

Abstract
The conserved N-domain of the STAT proteins has been implicated in several activities crucial to cytokine signaling including receptor recruitment and STAT activation, cooperative DNA binding and STAT-dependent gene expression. We evaluated the role of the STAT3 N-domain in the IL-6 signal transduction pathway leading to Socs3 gene expression, an essential mechanism that controls the quality and magnitude of IL-6-dependent transcriptional responses. Based on the model for STAT N-domain function in cooperative gene expression and the presence of tandem STAT binding motifs in the murine Socs3 promoter, we anticipated that stabilizing interactions between adjacent STAT3 dimers via N-domain sequences might be essential for Socs3 gene expression. This was underscored by the tight conservation in the location and sequence of the tandem STAT binding sites between the murine and human Socs3 promoters. Using reconstitution into Stat3−/− mouse embryonic fibroblasts (Stat3−/− MEFs), we find that a STAT3 N-domain deletion mutant (Δ133STAT3) is activated by tyrosine phosphorylation in response to IL-6 and then undergoes dephosphorylation with kinetics similar to full-length STAT3. These results highlight important differences compared to other STATs where the N-domain has been shown to mediate activation (STAT4) or dephosphorylation (STAT1). STAT3 binds predominantly to a single STAT consensus site in the Socs3 promoter, despite the presence of an adjacent STAT motif. Significantly, Δ133STAT3 stimulates expression of the endogenous Socs3 gene in Stat3−/− MEFs upon IL-6 treatment with an activity similar to reconstituted STAT3, demonstrating that the N-domain is dispensable for Socs3 gene expression. We propose that the Socs3 gene in its chromosomal context is activated by the IL-6/STAT3 pathway independent of STAT3 N-domain sequences.
Keywords: cytokines, IL-6, SOCS3, STAT3
Introduction
The transcription factor STAT3 plays a central role in numerous cytokine signaling pathways, demonstrating tissue-specific functions such as regulation of the hepatic acute phase response, control of macrophage deactivation, initiation of apoptosis during mammary gland involution and embryonic stem cell self renewal(1–4). Cytokine signaling via STAT3 is an inducible response pathway that is tightly regulated. STAT3 is stimulated by recruitment to an activated cytokine receptor complex via a central SH2 domain, tyrosine phosphorylation on a conserved C-terminal residue, and dimerization through reciprocal SH2/phosphotyrosine interactions. These events promote STAT3 nuclear accumulation, DNA binding and the rapid induction of cytokine-responsive target genes (5,6). STAT3 activity is downregulated by multiple mechanisms including dephosphorylation within the nucleus, loss of DNA binding activity and nuclear export (5–8). A balanced STAT3 response is essential, as alterations in this pathway have severe effects in vivo(1–3, 9–13).
Persistent activation of STAT3 has been observed in several diseases. Activated STAT3 is present in many solid tumors and tumor cell lines (14, 15), and a direct role in oncogenesis was demonstrated by use of an engineered STAT3 dimer with constitutive activity, which showed transforming activity in fibroblasts (13). Oncogenic tyrosine kinases such as v-Src can phosphorylate and activate STAT3 in a cytokine-independent manner and thereby contribute to tumor formation (16,17). STAT3 inhibitors are under development for the treatment of certain cancers including those of the head, neck and prostate (18–20). Furthermore, the STAT3 pathway may provide a therapeutic target for certain immune-mediated disorders including rheumatoid arthritis (1, 10, 12, 21). Constitutive activation of STAT3 in disease indicates an imbalance of stimulatory and downregulatory pathways; understanding the mechanics of these will contribute to the development of future therapies.
Several independent pathways regulate the cellular response to cytokines (22); their disruption can have severe to lethal consequences in vivo (23–28). Members of the SOCS protein family control a principal cytokine signal termination mechanism, operating at distinct steps in the receptor-signaling pathway (22,29). SOCS proteins competitively inhibit STAT recruitment to receptors, block receptor-associated JAK kinase activity and target cytokine-signaling intermediates for destruction via proteosomal degradation (22,29). Expression of certain SOCS genes, including Socs1 and Socs3, is stimulated rapidly by cytokines via a STAT-dependent pathway (29). Thus, STATs initiate a regulatory feedback loop to modulate cytokine signaling by the induction of SOCS protein expression.
The essential role of these regulatory pathways was highlighted by recent studies on STAT3 and its target gene Socs3 in the interleukin-6 (IL-6) signal transduction pathway. IL-6 is essential for the acute phase response of the immune system and is a significant factor in the pathogenesis of certain diseases such as multiple myeloma (30). Signal transduction by IL-6 requires the specific α chain (IL-6Rα) and the shared gp130 receptor subunit, which activate STAT1 and STAT3 in response to ligand binding(30). STAT3 controls the expression of many acute phase response genes as well as genes involved in cell growth and survival pathways(6, 30), while the role of STAT1 in IL-6 signaling is less clear. Cells lacking STAT3 or SOCS3 show an imbalanced signaling profile upon treatment with IL-6, with elevated and prolonged STAT3 and STAT1 activation (24, 26, 27). In response to IL-6, STAT3- or SOCS3-deficient cells induce many genes normally controlled by interferon-γ (IFNγ), which signals by a STAT 1-dependent pathway (24, 26, 31). Furthermore, mice lacking STAT3 or SOCS3 in the hematopoietic system throughout their development succumb to a severe inflammatory disease (10, 25).
The N-terminal region of the STAT proteins, which comprises approximately 130 residues that assemble into 8 α-helices separated by short linkers (32–34), has been implicated in diverse functions including STAT activation, deactivation and target gene expression (35–37). For example, the N-domain of STAT4 is required for cytokine-responsive activation via tyrosine phosphorylation (36, 38) by mediating the formation of STAT4 oligomers in the cytoplasm (35). By contrast, the N-domains of STAT1 and STAT5 control the kinetics of STAT deactivation; for STAT1 this is regulated by N-domain-dependent recruitment of the nuclear tyrosine phosphatase TC45 (37). Isolated STAT N-domains show potent homotypic interactions (35). STAT N-domains have been implicated in target gene regulation by providing an interface for stabilizing interactions between STAT dimers on adjacent DNA binding sites (37, 39, 40). The extent to which these functions are shared among all STAT family members is not clear.
Little is known about the role of the N-domain in the STAT3 signaling pathway. The presence of highly conserved, adjacent STAT binding motifs in the Socs3 promoter suggested that the STAT3 N-domain may be required for Socs3 gene regulation via a cooperative binding mechanism. To test this, we reconstituted Stat3−/− MEFs with wild type STAT3 or an N-terminal deletion mutant (Δ 133STAT3). We found that Δ133STAT3 was activated robustly by IL-6 treatment, and underwent nuclear localization and dephosphorylation with kinetics similar to full-length STAT3. Our studies showed that Δ133STAT3 stimulated IL-6-dependent SOCS3 expression in Stat3−/− MEFs as effectively as wild-type STAT3. STAT3 interacts preferentially with a single consensus binding motif in the Socs3 promoter, despite the presence of an adjacent conserved motif. These results highlight significant differences with N-domain function in other STAT proteins and indicate that STAT tetramer formation via the N-domain is not essential for all target gene responses.
Materials and methods
Plasmids, transfections and cell culture conditions
Δ133STAT3 was constructed by a polymerase chain reaction (PCR)-based strategy in which the forward primer encodes a Not I restriction site, the first six nucleotides of murine Stat3 (bold) and 21 nucleotides encoding STAT3 residues 134–139 (italics) (5′-ATAAGAAGCGGCCGCATGGCTGCCGCCGTAGTGACAGAGAAG-3′). The reverse primer encodes sequences surrounding a unique Bsu36I restriction site at position 1303 in murine Stat3 (5′-CACATCTCTGCTCCCTAAG-3′). The forward and reverse primers were used to amplify sequences from the murine Stat3 cDNA (pRc/STAT3; generously provided by Dr. Jim Darnell); the resulting PCR fragment was digested with Not I and Bsu36I and isolated following gel electrophoresis. The pRc/STAT3 plasmid, which contains the STAT3 coding region fused to a C-terminal FLAG epitope, was digested with Not I and Bsu36I to remove sequences encoding the STAT3 N-terminal region. The linearized plasmid was ligated to the purified PCR fragment. The resulting construct generated an internal deletion of STAT3 lacking residues 3–133 (termed Δ133STAT3). The C-terminal FLAG epitope was preserved in Δ133STAT3. Deletion of the N-domain coding region was confirmed by DNA sequencing and immunoblot analysis following transient transfection assays (data not shown). The Δ133STAT3 cDNA was subcloned into the retroviral vector pBABEpuro using the 5′ BamHI and 3′ ApaI restriction sites that flank STAT3 coding sequences. A pBABEpuro plasmid containing wild type STAT3 was generously provided by Dr. Curt Horvath.
NIH3T3 cells, a Stat3−/− mouse embryonic fibroblast cell line (Stat3−/− MEFs) and Stat3+/− cells (Stat3+/− MEFs) were grown in Dulbecco’s Modified Eagle’s medium containing 10% fetal calf serum (DME/FCS) in a humidified CO2 incubator. The derivation of Stat3−/− and Stat3+/− MEFs has been described elsewhere(41). 32D cells expressing ER-S3, an EpoR isoform engineered to activate STAT3 in place of STAT5, have been described previously (42,43).
To generate infectious retrovirus carrying STAT3 cDNAs, Phoenix packaging cells were transiently transfected with pBABE/STAT3 or pBABE/Δ133STAT3 plasmids and pCL-Eco packaging construct(44) using Fugene6 Transfection Reagent (Roche). Viral supernatants were collected at 48 h post-transfection and used immediately to infect NIH3T3 cells or Stat3−/− MEFs. Cells were incubated with undiluted viral supernatants for 5 hours at 37 °C in the presence of 6.5 μg/ml polybrene, cultured an additional 48 h in DME/FCS and then selected in media containing 6μg/ml (NIH3T3) or 3μg/ml (Stat3−/− MEFs) puromycin. Pooled cell lines were used in all experiments; expression of STAT3 or Δ133STAT3 was confirmed by FLAG immunoblot assays (data not shown).
Cytokine treatments, antibodies, immunoprecipitations and immunoblot assays
Cells were stimulated with IL-6 and soluble IL-6 receptor (sIL-6R) (R&D Systems) (25–50 ng/ml) as indicated. Antibodies specific for tyrosine-phosphorylated STAT3 (pSTAT3; Upstate Biotechnology, Cell Signaling), total STAT3 (Santa Cruz), and FLAG epitope sequences (Sigma) were obtained from the indicated commercial sources.
In non-denaturing immunoprecipitations, detergent cell extracts were prepared in isotonic Triton X-100 lysis buffer (1% Triton X-100, 150 mM NaCl, 50 mM Tris pH7.4, 1 mM EDTA) as described previously (42). Denaturing immunoprecipitations were performed with whole cell lysates prepared by solubilization of cells in 1% SDS, 150 mM NaCl, 50 mM Tris pH 7.4, 1 mM EDTA, followed by sonication and boiling. Whole cell SDS lysates were clarified by centrifugation and diluted 1:10 with isotonic Triton X–100 buffer. Antibody incubations were performed as described previously(42). Whole cell lysates for immunoblot analysis were prepared by solubilization of cells in 2× SDS-PAGE sample buffer (2% SDS, 80 mM Tris pH 6.8, 15% glycerol containing 1 % β-mercaptoethanol and bromphenol blue), as described previously(42). Nuclear extracts were prepared using a modification of the electrophoretic mobility shift assay (EMSA) extract preparation (42). Cells were washed in PBS, resuspended in ice-cold Buffer A (10mM Hepes pH 7.9, 50 mM KCl, 1.5 mM MgCl2, 1.0 mM EDTA, 3% glycerol), incubated 5 min on ice, pelleted by microcentrifugation at 3000rpm, resuspended in Buffer B (Buffer A containing 10 μl/ml 10% NP-40) and incubated for an additional 5 min on ice. Nuclei were pelleted by microcentrifugation at 3000 rpm, washed extensively in PBS, solubilized by sonication in 2× SDS-PAGE sample buffer and denatured by boiling.
Protein samples were separated by SDS-PAGE and transferred electrophoretically to nitrocellulose membranes (Protran). For immunoblots with pSTAT3 antibody, membranes were probed according to the antibody manufacturer’s instructions. For all other immunoblots, membranes were blocked in Tris-buffered saline (50 mM Tris pH 7.4,150 mM NaCl) containing 0.2% Tween 20 (TBS/T) and 4% milk for a minimum of 1 h at room temperature, then incubated overnight at 4 °C in blocking buffer containing the indicated primary antibody, using antibody dilutions recommended by the manufacturer. Membranes were washed with TBS/T for 30 min at room temperature, incubated with horseradish peroxidase (HRP)-conjugated secondary antibody (Amersham) for 60 min at room temperature and developed using enhanced chemiluminescent (ECL) reagents (Pierce) following the manufacturer’s instructions. To reprobe with a different antibody, membranes were incubated in 62mM Tris pH 6.8, 2% SDS and 0.7% β-mercaptoethanol for 30 min at 65 °C, washed extensively in TBS/T, blocked and incubated with appropriate primary and secondary antibody solutions. In some cases, identical samples were run on separate gels and individual membranes were probed in parallel with pSTAT3 or STAT3 antibodies.
Electrophoretic mobility shift assays (EMSAs)
Nuclear extracts were prepared as described in the previous section through the lysis and incubation steps in Buffer B. Following these, nuclei were isolated by centrifugation through an equal volume of Buffer C (10 mM Hepes pH 7.9, 1.5 mM MgCl2, 25% glycerol), washed in ice-cold PBS and extracts were generated by incubation in high salt lysis buffer (10 mM Hepes pH 7.9, 400 mM KCl, 1.5mM MgCl2, 1.0 mM EDTA). Protein concentrations were determined by commercial reagents (Pierce).
Binding reactions were performed with synthetic double-stranded radiolabeled oligonucleotides derived from the murine Socs3 promoter or the murine α2-macroglobulin promoter. Oligonucleotides that contain both STAT consensus motifs in the Socs3 promoter (−100 to −59, 5′-AAACATTACAAGAAGACCGGCCGGGCAGTTCCAGGAATCGGG-3′) or single STAT binding sites (proximal site, −79 to −59, 5′-CGGGCAGTTCCAGGAATCGGG-3′ or distal site, ′100 to ′80, 5′-AAACATTACAAGAAGACCGGC-3′) were used(45). Mutations (in italics) in the proximal (5′-CCACAGGAA-3′) and/or distal STAT binding site(s) (5′-CCACAAGAA-3′) were generated in the context of the −100 to −59, −100 to −80 and −79 to −59 oligonucleotides (see Fig. 5). In addition, an oligonucleotide containing adjacent STAT binding sites in the α2-macroglobulin promoter was utilized (5′-AGCAGTAACTGGAAAGTCCTTAATCCTTCTGGGAATTCTGG-3′) Binding reactions were incubated for 30 min at room temperature then separated on 6% non-denaturing polyacrylamide gels. Unlabeled competitor oligonucleotides (25 μM) or antibodies (1 μg anti-STAT1 or anti-STAT3 antibody; Santa Cruz) were included in a 15 min pre-incubation step in some reactions, as indicated in the figure legends.
Fig. 5.
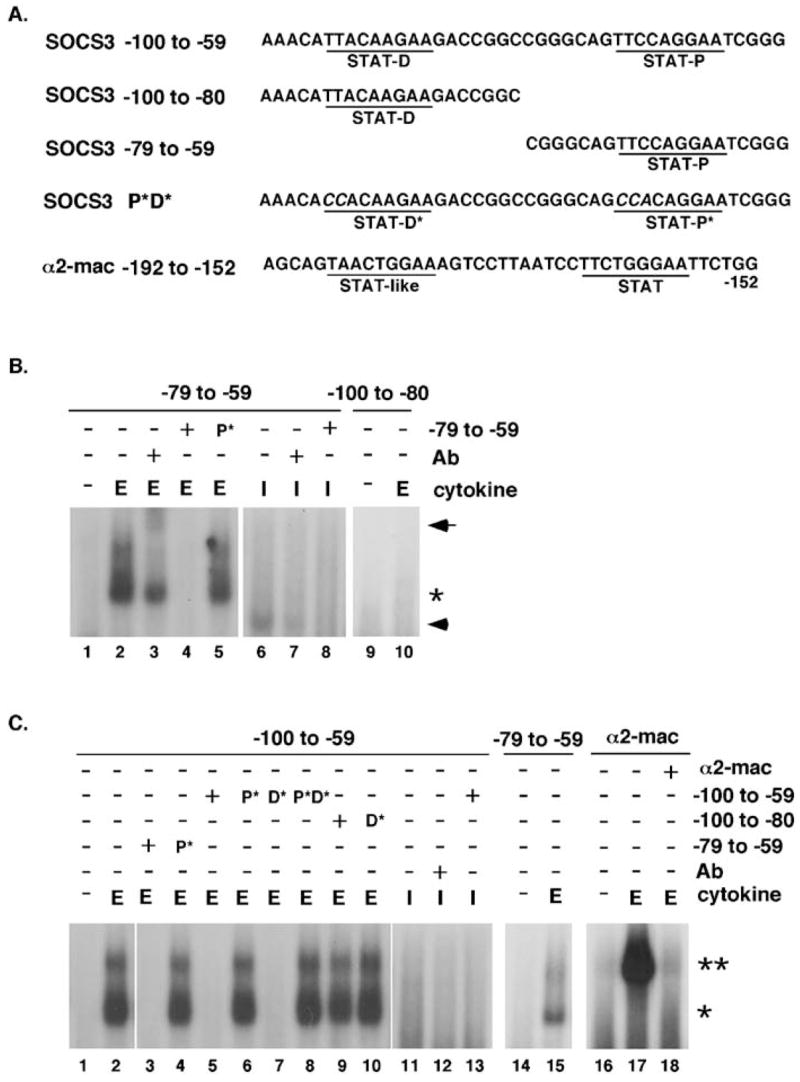
DNA binding activity on Socs3 and α2-macroglobulin promoter elements. (A) The sequences of oligonucleotides derived from the Socs3 or α2-macroglobulin promoters are shown. The proximal (STAT-P) and distal (STAT-D) STAT elements within the Socs3 promoter, as well as adjacent STAT motifs in the α2-macroglobulin promoter, are underlined. Mutations in Socs3 promoter STAT elements are shown in the context of the −100 to −59 oligonucleotide (mutant P*D*). Not shown are individual proximal or distal site mutations in the context of the −100 to −59 oligonucleotide (−100 to −59 P* or D*, respectively), or mutations in the context of the single site oligonucleotides (−79 to −59 P* or −100 to −80 D*); the sequence of the encoded mutations is the same as indicated for mutant P*D*. (B) and (C) ER-S3 cells were stimulated with 5U/ml Epo (E) or l00 U/ml IFNγ (I) for 30 min or left untreated (−), as indicated. Nuclear extracts were incubated with the indicated radiolabeled oligonucleotides alone or following a 15 min pre-incubation step with anti-STAT3 antibody (1 μg; Epo samples), anti-STATl antibody (1 μ IFNγ samples) or an excess (25 μM) of the indicated unlabeled oligonucleotide, as shown. DNA/protein complexes were resolved on 6% acrylamide gels and the migration positions of STAT3 dimers(*), STAT3 tetramers (**), supershifted STAT3 complexes (arrow) and IFNγ-inducible complexes (arrowhead) are shown.
Total RNA isolation, northern blot analysis and quantitative real time PCR
Total RNA was extracted with Trizol reagent (Molecular Research Center, Inc.), using conditions provided by the manufacturer. Denatured RNA (10 μg NIH3T3 cell lines; 20μg Stat3−/− or Stat3+/− MEFs) samples were separated on 1% agarose gels containing 6.4% formaldehyde, and transferred to nylon membranes in 10X SSC overnight. Cross-linking of RNA to the membrane was performed in a UV Stratalinker 2400 (Stratagene), using conditions provided by the manufacturer. Membranes were prehybridized in buffer containing 50% formamide, 5× Denhardt’s solution, 6 × SSC, 0.1% SDS and 50 μg/ml tRNA for 4–6 h at 42 °C. Denatured, radiolabeled cDNA probes were added (approximately 1 × 106cpm/ml) and membranes were hybridized for 20–24 h at 42 °C. Membranes were washed by incubation in 2× SSC/0.2% SDS for 30 min, followed by two washes in 0.2 × SSC/0.2% SDS for 30 min each and exposed to x-ray film at ×80 °C. For real time quantitative PCR, total RNA was reverse transcribed as described(46). SOCS3 expression was measured using the following primers (RL86: 5′-CGGAGATTTCGCTTCGGGAC-3′ RL87: 5′-AACTTGCTGTGGGTGACCATGG-3′) and aFAM-labeled probe (5′-AGCTCCCCGGGATGCGGT-3′). Expression of beta actin was determined with primers and real time PCR probes as described(47). cDNA samples were analyzed in duplicate and SOCS3 expression was normalized to beta actin as described previously(47).
Results
Δ133STAT3 signaling in response to IL-6
To examine STAT3 N-domain function, an internal deletion of residues 3–133 was generated in murine STAT3 (Δ133STAT3). This mutation removes the entire N-terminal region and fuses the initiator methionine and second alanine residue to the downstream coiled-coil domain. Based on crystal structure studies performed on STATs lacking N- and C-terminal domains, deletion of the N-domain is not expected to affect the structural integrity of the core protein(34, 48, 49). NIH3T3 cells, which contain endogenous STAT3, were engineered to stably express wild type STAT3 or Δ133STAT3 isoforms with a C-terminal FLAG epitope (3T3.STAT3 and 3T3. Δ133STAT3 cells, respectively).
Sequences in the N-domain of STAT1 have been shown to regulate nuclear TC45 phosphatase association and STAT1 dephosphorylation kinetics (37). By contrast, the N-domain of STAT4 is essential for cytokine-dependent activation by tyrosine phosphorylation (35, 36). To examine the potential for the STAT3 N-domain to mediate similar functions, we tested Δ133STAT3 signaling responses. Short-term stimulation with IL-6 (15 min) and FLAG immunoprecipitations showed that STAT3 and Δ133STAT3 were activated robustly when expressed ectopically in NIH3T3 cells (Fig. 1A). Significantly, both proteins underwent dephosphorylation with similar kinetics following IL-6 removal and chase treatments in medium lacking cytokine (Fig. 1 A). Furthermore, the tyrosine phosphorylation kinetics of Δ133STAT3 did not display significant differences with endogenous STAT3 (Fig. IB). Thus, FLAG-tagged STAT3 and Δ133STAT3 respond similarly to IL-6 treatment in the presence of endogenous STAT3 protein.
Fig. 1.

IL-6-dependent STAT3 activation in 3T3.STAT3 and 3T3. Δ133STAT3 cells. (A.) NIH3T3 cells expressing FLAG-tagged STAT3 (STAT3) or Δ133STAT3 (A133STAT3) were treated with 25ng/ml recombinant IL-6 and 25ng/ml recombinant soluble IL-6 receptor for 15min (+), followed by removal of IL-6-containing media and additional culture in DME/FCS for 1, 2 or 6 h, or were left untreated (−), as indicated. Denaturing immunoprecipitations from SDS-denatured whole cell lysates were performed with FLAG antibody and analyzed by immunoblotting with antibodies specific for tyrosine-phosphorylated STAT3 (pSTAT3) or total STAT3 (STAT3), upper and lower panels, respectively. (B) 3T3. Δ133STAT3 cells were stimulated for 2–15 min with recombinant IL-6 and soluble IL-6 receptor as described above, or left untreated, as indicated. Activated (pSTAT3 and pΔ133STAT3) STAT3 and total STAT3 isoforms were detected by immunoblotting.
IL-6-mediated activation of Δ133STAT3 in Stat3−/− MEFs
Recent studies have shown that STATs can form oligomeric complexes in unstimulated cells (35, 50–52) in addition to the SH2/phosphotyrosine dimers induced upon cytokine treatment. Therefore, endogenous STAT3 may influence the function of Δ133STAT3 through the formation of STAT3/Δ133STAT3 complexes. To circumvent this issue, Δ133STAT3 or STAT3 were stably expressed in STAT3-deficient mouse embryonic fibroblasts (Stat3−/− MEFs) by retroviral transduction (Δ133STAT3 or STAT3 MEFs, respectively) and tested for IL-6 responsiveness. The parental Stat3−/− MEFs lack detectable expression of STAT3 as judged by immunoblot analysis with N- or C-terminal specific antibodies (data not shown). Reconstituted MEFs were stimulated with IL-6 for 15 min then incubated in medium lacking cytokine for up to 6 h to examine the rate of STAT3 and Δ133STAT3 dephosphorylation. Importantly, STAT3 and Δ133STAT3 showed similar kinetics of dephosphorylation in cells lacking endogenous STAT3 (Fig. 2A). Furthermore, tyrosine-phosphorylated Δ133STAT3 accumulated rapidly in the nuclear fraction, similar to endogenous STAT3 or ectopically expressed FLAG-tagged STAT3, as judged by cellular fractionation and immunoblotting experiments (Fig. 2B; data not shown). Low levels of STAT3 were observed in nuclear extracts from unstimulated cells, which is consistent with a constitutive nuclear import and export process (7,53). The distribution of marker proteins histone H3 (nuclear) and HSP70 (cytoplasmic) indicated correct subcellular fractionation (Fig. 2C).
Fig. 2.

STAT3 activation and nuclear accumulation in MEFs treated with IL-6. (A) STAT3 or Δ133STAT3 MEFs were treated with 25ng/ml recombinant IL-6 and 25ng/ml recombinant soluble IL-6 receptor for 15 min (+), followed by removal of IL-6-containing media and culture in DME/FCS for 1–6 h, as indicated. Whole cell lysates were subjected to immunoblot analysis with pSTAT3 or total STAT3 antibodies, as indicated (upper and lower panels, respectively). The migration positions of STAT3 and Δ133STAT3 are shown, as well as a background band that appears with total STAT3 antibody (*). (B) Nuclear extracts were prepared from Stat3+/− or Δ133STAT3 MEFs following stimulation with 50ng/ml recombinant IL-6 and 50ng/ml recombinant soluble IL–6 receptor, as indicated. Samples were analyzed by immunoblotting with pSTAT3 or total STAT3 antibodies, as indicated (upper and lower panels, respectively). (C) Nuclear extracts from untreated or IL-6-treated cells were analyzed by immunoblotting with HSP70 or histone H3 antibodies (upper and lower panels, respectively). Curvature of some histone H3 bands was detected in samples run at the edge of the gel. Cytoplasmic extracts (C) were prepared from unstimulated cells and analyzed in parallel.
The comparison between NIH3T3 cells and MEFs enabled us to evaluate the potential role of endogenous STAT3 in Δ133STAT3 signaling. As indicated by the results in Fig. 2, endogenous STAT3 is not required for Δ133STAT3 activation, nuclear import or dephosphorylation. Furthermore, these functions are mediated by mechanisms that are independent of STAT3 N-domain sequences.
IL-6-dependent induction of SOCS3 by Δ133STAT3
Based on previous reports showing a requirement for STAT N-domain sequences in target gene expression(37, 39, 40), we speculated that the STAT3 N-domain was essential for transcriptional activation of the potent IL-6-responsive gene, Socs3. This was further suggested by the presence of adjacent STAT consensus binding motifs in the murine Socs3 promoter(45), which could potentially mediate STAT3 tetramer formation via N-domain interactions. A comparison of the human and murine Socs3 promoters revealed a striking conservation in the sequence and location of STAT motifs (Fig. 3), providing additional indication that stabilizing interactions between adjacent STAT3 dimers via their N-domains might be necessary for Socs3 expression.
Fig. 3.

Comparison of the murine and human Socs3 promoter sequences. Genomic sequences from the human and murine Socs3 promoter regions were obtained from the Ensembl database and aligned for comparison. The analysis extended to −160bp of upstream sequences since this has been shown to confer maximal activity of the murine promoter via gene reporter assays(45). The conserved STAT motifs (bold underline), TATA box (light underline) and murine transcriptional start site (45) (arrow) are indicated.
IL-6 treatment led to strong induction of SOCS3 in Stat3+/− MEFs and Stat3−/− MEFs reconstituted with STAT3, but not in Stat3−/− MEFs (Figs. 4A, B), thus confirming an indispensable role for STAT3 in IL-6-responsive Socs3 gene regulation. Surprisingly, reconstituted STAT3 and A133STAT3 stimulated SOCS3 expression in Stat3−/− MEFs with comparable kinetics and magnitude, as judged by Northern blot and quantitative real time PCR analyses, indicating similar functional activities of each STAT3 isoform (Figs. 4A, B). Thus, our results demonstrate that Δ133STAT3 mediates IL-6-dependent expression of the Socs3 gene in its chromosomal context.
Fig. 4.
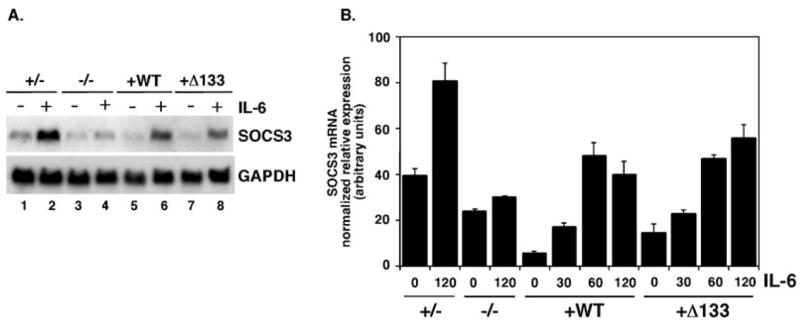
SOCS3 expression in MEFs. (A) Stat3+/− MEFs, Stat3 −/− MEFs, STAT3 MEFs (WT) and Δ133STAT3 MEFs (Δ133STAT3) were stimulated with 50ng/ml recombinant IL-6 and 50 ng/ml recombinant soluble IL-6 receptor for 2 h (+) or left untreated (−) as indicated. Total RNA was isolated and analyzed by Northern blotting with probes specific for murine SOCS3 (upper panel) or GAPDH (lower panel), as shown. (B) MEF cell lines were stimulated with IL-6 and soluble IL-6 receptor for 0–120 min, as indicated. Total RNA was isolated and analyzed by real time PCR for SOCS3 and actin mRNA expression. SOCS3 mRNA levels were normalized to actin for individual samples; the average of duplicate samples is shown.
STAT3 interacts with a single consensus binding site in the Socs3 proximal promoter
The adjacent position of conserved STAT binding motifs in the murine Socs3 promoter (Fig. 3)(45) raised the possibility that STAT3 dimers were recruited to each site, with the potential for cooperative binding via homotypic interactions between neighboring STAT3 N-domains. A previous study showed that deletion of both sites, or of the site closest to the mRNA initiation point, abrogated cytokine-responsive Socs3 promoter activity, while the function of the distal STAT site was not tested directly(45). Cooperative binding via STAT N-domain interactions has been delineated for other STAT-responsive genes; most significant to this study is STAT3-mediated activation of the α2-macroglobulin promoter via STAT3 tetramer formation (39). To examine STAT3 binding on Socs3 promoter elements, we analyzed a series of oligonucleotides containing either adjacent or single STAT motifs (Fig. 5A) in electrophoretic mobility shift assays (EMSAs). As a control for STAT3 tetramer formation, we included an oligonucleotide derived from the α2-macroglobulin gene, which contains its adjacent STAT binding motifs (Fig. 5A). Since IFNγ -activated STAT1 can induce SOCS3(54) and STAT1 is heavily tyrosine phosphorylated in Stat3−/− MEFs by IL-6 treatment (data not shown), we circumvented potential complications from hyperactivated STAT1 through the use of a chimeric erythropoietin receptor (ER-S3) that drives robust STAT3 activation due to the presence of engineered STAT3 binding sites derived from gp130 (42, 43).
Epo stimulation of 32D cells expressing ER-S3 resulted in rapid STAT3 tyrosine phosphorylation and enhanced DNA binding activity on the proximal STAT consensus sequence in the Socs3 promoter (−79 to −59 oligonucleotide) (Fig. 5B, lanes 1–2 and data not shown). This DNA binding activity was inhibited by excess unlabeled oligonucleotide and partially supershifted by an anti-STAT3 antibody, indicating that STAT3 interacts specifically with the proximal consensus site (Fig. 5B, lanes 2–4). In support of this, a corresponding oligonucleotide with a mutated STAT site failed to competitively inhibit STAT3 DNA binding activity (Fig. 5B, lane 5). Surprisingly, detectable STAT3 binding was not found at the distal STAT element in the Socs3 promoter (−100 to −80 oligonucleotide) (Fig. 5B, lanes 9–10). A comparison with IFNγ treatment was included to examine the potential for STAT1 interaction at these elements. IFNγ stimulated only modest levels of DNA binding activity on the proximal site oligonucleotide (Fig. 5B, lane 6), and no detectable binding to the distal site (data not shown). The IFNγ -responsive complex migrated with a faster mobility than the Epo-stimulated STAT3:oligonucleotide complex (Fig. 5B, compare lanes 6 and 2), suggesting it is comprised of STAT1 homodimers bound to the proximal site oligonucleotide. These results collectively indicate that activated STAT3 is recruited primarily to the proximal STAT motif in the Socs3 promoter.
EMSAs with an oligonucleotide containing both STAT binding sites in the Socs3 promoter (×100 to −59) showed the presence of two DNA binding complexes. The major species co-migrated with the complex formed on the proximal site oligonucleotide, suggesting it is comprised of a STAT3 dimer (Fig. 5C; compare lanes 2 and 15). The minor DNA binding species comigrated with the tetrameric STAT3 complex bound to the α2-macroglobulin promoter oligonucleotide, which has been shown previously to be stabilized by STAT3 N-domain interactions (39) (Fig. 5C, compare lanes 2 and 17). Competition with oligonucleotides containing an intact proximal STAT site effectively blocked STAT3 DNA binding activity on the Socs3 dual site oligonucleotide (Fig. 5C, lanes 3, 5, 7), while competition with oligonucleotides containing a mutated proximal site, or the distal site alone, failed to abrogate DNA binding (Fig. 5B, lanes 4,6,8–10). IFNγ treatment did not lead to significant DNA binding activity on the Socs3 dual site oligonucleotide (Fig. 5C, lanes 11–13). These results show that the proximal STAT site mediates STAT3 interaction with the Socs3 promoter. They also suggest the potential for STAT3 tetramer formation on Socs3 promoter sequences, which may be initiated or stabilized by STAT3 interaction at the proximal STAT motif. Nonetheless, induction of Socs3 by IL-6 is independent of the STAT3 N-domain, indicating that STAT3 tetramer formation via N-domain interactions is not essential for assembly of a stable transcriptional initiation complex on the endogenous Socs3 promoter.
Discussion
The highly conserved N-terminal domain of the STAT proteins has been shown to mediate several activities including cytokine receptor recruitment, target gene expression and STAT downregulation (33, 35–37, 39, 55). We examined the function of the N-domain of STAT3, an essential STAT family member with numerous roles in normal and disease signaling pathways. Deletion of the STAT3 N-domain did not impair IL-6-dependent tyrosine phosphorylation, nuclear import or dephosphorylation kinetics, indicating that this region is not essential for STAT3 recruitment to the IL-6 receptor complex, translocation to the nuclear compartment or downregulation. These results point to significant differences with N-domain function in other STAT proteins, despite the structural similarity of this region. For example, the STAT4 N-domain is essential for activation through cytokine receptors, an event that requires N-domain-dependent dimerization between non-phosphorylated STAT4 molecules in unstimulated cells (35, 36). In STAT1, the N-domain appears to regulate association with the nuclear phosphatase TC45 and subsequent STAT1 dephosphorylation (37, 55). By contrast, deletion of the STAT5a N-domain does not abrogate cytokine-responsive tyrosine phosphorylation, dimerization or dimer DNA binding yet appears to render constitutive activation, indicating a negative regulatory function (56). Thus, the N-domain may participate in unique signaling responses for individual members of the STAT protein family.
STAT N-domains were initially found to have an important role in stabilizing contacts between adjacent STAT dimers on tandem DNA binding sites, which appears to be required for the expression of certain STAT target genes (37, 39, 40). Isolated STAT N-domains demonstrate highly specific, homotypic interactions in overexpression systems, yet do not bind the N-domains of distinct STAT family members (35). This suggests that STAT N-domains have a shared function in stabilizing STAT oligomeric complexes, either on adjacent promoter elements within the nucleus or in the cytoplasm of unstimulated cells (32, 33, 36, 37, 39). We found that the tandem STAT consensus motifs in the murine Socs3 promoter were highly conserved with the human promoter in sequence and in location relative to the transcriptional initiation site. This suggested an important function in STAT3 recruitment and Socs3 gene regulation. In light of these collective results, we speculated that STAT3 N-domain sequences might be required to maintain an efficient STAT3 activator complex on the Socs3 promoter. Surprisingly, however, IL-6-dependent SOCS3 expression was stimulated by Δ133STAT3 as effectively as STAT3 in reconstituted Stat3 −/− MEFs, which show a tight requirement for STAT3 protein. Furthermore, STAT3 DNA binding activity on the Socs3 promoter is mediated primarily through the proximal STAT motif. Taken together, our studies indicate that Socs3 gene expression does not require STAT3 tetramer formation by N-domain sequences, rather STAT3 interaction at the proximal motif in the Socs3 promoter may be sufficient to stabilize an effective transcriptional initiation complex via an N-domain-independent mechanism.
STAT3 showed potential ability to mediate tetramer formation on the Socs3 promoter dual site oligonucleotide as judged by EMSAs. A dramatic difference was observed in the efficiency of apparent tetramer assembly on individual promoter sequences, however, with strong tetramer formation on the α2-macro globulin promoter relative to the Socs3 promoter. This may reflect differences in the relative affinities of STAT binding motifs or may be a function of spacing between adjacent STAT elements in these promoter regions (12 bp spacing versus 14 bp in α2-macroglobulin and Socs3 promoters, respectively) (39, 45). It is important to point out, however, that the apparent STAT3 tetramers observed on the Socs3 promoter sequences were detected in nuclear extracts but have not been confirmed with purified STAT3 protein, thus leaving open the possibility that these complexes contain additional cofactors and are not comprised of STAT3 tetramers exclusively. Importantly, EMSAs with oligonucleotides encoding individual STAT consensus sites from the murine Socs3 promoter indicated that the proximal site centered at ×70bp was the principal STAT3 recruitment sequence, while the distal STAT motif did not show considerable affinity for STAT3. These findings are reminiscent of the α2-macroglobulin enhancer in which a high affinity STAT binding site regulates STAT3 tetramer assembly (39). Presumably, the tight conservation in the human and murine Socs3 promoters is necessary for maximal expression of this critical negative regulatory gene, although little is understood about the mechanisms regulating inducible transcription beyond the requirement for STAT3.
Recent studies have demonstrated that Socs3 gene expression can be stimulated by a STAT1-dependent pathway in response to IFNγ (54). Moreover, STAT1 and STAT3 were found to have interchangeable functions in IFNγ-dependent regulation of SOCS3(57). These results raised the possibility that Socs3 gene induction was regulated via a STAT1-mediated pathway in Stat3−/− MEFs, regardless of STAT3 or Δ133STAT3 expression. Our data, however, argue against this scenario. Enforced expression of STAT3 or Δ133STAT3 rendered significant IL-6-inducible SOCS3 expression in Stat3−/− MEFs, while cells lacking STAT3 did not show induction of SOCS3 by IL-6 (Fig. 4), despite containing high levels of activated STAT1 (data not shown). In addition, IFNγ-activated STAT1 showed only modest DNA binding on Socs3 promoter sequences. These data collectively indicate that STAT3 or Δ133STAT3 represent the major IL-6-inducible DNA binding species on the Socs3 promoter in MEFs, with their recruitment mediated by the proximal STAT motif. We did confirm IFNγ -dependent regulation of SOCS3 in all MEF cell lines, indicating that SOCS3 regulation via a STAT1 response was intact (data not shown). Presently, it is unclear why STAT1 activation by IL-6 in Stat3-null cells does not lead to significant induction of SOCS3. Perhaps specific STAT1 post-translational modifications, nuclear localization mechanisms or protein co-factors are required for efficient SOCS3 regulation by STAT1, which are lacking in the IL-6 pathway on the Stat3-null background.
In summary, we have demonstrated that IL-6-dependent STAT3 activation, nuclear accumulation and Socs3 gene expression are regulated by a STAT3 N-domain deletion mutant, Δ133STAT3. This indicates that the N-domain is dispensable for IL-6 stimulation of the STAT3/SOCS3 pathway, an important negative regulatory response in vivo. Our results are significant in light of the essential role of STAT4 N-domain sequences in cytokine activation (35, 36), downregulation of STAT1 and STAT5 activity by the N-domain(37, 56), and the function of the N-domain in cooperative DNA binding and gene transcription in other systems, including STAT3-mediated expression of the α2-macroglobulin gene (39), since they reinforce the concept that the STAT N-domains may be involved in unique signaling responses. This may be particularly relevant in light of the recent finding that STAT5 tetramer formation via N-domain sequences is linked with leukemogenesis (56). To our knowledge, mutations in the STAT N-domain have not yet been associated with human disease, although this remains to be explored further due to the critical role for the N-domain in several pathways. Furthermore, our findings highlight the potential for STAT proteins lacking the N-domain to mediate certain biological functions, which may be significant with genetically engineered animals that retain a portion of the STAT coding sequence (56, 58). With STAT3, the N-domain appears to show selective activity on target gene promoters. This reveals additional complexity in STAT3 transcriptional control mechanisms and underscores the idea that mutations in STAT N-domains may impact some but not all biologic functions.
Acknowledgments
We thank Merav Socolovsky for her generous advice on retroviral production and infection. We greatly appreciate the discussions and insights provided by Xiaomin Chen, Sharon Dent, Brad McIntyre, Miles Wilkinson and Athanasia Panopoulos during this study. We thank Xiaomin Chen and Athanasia Panopoulos for their critical review of the manuscript. This work was supported by a predoctoral NIH training grant (DBB) and grants from the NIH (RO1 CA77447), AHA Texas Affiliate (0455143Y) and Gillson Longenbaugh Foundation to SSW.
References
- 1.Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, Forster I, Akira S. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999;10:39–49. doi: 10.1016/s1074-7613(00)80005-9. [DOI] [PubMed] [Google Scholar]
- 2.Chapman RS, Lourenco PC, Tonner E, Flint DJ, Selbert S, Takeda K, Akira S, Clarke AR, Watson CJ. Suppression of epithelial apoptosis and delayed mammary gland involution in mice with a conditional knockout of Stat3. Genes Dev. 1999;13:2604–2616. doi: 10.1101/gad.13.19.2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alzoni T, Maritano D, Gorgoni B, Rizzuto G, Libert C, Poli V. Essential role of STAT3 in the control of the acute-phase response as revealed by inducible gene inactivation in the liver. Mol Cell Biol. 2001;21:1621–1632. doi: 10.1128/MCB.21.5.1621-1632.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Niwa H, Burdon T, Chambers I, Smith A. Self-renewal of pluripotent embryonic stem cells is mediated via activation of Stat3. Genes Dev. 1998;12:2048–2060. doi: 10.1101/gad.12.13.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Darnell JE., Jr STATs and gene regulation. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 6.Kisseleva T, Bhattacharya S, Braunstein J, Schindler CW. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene. 2002;285:1–24. doi: 10.1016/s0378-1119(02)00398-0. [DOI] [PubMed] [Google Scholar]
- 7.Bhattacharya S, Schindler C. Regulation of Stat3 nuclear export. J Clin Invest. 2003;111:553–559. doi: 10.1172/JCI15372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.ten Hoeve J, de Jesus Ibarra-Sanchez M, Fu Y, Zhu W, Tremblay M, David M, Shuai K. Identification of a nuclear Stat1 protein tyrosine phosphatase. Mol Cell Biol. 2002;22:5662–5668. doi: 10.1128/MCB.22.16.5662-5668.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee CK, Raz R, Gimeno R, Gertner R, Wistinghausen B, Takeshita K, DePinho RA, Levy DE. STAT3 is a negative regulator of granulopoiesis but is not required for G–CSF–dependent differentiation. Immunity. 2002;17:63–72. doi: 10.1016/s1074-7613(02)00336-9. [DOI] [PubMed] [Google Scholar]
- 10.Welte T, Zhang SS, Wang T, Zhang Z, Hesslein DG, Yin Z, Kano A, Iwamoto Y, Li E, Craft JE, Bothwell AL, Fikrig E, Koni PA, Flavell RA, Fu XY. STAT3 deletion during hematopoiesis causes Crohn’s disease–like pathogenesis and lethality: A critical role of STAT3 in innate immunity. Proc Natl Acad Sci USA. 2003;100:1879–1884. doi: 10.1073/pnas.0237137100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sano S, Itami S, Takeda K, Tarutani M, Yamaguchi Y, Miura H, Yoshikawa K, Akira S, Takeda J. Keratinocyte-specific ablation of Stat3 exhibits impaired skin remodeling, but does not affect skin morphogenesis. EMBO J. 1999;18:4657–4668. doi: 10.1093/emboj/18.17.4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng F, Wang H-W, Cuenca A, Huang M, Ghansah T, Brayer J, Kerr WG, Takeda K, Akira S, Schoenberger SP, Yu H, Jove R, Sotomayer EM. A critical role for STAT3 signaling in immune tolerance. Immunity. 2003;19:425–436. doi: 10.1016/s1074-7613(03)00232-2. [DOI] [PubMed] [Google Scholar]
- 13.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE., Jr Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 14.Benekli M, Baer MR, Baumann H, Wetzler M. Signal transducer and activator of transcription proteins in leukemias. Blood. 2003;101:2940–2954. doi: 10.1182/blood-2002-04-1204. [DOI] [PubMed] [Google Scholar]
- 15.Frank DA. STAT signaling in the pathogenesis and treatment of cancer. Mol Med. 1999;5:432–456. [PMC free article] [PubMed] [Google Scholar]
- 16.Bromberg JF, Horvath CM, Besser D, Lathem WW, Darnell JE., Jr Stat3 activation is required for cellular transformation by v-src. Mol Cell Biol. 1998;18:2553–2558. doi: 10.1128/mcb.18.5.2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Turkson J, Bowman T, Garcia R, Caldenhoven E, De Groot RP, Jove R. Stat3 activation by Src induces specific gene regulation and is required for cell transformation. Mol Cell Biol. 1998;18:2545–2552. doi: 10.1128/mcb.18.5.2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Turkson J, Ryan D, Kim JS, Zhang Y, Chen Z, Haura E, Laudano A, Sebti S, Hamilton AD, Jove R. Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. J Biol Chem. 2001;276:45443–45455. doi: 10.1074/jbc.M107527200. [DOI] [PubMed] [Google Scholar]
- 19.Ren Z, Cabell LA, Schaefer TS, McMurray JS. Identification of a high-affinity phosphopeptide inhibitor of Stat3. Bioorg Med Chem Lett. 2003;13:633–636. doi: 10.1016/s0960-894x(02)01050-8. [DOI] [PubMed] [Google Scholar]
- 20.Leong PL, Andrews GA, Johnson DE, Dyer KF, Xi S, Mai JC, Robbins PD, Gadiparthi S, Burke NA, Watkins SF, Grandis JR. Targeted inhibition of Stat3 with a decoy oligonucleotide abrogates head and neck cancer cell growth. Proc Natl Acad Sci USA. 2003;100:4138–4143. doi: 10.1073/pnas.0534764100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shouda T, Yoshida T, Hanada T, Wakioka T, Oishi M, Miyoshi K, Komiya S, Kosai K, Hanakawa Y, Hashimoto K, Nagata K, Yoshimura A. Induction of the cytokine signal regulator SOCS3/CIS3 as a therapeutic strategy for treating inflammatory arthritis. J Clin Invest. 2001;108:1781–1788. doi: 10.1172/JCI13568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wormald S, Hilton DJ. Inhibitors of cytokine signal transduction. J Biol Chem. 2004;279:821–824. doi: 10.1074/jbc.R300030200. [DOI] [PubMed] [Google Scholar]
- 23.Shultz LD, Rajan TV, Greiner DL. Severe defects in immunity and hematopoiesis caused by SHP-1 protein-tyrosine-phosphatase deficiency. Trends Biotechnol. 1997;15:302–307. doi: 10.1016/S0167-7799(97)01060-3. [DOI] [PubMed] [Google Scholar]
- 24.Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, Robb L, Greenhalgh CJ, Forster I, Clausen BE, Nicola NA, Metcalf D, Hilton DJ, Roberts AW, Alexander WS. SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol. 2003;4:540–545. doi: 10.1038/ni931. [DOI] [PubMed] [Google Scholar]
- 25.Croker BA, Metcalf D, Robb L, Wei W, Mifsud S, DiRago L, Cluse LA, Sutherland KD, Hartley L, Williams E, Zhang JG, Hilton DJ, Nicola NA, Alexander WS, Roberts AW. SOCS3 is a critical physiological negative regulator of G-CSF signaling and emergency granulopoiesis. Immunity. 2004;20:153–165. doi: 10.1016/s1074-7613(04)00022-6. [DOI] [PubMed] [Google Scholar]
- 26.Lang R, Pauleau AL, Parganas E, Takahashi Y, Mages J, Ihle JN, Rutschman R, Murray PJ. SOCS3 regulates the plasticity of gp130 signaling. Nat Immunol. 2003;4:546–550. doi: 10.1038/ni932. [DOI] [PubMed] [Google Scholar]
- 27.Yasukawa H, Ohishi M, Mori H, Murakami M, Chinen T, Aki D, Hanada T, Takeda K, Akira S, Hoshijima M, Hirano T, Chien KR, Yoshimura A. IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nat Immunol. 2003;4:551–556. doi: 10.1038/ni938. [DOI] [PubMed] [Google Scholar]
- 28.Alexander WS, Starr R, Fenner JE, Scott CL, Handman E, Sprigg NS, Corbin JE, Cornish AL, Darwiche R, Owczarek CM, Kay TW, Nicola NA, Hertzog PJ, Metcalf D, Hilton DJ. SOCS1 is a critical inhibitor of interferon gamma signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell. 1999;98:597–608. doi: 10.1016/s0092-8674(00)80047-1. [DOI] [PubMed] [Google Scholar]
- 29.Krebs DL, Hilton DJ. SOCS proteins: negative regulators of cytokine signaling. Stem Cells. 2001;19:378–387. doi: 10.1634/stemcells.19-5-378. [DOI] [PubMed] [Google Scholar]
- 30.Kishimoto T, Akira S, Narazaki M, Taga T. Interleukin-6 family of cytokines and gp130. Blood. 1995;86:1243–1254. [PubMed] [Google Scholar]
- 31.Costa-Pereira AP, Tininini S, Strobl B, Alonzi T, Schlaak JF, Is’harc H, Gesualdo I, Newman SJ, Kerr IM, Poli V. Mutational switch of an IL-6 response to an interferon-gamma-like response. Proc Natl Acad Sci USA. 2002;99:8043–8047. doi: 10.1073/pnas.122236099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen X, Bhandari R, Vinkemeier U, Van Den Akker F, Darnell JE, Jr, Kuriyan J. A reinterpretation of the dimerization interface of the N– terminal domains of STATs. Protein Sci. 2003;12:361–365. doi: 10.1110/ps.0218903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vinkemeier U, Moarefi I, Darnell JE, Jr, Kuriyan J. Structure of the amino-terminal protein interaction domain of STAT-4. Science. 1998;279:1048–1052. doi: 10.1126/science.279.5353.1048. [DOI] [PubMed] [Google Scholar]
- 34.Mao X, Ren Z, Parker GN, Sondermann H, Pastorello MA, Wang W, McMurray JS, Demeler B, Darnell JE, Jr, Chen X. Structural bases of unphosphorylated STAT1 association and receptor binding. Mol Cell. 2005;17:761–771. doi: 10.1016/j.molcel.2005.02.021. [DOI] [PubMed] [Google Scholar]
- 35.Ota N, Brett TJ, Murphy TL, Fremont DH, Murphy KM. N-domain-dependent nonphosphorylated STAT4 dimers required for cytokine-driven activation. Nat Immunol. 2004;5:208–215. doi: 10.1038/ni1032. [DOI] [PubMed] [Google Scholar]
- 36.Murphy TL, Geissal ED, Farrar JD, Murphy KM. Role of the Stat4 N domain in receptor proximal tyrosine phosphorylation. Mol Cell Biol. 2000;20:7121–7131. doi: 10.1128/mcb.20.19.7121-7131.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meyer T, Hendry L, Begitt A, John S, Vinkemeier U. A single residue modulates tyrosine dephosphorylation, oligomerization, and nuclear accumulation of stat transcription factors. J Biol Chem. 2004;279:18998–19007. doi: 10.1074/jbc.M400766200. [DOI] [PubMed] [Google Scholar]
- 38.Chang HC, Zhang S, Oldham I, Naeger L, Hoey T, Kaplan MH. STAT4 requires the N-terminal domain for efficient phosphorylation. J Biol Chem. 2003;278:32471–32477. doi: 10.1074/jbc.M302776200. [DOI] [PubMed] [Google Scholar]
- 39.Zhang X, Darnell JE., Jr Functional importance of Stat3 tetramerization in activation of the alpha 2-macroglobulin gene. J Biol Chem. 2001;276:33576–33581. doi: 10.1074/jbc.M104978200. [DOI] [PubMed] [Google Scholar]
- 40.Xu X, Sun YL, Hoey T. Cooperative DNA binding and sequence-selective recognition conferred by the STAT amino-terminal domain. Science. 1996;273:794–797. doi: 10.1126/science.273.5276.794. [DOI] [PubMed] [Google Scholar]
- 41.Yang J, Chatterjee-Kishore M, Staugaitis SM, Nguyen H, Schlessinger K, Levy DE, Stark GR. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res. 2005;65:939–947. [PubMed] [Google Scholar]
- 42.Wooten DK, Xie X, Bartos D, Busche RA, Longmore GD, Watowich SS. Cytokine signaling through Stat3 activates integrins, promotes adhesion, and induces growth arrest in the myeloid cell line 32D. J Biol Chem. 2000;275:26566–26575. doi: 10.1074/jbc.M003495200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Panopoulos AP, Bartos D, Zhang L, Watowich SS. Control of myeloid-specific integrin αMβ2 (CD11b/CDl8) expression by cytokines is regulated by Stat3-dependent activation of PU.1. J Biol Chem. 2002;277:19001–19007. doi: 10.1074/jbc.M112271200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Naviaux RK, Costanzi E, Haas M, Verma IM. The pCL vector system: rapid production of helper-free, high-titer, recombinant retroviruses. J Virol. 1996;70:5701–5705. doi: 10.1128/jvi.70.8.5701-5705.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Auemhammer CJ, Bousquet C, Melmed S. Autoregulation of pituitary corticotroph SOCS-3 expression: characterization of the murine SOCS-3 promoter. Proc Natl Acad Sci U S A. 1999;96:6964–6969. doi: 10.1073/pnas.96.12.6964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lang R, Rutschman RL, Greaves DR, Murray PJ. Autocrine deactivation of macrophages in transgenic mice constitutively overexpressing IL-10 under control of the human CD68 promoter. J Immunol. 2002;168:3402–3411. doi: 10.4049/jimmunol.168.7.3402. [DOI] [PubMed] [Google Scholar]
- 47.Lang R, Patel D, Morris JJ, Rutschman RL, Murray PJ. Shaping gene expression in activated and resting primary macrophages by IL-10. J Immunol. 2002;169:2253–2263. doi: 10.4049/jimmunol.169.5.2253. [DOI] [PubMed] [Google Scholar]
- 48.Chen X, Vinkemeier U, Zhao Y, Jeruzalmi D, Darnell JE, Jr, Kuriyan J. Crystal structure of a tyrosine phosphorylated STAT-1 dimer bound to DNA. Cell. 1998;93:827–839. doi: 10.1016/s0092-8674(00)81443-9. [DOI] [PubMed] [Google Scholar]
- 49.Becker S, Groner B, Muller CW. Three-dimensional structure of the Stat3beta homodimer bound to DNA. Nature. 1998;394:145–151. doi: 10.1038/28101. [DOI] [PubMed] [Google Scholar]
- 50.Schroder M, Kroeger KM, Volk HD, Eidne KA, Grutz G. Preassociation of nonactivated STAT3 molecules demonstrated in living cells using bioluminescence resonance energy transfer: a new model of STAT activation? J Leukoc Biol. 2004;75:792–797. doi: 10.1189/jlb.1003496. [DOI] [PubMed] [Google Scholar]
- 51.Novak U, Ji H, Kanagasundaram V, Simpson R, Paradiso L. STAT3 forms stable homodimers in the presence of divalent cations prior to activation. Biochem Biophys Res Commun. 1998;247:558–563. doi: 10.1006/bbrc.1998.8829. [DOI] [PubMed] [Google Scholar]
- 52.Kretzschmar AK, Dinger MC, Henze C, Brocke-Heidrich K, Horn F. Analysis of Stat3 (signal transducer and activator of transcription 3) dimerization by fluorescence resonance energy transfer in living cells. Biochem J. 2004;377:289–297. doi: 10.1042/BJ20030708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pranada AL, Metz S, Herrmann A, Heinrich PC, Muller-Newen G. Real time analysis of STAT3 nucleocytoplasmic shuttling. J Biol Chem. 2004;279:15114–15123. doi: 10.1074/jbc.M312530200. [DOI] [PubMed] [Google Scholar]
- 54.Gatto L, Berlato C, Poli V, Tininini S, Kinjyo I, Yoshimura A, Cassatella MA, Bazzoni F. Analysis of SOCS3 promoter responses to interferon gamma. J Biol Chem. 2004;279:13746–13754. doi: 10.1074/jbc.M308999200. [DOI] [PubMed] [Google Scholar]
- 55.Strehlow I, Schindler C. Amino-terminal signal transducer and activator of transcription (STAT) domains regulate nuclear translocation and STAT deactivation. J Biol Chem. 1998;273:28049–28056. doi: 10.1074/jbc.273.43.28049. [DOI] [PubMed] [Google Scholar]
- 56.Moriggl R, Sexl V, Kenner L, Duntsch C, Stangl K, Gingras S, Hoffmeyer A, Bauer A, Piekorz R, Wang D, Bunting KD, Wagner EF, Sonneck K, Valent P, Ihle JN, Beug H. Stat5 tetramer formation is associated with leukemogenesis. Cancer Cell. 2005;7:87–99. doi: 10.1016/j.ccr.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 57.Qing Y, Stark GR. Alternative activation of STAT1 and STAT3 in response to interferon-γ. J Biol Chem. 2004;279:41679–41685. doi: 10.1074/jbc.M406413200. [DOI] [PubMed] [Google Scholar]
- 58.Cui Y, Riedlinger G, Tang W, Li C, Deng C-X, Robinson GW, Hennighausen L. Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation. Mol Cell Biol. 2004;24:8037–8047. doi: 10.1128/MCB.24.18.8037-8047.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]