

Summary
Successful liposomal-mediated gene therapy is often limited by poor transfection efficiencies. One method previously shown to increase the efficiency of liposomal gene delivery is through the administration of a non-therapeutic dose of the chemotherapeutic drug cisplatin prior to lipofection. The currents study aims to utilise this method to deliver lipoplexes containing the p53 tumour suppressor gene with the aim of increasing therapeutic effect of the p53 gene on a solid tumour in vivo. Rats, implanted with solid salivary adenocarcinomas, were pre-treated with a low dose of cisplatin seven days prior to liposomal mediated p53 treatment. Following treatment with p53, tumour growth, p53 expression and levels of apoptosis were examined and compared to animals treated with p53 without cisplatin pre-treatment and a saline control. Tumours that had been pre-treated with cisplatin prior to p53-lipofection were significantly smaller than both the saline control and the non-cisplatin treated tumours. Saline treated tumours increased in size by an average of 164% over a 96-hour period compared to 64% and 101% for the cisplatin and non-cisplatin p53-liposome treated tumours. The cisplatin pre-treated tumours resulted in significantly higher levels of apoptosis surrounding the treatment site and exhibited prolonged p53 expression when compared to the non-cisplatin pre-treated tumours. The results suggest that the use of cisplatin to pre-sensitise tumours to lipofection has significant benefits when used in conjunction with p53.
Keywords: liposomes, cisplatin, gene therapy, p53, chemotherapy
I. Introduction
Liposomal vectors have a number of advantages over viral vectors. They have been shown to have the ability to transfect cells without the need for specific receptors, are able to carry large DNA molecules and generally display low levels of cytotoxicity and immunogenicity (Rolland, 1998). The low levels of cytotoxicity and immunogenicity make liposomal vectors generally safer for human gene therapy studies. Possibly the greatest drawback to the use of liposomal vectors is found in their lack of transfection efficiency when compared to viral vectors (Audouy et al, 2002). One method found to increase the transfection efficiency of liposomal vectors is to pre-treat the target cells with chemotherapeutic agents, such as cisplatin, colchicines, mechlorethamine (nitrogen mustard) or paclitaxel (Son and Huang, 1996; Son, 1997, 1999; Nair et al, 2002). The use of chemotherapeutic agents to increase the transfection efficiency of liposomal gene therapy may have significant benefits in gene therapy protocols to treat cancer, as these patients may be already receiving chemotherapy as part of their current treatment.
Genetic alteration to the p53 tumour suppressor gene is the most commonly observed abnormality in human cancers, making it an ideal target for gene therapy trials (Fisher, 2001). p53 plays a significant role in cell cycle regulation and the induction of apoptosis in response to DNA damage. Its mutation has been implicated with poor clinical prognosis for various types of malignancies as well as the induction of chemo-resistance (Rahko et al, 2003). A number of studies have previously shown that the reintroduction of p53 via gene therapy can result in a return of chemo-sensitivity in cells which were previously chemo-resistant to cisplatin (Miyake et al, 1998; Weinrib et al, 2001). These cells treated with cisplatin following p53 gene therapy exhibited increased levels of apoptosis and tumour reductions compared to either treatment alone. The current study differs from these previous studies by using a non-therapeutic dose of cisplatin seven days prior to liposomal delivered p53 gene therapy. The function of the cisplatin is to sensitise the tumour to increased lipofection rather than induce a therapeutic response. Any tumour reduction would occur as a result of increased transfection of the p53 tumour suppressor gene.
In this study we examine whether the pre-treatment of a tumour, in vivo, with cisplatin can result in both an increased anti-tumoural response as well as increased levels of p53 transfection.
II. Materials and methods
A. p53 DNA preperation
A 1.85 kb XbaI-bound human p53 cDNA cloned into the polylinker of the pRcCMV eukaryotic expression vector was kindly provided by The Children’s Medical Research Institute, Sydney, Australia. The plasmid was amplified in JM109 E.Coli cells and purified with QIAprepì Spin Miniprep Kit (QIAGEN). Quantification of plasmid was performed by gel electrophoresis prior to binding with liposomes (Sambrook & Russell 2000).
B. Animals
Syngeneic DA rats were housed 3–5 per cage, sex segregated, in temperature and humidity controlled rooms. The lighting conditions were based off a 12 hour light/dark cycle. Food and tap water were given ad libitum through wire roofed plastic cages. For experimental work, all animals were over 10 weeks old and were randomised by weight and sex into designated groups (six animals per group). All experiments were conducted with the approval of the Charles Sturt University Animal Care and Ethics Committee.
C. Tumour implantation
Solid tumours were established from a transplantable rat salivary adenocarcinoma (CSU-SA1). CSU-SA1 has been shown to have a mutation to the p53 tumour suppressor gene (unpublished results). The tumours were grown on the lateral aspect of the hind limbs of DA rats. A small incision was made through the skin and a 1mm3 piece of healthy tumour was implanted subcutaneously. Tumour growth was assessed daily using calibrated vernier callipers and expressed as the product of the minimal and maximal axises of the tumour. This method has been used by this and other groups (Burton et al, 1990; Napoli et al, 1992; Walker et al, 1996).
D. Cisplatin dose determination
A preliminary study was performed to determine a non-therapeutic dose of cisplatin. Five animals per group were implanted with tumours, as above. Six days post-implantation of the tumour tissues the animals were treated with a single intra-peritoneal injection of either 5mg/kg or 1mg/kg of cisplatin. The animals were examined at 24-hour intervals for the next 5 days for tumour growth as well as signs of cytotoxicity. The dose of cisplatin, which exhibited no significant anti-tumour effect, or signs of toxicity was chosen for the p53 study.
E. Preparation of liposomes
Liposomes consisting of small unilamellar vesicles (SUV) were prepared by the injection of an ethanoic solution of lipids into an aqueous solution as previously described (Campbell, 1995). Briefly, eight milligrams of dimethyldioctadecyl ammonium bromide (DDAB) and four milligrams of Dioleoyl-L-phosphatidylethanolamine (DOPE) were dissolved in 1.0mL absolute ethanol to give mass ratios of 2:1 of DDAB:DOPE. 50 μl of the ethanoic lipid mix was rapidly injected (over 0.5s) into vortexing water. This method results in spontaneous rearrangement of the lipids into SUV. The liposome mix was filtered through 0.2 μm polycarbonate filters to size liposomes to a maximum diameter of 0.2 μm.
F. Preparation of p53-liposome complexes
The p53-liposome complexes were prepared by adding 35 μg of p53 plasmid to 35nmol of DDAB:DOPE liposomes made up to a total volume of 150 μl with saline. The formulation was mixed gently and incubated at room temperature for 15–30 minutes to allow complexing of DNA to the liposomes. Plasmid-liposome complexes were made on the day of the treatment and kept on ice until 5 minutes before use, at which point they were they were warmed to 37°C for injection.
G. Injection protocol
On day six (post-tumour implantation) a dose of 1mg/kg of cisplatin was injected intra-peritoneally into the treatment group of DA rats (6 animals per treatment group). This was the dose of cisplatin shown not to produce a tumour response from the dose determination study. On day 13, the treatment and control groups received two 150 μl intratumoural injections of either saline (for saline control) or p53-liposome complexes (for p53 control and treatment groups). The injections were performed over a 3 second period and the needle was left in the tumour for up to 5 minutes to prevent back-flushing of the treatment. The animals were sacrificed 96 hours later and their tumours examined histologically for signs of apoptosis.
H. p53 expression detection
For this study, tumours were examined for p53 expression 12, 36, or 96 hours post-treatment. Tumours from 4 animals per time point were analysed for each of the treatments. The tumour tissue was homogenised in phosphate buffered saline (PBS), lysed with 2% sodium dodecyl sulfate (SDS) and pelleted by centrifugation (12 000 g, 10 min). A 50μl aliquot of the resulting supernatant was added to 50μl of sample buffer (0.05M Tris (pH 6.8), 3% SDS, 20% Glycerol, 6% 2-Mercaptoethanol and 0.001% Bromophenol blue) and boiled for 3 minutes. The resulting mix was loaded on a SDS/Polyacrylamide gel (3% stacking, 12% running gel) and electrophoresed at 15mA for 10 hours. The protein was transferred to a nitrocellulose membrane using established techniques (Napoli et al, 1992). The blotted proteins were immunologically probed using a human monoclonal anti-p53 antibody (1:1,000 (vol/vol); Pab-1801, Santa Cruz) and an anti-goat immunoglobulin G conjugated with alkaline phosphatase (1:1,000 (vol/vol); Sigma). The membrane was then washed and developed colorimetrically with FAST Red TR/Naphthol AS-MX (4-chloro-2-methylbenzene diazonium/3-hydroxy-2-naphthoic acid 2,4-dimethylanilide phosphate; Sigma). The membrane was scanned, and density analysis was performed with the Sigma Gel program (SPSS, Inc.). All densitometry readings were normalized to nanograms of total protein loaded onto the gel.
I. Histology
Tumours were excised from the hind limb and fixed in 10% neutral buffered formalin for a minimum of 48 hours before being wax-embedded. Serial sections were cut and stained with Haematoxylin and Eosin. Sections were examined for histological signs of apoptosis, namely the presence of apoptotic bodies. The level of apoptosis at the treatment site of the tumour was determined as the average number of apoptotic bodies per 1000 cells taken from 10 sections of the tumour. This was repeated for each of the 6 tumours per group. Slides were examined randomly to avoid microscopist bias.
J. Statistical evaluation
Statistical evaluation of the differences between each of the treatments was determined using the one-way analysis of variance test followed by the Student-Kewman-Keuls multiple comparison procedure. Statistical analysis was performed at 95% confidence level (p=0.05).
III. Results
A. Cisplatin dose determination
This experiment was designed to determine the dose of cisplatin to be used for pre-sensitisation in the p53 tumour response assays. Two doses of cisplatin were examined, 1mg/kg and 5mg/kg delivered intra-peritoneally (Figure 1). The 5mg/kg dose resulted in a significant anti-tumour response with tumours exhibiting regression in size (p<0.01). The mean size of the tumours (at the completion of the study) treated with the 5mg/kg was 25mm2 compared to the saline treated control tumours that were over 200mm2. Four of the five rats given 5mg/kg cisplatin also exhibited some signs of toxicity. Animals receiving 1mg/kg exhibited no signs of toxicity and no significant reduction in tumour size or tumour growth when compared to the saline treated control (p>0.05). The 1mg/kg dose of cisplatin was chosen for the pre-sensitisation studies.
Figure 1.
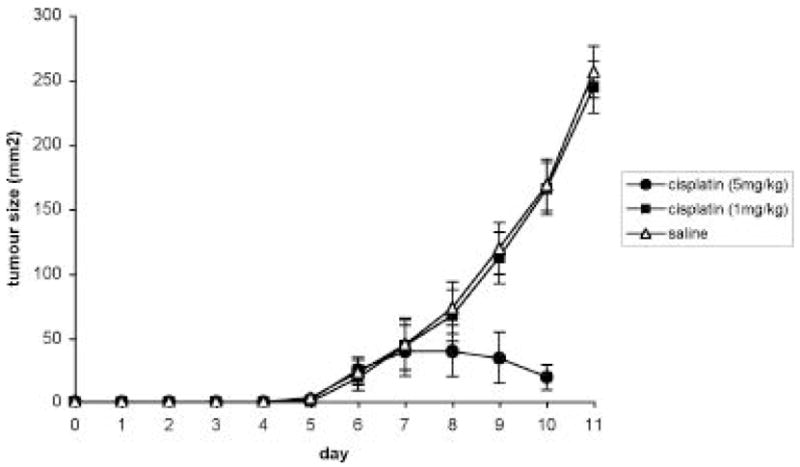
Tumour growth curve for cisplatin dose determination. Five rats per group were treated with a single intra-peritoneal injection of saline, cisplatin at 5mg/kg or cisplatin at 1mg/kg on day 6. Tumour growth was evaluated daily. Each time point is the average size of 5 tumours +/− the SD of the mean.
B. In vivo tumour response
In vivo experiments were designed to test the efficiency of p53-liposome complexes on the growth of CSU-SA1 cells following a non-therapeutic dose of cisplatin. A dose of 1mg/kg of cisplatin was delivered i.p. into the treatment group seven days prior to liposome transfection. Following p53 lipofection, the cisplatin treated group was compared to non-cisplatin treated groups (saline control, empty liposomes and p53-liposome control) for differences in growth kinetics (Figure 2). Both the p53-liposome control and the cisplatin treated p53-liposome group caused significant tumour growth retardation (p<0.05) when compared to the saline and liposome controls. The saline and the liposome treated tumours increased in size by greater than 164% over a 96-hour period compared to 64% and 101% for the cisplatin and non-cisplatin p53-liposome treated tumours over the same time period. The cisplatin p53-liposome treated tumours showed a maximum therapeutic response 24–48 hours post p53-liposome injection. During this period these tumours increased in size by only 2% compared to 29% for saline and 27% for the non-cisplatin treated tumours. At the 48-hour time point the cisplatin treated tumours were significantly smaller than either of the non-cisplatin treated tumours (p<0.05). The tumours of the cisplatin treated tumours remained significantly smaller than the non-cisplatin tumours for the remainder of the study (p<0.05). By the 72-hour time point the liposome delivered p53 treated tumours without cisplatin, were exhibiting significant anti-tumour effect when compared to the saline control however these tumours remained larger than the cisplatin pre-treated tumours.
Figure 2.
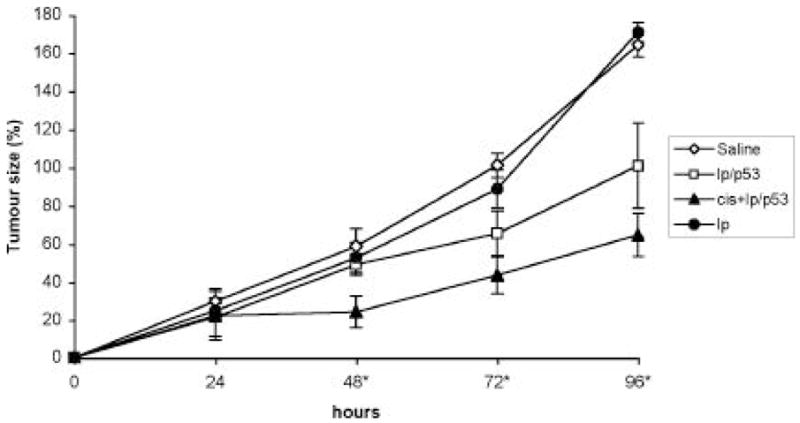
Tumour growth curve following treatment with p53 on liposomes (lp/p53), saline or empty liposomes. Four treatment groups were evaluated: a saline control, a liposome control, a lp/p53 control and cisplatin pre-treated lp/p53 (cis+lp/p53). The cisplatin pre-treated tumours were given a dose of cisplatin at 1mg/kg seven days prior to treatment with lp/p53. Each time point represents the average tumour size increase of 6 tumours +/− the SD of the mean. * indicates where cis+lp/p53 treated tumours are significantly smaller than other treatments.
C. Protein expression
p53 protein expression was determined by SDS-PAGE followed by “Western Blotting”. A human specific p53 antibody (not cross reactive with rat p53) was used to detect the p53 expressed in the tumour (Figure 3). As human p53 was used in this experiment, all p53 detected with this antibody was as a direct result of liposome transfection. Three time points were chosen in which 4 tumours per group were analysed for p53 expression. The first analysis was performed 12 hours post p53-liposome treatment, the second 36 hours post treatment and the final 96 hours post treatment (Table 1). The level of p53 expression 12 hours post treatment does not reveal any significant difference in the protein expression levels between the cisplatin and non-cisplatin treated tumours. By the 36 hour time point a significant difference in the level of p53 protein expression between the cisplatin and non-cisplatin treated groups could be noted (p<0.05), with the cisplatin treated group producing 76% more p53 protein than the non-cisplatin treated tumours. This trend increased by the 96 hour time point where the cisplatin treated tumours were producing 8-fold more p53 protein than the non-cisplatin treated tumours. Whilst there was no difference in the maximum levels of p53 expression between cisplatin and non-cisplatin treated groups there was a significant difference in the levels of p53 over time. p53 expression for both the cisplatin and non cisplatin treated tumours fell significantly over the time course of the experiment, however the cisplatin treated tumours continued to express p53 at higher levels than non cisplatin treated tumours for the course of the experiment.
Figure 3.

Western blot of human p53 expression within rat tumours following treatment with human p53, with or without cisplatin pre-treatment, and saline. Lanes 1, 2, 3 and 7, 8, 9 represent the expression of p53 following cisplatin treatment 12, 36 and 96 hours respectively. Lanes 4, 5, 6 and 10, 11, 12 represent the expression of p53 without cisplatin treatment and lanes 13, 14, represents the p53 expression following treatment with a saline control.
Table 1.
p53 protein expression following treatment with lp/p53 with (cis+Lp/p53) and without (Lp/p53) cisplatin pre-treatment or a saline control. Protein determination was examined immunologically using a human p53 specific antibody, which did not cross react with rat p53, followed by western blotting. Each time point represents the average western blot band intensity of four tumours +/− the SD of the mean.
| Treatment | 12 hours | 36 hours | 96 hours |
|---|---|---|---|
| Cis+Lp/p53 | 10653 +/− 471 | 6595 +/− 389 | 2281 +/− 286 |
| Lp/p53 | 11140 +/− 383 | 3748 +/− 147 | 285 +/− 67 |
| Saline control | 4 +/− 3 | 3 +/− 1 | 7 +/− 3 |
D. Histological analysis
As apoptosis is one of the primary ways in which p53 functions to induce tumour suppression, increased levels of apoptosis is an indicator of the functionality and presence of the p53 in this rat model. Histological evaluation of the tumours revealed the presence of significantly more apoptotic bodies around the p53 treatment site than that of the saline control tumours (p<0.05) (Figure 4). At the treatment site, p53 treated tumours showed levels of apoptosis more than 50-fold greater than the level of apoptosis in the saline control tumours, however this high level of apoptosis was limited to areas close to the injection site and the needle tract (data not shown). Apoptosis levels at a distance to this site were not significantly different to the saline control.
Figure 4.
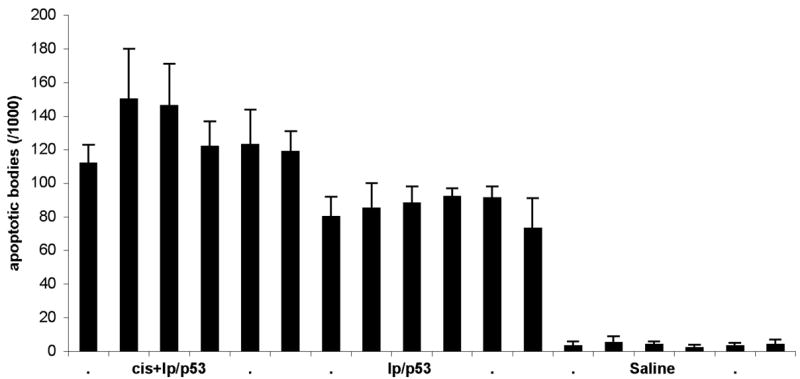
Apoptotic bodies/1000 cells following treatment with lp/p53 (with or without cisplatin pre-treatment) or saline. The cisplatin pre-treated tumours were given a dose of cisplatin at 1mg/kg seven days prior to treatment with lp/p53. Animals were sacrificed 96 hours post-treatment and the tumours examined histologically for the numbers of apoptotic bodies present surrounding the treatment site. Each bar represents the average number of apoptotic bodies present in ten sections per tumour +/− the SD of the mean. Six animals were used for each treatment.
The restriction of apoptosis to areas close to the injection site was not however a result of the lack of functional status of the p53 but as a consequence of limited treatment distribution as a result of the use of liposomes. (Nomura et al, 1997). On comparing the cisplatin pre-treated tumours with those not pre-treated with cisplatin we found that there was a statistical difference between the numbers of apoptotic bodies found following p53 lipofection (p<0.05), with an average of 129 apoptotic cells/1000 cells +/− 15 for the cisplatin treated tumours and 85 apoptotic cells/1000 +/− 7 for non-cisplatin treated tumours. Once again apoptosis was primary limited to the injection site and needle tract indicating that the cisplatin pre-treatment did not increase the distribution of the liposomes throughout the tumour.
IV. Discussion
The primary aim of this study was to attempt to increase the efficiency of liposomal vectors by the pre-treatment of the tumour with a low dose of the chemotherapeutic drug cisplatin (prior to lipofection) such that a greater anti-tumoural response could be achieved. We showed that this was indeed possible with animals pre-treated with the cisplatin having 36% smaller tumours after lipofection with p53 compared to those animals without cisplatin pre-treatment. These animals showed a characteristic pattern of tumour response in which maximum anti-tumoural effects occurred 24–48 hours post liposome p53 treatment. This correlated to a time point in which the cisplatin treated tumours were expressing over 75% more p53 protein than that of the non-cisplatin pre-treated tumours. Interesting both the cisplatin and non-cisplatin treated tumours exhibited the highest level of p53 protein expression after the first 12 hours at which point there was no statistical difference between the expression levels. However this time point correlated with no significant growth reduction for either p53 treated group when compared to the saline controls. At this early post-treatment stage the p53 protein would be building up within cells prior to apoptosis so it is reasonable to have little anti-tumour effect at this point. By the completion of the study (96-hours post-treatment) the level of p53 expression in both the cisplatin and non-cisplatin pre-treated tumours had fallen significantly. The cisplatin pre-treated tumours however had significantly higher levels of p53 protein detected indicating that these tumours exhibited prolonged expression. At this time point the cisplatin pre-treated tumours also displayed significantly higher levels of apoptosis.
While the exact mechanism for the increased liposomal efficiency is unknown, we believe that the ability of the cisplatin to cause cross-linking of the DNA may induce a number of biological changes occur to the cell to aid in the repair of the DNA damage or to induce apoptosis. One of these changes known to occur is an increase in intracellular trafficking and its associated modification to membrane transport (Son & Huang 1994). The changes in intracellular trafficking may aid in the escape of the liposomal vectors from the endosome (a limiting factors to successful liposomal transfection: (Rolland 1998)) or increase the efficiency of transport of the liposomes from the cytoplasm into the nucleus (another limiting factor to lipofection: (Zabner et al. 1995)). The altering of intracellular trafficking may also be responsible for the prolonged protein expression and the resulting prolonged tumour response, this however is yet to be confirmed.
The use of liposomes as a gene vector for the treatment of cancer has been shown to be a safe clinical option resulting in little toxicity. Whilst the use of liposomes in vitro have shown efficient transfection rates, this has not transcribed into in vivo studies which have shown low transfection efficiencies. This study has shown that the transfection efficiencies of liposomes can be increased through the use of cisplatin resulting in increase tumour response when combined with the tumour suppressor gene p53.
The use of cisplatin as a chemotherapeutic agent is often limited by a build-up of chemo-resistance. The return of functional p53 following cisplatin administration would not only induce tumour suppression but also return chemo-sensitivity allowing an increased effectiveness of cisplatin as a chemotherapeutic agent. A successful treatment regime may involve the pre-treatment of tumour cells with a dose of cisplatin, followed by liposomal delivered p53, followed by a second dose of cisplatin and a second dose of p53 and so on, with each cycle of treatment positively influencing the next treatment.
Abbreviations
- DDAB
dimethyldioctadecyl ammonium bromide
- DOPE
Dioleoyl-L-phosphatidylethanolamine
- (PBS)
phosphate buffered saline
- (QIAGEN)
QIAprep™ Spin Miniprep Kit
- (CSU-SA1)
salivary adenocarcinoma
- (SUV)
small unilamellar vesicles
- (SDS)
sodium dodecyl sulfate
References
- Audouy S, de Leij L, Hoekstra D, Molema G. In vivo characteristics of cationic liposomes as delivery vectors for gene therapy. Pharm Res. 2002;19:1599. doi: 10.1023/a:1020989709019. [DOI] [PubMed] [Google Scholar]
- Burton M, Jones C, Trotter J, Gray B, Codde J. Efficacy of ion-exchange resins for anti-tumour drug delivery. Regional Cancer Treatments. 1990;3:36. [Google Scholar]
- Campbell M. Lipofection reagents prepared by a simple ethanol injection technique. Biotechniques. 1995;18:1027. [PubMed] [Google Scholar]
- Fisher D. The p53 tumor suppressor, critical regulator of life & death in cancer. Apoptosis. 2001;6:7. doi: 10.1023/a:1009659708549. [DOI] [PubMed] [Google Scholar]
- Kigawa J, Sato S, Shimada M, Kanamori Y, Itamochi H, Terakawa N. Effect of p53 gene transfer and cisplatin in a peritonitis carcinomatosa model with p53-deficient ovarian cancer cells. Gynecol Oncol. 2002;84:210. doi: 10.1006/gyno.2001.6488. [DOI] [PubMed] [Google Scholar]
- Miyake H, Hara I, Gohji K, Yamanaka K, Arakawa S, Kamidono S. Enhancement of chemosensitivity in human bladder cancer cells by adenoviral-mediated p53 gene transfer. Anticancer Res. 1998;18:3087. [PubMed] [Google Scholar]
- Nair R, Rodgers J, Schwarz L. Enhancement of transgene expression by combining glucocorticoids and anti-mitotic agents during transient transfection using DNA-cationic liposomes. Mol Ther. 2002;5:455. doi: 10.1006/mthe.2002.0567. [DOI] [PubMed] [Google Scholar]
- Napoli S, Burton M, Martins I, Chen Y, Codde J, Gray B. Dose response and toxicity of doxorubicin microspheres in a rat tumor model. Anticancer Drugs. 1992;3:47. doi: 10.1097/00001813-199202000-00009. [DOI] [PubMed] [Google Scholar]
- Nomura T, Nakajima S, Kawabata K, Yamashita F, Takakura Y, Hashida M. Intratumoural pharmacokinetics and in vivo gene expression of naked plasmid DNA and its cationic liposome complexes after direct gene transfer. Cancer Res. 1997;57:2681. [PubMed] [Google Scholar]
- Rahko E, Blanco G, Soini Y, Bloigu R, Jukkola A. A mutant TP53 gene status is associated with a poor prognosis and anthracycline-resistance in breast cancer patients. Eur J Cancer. 2003;39:447. doi: 10.1016/s0959-8049(02)00499-9. [DOI] [PubMed] [Google Scholar]
- Rolland JA. From genes to gene medicines, Recent advances in nonviral gene delivery. Crit Rev Ther Drug Carrier Syst. 1998;15:143. doi: 10.1615/critrevtherdrugcarriersyst.v15.i2.20. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russell D. Molecular Cloning, a Laboratory Manual. 3. New York: Cold Spring Harbor Laboratory Press; 2000. [Google Scholar]
- Son K. Cisplatin-based interferon gamma gene therapy of murine ovarian carcinoma. Cancer Gene Ther. 1997;4:391. [PubMed] [Google Scholar]
- Son K. Chemical toxicants activate murine ovarian ascitic tumor cells for in situ lipofection. Drug Deliv. 1999;6:75. [Google Scholar]
- Son K, Huang L. Exposure of human ovarian carcinoma to cisplatin transiently sensitizes the tumor cells for liposome-mediated gene transfer. Proc Natl Acad Sci U S A. 1994;91:12669. doi: 10.1073/pnas.91.26.12669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son K, Huang L. Factors influencing the drug sensitization of human tumor cells for in situ lipofection. Gene Ther. 1996;3:630. [PubMed] [Google Scholar]
- Walker T, White J, Esdale W, Burton M, De CE. Tumour cells surviving in vivo cisplatin chemotherapy display elevated c-myc expression. Br J Cancer. 1996;73:610. doi: 10.1038/bjc.1996.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinrib L, Li J, Donovan J, Huang D, Liu F. Cisplatin chemotherapy plus adenoviral p53 gene therapy in EBV-positive and -negative nasopharyngeal carcinoma. Cancer Gene Ther. 2001;8:352. doi: 10.1038/sj.cgt.7700319. [DOI] [PubMed] [Google Scholar]
- Zabner J, Fasbender AJ, Moninger T, Poellinger KA, Welsh MJ. Cellular and molecular barriers to gene transfer by a cationic lipid. J Biol Chem. 1995;270:8997. doi: 10.1074/jbc.270.32.18997. [DOI] [PubMed] [Google Scholar]