

Abstract
The acidic oligosaccharides of human milk are predominantly sialyloligosaccharides. Pathogens that bind sialic acid-containing glycans on their host mucosal surfaces may be inhibited by human milk sialyloligosaccharides, but testing this hypothesis requires their reliable quantification in milk. Sialyloligosaccharides have been quantified by anion exchange HPLC, reverse or normal phase HPLC, and capillary electrophoresis (CE) of fluorescent derivatives; in milk, these oligosaccharides have been analyzed by high pH anion exchange chromatography with pulsed amperometric detection, and, in our laboratory, by CE with detection at 205 nm. The novel method described herein uses a running buffer of aqueous 200 mM NaH2PO4 at pH 7.05 containing 100 mM SDS made 45% (v/v) with methanol to baseline resolve five oligosaccharides, and separate all 12. This allows automated simultaneous quantification of the 12 major sialyloligosaccharides of human milk in a single 35-minute run. This method revealed differences in sialyloligosaccharide concentrations between less and more mature milk from the same donors. Individual donors also varied in expression of sialyloligosaccharides in their milk. Thus, the facile quantification of sialyloligosaccharides by this method is suitable for measuring variation in expression of specific sialyloligosaccharides in milk and their relationship to decreased risk of specific diseases in infants.
Keywords: human milk oligosaccharides, sialyloligosaccharides, capillary electrophoresis
INTRODUCTORY STATEMENT
Human milk contains a large number of compounds that provide nutritional support and contribute toward defense of the newborn. Human milk oligosaccharides (HMOS), a principal component of human milk, are found in concentrations of 6 to 12 g/L, and these oligosaccharides vary in size, structure, and abundance [1]. The oligosaccharides of milks from other species vary widely, and the composition of oligosaccharides from human milk is unique [2].
HMOS play a potentially protective role to nursing infants. Breastfed infants have a different microbiota than infants fed artificially, and human milk glycans are thought to act as prebiotics that promote the growth of Bifidobacterium bifidus [3]. Breast-fed infants have fewer or less severe gastrointestinal and respiratory infections during the first year of life than formula-fed infants, and oligosaccharides in human milk contribute significantly to this phenomenon [4]. The adhesion of microbes to mucosal epithelial cells, an essential first step for pathogenesis by Campylobacter jejuni, Escherichia coli, Vibrio cholerae, and many rotavirus and norovirus strains, can be prevented by specific inhibition by HMOS. Such inhibition can result from homology between epithelial cell surface receptor glycans and HMOS [5, 6]. HMOS may also modify glycan expression by intestinal epithelium. Exposure of Caco-2 intestinal epithelial cells to 3'-sialyllactose, one of the predominant sialylated HMOS, changed the micro-array glycan profiling of Caco-2 cells, and reduced the adhesion of enteropathogenic E. coli by 50% [7].
HMOS are composed of 5 monosaccharides: D-glucose (Glc), D-galactose (Gal), N-acetylglucosamine (GlcNAc), L-fucose (Fuc), and sialic acid (N-acetyl neuraminic acid, Neu5Ac, NANA) [8]. HMOS are usually divided into two groups according to their chemical structures: neutral compounds containing Glc, Gal, GlcNAc, and often Fuc, linked to a lactose (Galβ1-4Glc) core, and acidic compounds including the same sugars, and often the same core structures, plus NANA [9]. More than 130 different HMOS have been identified [8]. However, the amounts of HMOS expressed in human milk change during the course of lactation and vary among individuals [10, 11]. To observe these differences, it is critical to have efficient, simple, quantitative methods for routine measurement of HMOS in large numbers of samples.
Several methods for the separation and quantification of neutral compounds in human milk have been described, including high-performance liquid chromatography (HPLC) [12-15] and capillary electrophoresis (CE) of derivatized HMOS [16], and HPLC with high pH amperometric detection of underivatized HMOS [17]. Acidic oligosaccharides have been resolved by anion exchange HPLC of radiolabelled derivatives [18], derivatization of the oligosaccharides followed by ion exchange LC with absorption at 200 nm [19] and capillary electrophoresis of fluorescent derivatives [20, 21]. Native acidic oligosaccharides of milk have been resolved into monosialyl-, disialyl-, and trisialyloligosaccharide mixtures by medium-pressure anion-exchange chromatography with absorption at 214 nm [22], but further resolution was not achieved. A technique more commonly employed for native milk oligosaccharide resolution and quantification is high pH anion exchange chromatography with pulsed amperometric detection [17, 23-26], but the electrochemical response factors for anionic sialyloligosaccharides by this technique are not consistent, and differ by the linkage position of the sialic acid [23]. Reverse- or normal-phase chromatography was able to resolve derivatized or reduced acidic oligosaccharides, with detection by UV absorbance or spectrofluorimetry as the basis for quantification [27, 28]. In our laboratory, capillary electrophoresis with detection at 205 nm was shown to be able to resolve human milk sialyloligosaccharides effectively, and to have a linear response factor over a wide range of concentrations. However, under conditions that resolved four sialyloligosaccharide standards, this technique only partially resolved the three isomers of sialyllacto-N-tetraose. Conversely, conditions that fully resolved the sialyllacto-N-tetraose isomers would only partially resolve the other four sialyloligosaccharides; to quantify all seven of these glycans required two runs. At that time only 7 acidic oligosaccharide standards were available to validate the resolution and quantification of the milk acidic oligosaccharides [9].
Our objective was to develop, in a single assay with modest run time, a technique that would be able to resolve, identify, and quantify 12 major acidic oligosaccharides simultaneously in human milk. Twelve authentic standards were used to test for reproducibility, accuracy, and sensitivity, both independently of and within the matrix of human milk. The utility of the method was then confirmed by its ability to detect variation in sialyloligosaccharide expression between less and more mature milk of the same mother, and variation among milks from different mothers.
MATERIALS AND METHODS
Reference standards
Monosialyl, monofucosyllacto-N-neohexaose, monosialyllacto-N-neohexaose I, monosialyl, monofucosyllacto-N-hexaose I, sialyllacto-N-fucopentaose II, sialyllacto-N-tetraose b, sialyllacto-N-tetraose c, sialyllacto-N-tetraose a, disialomonofucosyllacto-N-neohexaose, 3'-sialyl-3-fucosyllactose, 3'-sialyllactose, and disialyllacto-N-tetraose were obtained from Glycoseparations (Moscow, Russia). Authentic 6'-sialyllactose was purchased from Sigma Chemical Company (St. Louis, MO, USA). Abbreviations and structures of the above twelve sialyloligosaccharides are shown in Table 1. Monosodium phosphate (NaH2PO4) was obtained from Sigma-Aldrich (Milwaukee, WI, USA), sodium dodecyl sulfate (SDS) from Bio-Rad Laboratories (Richmond, CA, USA) and methanol (MeOH) and ethanol (EtOH) from Fisher Scientific (Waltham, MA, USA). MilliQ deionized water was used in this study.
Table 1.
Structures of sialyloligosaccharides quantified in this study
| Abbreviation | Trivial name | Structure |
|---|---|---|
| MSMFLNnH | Monosialyl, monofucosyllacto-N-neohexaose |
NANAα2-6Galβ1-4GlcNAcβ1-3(Galβ1-4GlcNAcβ1-6)Galβ1-4Glc |
| MSLNnH I | Monosialyllacto-N-neohexaose I | NANAα2-6Galβ1-3GlcNAcβ1-3(Galβ1-4GlcNAcβ1-6)Galβ1-4Glc |
| MSMFLNH I | Monosialyl, monofucosyllacto-N-hexaose I |
NANAα2-6Galβ1-4GlcNAcβ1-6(Fucα1-2Galβ1-3GlcNAcβ1-3) Galβ1-4Glc |
| SLNFP II | Sialyllacto-N-fucopentaose II | NANAα2-3Galβ1-3(Fucβ1-4)GlcNAcβ1-3Galβ1-4Glc |
| LST b | Sialyllacto-N-tetraose b | NANAα2-6(Galβ1-3)GlcNAcβ1-3Galβ1-4Glc |
| LST c | Sialyllacto-N-tetraose c | NANAα2-6Galβ1-4GlcNAcβ1-3Galβ1-4Glc |
| LST a | Sialyllacto-N-tetraose a | NANAα2-3Galβ1-3GlcNAcβ1-3Galβ1-4Glc |
| DSMFLNH | Disialyl, monofucosyllacto-N-neohexaose |
NANAα2-3Galβ1-3(NANAα2-6)GlcNAcβ1-3(Galβ1-4(Fucα1-) GlcNAcβ1-6)Galβ1-4Glc |
| 3′-S-3-FL | 3′-Sialyl-3-fucosyllactose | NANAα2-3Galβ1-4(Fucα1-3)Glc |
| 6′-SL | 6′-Sialyllactose | NANAα2-6Galβ1-4Glc |
| 3′-SL | 3′-Sialyllactose | NANAα2-3Galβ1-4Glc |
| DSLNT | Disialyllacto-N-tetraose | NANAα2-6Galβ1-3(NANAα2-6) GlcNAcβ1-3Galβ1-4Glc |
Milk
Surplus milk from Massachusetts mothers was used for validation of the method, as approved by the IRB of Massachusetts General Hospital. Human milk was collected with a breast pump from 13 healthy donors. Milk samples were stored at −20°C until use. Pooled milk used for method development was a mixture of random amounts of milk samples from these 13 donors at different stages of lactation, and these milks were analyzed individually after the method had been standardized to determine variation of expression.
Preparation of Milk oligosaccharides
Milk samples were thawed and an aliquot of 550 μL was centrifuged at 4,000 × g for 15 minutes at 4°C. While avoiding the upper, creamy layer, 400 μL of liquid was pipetted from the lower, aqueous layer into a clean test tube. EtOH (800 μL) was added to each sample (to give 66.7% EtOH), mixed and kept at 4°C overnight. The precipitate, containing mostly proteins, was removed by centrifugation at 12,000 × g for 10 minutes at 4°C. The clear supernatant, consisting primarily of oligosaccharides and lactose, was transferred to another test tube and dried under nitrogen at room temperature. The dried residue was reconstituted with 100 μL of fresh 30% MeOH solution and stored at −20°C until CE analysis.
CE
Analysis was performed on a Hewlett Packed 3D capillary electrophoresis apparatus (Waldbronn, German); parameters were controlled by Agilent Chemstation software, which also acquired and processed data. Capillary dimensions were 56 cm effective length and 50 μm I.D. with extended light path geometry. Detection was by absorbance at 205 nm, and applied voltage was 30 kV with normal polarity (sample was loaded at the anode and detected at the cathode). Temperature was maintained at 25°C. In a typical run, injection was at 50 mbar for 6 s, loading approximately 2 nL into the column. The running buffer was 55% aqueous buffer (200 mM NaH2PO4 at pH 7.05 with 100 mM SDS) mixed with 45% (v/v) pure MeOH as the organic modifier.
Standard Curves
Primary stock solutions of the 12 oligosaccharide standards were dissolved in 30% MeOH at 20 mg/mL. Each stock solution was serially diluted with 30% MeOH to 2500, 1250, 625, 312, 156, 78, and 39 μg/mL to construct standard curves. The oligosaccharide peak area (Y) was regressed against the nominal concentration of the test oligosaccharide (X, μg/mL) with weighting of the reciprocal concentration (1/X) to create each standard curve, and its closeness of fit was calculated as the regression coefficient r.
Method Validation
To determine the precision and accuracy of the method, 30% MeOH solutions of each of the 12 test oligosaccharides were analyzed individually 5 times at three different nominal concentrations (156, 625, and 1250 μg/mL). The measured value was calculated from the standard curve (previously obtained as the regression equation), expressed as the mean of the 5 values. The accuracy was measured as the difference between the nominal value and measured value expressed as a percentage of the nominal value. The precision was expressed as the coefficient of variation (CV), i.e., the standard deviation divided by the mean value multiplied by 100.
Recovery from milk
To evaluate the recovery of each test oligosaccharide from human milk, known amounts of each authentic standard (156, 625, and 1250 μg/mL) were added individually to pooled human milk. The measured concentration after addition of the standard to milk, minus the measured value in the milk without the addition of standard, all divided by the value of the standard run alone, multiplied by 100 gave the percent recovery at each concentration of each standard.
Measuring individual variation
Colostrum (d 2 to 4) samples were available from 5 mothers, milk (d 12 to 67) samples were available from another 5 women, and matched less and more mature milk samples were available from three mothers (d 4, 21; d 3, 15; d 5, 9). The acidic oligosaccharides were measured in each sample to assess whether this method is able to detect natural variation in the amount of sialyloligosaccharides expressed in human milk by different mothers, and in the amount expressed in the same mother at different stages of lactation.
RESULTS
Method
The CE running conditions using a Tris-based buffer system that we had described previously only marginally separated 3'-SL from 6'-SL, and did not afford separation of the LST isomers or some of the other sialyloligosaccharides for which we now have standards. A phosphate-based buffer system was explored for its ability to simultaneously separate these 12 sialyloligosaccharides of human milk, and 200 mM NaH2PO4 brought to pH 7.05 provided a promising point of departure. However, without further modification, this system could not resolve peaks 4 and 5 (SLNFP II and LST b), peaks 6 and 7 (LST c and LST a), peaks 8 and 9 (DSMFLNH and) and peaks 10 and 11 (6'-SL and 3'-SL). Incremental addition of MeOH to the phosphate running buffer afforded increased resolution of peaks 4/5, 8/9, and 10/11. Figure 1 illustrates that the best resolution in the least time occurred when 55% buffer was mixed with 45% MeOH, the organic modifier. Addition of higher amounts of methanol caused increased running time, broader peaks, and less resolution of peak 9 (3'-S-3-FL) from peak 10 (6'-SL). Addition of methanol at any concentration did not improve the resolution of peaks 6/7 (LST c and LST a).
Figure 1.
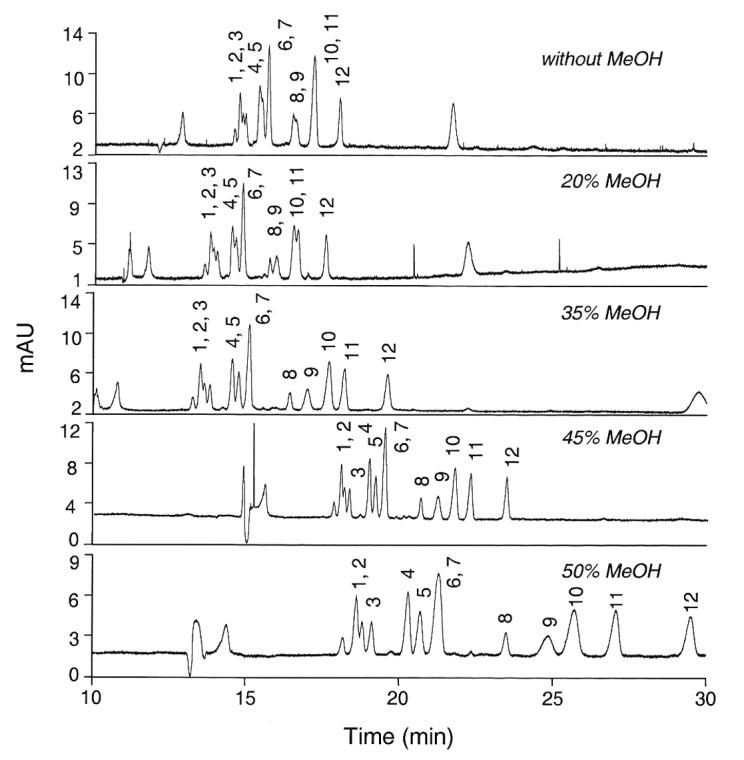
Resolution of twelve sialyloligosaccharides with increasing concentrations of the organic modifier methanol. The running buffer consisted of 200 mM NaH2PO4 (pH 7.05) containing up to 50% MeOH as an organic modifier. 1, MSMFLNnH; 2, MSLNnH I; 3, MSMFLNH I; 4, SLNFP II; 5, LST b; 6, LST c; 7, LST a; 8, DSMFLNH; 9, 3'-S-3-FL; 10, 6'-SL; 11, 3'-SL; 12, DSLNT.
Further modification of the running buffer with incremental concentrations of the ionic detergent SDS resulted in the changes in resolution depicted in Figure 2. Increasing amounts of SDS increased the resolution of peaks 6/7 (LST c and LST a), but at the expense of decreasing the resolution of peaks 4/5 (SLNFP II and LST b) and peaks 9/10 (3'-S-3-FL and 6'-SL). To be able to measure all of these sialyloligosaccharides simultaneously, the most effective compromise seemed to be a concentration of 100 mM SDS.
Figure 2.
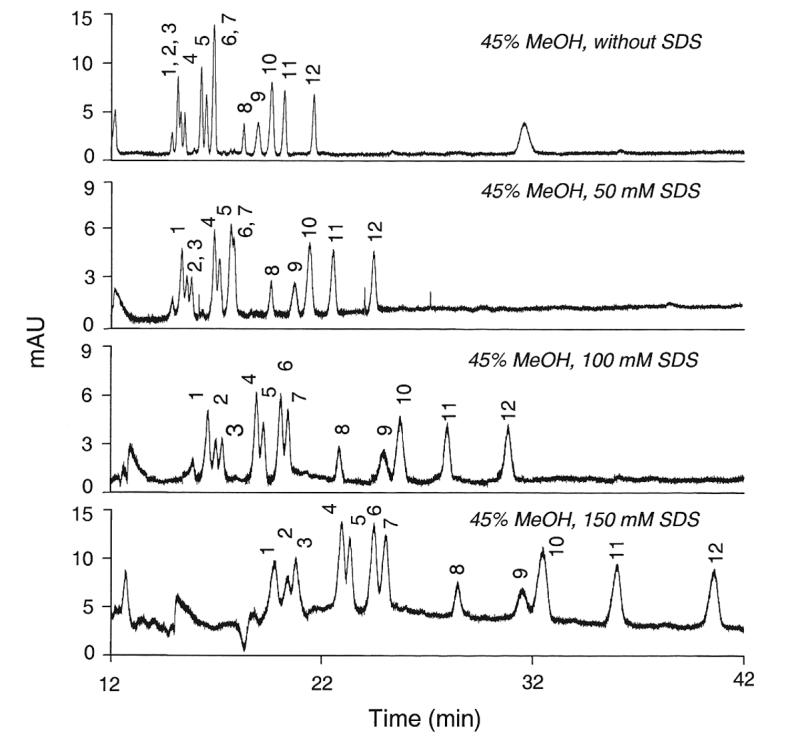
Effect of sodium dodecyl sulfate (SDS) on the resolution of twelve sialyloligosaccharides. The running buffer was the mixture of 200 mM NaH2PO4 (pH 7.05) and MeOH (55:45, v/v) containing up to 150 mM SDS. 1, MSMFLNnH; 2, MSLNnH I; 3, MSMFLNH I; 4, SLNFP II; 5, LST b; 6, LST c; 7, LST a; 8, DSMFLNH; 9, 3'-S-3-FL; 10, 6'-SL; 11, 3'-SL; 12, DSLNT.
The result of using these conditions on standards and on milk samples is demonstrated in Figure 3. The twelve standards (electropherogram A, figure 3) are well resolved, but not all to baseline. The three monosialylhexoses (peaks 1, 2, and 3) are resolved close to baseline. Sialyllacto-N-fucopentaose II (peak 4) did not totally separate from sialyllacto-N-tetraose b (peak 5), nor did the sialyllacto-N-tetraose c (peak 6) fully separate from its isomer, sialyllacto-N-tetraose a (peak 7). All of the remaining standards were baseline resolved. However, in typical colostrum (electroperogram B, figure 3) and milk samples (electropherogram C, figure 3) this resolution results in individual peaks that are readily quantifyable.
Figure 3.
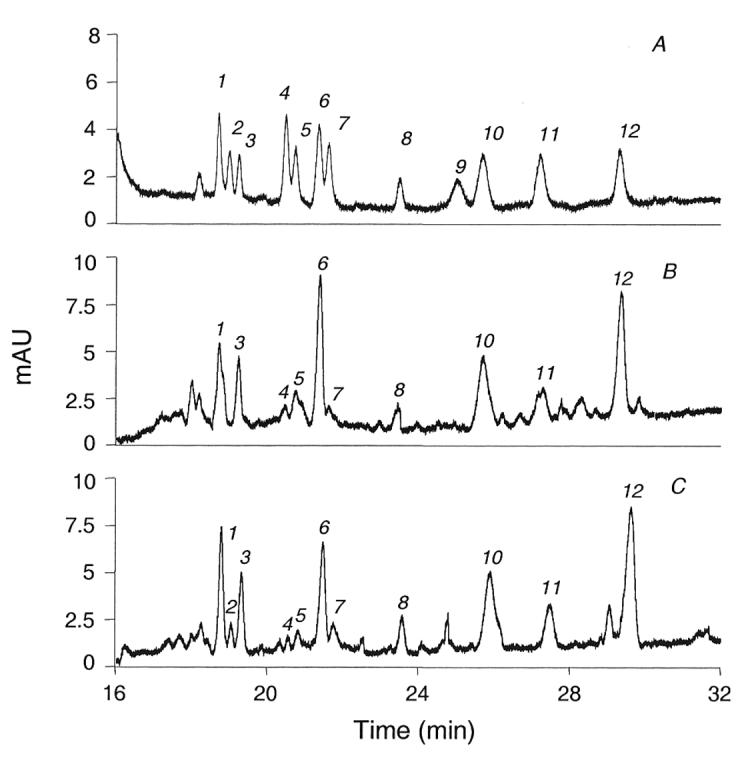
Electropherograms of sialyloligosaccharides from: (A) mixture of 12 standard sialyloligosaccharides; (B) colostrum sample from one mother in Boston area; (C) mature milk sample from one mother in Boston area. 1, MSMFLNnH; 2, MSLNnH I; 3, MSMFLNH I; 4, SLNFP II; 5, LST b; 6, LST c; 7, LST a; 8, DSMFLNH; 9, 3'-S-3-FL; 10, 6'-SL; 11, 3'-SL; 12, DSLNT.
Validation
The regression equations vary for the twelve standards, as seen in Table 2. However, the relationship between concentration and response for each of the twelve standards is remarkably linear over the entire range tested, between 32 and 64-fold (Table 2). The sensitivity (signal to noise ratio ≥ 3) is between 39 and 78 μg/mL. The extreme linearity of the curve over the entire dynamic range (to 2500 μg/mL) is evident by the correlation coefficient (r) approaching 1 for all twelve standards.
Table 2.
Linearity of peak areas obtained for various concentrations of sialyloligosaccharides
| Oligosaccharide | Regression equationa |
Correlation coefficient (r) |
Dynamic range (μg/mL) |
|---|---|---|---|
| MSMFLNnH | Y = 0.217 X + 0.79 | 0.999 | 39 ∼ 2500 |
| MSLNnH I | Y = 0.090 X + 2.27 | 0.999 | 78 ∼ 2500 |
| MSMFLNH I | Y = 0.092 X + 1.47 | 0.999 | 78 ∼ 2500 |
| SLNFP II | Y = 0.127 X + 1.70 | 0.999 | 78 ∼ 2500 |
| LST b | Y = 0.155 X + 2.88 | 0.998 | 39 ∼ 2500 |
| LST c | Y = 0.239 X − 0.59 | 0.999 | 39 ∼ 2500 |
| LST a | Y = 0.170 X + 2.66 | 0.999 | 39 ∼ 2500 |
| DSMFLNH | Y = 0.070 X + 1.50 | 0.998 | 78 ∼ 2500 |
| 3′-S-3-FL | Y = 0.067 X + 4.45 | 0.996 | 78 ∼ 2500 |
| 6′-SL | Y = 0.128 X − 1.02 | 0.999 | 78 ∼ 2500 |
| 3′-SL | Y = 0.293 X − 1.69 | 0.999 | 39 ∼ 2500 |
| DSLNT | Y = 0.072 X + 0.95 | 0.999 | 78 ∼ 2500 |
Y, peak area
X, nominal standard concentration (μg/mL).
The precision and accuracy of the method were tested at three different concentrations, representing low, intermediate, and high portions of the dynamic range for each standard (Table 3). Each measurement for each standard was repeated in 5 independent analyses, and the precision expressed as the coefficient of variation (CV), i.e., the SD/Mean × 100%. The range of CVs for all of the concentrations of standards is between 2 to 9 %. The accuracy, a comparison between the nominal and measured concentrations, was measured at low, intermediate, and high concentrations, and ranges from 93 to 109%, but with a strong central tendency about 100%.
Table 3.
Precision and accuracy of sialyloligosaccharide analyses (n = 5)
| Oligosaccharide | Low (156 μg/mL) |
Intermediate (625 μg/mL) |
High (1250 μg/mL) |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Measa | Precb | Accc | Meas | Prec | Acc | Meas | Prec | Acc | |
| MSMFLNnH | 154 ± 5 | 4 | 99 | 643 ± 28 | 4 | 103 | 1178 ± 83 | 7 | 94 |
| MSLNnH I | 159 ± 11 | 7 | 102 | 623 ± 38 | 6 | 100 | 1219 ± 45 | 4 | 98 |
| MSMFLNH I | 157 ± 3 | 2 | 100 | 646 ± 45 | 4 | 103 | 1352 ± 43 | 3 | 108 |
| SLNFP II | 158 ± 10 | 6 | 101 | 665 ± 27 | 4 | 106 | 1220 ± 22 | 2 | 98 |
| LST b | 171 ± 16 | 9 | 109 | 621 ± 37 | 6 | 99 | 1325 ± 20 | 2 | 106 |
| LST c | 153 ± 10 | 6 | 98 | 582 ± 33 | 6 | 93 | 1202 ± 76 | 6 | 96 |
| LST a | 167 ± 13 | 8 | 107 | 640 ± 33 | 5 | 102 | 1294 ± 70 | 5 | 103 |
| DSMFLNH | 148 ± 11 | 8 | 95 | 676 ± 33 | 5 | 108 | 1172 ± 90 | 8 | 94 |
| 3′-S-3-FL | 170 ± 3 | 2 | 108 | 667 ± 49 | 7 | 106 | 1330 ± 80 | 6 | 106 |
| 6′-SL | 169 ± 9 | 6 | 108 | 617 ± 35 | 6 | 99 | 1249 ± 105 | 8 | 100 |
| 3′-SL | 161 ± 8 | 5 | 103 | 590 ± 47 | 8 | 95 | 1161 ± 107 | 9 | 93 |
| DSLNT | 163 ± 14 | 9 | 104 | 676 ± 26 | 4 | 108 | 1215 ± 111 | 9 | 97 |
Meas, measured concentration (μg/mL)
Prec, precision (CV = SD/Mean × 100%)
Acc, accuracy (% of nominal authentic standard oligosaccharide).
Authentic standards were added to a normalized pooled milk sample to measure any effects of milk matrix on the response factor, and any loss of oligosaccharide upon extraction from milk. The data in Table 4 indicate that there is little interference by milk components after extraction of the oligosaccharides from the milk, and that the recovery is approximately 90 to 100%.
Table 4.
Recovery of sialyloligosaccharides from pooled samples of human milka
| Oligosaccharide | Recovery (%) |
|||
|---|---|---|---|---|
| Low (156 μg/mL) |
Intermed. (625 μg/mL) |
High (1250 μg/mL) |
Mean | |
| MSMFLNnH | 97 | 94 | 101 | 97 |
| MSLNnH I | 98 | 94 | 96 | 96 |
| MSMFLNH I | 93 | 89 | 91 | 91 |
| SLNFP II | 98 | 96 | 98 | 97 |
| LST b | 95 | 95 | 99 | 96 |
| LST c | 87 | 93 | 98 | 93 |
| LST a | 91 | 92 | 94 | 92 |
| DSMFLNH | 98 | 97 | 108 | 101 |
| 3′-S-3-FL | 94 | 99 | 93 | 95 |
| 6′-SL | 96 | 86 | 89 | 90 |
| 3′-SL | 94 | 97 | 103 | 98 |
| DSLNT | 97 | 94 | 98 | 96 |
Standards were added to pooled milk from 13 donors.
Ability of method to discern natural variation of human milk sialyloligosaccharides
Three sets of matched milks from the same mothers, one from days 4 and 21 of lactation, one from days 3 and 15, and one from days 5 and 9, were used to determine whether this technique discern differences in sialyloligosaccharide expression from less and more mature milk samples. The data in table 5 indicate that the measured concentrations of some sialyloligosaccharides were consistently lower in the more mature milks, including MSMFLNH I, LST b, LST c, LST a, 3'-SL, and DSLNT. In contrast, MSMFLNnH, DSMFLNH, SLNFP II, and 6'-SL were found to increase in concentration in the more mature milk.
Table 5.
Concentrations of sialyloligosaccharides from three donors (μg/mL)
| Oligosaccharide | Donor 1 |
Donor 2 |
Donor 3 |
Mean |
||||
|---|---|---|---|---|---|---|---|---|
| d 4a | d 21 | d 3 | d 15 | d 5 | d 9 | d 3-d 5 | d 9-d 21 | |
| MSMFLNnH | 171 | 212 | 129 | 198 | 97 | 164 | 133 ± 37 | 191 ± 25 |
| MSLNnH I | n.d.b | n.d. | n.d. | n.d. | 104 | 28 | -- | -- |
| MSMFLNH I | 139 | 30 | 243 | 128 | 176 | 94 | 186 ± 53 | 84 ± 50 |
| SLNFP II | n.d. | 9 | 21 | 44 | 19 | 22 | -- | 25 ± 18 |
| LST b | 202 | 49 | 113 | 100 | 78 | 71 | 131 ± 64 | 74 ± 26 |
| LST c | 207 | 81 | 298 | 196 | 191 | 168 | 232 ± 58 | 148 ± 60 |
| LST a | 13 | n.d. | 33 | 13 | 30 | 10 | 26 ± 11 | -- |
| DSMFLNH | 162 | 202 | 59 | 97 | 96 | 116 | 106 ± 52 | 139 ± 56 |
| 3′-S-3-FL | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | -- | -- |
| 6′-SL | 297 | 337 | 349 | 441 | 358 | 410 | 335 ± 33 | 396 ± 54 |
| 3′-SL | 140 | 92 | 71 | 68 | 80 | 68 | 97 ± 38 | 76 ± 14 |
| DSLNT | 1882 | 623 | 1167 | 1061 | 771 | 702 | 1274 ± 563 | 795 ± 234 |
d, days after the delivery
n.d., not detected.
Five random samples of colostrums and 5 random samples of milks from 10 unrelated mothers were used to determine if this technique could measure individual variation in sialyloligosaccharide expression in milk samples. Table 6 illustrates that there is variation in expression of sialyloligosaccharides among mothers that is readily measured by this technique. Even within the colostrum samples and within the mature milk samples, there was high variation in sialyloligosaccharides that could not be accounted for by the variation intrinsic to this method, suggesting significant biological differences in expression of acidic milk oligosaccharides. Thus, this method shows promise for defining variation in milk sialyloligosaccharide expression and its biological consequences.
Table 6.
Concentrations of sialyloligosaccharides from ten donors (μg/mL)
| Oligosaccharide | Colostrum (day of lactation) |
Milk (day of lactation) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| d 2a | d 2 | d 2 | d 3 | d 4 | mean | d 12 | d 18 | d 21 | d 49 | d 67 | mean | |
| MSMFLNnH | 50 | 96 | 63 | 123 | 140 | 94 ± 38 | 148 | 127 | 125 | 121 | 28 | 110 ± 47 |
| MSLNnH I | n.d. | 21 | n.d. | n.d. | n.d. | -- | 46 | n.d. | 182 | n.d. | n.d. | -- |
| MSMFLNH I | 112 | 101 | 74 | 134 | 122 | 108 ± 23 | 209 | 144 | 68 | 71 | 26 | 104 ± 73 |
| SLNFP II | n.d. | n.d. | n.d. | n.d. | n.d. | -- | 20 | 14 | 22 | 11 | n.d. | -- |
| LST b | 10 | 9 | 36 | 25 | 104 | 37 ± 39 | 18 | 68 | 37 | 91 | 10 | 44 ± 34 |
| LST c | 138 | 146 | 156 | 199 | 229 | 174 ± 39 | 129 | 123 | 60 | 79 | 12 | 80 ± 48 |
| LST a | n.d. | 26 | 10 | 13 | 33 | -- | 30 | n.d. | n.d. | 8 | n.d. | -- |
| DSMFLNH | n.d. | 39 | 18 | 68 | 53 | -- | 146 | 96 | 112 | 49 | 39 | 89 ± 45 |
| 3'-S-3-FL | n.d. | n.d. | n.d. | n.d. | n.d. | -- | n.d. | n.d. | n.d. | n.d. | n.d. | -- |
| 6'-SL | 149 | 356 | 215 | 270 | 391 | 276 ± 99 | 399 | 383 | 426 | 277 | 43 | 306 ± 157 |
| 3'-SL | 61 | 75 | 73 | 71 | 133 | 82 ± 29 | 69 | 68 | 57 | 78 | 42 | 63 ± 14 |
| DSLNT | 632 | 570 | 500 | 931 | 1250 | 777 ± 312 | 910 | 930 | 584 | 639 | 236 | 660 ± 283 |
d, days after the delivery
n.d., not detected.
DISCUSSION
The CE analytical technique described herein is a major advance over our previous CE method for analysis of human milk sialyloligosaccharides. The original method could not simultaneously resolve and quantify LST a from LST c and 3'-SL from 6'-SL. Therefore we developed two separate conditions for CE: The first condition could resolve 3'-SL from 6'-SL but not the isomers LST a, b, and c (originally labeled SLNT a, b, and c) [9]. The second condition could resolve the isomers LST a, b, and c, but not to baseline, and could not simultaneously resolve 3'-SL from 6'-SL [29]. This report describes new CE conditions that simultaneously resolve not only these two pairs of closely eluting sialyloligosaccharides, but also the rest of the 12 human milk sialyloligosaccharides for which we now have standards.
The original method used Tris buffer at 376 mM, the new method is based on phosphate buffer at 200 mM; this lower concentration of inorganic buffer allowed resolution of the 12 sialyloligosaccharides within a shorter run time. The pH of 7.05 used in the current running buffer conditions provided superior resolution from the 7.9 that was used previously. Using this inorganic buffer, however, required 45% methanol as an organic modifier with 100 mM SDS to achieve the separation of the 12 oligosaccharides. In our former method, 6% of methanol with 150 mM SDS separated 7 oligosaccharides, but required two independent runs using different conditions.
More MeOH as an organic modifier in the running buffer increased resolution between most oligosaccharides, but reduced the separation between 3'-S-3-FL and 6'-SL; the addition of 45% MeOH to the aqueous buffer seemed to be the best balance for overall performance. Addition of the surfactant SDS in the running buffer improved resolution for LST c and LST a, but degraded other resolutions; 100 mM of SDS represents the compromise to balance these resolutions. Note that the aqueous buffer was 200 mM NaH2PO4 at pH 7.05 containing 100 mM SDS before mixing this buffer with MeOH to create the mixture of 55% buffer to 45% (v/v) MeOH for formulating the final running buffer. Further systematic modifications of the buffer did not improve resolution in a 35 minute run.
This new method uses different separation principals from methods previously described from other laboratories for analysis of human milk oligosaccharides. In 1987, Parkkinen and Finne isolated acidic oligosaccharides from bovine milk and human urine by DEAE ion exchange followed by paper chromatography, and quantified the isolated oligosaccharides by gravimetry [30]. This method was quite effective for samples of 2 liters or more. In 1999, Charlwood et al. analyzed acidic and neutral human milk oligosaccharides by reverse and normal phase chromatography of their 2-aminoacridone derivatives [27]. Although this method allowed simultaneous analysis of both acidic and neutral species, many of the individual oligosaccharides could not be resolved. Martin-Sosa and associates described a method in 2003 of desialylating sialyloligosaccharides with neuraminidase, resolving them on an amino column, and detecting them at 206 nm [28]. Eight peaks were detected and quantified, but isomers of sialic acid attachment could not be resolved after desialylation, and the samples required extensive manipulation before analysis. Anion exchange with pulsed amperometric detection (HPAEC-PAD) has been used for analysis of acidic oligosaccharides by three groups. Townsend et al. (1988) separated the oligosaccharides on the basis of the number of sialic acids per molecule at pH 4.6, and further resolved them by desialylation and resolution of the resulting neutral oligosaccharides at pH 13 [23]. This two-step process overcame the inconsistent response factors for sialyloligosaccharides with different linkages by the electrochemical detector. In 1996 an HPAEC-PAD method for acidic oligosaccharides was described that utilized a two step sample work-up of ion exchange and size exclusion chromatography and required 5-10 mL of milk [26]. Musumeci et al. (2006) used a similar technique, but with a modified mobile phase [17]. The CE analytical method that we describe herein improves upon these methods: sample preparation is simple, and the method resolves and quantifies many more milk oligosaccharides. The method was validated for the twelve major sialyloligosaccharides of human milk for which standards are available; this is more than any of the preceding methods.
Concentrations of sialyloligosaccharides in human milk measured by the CE method described herein were compared with the concentrations reported in prior publications using other methods. The range of our CE results overlaps with the range of oligosaccharides reported by Kunz et al. [8] for LST c, 6′-SL, and DSLNT (Table 7). Our range of results also overlaps with the results of the report of Martin-Sosa et al. [28] for 6′-SL, 3′-SL, and DSLNT. There was a wide discrepancy between the values for LST a between these other two reports. Thus, our CE method is in agreement with many of the previously published values for the human milk oligosaccharides. A notable exception is 3′-S-3-SL, which was reported to be between 2 and 5 g/L of milk by Martin-Sosa et al., but which our CE analysis indicated was less than 10 mg/L in samples from donors in the Boston area.
Table 7.
Concentrations of oligosaccharides from three donors (μg/mL)
The CE method described herein was tested for its ability to detect and quantify acidic oligosaccharide variance in milk of human populations. The expression of MSMFLNH I, LST a, and possibly DSLNT, LST b, LST c, and 3′-SL decreased as lactation progressed from colostrum to mature milk. There was also a tendency for 6′-SL, DSMFLNH, SLNFP II, and MSMFLNnH expression in milk to increase as lactation progressed. A comparison of five random samples of colostrum with five random samples of more mature milk demonstrated that expression of acidic oligosaccharides in milk varies by individual. These data indicate that populations of lactating mothers exhibit individual variation in their expression of acidic oligosaccharides, and that our CE method can measure this variation. The relatively straightforward sample preparation and the throughput of this CE method are sufficient to allow of this variation to be fully studied and documented in future studies on larger human cohorts. These may allow the biological significance of the variation in silayloligosaccharide expression in milk to be studied in relation to medical outcomes in nursing infants.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Newburg DS, Neubauer SH. Carbohydrates in milks: analysis, quantities, and significance. In: Jensen RG, editor. Handbook of Milk Composition. Academic Press; San Diego: 1995. pp. 273–350. [Google Scholar]
- 2.Warren CD, Chaturvedi P, Newburg AR, Oftedal OT, Tilden CD, Newburg DS. Comparison of oligosaccharides in milk specimens from humans and twelve other species. In: Newburg DS, editor. Bioactive Components of Human Milk. Kluwer Academic/Plenum Publishers; New York: 2001. pp. 325–339. [DOI] [PubMed] [Google Scholar]
- 3.Gyorgy P, Norris RF, Rose CS. Bifidus factor I. A variant of Lactobacillus bifidus requiring a special growth factor. Arch. Biochem. Biophys. 1954;48:193–201. doi: 10.1016/0003-9861(54)90323-9. [DOI] [PubMed] [Google Scholar]
- 4.Newburg DS. Oligosaccharides in human milk and bacterial colonization. J. Pediatr. Gastroenterol. Nutr. 2000;30:S8–S17. [PubMed] [Google Scholar]
- 5.Sharon N. Carbohydrate-lectin interactions in infectious disease. Adv. Exp. Med. Biol. 1996;408:1–8. [PubMed] [Google Scholar]
- 6.Newburg DS, Ruiz-Palacios GM, Morrow AL. Human milk glycans protect infants against enteric pathogens. Annu. Rev. Nutr. 2005;25:37–58. doi: 10.1146/annurev.nutr.25.050304.092553. [DOI] [PubMed] [Google Scholar]
- 7.Angeloni S, Ridet JL, Kusy N, Gao H, Crevoisier F, Guinchard S, Kochhar S, Sigrist H. Glycoprofiling with micro-arrays of glycoconjugates and lectins. Glycobiology. 2005;15:31–41. doi: 10.1093/glycob/cwh143. [DOI] [PubMed] [Google Scholar]
- 8.Kunz C, Rudloff S, Baier W, Klein N, Strobel S. Oligosaccharides in human milk: structural, functional, and metabolic aspects. Annu. Rev. Nutr. 2000;20:699–722. doi: 10.1146/annurev.nutr.20.1.699. [DOI] [PubMed] [Google Scholar]
- 9.Shen Z, Warren CD, Newburg DS. High-performance capillary electrophoresis of sialylated oligosaccharides of human milk. Anal. Biochem. 2000;279:37–45. doi: 10.1006/abio.1999.4448. [DOI] [PubMed] [Google Scholar]
- 10.Chaturvedi P, Warren CD, Altaye M, Morrow AL, Ruiz-Palacios G, Pickering LK, Newburg DS. Fucosylated human milk oligosaccharides vary between individuals and over the course of lactation. Glycobiology. 2001;11:365–372. doi: 10.1093/glycob/11.5.365. [DOI] [PubMed] [Google Scholar]
- 11.Newburg DS, Ruiz-Palacios G, Pickering LK, Altaye M, Chaturvedi P, Meinzen-Derr J, de Lourdes Guerrero M, Morrow AL. Innate protection conferred by fucosylated oligosaccharides of human milk against diarrhea in breastfed infants. Glycobiology. 2004;14:253–263. doi: 10.1093/glycob/cwh020. [DOI] [PubMed] [Google Scholar]
- 12.Chaturvedi P, Warren CD, Ruiz-Palacios GM, Pickering LK, Newburg DS. Profiling of milk oligosaccharides using reversed phase HPLC of their perbenzoylated derivatives. Anal. Biochem. 1997;251:89–97. doi: 10.1006/abio.1997.2250. [DOI] [PubMed] [Google Scholar]
- 13.Warren CD, Chaturvedi P, Newburg AR, Oftedal OT, Tilden CD, Newburg DS. Comparison of oligosaccharides in milk specimens from humans and twelve other species. Adv. Exp. Med. Biol. 2001;501:325–332. doi: 10.1007/978-1-4615-1371-1_40. [DOI] [PubMed] [Google Scholar]
- 14.Sumiyoshi W, Urashima T, Nakamura T, Arai I, Saito T, Tsumura N, Wang B, Brand-Miller J, Watanabe Y, Kimura K. Determination of each neutral oligosaccharide in the milk of Japanese women during the course of lactation. Br. J. Nutr. 2003;89:61–69. doi: 10.1079/BJN2002746. [DOI] [PubMed] [Google Scholar]
- 15.Asakuma S, Urashima T, Akahori M, Obayashi H, Nakamura T, Kimura K, Watanabe Y, Arai I, Sanai Y. Variation of major neutral oligosaccharides levels in human colostrums. Eur. J. Clin. Nutr. 2007:1–7. doi: 10.1038/sj.ejcn.1602738. [DOI] [PubMed] [Google Scholar]
- 16.Song JF, Weng MQ, Wu SM, Xia QC. Analysis of neutral saccharides in human milk derivatized with 2-aminoacridone by capillary electrophoresis with laser-induced fluorescence detection. Anal. Biochem. 2002;304:126–129. doi: 10.1006/abio.2001.5589. [DOI] [PubMed] [Google Scholar]
- 17.Musumeci M, Simpore J, D'Agata A, Sotgiu S, Musumeci S. Oligosaccharides in colostrums of Italian and Burkinabe women. J. Pediatr. Gastroenterol. Nutr. 2006;43:372–378. doi: 10.1097/01.mpg.0000228125.70971.af. [DOI] [PubMed] [Google Scholar]
- 18.Baenziger JU, Natowicz M. Rapid separation of anionic oligosaccharide species by high-performance liquid chromatography. Anal. Biochem. 1981;112:357–361. doi: 10.1016/0003-2697(81)90305-5. [DOI] [PubMed] [Google Scholar]
- 19.Cardon P, Parente JP, Leroy Y, Montreuil J, Fournet B. Separation of sialyl-oligosaccharides by high-performance liquid chromatography. Application to the analysis of mono-, di-, tri- and tetrasialyl-oligosaccharides obtained by hydrazinolysis of alpha 1-acid glycoprotein. J. Chromatogr. 1986;356:135–146. doi: 10.1016/s0021-9673(00)91473-0. [DOI] [PubMed] [Google Scholar]
- 20.Liu JP, Shirota O, Wiesler D, Novotny M. Ultrasensitive fluorometric detection of carbohydrates as derivatives in mixtures separated by capillary electrophoresis. Proc. Natl. Acad. Sci. 1991;88:2302–2306. doi: 10.1073/pnas.88.6.2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stefansson M, Novotny M. Separation of complex oligosaccharide mixtures by capillary electrophoresis in the open-tubular format. Anal. Chem. 1994;66:1134–1140. doi: 10.1021/ac00079a031. [DOI] [PubMed] [Google Scholar]
- 22.van Pelt J, Damm JB, Kamerling JP, Vliegenthart JF. Separation of sialyl-oligosaccharides by medium pressure anion-exchange chromatography on Mono Q. Carbohydr. Res. 1987;169:43–51. doi: 10.1016/0008-6215(87)80241-0. [DOI] [PubMed] [Google Scholar]
- 23.Townsend RR, Hardy MR, Hindsgaul O, Lee YC. High-performance anion-exchange chromatography of oligosaccharides using pellicular resins and pulsed amperometric detection. Anal. Biochem. 1988;174:459–470. doi: 10.1016/0003-2697(88)90044-9. [DOI] [PubMed] [Google Scholar]
- 24.Townsend RR, Hardy MR, Cumming DA, Carver JP, Bendiak B. Separation of branched sialylated oligosaccharides using high-pH anion-exchange chromatography with pulsed amperometric detection. Anal. Biochem. 1989;182:1–8. doi: 10.1016/0003-2697(89)90708-2. [DOI] [PubMed] [Google Scholar]
- 25.Watson E, Bhide A, Kenney WC, Lin FK. High-performance anion-exchange chromatography of asparagines-linked oligosaccharides. Anal. Biochem. 1992;205:90–95. doi: 10.1016/0003-2697(92)90583-s. [DOI] [PubMed] [Google Scholar]
- 26.Kunz C, Rudloff S, Hintelmann A, Pohlentz G, Egge H. High-pH anion-exchange chromatography with pulsed amperometric detection and molar response factors of human milk oligosaccharides. J. Chromatogr. B. 1996;685:211–221. doi: 10.1016/s0378-4347(96)00181-8. [DOI] [PubMed] [Google Scholar]
- 27.Charlwood J, Tolson D, Dwek M, Camilleri P. A detailed analysis of neutral and acidic carbohydrates in human milk. Anal. Biochem. 1999;273:261–277. doi: 10.1006/abio.1999.4232. [DOI] [PubMed] [Google Scholar]
- 28.Martin-Sosa S, Martin MJ, Garcia-Pardo LA, Hueso P. Sialyloligosaccharides in human and bovine milk and infant formulas: variations with the progression of lactation. J. Dairy Sci. 2003;86:52–59. doi: 10.3168/jds.S0022-0302(03)73583-8. [DOI] [PubMed] [Google Scholar]
- 29.Shen Z, Warren CD, Newburg DS. Resolution of structural isomers of sialylated oligosaccharides by capillary electrophoresis. J. Chromatogr. A. 2001;921:315–321. doi: 10.1016/s0021-9673(01)00872-x. [DOI] [PubMed] [Google Scholar]
- 30.Parkkinen J, Finne J. Isolation of sialyl oligosaccharides and sialyl oligosaccharide phosphate from bovine colostrum and human urine. Methods Enzymol. 1987;138:289–300. doi: 10.1016/0076-6879(87)38024-3. [DOI] [PubMed] [Google Scholar]