. Proc Natl Acad Sci USA. 1992;89:10016–10020. doi: 10.1073/pnas.89.21.10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Johnson LV, Leitner WP, Rivest AJ, Staples MK, Radeke MJ, Anderson DH. The Alzheimer's A beta-peptide is deposited at sites of complement activation in pathologic deposits associated with aging and age-related macular degeneration. Proc Natl Acad Sci USA. 2002;99:11830–11835. doi: 10.1073/pnas.192203399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tobe T, Ortega S, Luna JD, Ozaki H, Okamoto N, Derevjanik NL, Vinores SA, Basilico C, Campochiaro PA. Targeted disruption of the FGF2 gene does not prevent choroidal neovascularization in a murine model. Am J Pathol. 1998;153:1641–1646. doi: 10.1016/S0002-9440(10)65753-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bora PS, Sohn JH, Cruz JM, Jha P, Nishihori H, Wang Y, Kaliappan S, Kaplan HJ, Bora NS. Role of complement and complement membrane attack complex in laser-induced choroidal neovascularization. J Immunol. 2005;174:491–497. doi: 10.4049/jimmunol.174.1.491. [DOI] [PubMed] [Google Scholar]
- 51.Bora NS, Kaliappan S, Jha P, Xu Q, Sohn JH, Dhaulakhandi DB, Kaplan HJ, Bora PS. Complement activation via alternative pathway is critical in the development of laser-induced choroidal neovascularization: role of factor B and factor H. J Immunol. 2006;177:1872–1878. doi: 10.4049/jimmunol.177.3.1872. [DOI] [PubMed] [Google Scholar]
- 52.Coffey PJ, Gias C, McDermott CJ, Lundh P, Pickering MC, Sethi C, Bird A, Fitzke FW, Maass A, Chen LL, Holder GE, Luthert PJ, Salt TE, Moss SE, Greenwood J. Complement factor H deficiency in aged mice causes retinal abnormalities and visual dysfunction. Proc Natl Acad Sci USA. 2007;104:16651–16656. doi: 10.1073/pnas.0705079104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bora NS, Kaliappan S, Jha P, Xu Q, Sivasankar B, Harris CL, Morgan BP, Bora PS. CD59, a complement regulatory protein, controls choroidal neovascularization in a mouse model of wet-type age-related macular degeneration. J Immunol. 2007;178:1783–1790. doi: 10.4049/jimmunol.178.3.1783. [DOI] [PubMed] [Google Scholar]
- 54.Chan CC, Li Q. Immunopathology of uveitis. Br J Ophthalmol. 1998;82:91–96. doi: 10.1136/bjo.82.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith JR, Hart PH, Williams KA. Basic pathogenic mechanisms operating in experimental models of acute anterior uveitis. Immunol Cell Biol. 1998;76:497–512. doi: 10.1046/j.1440-1711.1998.00783.x. [DOI] [PubMed] [Google Scholar]
- 56.Adamus G, Chan CC. Experimental autoimmune uveitides: multiple antigens, diverse diseases. Int Rev Immunol. 2002;21:209–229. doi: 10.1080/08830180212068. [DOI] [PubMed] [Google Scholar]
- 57.Ooi KG, Galatowicz G, Calder VL, Lightman SL. Cytokines and chemokines in uveitis: is there a correlation with clinical phenotype? Clin Med Res. 2006;4:294–309. doi: 10.3121/cmr.4.4.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lopez PF, Grossniklaus HE, Lambert HM, Aaberg TM, Capone A, Jr, Sternberg P, Jr, L'Hernault N. Pathologic features of surgically excised subretinal neovascular membranes in age-related macular degeneration. Am J Ophthalmol. 1991;112:647–656. doi: 10.1016/s0002-9394(14)77270-8. [DOI] [PubMed] [Google Scholar]
- 59.Forrester JV. Macrophages eyed in macular degeneration. Nat Med. 2003;9:1350–1351. doi: 10.1038/nm1103-1350. [DOI] [PubMed] [Google Scholar]
- 60.Kuziel WA, Morgan SJ, Dawson TC, Griffin S, Smithies O, Ley K, Maeda N. Severe reduction in leukocyte adhesion and monocyte extravasation in mice deficient in CC chemokine receptor 2. Proc Natl Acad Sci USA. 1997;94:12053–12058. doi: 10.1073/pnas.94.22.12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lu B, Rutledge BJ, Gu L, Fiorillo J, Lukacs NW, Kunkel SL, North R, Gerard C, Rollins BJ. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J Exp Med. 1998;187:601–608. doi: 10.1084/jem.187.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bojanowski CM, Shen D, Chew EY, Ning B, Csaky KG, Green WR, Chan CC, Tuo J. An apolipoprotein E variant may protect against age-related macular degeneration through cytokine regulation. Environ Mol Mutagen. 2006;47:594–602. doi: 10.1002/em.20233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ambati J, Anand A, Fernandez S, Sakurai E, Lynn BC, Kuziel WA, Rollins BJ, Ambati BK. An animal model of age-related macular degeneration in senescent Ccl-2- or Ccr-2-deficient mice. Nat Med. 2003;9:1390–1397. doi: 10.1038/nm950. [DOI] [PubMed] [Google Scholar]
- 64.Csaky K, Baffi J, Chan CC, Byrnes GA. Clinicopathologic correlation of progressive fibrovascular proliferation associated with occult choroidal neovascularization in age-related macular degeneration. Arch Ophthalmol. 2004;122:650–652. doi: 10.1001/archopht.122.4.650. [DOI] [PubMed] [Google Scholar]
- 65.Kamei M, Yoneda K, Kume N, Suzuki M, Itabe H, Matsuda K, Shimaoka T, Minami M, Yonehara S, Kita T, Kinoshita S. Scavenger receptors for oxidized lipoprotein in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2007;48:1801–1807. doi: 10.1167/iovs.06-0699. [DOI] [PubMed] [Google Scholar]
- 66.Suzuki M, Kamei M, Itabe H, Yoneda K, Bando H, Kume N, Tano Y. Oxidized phospholipids in the macula increase with age and in eyes with age-related macular degeneration. Mol Vis. 2007;13:772–778. [PMC free article] [PubMed] [Google Scholar]
- 67.Espinosa-Heidmann DG, Suner IJ, Hernandez EP, Monroy D, Csaky KG, Cousins SW. Macrophage depletion diminishes lesion size and severity in experimental choroidal neovascularization. Invest Ophthalmol Vis Sci. 2003;44:3586–3592. doi: 10.1167/iovs.03-0038. [DOI] [PubMed] [Google Scholar]
- 68.Sakurai E, Anand A, Ambati BK, van RN, Ambati J. Macrophage depletion inhibits experimental choroidal neovascularization. Invest Ophthalmol Vis Sci. 2003;44:3578–3585. doi: 10.1167/iovs.03-0097. [DOI] [PubMed] [Google Scholar]
- 69.Apte RS, Richter J, Herndon J, Ferguson TA. Macrophages inhibit neovascularization in a murine model of age-related macular degeneration. PloS Med. 2006;3:e310. doi: 10.1371/journal.pmed.0030310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tsutsumi C, Sonoda KH, Egashira K, Qiao H, Hisatomi T, Nakao S, Ishibashi M, Charo IF, Sakamoto T, Murata T, Ishibashi T. The critical role of ocular-infiltrating macrophages in the development of choroidal neovascularization. J Leukoc Biol. 2003;74:25–32. doi: 10.1189/jlb.0902436. [DOI] [PubMed] [Google Scholar]
- 71.Yamada K, Sakurai E, Itaya M, Yamasaki S, Ogura Y. Inhibition of laser-induced choroidal neovascularization by atorvastatin by downregulation of monocyte chemotactic protein-1 synthesis in mice. Invest Ophthalmol Vis Sci. 2007;48:1839–1843. doi: 10.1167/iovs.06-1085. [DOI] [PubMed] [Google Scholar]
- 72.Chen J, Connor KM, Smith LE. Overstaying their welcome: defective CX3CR1 microglia eyed in macular degeneration. J Clin Invest. 2007;117:2758–2762. doi: 10.1172/JCI33513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 74.Dick AD, Forrester JV, Liversidge J, Cope AP. The role of tumour necrosis factor (TNF-alpha) in experimental autoimmune uveoretinitis (EAU) Prog Retin Eye Res. 2004;23:617–637. doi: 10.1016/j.preteyeres.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 75.Dick AD, Ford AL, Forrester JV, Sedgwick JD. Flow cytometric identification of a minority population of MHC class II positive cells in the normal rat retina distinct from CD45lowCD11b/c+CD4low parenchymal microglia. Br J Ophthalmol. 1995;79:834–840. doi: 10.1136/bjo.79.9.834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rao NA, Kimoto T, Zamir E, Giri R, Wang R, Ito S, Pararajasegaram G, Read RW, Wu GS. Pathogenic role of retinal microglia in experimental uveoretinitis. Invest Ophthalmol Vis Sci. 2003;44:22–31. doi: 10.1167/iovs.02-0199. [DOI] [PubMed] [Google Scholar]
- 77.Langmann T. Microglia activation in retinal degeneration. J Leukoc Biol. 2007;81:1345–1351. doi: 10.1189/jlb.0207114. [DOI] [PubMed] [Google Scholar]
- 78.Roque RS, Rosales AA, Jingjing L, Agarwal N, Al-Ubaidi MR. Retina-derived microglial cells induce photoreceptor cell death in vitro. Brain Res. 1999;836:110–119. doi: 10.1016/s0006-8993(99)01625-x. [DOI] [PubMed] [Google Scholar]
- 79.Fong AM, Robinson LA, Steeber DA, Tedder TF, Yoshie O, Imai T, Patel DD. Fractalkine and CX3CR1 mediate a novel mechanism of leukocyte capture, firm adhesion, and activation under physiologic flow. J Exp Med. 1998;188:1413–1419. doi: 10.1084/jem.188.8.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Silverman MD, Zamora DO, Pan Y, Texeira PV, Baek SH, Planck SR, Rosenbaum JT. Constitutive and inflammatory mediator-regulated fractalkine expression in human ocular tissues and cultured cells. Invest Ophthalmol Vis Sci. 2003;44:1608–1615. doi: 10.1167/iovs.02-0233. [DOI] [PubMed] [Google Scholar]
- 81.Tuo J, Smith BC, Bojanowski CM, Meleth AD, Gery I, Csaky KG, Chew EY, Chan CC. The involvement of sequence variation and expression of CX3CR1 in the pathogenesis of age-related macular degeneration. FASEB J. 2004;18:1297–1299. doi: 10.1096/fj.04-1862fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chan CC, Tuo J, Bojanowski CM, Csaky KG, Green WR. Detection of CX3CR1 single nucleotide polymorphism and expression on archived eyes with age-related macular degeneration. Histol Histopathol. 2005;20:857–863. doi: 10.14670/hh-20.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McDermott DH, Fong AM, Yang Q, Sechler JM, Cupples LA, Merrell MN, Wilson PW, D'Agostino RB, O'Donnell CJ, Patel DD, Murphy PM. Chemokine receptor mutant CX3CR1-M280 has impaired adhesive function and correlates with protection from cardiovascular disease in humans. J Clin Invest. 2003;111:1241–1250. doi: 10.1172/JCI16790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Moatti D, Faure S, Fumeron F, Amara M, Seknadji P, McDermott DH, Debre P, Aumont MC, Murphy PM, de PD, Combadiere C. Polymorphism in the fractalkine receptor CX3CR1 as a genetic risk factor for coronary artery disease. Blood. 2001;97:1925–1928. doi: 10.1182/blood.v97.7.1925. [DOI] [PubMed] [Google Scholar]
- 85.Combadiere C, Feumi C, Raoul W, Keller N, Rodero M, Pezard A, Lavalette S, Houssier M, Jonet L, Picard E, Debre P, Sirinyan M, Deterre P, Ferroukhi T, Cohen SY, Chauvaud D, Jeanny JC, Chemtob S, Behar-Cohen F, Sennlaub F. CX3CR1-dependent subretinal microglia cell accumulation is associated with cardinal features of age-related macular degeneration. J Clin Invest. 2007;117:2920–2928. doi: 10.1172/JCI31692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xu H, Chen M, Manivannan A, Lois N, Forrester JV. Age-dependent accumulation of lipofuscin in perivascular and sub-retinal microglia in experimental mice. Aging Cell. 2007;7:58–68. doi: 10.1111/j.1474-9726.2007.00351.x. [DOI] [PubMed] [Google Scholar]
- 87.Kalayoglu MV, Galvan C, Mahdi OS, Byrne GI, Mansour S. Serological association between Chlamydia pneumoniae infection and age-related macular degeneration. Arch Ophthalmol. 2003;121:478–482. doi: 10.1001/archopht.121.4.478. [DOI] [PubMed] [Google Scholar]
- 88.Robman L, Mahdi O, McCarty C, Dimitrov P, Tikellis G, McNeil J, Byrne G, Taylor H, Guymer R. Exposure to Chlamydia pneumoniae infection and progression of age-related macular degeneration. Am J Epidemiol. 2005;161:1013–1019. doi: 10.1093/aje/kwi130. [DOI] [PubMed] [Google Scholar]
- 89.Kalayoglu MV, Bula D, Arroyo J, Gragoudas ES, D'Amico D, Miller JW. Identification of Chlamydia pneumoniae within human choroidal neovascular membranes secondary to age-related macular degeneration. Graefes Arch Clin Exp Ophthalmol. 2005;243:1080–1090. doi: 10.1007/s00417-005-1169-y. [DOI] [PubMed] [Google Scholar]
- 90.Kessler W, Jantos CA, Dreier J, Pavlovic S. Chlamydia pneumoniae is not detectable in subretinal neovascular membranes in the exudative stage of age-related macular degeneration. Acta Ophthalmol Scand. 2006;84:333–337. doi: 10.1111/j.1600-0420.2005.00591.x. [DOI] [PubMed] [Google Scholar]
- 91.Robman L, Mahdi OS, Wang JJ, Burlutsky G, Mitchell P, Byrne G, Guymer R, Taylor H. Exposure to Chlamydia pneumoniae infection and age-related macular degeneration: the Blue Mountains Eye Study. Invest Ophthalmol Vis Sci. 2007;48:4007–4011. doi: 10.1167/iovs.06-1434. [DOI] [PubMed] [Google Scholar]
- 92.Abdelrahman YM, Belland RJ. The chlamydial developmental cycle. FEMS Microbiol Rev. 2005;29:949–959. doi: 10.1016/j.femsre.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 93.Yamaguchi H, Haranaga S, Widen R, Friedman H, Yamamoto Y. Chlamydia pneumoniae infection induces differentiation of monocytes into macrophages. Infect Immun. 2002;70:2392–2398. doi: 10.1128/IAI.70.5.2392-2398.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.MacIntyre A, Abramov R, Hammond CJ, Hudson AP, Arking EJ, Little CS, Appelt DM, Balin BJ. Chlamydia pneumoniae infection promotes the transmigration of monocytes through human brain endothelial cells. J Neurosci Res. 2003;71:740–750. doi: 10.1002/jnr.10519. [DOI] [PubMed] [Google Scholar]
- 95.Yoshida T, Koide N, Mori I, Ito H, Yokochi T. Chlamydia pneumoniae infection enhances lectin-like oxidized low-density lipoprotein receptor (LOX-1) expression on human endothelial cells. FEMS Microbiol Lett. 2006;260:17–22. doi: 10.1111/j.1574-6968.2006.00286.x. [DOI] [PubMed] [Google Scholar]