

Abstract
Context
In addition to trauma exposure, other factors contribute to risk for development of posttraumatic stress disorder (PTSD) in adulthood. Both genetic and environmental factors are contributory, with child abuse providing significant risk liability.
Objective
To increase understanding of genetic and environmental risk factors as well as their interaction in the development of PTSD by gene × environment interactions of child abuse, level of non–child abuse trauma exposure, and genetic polymorphisms at the stress-related gene FKBP5.
Design, Setting, and Participants
A cross-sectional study examining genetic and psychological risk factors in 900 non psychiatric clinic patients (762 included for all genotype studies) with significant levels of childhood abuse as well as non–child abuse trauma using a verbally presented survey combined with single-nucleotide polymorphism (SNP) genotyping. Participants were primarily urban, low-income, black (>95%) men and women seeking care in the general medical care and obstetrics-gynecology clinics of an urban public hospital in Atlanta, Georgia, between 2005 and 2007.
Main Outcome Measures
Severity of adult PTSD symptomatology, measured with the modified PTSD Symptom Scale, non–child abuse (primarily adult) trauma exposure and child abuse measured using the traumatic events inventory and 8 SNPs spanning the FKBP5 locus.
Results
Level of child abuse and non–child abuse trauma each separately predicted level of adult PTSD symptomatology (mean [SD], PTSD Symptom Scale for no child abuse, 8.03 [10.48] vs ≥2 types of abuse, 20.93 [14.32]; and for no non–child abuse trauma, 3.58 [6.27] vs ≥4 types, 16.74 [12.90]; P<.001). Although FKBP5 SNPs did not directly predict PTSD symptom outcome or interact with level of non–child abuse trauma to predict PTSD symptom severity, 4 SNPs in the FKBP5 locus significantly interacted (rs9296158, rs3800373, rs1360780, and rs9470080; minimum P=.0004) with the severity of child abuse to predict level of adult PTSD symptoms after correcting for multiple testing. This gene × environment interaction remained significant when controlling for depression severity scores, age, sex, levels of non–child abuse trauma exposure, and genetic ancestry. This genetic interaction was also paralleled by FKBP5 genotype-dependent and PTSD-dependent effects on glucocorticoid receptor sensitivity, measured by the dexamethasone suppression test.
Conclusions
Four SNPs of the FKBP5 gene interacted with severity of child abuse as a predictor of adult PTSD symptoms. There were no main effects of the SNPs on PTSD symptoms and no significant genetic interactions with level of non–child abuse trauma as predictor of adult PTSD symptoms, suggesting a potential gene-childhood environment interaction for adult PTSD.
Posttraumatic stress disorder (PTSD) is a debilitating stress-related psychiatric disorder, with prevalence rates of at least 7% to 8% in the US population, and with much higher rates among combat veterans and those living in high-violence areas.1–3 Initially viewed as a potentially normative response to traumatic exposure,4 it became clear that not everyone experiencing trauma develops PTSD. Thus, a central question in research on PTSD is why some individuals are more likely than others to develop the disorder in the face of similar levels of trauma exposure.5–8 Although PTSD is the single disorder within the Diagnostic and Statistical Manual of Mental Disorders (Fourth Edition) (DSM-IV)9 that requires a specific environmental insult within its diagnostic criteria, it is becoming increasingly clear that there are critical roles for predisposing genetic and environmental influences in differentially mediating psychological risk to the traumatized individual.10–13
Child abuse occurs at disturbingly high rates and is a major public health problem.14 Despite the resilience of many abused children, child abuse significantly increases risk for impaired physical and psychological health and decreases adaptive functioning in adulthood.15–17 A matter of central importance to public health is the identification of factors related to risk and resilience in the wake of child abuse. Of particular relevance to this study is the well-established relationship of child abuse with adult PTSD.18–22 A number of factors may account for this relationship. PTSD in adults may represent a prolonged symptomatic reaction to traumatic child abuse.
The experience of child abuse is associated with an increased number of traumatic experiences across the lifespan. Child abuse may also increase vulnerability for the later development of PTSD by altering psychological (eg, attachment) and biological (eg, hypothalamic-pituitary-adrenal [HPA] axis function) developmental processes, including interaction with genetic factors. Although other non–child abuse types of traumatic experiences in childhood23 (eg, a house fire or being in a vehicle crash) might be expected to negatively affect development, the most robust research to date points to child abuse and related family/interpersonal stressful life events in predicting a wide range of later psychological and physical health problems.
The reasons for this are not fully understood but some possible explanations are (1) compared with other types of traumatic events, child abuse is more likely to occur in the family context24; (2) any 1 type of child abuse is associated with an increased likelihood of exposure to other types of abuse and to increased levels of family-related stressful events/parental dysfunction (eg, parental substance abuse)25; and (3) compared with some other types of trauma exposure, child abuse may be more likely a repeated experience rather than a single event (eg, multiple incidents of sexual abuse by the same perpetrator over a number of years).25 Our study focuses on the interplay between child abuse and polymorphisms in the FKBP5 gene, which is involved in the glucocorticoid-mediated stress response, in the prediction of adult PTSD.
The literature to date on the genetics of PTSD has recently been reviewed,12,13 with the resulting suggestion that gene × environment studies are needed to focus more on distinct endophenotypes and influences from environmental factors. A number of studies suggest that genetic factors contribute to the development of PTSD, with heritability estimates ranging from 30% to 40%.11,26–29 Candidate gene studies, however, have been inconclusive so far, usually limited by extremely low power to detect any but the strongest possible genetic effects (current published studies include sample sizes of <100).
A recent review12 covering candidate genes in the serotonin, dopamine, glucocorticoid receptor (GR), γ-aminobutyric acid, apolipoprotein E, brain-derived neurotrophic factor, and the neuropeptide Y system finds that (1) the support for a relationship between the serotonin transporter gene and PTSD exists only in research on the interaction of this system with stressful life events in predicting depressive symptoms; (2) results on the dopamine system are inconsistent; (3) there is a lack of evidence for relationships with brain-derived neurotrophic factor, neuropeptide Y, or GR polymorphisms and PTSD with the exception of a finding between GR genotype and basal cortisol levels in a subgroup of patients with PTSD30; and finally, (4) limited evidence for the γ-aminobutyric acid system and the apolipoprotein E system.
One of the first studies finding an interaction between a genetic polymorphism and child abuse in predicting psychopathology was the study by Caspi et al,31 which found that maltreated children with a monoamine oxidase A (MAOA) genotype conferring low levels of MAOA expression were more likely to develop conduct disorder and antisocial-personality disorder and to commit violent crimes as adults compared with those children with the high-activity MAOA genotype. A recent study32 has replicated this result, and a second study33 replicated the effect in white participants but not in other participants (blacks, Hispanics, American Indians, Pacific Islanders, and others) in the sample. The largest group of genotype × environment studies has examined the interaction between variation at the 5HTTLPR (a complex-repeat polymorphism in the 5′ upstream region of SLC6A4, which encodes the serotonin transporter the 5HTTLPR) and stressful life events, including child abuse, in predicting depression.34–40 Very recently, Kilpatrick et al41 reported an interaction of this polymorphism with severity of trauma and level of social support with the development of PTSD following hurricane exposure as outcome, supporting the relevance of gene × environment interactions for this disease.
From a developmental perspective, HPA axis genes are strong candidates with respect to altering susceptibility to PTSD. Exposure to trauma and stress increase HPA axis activity, and PTSD has been associated with long-lasting alterations in HPA axis reactivity42,43 and specifically higher GR sensitivity.44,45 Polymorphisms in genes regulating GR activity may impact the acute effects of trauma on the HPA axis and thereby possibly impact long-term HPA axis regulation affecting the development of PTSD. A number of studies suggest that child abuse and neglect affect HPA axis functioning (reviewed by Watts-English et al46). Several studies suggest that the depression-related HPA axis hyperactivity may be related to early life stress. For example, plasma corticotropin and cortisol, as well as cerebrospinal fluid corticotropin-releasing hormone (CRH) concentrations, correlate with perceived early life stress more than with current depression.47,48 Preclinical studies indicate that the persistent hyperactivity of the HPA axis associated with early life stress is mediated by a hyperactive CRH receptor 1 (CRHR1) system, with chronic over activity of CRHR1 in limbic brain regions.49,50
FKBP5 is a co-chaperone of hsp90. It directly interacts with hsp90, which binds to the GR. FKBP5 also has been shown to regulate GR sensitivity. FKBP5 is part of the mature GR heterocomplex.51 On hormone binding, FKBP5 is replaced by FKBP4, which then recruits dynein into the complex, allowing translocation into the nucleus where the complex regulates expression of glucocorticoid-responsive genes by functioning as a transcription factor.52 FKBP5 expression is induced by glucocorticoids as part of an intracellular ultrashort negative feedback loop for GR activity.53
Overexpression of human FKBP5 in vitro reduces hormone binding affinity54 and nuclear translocation of GR.55 Naturally occurring overexpression of FKBP5 causes GR resistance in New World monkeys,54,56 which is accompanied by increased plasma cortisol levels. Furthermore, potentially functional single-nucleotide polymorphisms (SNPs), or SNPs in very strong linkage disequilibrium with a functional variant, appear to alter FKBP5 function. The rare homozygous genotypes of FKBP5 SNPs (rs4713916, rs1360780, and rs3800373) were associated with higher FKBP5 expression in human blood monocytes as well as with a stronger induction of FKBP5 messenger RNA (mRNA) by cortisol. This was accompanied by less corticotropin release measured in patients who were depressed with the combined dexamethasone-CRH test.57
The same alleles of rs3800373 and rs1360780 were associated with increased peritraumatic dissociation in children after medical trauma.58 Higher levels of peritraumatic dissociation have been shown to be predictors of PTSD in adults.10 In addition, the extent of up-regulation of FKBP5 mRNA in peripheral blood mononuclear cells only hours after a trauma has been shown to correlate with the development of PTSD at 4 months.59 These data suggest that FKBP5 could be an important candidate gene in trauma-related HPA axis disturbances. We therefore hypothesized that the putative functional SNPs in FKBP5 moderate the development of PTSD. Because early trauma, PTSD, and FKBP5 SNPs have all been shown to influence GR resistance, we also hypothesize that variants in this gene may alter the impact of early trauma or PTSD on GR sensitivity and address this by investigating the dexamethasone suppression test (DST) in a subsample of individuals.44,57,60
Our study addresses the role of polymorphisms in FKBP5 in predicting PTSD, as well as the PTSD symptom–associated changes in GR sensitivity, in a highly traumatized, inner city sample. Specifically, we address whether these polymorphisms interact with increasing levels of child abuse and increasing levels of non–child abuse trauma exposure to predict PTSD symptomatology during adulthood.
METHODS
Sample, Recruitment, and Procedure
Data were collected to investigate the roles of genetic and environmental factors in predicting the development of PTSD in a population of urban, low-income, predominantly black men and women. To determine the race/ethnicity composition of the sample as part of the screening procedures, we asked participants to self-identify their race/ethnicity. Their response was coded into 5 common categories (black, white, Hispanic or Latino, Asian, mixed) or other (participants checked “other” when they thought their race/ethnicity was not included in the other 5 categories) (Table 1). The mean (SD) age in the sample was 40.8 (13.8) years, ranging from 18 to 81 years.
Table 1.
Sample Demographics
| Demographics | No. (%) of Participants |
|---|---|
| Sex (n = 900) | |
| Male | 384 (42.7) |
| Female | 516 (57.3) |
| Self-identified race/ethnicity (n = 898) | |
| Black | 855 (95.2) |
| White | 20 (2.2) |
| Hispanic or Latino | 5 (0.6) |
| Asian | 1 (0.1) |
| Mixed | 8 (0.9) |
| Othera | 9 (1.0) |
| Education (n = 897) | |
| <12th grade | 245 (27.3) |
| High school graduate | 324 (36.1) |
| Graduate equivalency diploma | 52 (5.8) |
| Some college/technical school | 176 (19.6) |
| College/technical school graduate | 85 (9.5) |
| Graduate school | 15 (1.7) |
| Employment status (n = 898) | |
| Currently unemployed | 606 (67.5) |
| Currently employed | 292 (32.5) |
| Disability status (n = 896) | |
| Not currently receiving disability | 692 (77.2) |
| Currently receiving disability | 204 (22.8) |
| Household monthly income, US $ (n = 884) | |
| 0–249 | 278 (31.4) |
| 250–499 | 75 (8.5) |
| 500–999 | 238 (26.9) |
| 1000–1999 | 205 (23.2) |
| ≥2000 | 88 (10.0) |
Because race/ethnicity was self-identified, participants checked “other” when they thought their race/ethnicity was not included in the other 5 categories (eg, American Indian).
Screen interviews were completed with 900 participants approached while in the waiting rooms of primary care or obstetrical-gynecological clinics of Grady Memorial Hospital in Atlanta, Georgia, between 2005 and 2007. Approximately 58% of those approached to participate in the study agreed to do so. Participants completed a battery of self-report measures that took 45 to 75 minutes to complete (dependent in large part on the extent of the participant’s trauma history and symptoms). All measures were obtained by verbal interview. Each person was paid US $15.00 for participation in this phase of the study. Each participant also provided a saliva sample for DNA extraction (participants were paid the same amount for completion of screening data even if they declined to provide a DNA sample). Written and verbal informed consent was obtained for all participants, and all procedures in this study were approved by the institutional review boards of Emory University School of Medicine and Grady Memorial Hospital, Atlanta, Georgia.
Main Outcome Measures
Modified PTSD Symptom Scale
The modified PTSD Symptom Scale (PSS) is a psychometrically valid 17-item self-report scale assessing PTSD symptomatology61–65 over the prior 2 weeks. Consistent with prior literature, we summed the PSS frequency items (0 indicates not at all to 3 indicates ≥5 times a week) to obtain a continuous measure of PTSD symptom severity ranging from 0 to 51. For this sample, the PSS frequency items had standardized α=.90 (mean [SD], 13.81 [11.96]). No clearly established PSS cutoff score for PTSD diagnosis has been established; however, DSM-IV criteria for PTSD can be applied to PSS frequency items to create a proxy variable for PTSD diagnostic status.
Clinician Administered PTSD Scale
The Clinician Administered PTSD Scale (CAPS) was also administered to a subset of 240 participants within 2 to 6 weeks after completing the screening assessment. We found a significant difference (F1,239=56.55, P < .001) between average PSS score (mean [SD], 18.20 [12.82]) for participants positive for current PTSD based on the CAPS (applying DSM-IV decision rules with a symptom considered as present with a CAPS frequency score of ≥1 and intensity score of ≥2) compared with those participants not meeting current CAPS PTSD criteria (mean [SD], 7.51 [9.23]). In addition, 70% of those participants identified as PTSD positive by using the PSS-based proxy variable were also positive for current PTSD at the time of CAPS administration (the criterion A traumatic experiences used for CAPS diagnosis was not necessarily the same one used for obtaining PSS data). Study participants were asked to respond to the PSS items based on the trauma exposure (inclusive of child physical and sexual abuse and other life trauma exposure) that they believed had impacted them the most. Because we recorded data on the type of trauma identified for PSS score but not the age at which it occurred and because many study participants reported several incidents of the same type of trauma across the lifespan, we were unable to determine if the PSS data was based on a traumatic event occurring in childhood vs adulthood.
Beck Depression Inventory
The 21-item Beck Depression Inventory (BDI)66 is a psychometrically validated, commonly used measure of depressive symptoms.67 In our sample, the BDI had a standardized α coefficient of .99 and a mean (SD) of 14.4 (13.2).
Traumatic Events Inventory
The traumatic events inventory (TEI)64,65 assesses lifetime history of trauma exposure and is our primary measure of both child abuse and non–child abuse trauma. The TEI assesses past experience and frequency of 13 separate types of traumatic events as well as feelings of terror, horror, and helplessness with such events.
For the measure of child abuse, 2 of the TEI questions assessed physical abuse and sexual abuse occurring before age 14 years. Based on these questions, 17.6% of the sample reported a history of childhood physical abuse and 18.8% reported a history of childhood sexual abuse. With these data, we created a 3-level categorical variable reflecting number of types of child abuse: no child abuse (70.5% of sample), 1 type of either physical or sexual abuse (22.7%), or 2 types of both physical and sexual abuse (6.8%).
For the measure of non–child abuse trauma, the remaining TEI questions addressed other types of trauma exposure (Table 2). To summarize level of exposure to trauma other than child abuse, we summed total number of different types of non–child abuse trauma exposure reported by each participant. The mean number of types of non–child abuse trauma reported in our sample was 2.46 types (SD, 1.92). Using the same data, we created a 4-level categorical variable reflecting number of types of non–child abuse trauma experienced reported with 19.9% of participants in the no non–child abuse trauma exposure category, 22.3% of participants reporting 1 type, 31.7% of participants reporting 2 to 3 types, and 26.7% of participants reporting 4 or more types of non–child abuse trauma. The total number or types of trauma exposure variable was chosen because in our prior research64,65 and in other research on impact of trauma exposure68 it relates in a predictable and consistent manner with a number of measures of adaptive functioning and trauma-related functioning.
Table 2.
Percentage of Sample Reporting Exposure to Non–Child Abuse Traumatic Experiences Assessed by Traumatic Events Inventorya
| Trauma Type Experienced | No. (%) of Participants | ||
|---|---|---|---|
| Total Sample (N = 900) | Male (n = 384) | Female (n = 516) | |
| Natural disaster | 170 (19.4) | 101 (26.9) | 69 (13.7) |
| Serious accident or injury | 351 (42.9) | 183 (53.0) | 168 (35.4) |
| Sudden life-threatening illness | 226 (25.9) | 113 (30.1) | 113 (22.7) |
| Military combat | 28 (3.2) | 26 (7.0) | 2 (0.4) |
| Attacked with knife, gun, or other weapon by someone other than spouse, romantic partner, or boyfriend/girlfriend | 324 (37.2) | 204 (54.5) | 120 (24.2) |
| Attacked without a weapon by someone other than spouse, romantic partner, or boyfriend/girlfriend | 261 (30.5) | 142 (38.5) | 119 (24.4) |
| Witness of murder of friend or family member | 79 (9.2) | 43 (11.6) | 36 (7.3) |
| Forced sexual contact ≥14 y of age | 132 (15.5) | 18 (5.1) | 114 (23.9) |
| Any significant trauma | 746 (84.9) | 337 (89.6) | 409 (81.3) |
Because some participants declined to answer some questions, the total number in each trauma-type category may vary slightly. A subset of the trauma types queried within the Traumatic Events Inventory are listed.
Although the non–child abuse variable assesses traumatic experience other than child abuse across the lifespan, data from our sample suggests that these non–child abuse traumatic experiences primarily occur in adulthood. Specifically, we have data from a subset of study participants (n=322) asking them the earliest age at which all of the traumatic events assessed by the TEI occurred. Averaged across all of the non–child abuse trauma categories, the mean (SD) for youngest age of trauma exposure in the sample was 25.1 (10.6) years and the median youngest of trauma exposure was 23 years.
Childhood Trauma Questionnaire
The Childhood Trauma Questionnaire (CTQ)69 is a 28-item psychometrically validated, self-report inventory assessing self-reported level of child abuse and neglect.70 We used the total score from this scale in our secondary analyses as a separate continuous measure of child abuse exposure.
Dexamethasone Suppression Test
A total of 80 participants were characterized using a low-dose DST.60 Blood samples in the fasting state were obtained for baseline serum cortisol concentration and were drawn between 8 AM and 9 AM on day 1. Participants then received 0.5-mg dexamethasone by mouth at 11 PM before the second day blood draw, and blood samples for serum cortisol concentration was collected again in the fasting state between 8 AM and 9 AM for the day 2 collection. Using the cortisol concentration values from day 1 and day 2, the percentage suppression was calculated by 100×[(cortisol day 1-cortisol day 2)/cortisol day 1]. Serum cortisol concentration was measured by using commercially available radioimmunoassay kits (Diagnostic Systems Laboratories, Webster, Texas) from replicate samples and with interassay quality control measures. Significance of the interaction effect of FKBP5 SNPs and PTSD on percentage suppression was determined by using permutation-based methods with age and sex as covariates (10 000 permutations) to avoid inflated P values due to outlier or small cell sizes. In addition, we used repeated measures analysis of variance to examine the interaction between probable PTSD diagnosis and FKBP5 genotypes on the change in serum cortisol suppression from day 1 to day 2. For cortisol analyses, a probable PTSD diagnosis was determined from the DSM-IV criteria applied to the modified PSS interview, as described previously.64,65
The individuals with available DST present only a subset of the whole sample. However, these 80 individuals are not different compared with the rest of the sample in PSS total score for samples with and without DST (mean [SD], 11.30 [11.5] vs 10.15 [11.4]; P=.40), BDI total score (13.55[12.1]vs14.57[12.1], P=.45), race (93.7% vs 96.0% black, P=.92), degree of childhood trauma (28.8% and 10.0% in the 1 and ≥2 types of severe child abuse vs 23.7% and 6.7%, respectively; P=.28), and also non–child abuse trauma and sex. We also found no differences in the monthly household income and relationship status. However, the endocrinologically characterized individuals are older (mean age[SD],43.3[11.1]vs39.3[11.1]; P=.01). Due to the high comparability of all other variables and because all our analyses are corrected for age and sex, this sub sample is representative of the sample as a whole.
Genetic Data
DNA Extraction
DNA was extracted from saliva collected into Scope mouthwash (n=46) or into Oragene saliva kits (DNA Genotek, Ottawa, Ontario, Canada) by using the Qiagen M48 automated extraction system. DNA was available for 762 individuals. One hundred thirty-eight individuals with no genotype information consisted of 11 in which DNA was not collected, 88 in which we attempted to collect DNA using Oragene-spit samples but DNA extraction failed completely in 2 separate extraction trials (these failures were either due to noncompliance of the study participants or failure to correctly break the seal that releases the stabilizing solution in the Oragene saliva kits), and 39 for which DNA had been collected but not extracted at the time of analysis. When comparing the 762 individuals with genotypes to those 138 participants without, we found no significant difference in age, sex, self-described race, income, employment status, relationship status, child abuse, and non–child abuse trauma exposure as well as PTSD and depression severity.
SNP Genotyping
Eight SNPs within FKBP5 were selected from dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/) to include the 3 potentially functional SNPs (rs4713916, rs1360780, and rs3800373)57; the other 5 SNPs were selected to span 120 kb covering the remainder of the FKBP5 locus (NM_004117). We also genotyped 3SNPs with in the CRHR1 gene (rs110402, rs242924, and rs7209436), which have been recently reported to show strong interaction with childhood abuse on adult depression symptoms.71 All SNPs were genotyped using a TaqMan allelic discrimination assay72 on an ABI 7900HT instrument (Applied Biosystems, Foster City, California), using predesigned and validated TaqMan assay reagent kits containing 1 pair of polymerase chain reaction primers and 1 pair of fluorescently labeled probes (Applied Biosystems). Polymerase chain reactions were performed in 5-μL reaction volumes in a 384-well plate and contained 5 ng of DNA. The standard protocol that was provided with the kit was followed. Thermal cycler conditions were 95°C for 10 minutes, 40 cycles of 92°C for 15 seconds, and 60°C for 1 minute. The SDS version 2.2 software (Applied Biosystems) was used for allelic discrimination. For quality control, 9% of the samples were genotyped as duplicates across and within a 384-well plate. Only 0.03% of discordances were recorded and excluded from the analyses. Call rates for SNPs ranged from 93% to 97%, which included some samples that failed all assays and for which DNA quality was inferior. We used Haploview73 to determine the linkage-disequilibrium structure of the SNPs within the FKBP5 gene and to test for Hardy-Weinberg Equilibrium. The SNP identifications, their location, the Hardy-Weinberg Equilibrium test P values, minor allele frequencies, and call rate percentages are shown in Table 3.
Table 3.
List of Tested FKBP5 SNPs, Their Positions on Human Chromosome 6 According to University of California Santa Cruz Genome Browser Version hg17, Hardy-Weinberg Equilibrium Test P Value, Minor Allele Frequency, and Call Rate
| dbSNP ID | Position | Hardy-Weinberg Equilibrium Test P Value | Minor Allele Frequency | Call Rate, % |
|---|---|---|---|---|
| rs3800373 | 35650460 | .45 | 0.45 | 95.0 |
| rs992105 | 35663160 | .94 | 0.18 | 97.1 |
| rs9296158 | 35675060 | .60 | 0.49 | 95.3 |
| rs737054 | 35683460 | >.99 | 0.07 | 97.3 |
| rs1360780 | 35715550 | .51 | 0.42 | 97.5 |
| rs1334894 | 35723110 | >.99 | 0.02 | 96.3 |
| rs9470080 | 35754410 | .34 | 0.47 | 93.3 |
| rs4713916 | 35777960 | .53 | 0.11 | 94.5 |
Abbreviation: SNPs, single-nucleotide polymorphisms.
Ancestry Informative Markers
Population stratification is a potential confounder in association studies of complex traits. If such allele-frequency heterogeneity within a population is coupled to similar population heterogeneity in the outcome of interest, naive tests of interaction could yield spurious results if the resulting confounding is neglected in analysis. Because 95.2% of our sample participants were self-identified as being of black race (which is known to contain varying degrees of genetic admixture of sub-Saharan African, European, and Native American ancestry), we took steps to ensure our results were robust to potential confounding from population stratification by genotyping 134 ancestry informative markers74,75 in a subset of 280 individuals from our sample using single-base primer extension and a Beckman SNP stream instrument (Beckman-Coulter, Fullerton, California). Genotyping of ancestry informative markers and data analysis for estimates of ancestry were performed by DNA Print Genomics, Sarasota, Florida (http://www.dnaprint.com). Maximum likelihood estimates of individual biogeographical ancestry admixture were determined by using methods described previously76 and by using a 4-continental population model. The choice of a 4-continential population model was based on previously published hypothesis-free cluster analyses of worldwide populations.77,78 This model lent itself to use the terms European (genetic ancestry shared among Europeans, Middle Eastern, and to a lesser extent South Asians), sub-Saharan African, East Asian, and Indigenous American (genetic ancestry shared among American Indians, Latin and South American Indians, and certain Central Asian populations). The names of these parental populations were chosen to describe extant elements of genetic structure and are arbitrary in that they are reflective of modern population distributions—not necessarily the distributions of the original parental populations 15 000 to 50 000 years ago. Individuals with more than 35 failed ancestry informative marker genotypes were excluded from the analysis (7%), because their individual biogeographical ancestry admixture could not be estimated with sufficient certainty.
Statistical Analyses
Primary, Descriptive, and Secondary Analyses
Our primary analyses were the main FKBP5 SNP effects on PTSD symptom severity and their interaction effects with child abuse and non–child abuse trauma (690 participants for interaction with non–child abuse trauma and 678 participants for interaction with child abuse). Secondary analyses included BDI scores as outcome (number same as for PSS outcome), FKBP5 SNP genotype dependent correlation of child abuse severity and PTSD symptom severity (n=728), and effects on the DST (n=80), as well as analysis using CRHR1 genotypes(number same as for FKBP5 SNP interactions). Descriptive analyses were run on a sample of 900 individuals with valid PTSD measures.
Regression Analyses
We used a variation of linear regression to examine the effects of trauma exposure, FKBP5 genotypes, and their potential interaction on PSS scores. For FKBP5 genotype, we modeled a participant’s genotype at each SNP under an additive model that is equivalent to coding of the number of copies of a reference allele that the participant possesses. For trauma exposure, we included both child abuse (the 3-level child abuse TEI score) and non–child abuse trauma exposure (using the 4-level non–child abuse trauma TEI score) as predictors. We chose to separate these 2 types of trauma exposure based on the above described research, suggesting that exposure to child abuse increases risk for development of PTSD both as a response to the child abuse itself and in response to other non–child abuse stressors. Because child abuse is related to increased likelihood of subsequent exposure to additional traumatic stressors, we wanted to control for the possibility that effects associated with our child abuse variables were not artifacts of increased overall trauma exposure alone. Within the regression model, we further modeled the potential confounding effects of age, sex, level of depressive symptoms (BDI total score), total number of experienced non–child abuse trauma types, and genetic ancestry (by incorporating the ancestry estimates based on analysis of the ancestry informative markers). Due to missing phenotype and genotype information on some participants, a maximum number of 676 individuals and a minimum of 633 individuals were informative in the regression models for the interaction effects. The individuals in the group with 676 and 633 that actually entered the analyses were not different in age, sex, self-described race, income, employment status, relationship status, child abuse trauma exposure, as well as PTSD and depression severity from the remainder of the total 900 individuals. They did, however, have higher child abuse rates. In the individuals entering the analyses, the no abuse group size ranged from 67.5% to 68.6% (and were not different) vs 77.1% to 79.0% in the excluded individuals (P=.01 to P=.14).
Permutation Analyses
As the non-normality of PSS and possible sparse-cell counts in the FKBP5-trauma interaction strata could each invalidate the asymptotic P values produced by regression models, we established the significance of main environment, genotype, or genotype-trauma interaction effects using permutation procedures that randomly assigned the sample PSS scores to participants (sampled without replacement), while holding each participant’s genotype and environmental variables fixed.79,80 For each analysis, we based inference on 10 000 to 100 000 permutations. We conducted these analyses by using appropriate components of SAS version 9.1 (SAS Institute, Cary, North Carolina). Due to smaller cell sizes, we also used these permutation-based analyses to establish the main genotype or genotype and PTSD interaction effects on the DST.
Correlation Analyses
For descriptive and quantitative examination of genotype × continuous environment (CTQ total score) interaction of PTSD symptom severity, we used partial correlation controlling for age and sex, correlating the semi quantitative child abuse measures of the CTQ total score with the current continuous PSS score in the whole sample as well as the sample stratified by FKBP5 SNP genotype. The significance of the interaction ofFKBP5 SNP genotype and CTQ total scores on PSS was established using permutation-based methods, and the post hoc genotype-dependent differences of the correlation coefficients were established by converting the correlation coefficients of each genotype group to Fisher Z scores by Z=ln[(r+1)/(r−1)]/2. We then estimated the standard error of difference between the 2 correlations by SE={[1/(n1−3)] + [1/(n2−3)]}1/2 and divided the difference between the 2 Z scores by the standard error. If the Z value for the difference was 1.96 or higher, the difference in the correlation was significant at P =.05 level. If the difference was 2.58 or higher, the difference in the correlation was significant at P =.01 level.81
Correction for Multiple Testing
To ensure an overall significance level of P ≤ .05 for the primary analyses (main FKBP5 SNP effects on PTSD symptoms, interaction of FKBP5 SNPs with child abuse or non–child abuse trauma exposure on adult PTSD symptoms), we corrected for the examination of 24 tests (8 tests of the main effects of the FKBP5 SNPs, 8 tests of interaction between each SNP and non–child abuse trauma exposure, and 8 tests of interaction between each SNP and categorical measure of child abuse). We used the conservative Bonferroni method for such a multiple-testing correction, which yielded a significance threshold of α=.002. In an exploratory approach, we also genotyped 3CRHR1SNPs based on previous results from our group in examining depression.71 When we corrected for this additional set of 9 tests (3 CRHR1SNPs, main effect and interaction of SNP × child abuse or non–child abuse trauma exposure), the α level after correction was .05/33=.0015.
RESULTS
Descriptive Analyses
Non–Child Abuse Trauma Exposure and PTSD Symptoms
To analyze the differential roles of gene × environment interaction with PTSD in adult participants, we first had to demonstrate that within our participant population, the level of total non–child abuse traumatic exposure contributes significantly to current PTSD symptoms. To perform these analyses, we first used a general linear model (controlling for age and sex), using the PSS total score as the dependent variable and the TEI categorical variable representing number of types of non–child abuse traumatic experiences as the independent variable. We found a significant sex effect (F1,818 = 29.5, P < .001) and a very robust non–child abuse trauma exposure effect (F 3,818 = 61.9, P < .001) (Table 4). In the zero types of non–child abuse trauma group, the mean (SD) PSS score was 3.58 (6.27) and in the 4 or more types of trauma group, we found continuous increases by more than 5-fold to 16.74 (12.90).
Table 4.
Non–Child Abuse Trauma Exposure and PTSD Symptoms
| Level of Non–Child Abuse Traumaa |
||||
|---|---|---|---|---|
| PTSD Symptom Scale | None (n = 159) | 1 Type (n = 183) | 2–3 Types (n = 265) | ≥4 Types (n = 215) |
| Mean (SD) [95% CI] | 3.58 (6.27) [2.60–4.56]b,c | 7.30 (10.04) [5.83–8.76]c,d | 11.57 (11.66) [10.16–12.98]c | 16.74 (12.90) [15.00–18.47]c |
Abbreviations: CI, confidence interval; PTSD, posttraumatic stress disorder.
No. of types of non–child abuse (primarily adult) trauma experienced.
P < .005 difference in PTSD Symptom Scale from 1 type of trauma.
P < .001 difference in PTSD Symptom Scale from other groups.
P < .005 difference in PTSD Symptom Scale from no trauma.
Child Abuse Trauma Exposure and PTSD Symptoms
We next examined whether child abuse exposure also predicted the level of current PTSD symptomatology (Table 5). We performed the same analyses as above with the categorical child abuse variable (none, 1 type [physical or sexual], and 2 types [physical and sexual]). Similar to the effect of non–child abuse trauma, we also found a significant effect in the presence of child abuse (F2,806=50.9, P < .001). The experience of child abuse increased the mean (SD) PSS scores from 8.03 (10.48) in the no child abuse group to 14.65 (11.92) in the 1 type and 20.93 (14.32) in the 2 types of child abuse group.
Table 5.
Child Abuse Trauma Exposure and PTSD Symptoms
| Level of Child Abuse Traumaa |
|||
|---|---|---|---|
| PTSD Symptom Scale | None (n = 566) | 1 Type (n = 189) | 2 Types (n = 54) |
| Mean (SD) [95% CI] | 8.03 (10.48) [7.17–8.90]b | 14.65 (11.92) [12.94–16.36]c | 20.93 (14.32) [17.02–24.84]d |
Abbreviations: CI, confidence interval; PTSD, posttraumatic stress disorder.
No. of types of child abuse trauma experienced.
P < .001 difference in PTSD Symptom Scale from other groups.
P < .001 for both differences in PTSD Symptom Scale from no abuse and 2 types.
P < .001 for both differences in PTSD Symptom Scale from no abuse and 1 type.
Non–Child Abuse Trauma Exposure, Child Abuse, and PTSD Symptoms
Using a general linear model that included both non–child abuse trauma and child abuse trauma (Table 6) and their interaction term as predictors of adult PTSD symptoms, we observed significant main effects of the 2 terms (non–child abuse trauma: F 3,806 = 16.2, P < .001; child abuse trauma: F2,806=14.4, P < .001) and sex (F1,806 = 20.2, P < .001) as described above, but no significant interaction (F6,806=0.51, P =.79) between the 2 main predictors. The presence of 1 type of child abuse increases the PSS score at each non–child abuse trauma level by 4.58 points on average (range, 2.03–6.51) and the presence of 2 types of child abuse increases the PSS score at each non–child abuse trauma level by 9.04 points on average (range, 7.79–10.20), suggesting that exposure to child abuse increases risk for higher levels of PTSD symptoms in response to non–child abuse trauma exposure.
Table 6.
Interaction of Child Abuse Trauma and Non–Child Abuse Traumaa
| Non-Child abuse Trauma | PTSD Symptom Scale, Mean (SEM)b | ||
|---|---|---|---|
| No Child Abuse | 1 Type of Child Abuse | 2 Types of Child Abuse | |
| None | 3.18 (0.50) | 5.24 (1.73) | 11.00 (5.57) |
| 1 Type | 5.80 (0.78) | 12.32 (1.98) | 14.44 (2.21) |
| 2–3 Types | 9.90 (0.81) | 15.78 (1.42) | 18.00 (4.49) |
| ≥4 Types | 13.77 (1.15) | 17.70 (1.45) | 23.97 (2.49) |
Abbreviation: PTSD, posttraumatic stress disorder.
Note that significant effects are observed for both non–child abuse trauma (F3,806=16.2, P < .001) and child abuse trauma (F2,806=14.4, P < .001).
The PTSD Symptom Scale ranges from 0 to 51.
Primary Genetic Analyses
Main Effect of FKBP5 Polymorphisms on PTSD Symptoms
Based on previous data supporting a clear role of FKBP5 in modulating the glucocorticoid response to stress, as well as evidence supporting association of FKBP5 variants with risks for and rate of recovery from affective disorders,52 we hypothesized that genetic variation at FKBP5 may influence liability to PTSD. We examined 8 SNPs spanning 120 kb of the FKBP5 locus (Table 3) but found no significant main effect of FKBP5 genotypes on PSS total score (Figure 1).
Figure 1. FKBP SNPs and Main Genetic Effect on PTSD Symptoms and Interaction Effects With Non–Child Abuse Trauma Levels and Child Abuse.
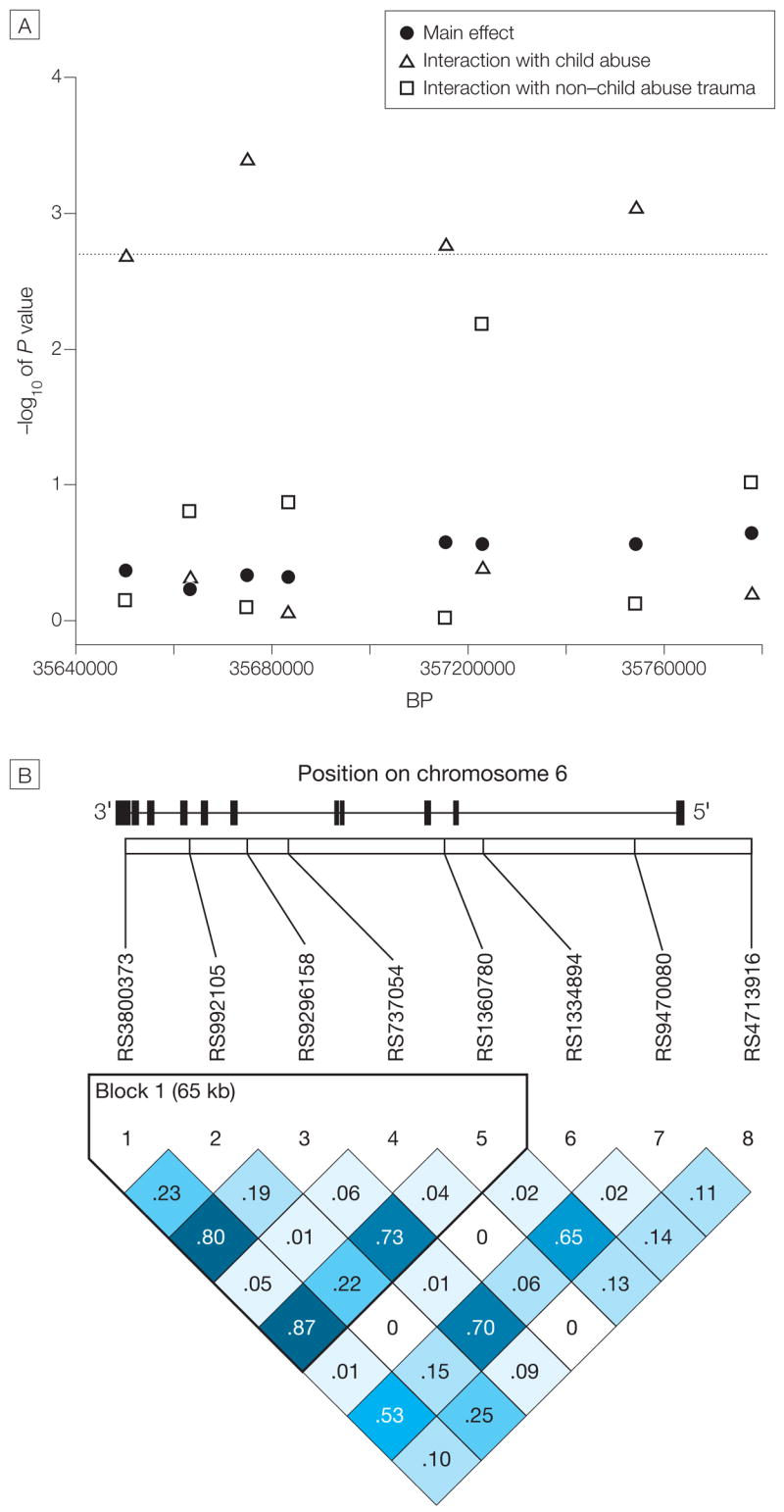
SNPs indicate single-nucleotide polymorphisms; PTSD, posttraumatic stress disorder. A, The plot shows the negative log10 of the P value for the main genetic effect (filled circles), the interaction of FKBP5 SNP genotypes and non–child abuse trauma level (open squares), and the interaction of FKBP5 SNP genotypes and child abuse (open triangles) to predict adult PTSD symptoms. The x-axis shows the position of the SNPs on chromosome 6 and the y-axis shows the P value for the respective effects, plotted as the negative log10 of the P value. Dotted line indicates P < .002. B, The position of the FKBP5 gene and its exons (filled rectangles) on chromosome 6 as well as a linkage disequilibrium (LD) plot of all tested SNPs using r2 as the measure of LD is also shown. r2=1 indicates complete LD and is depicted by the darkest shade of blue. r2<1.0 are printed in the respective square for compared SNPs, with darker shades of blue representing higher levels of LD.
Interaction of FKBP5 Polymorphisms With Non–Child Abuse Trauma to Predict PTSD Symptoms
We next examined whether FKBP5 polymorphisms interact with increasing levels of non–child abuse (primarily adult) trauma to predict adult PTSD symptoms. We regressed PSS scores on FKBP5 genotype, non–child abuse trauma (using the 4-level variable), and the interaction between genotype and non–child abuse trauma, adjusting for age and sex. We did not find any significant interaction effects using permutation-based methods (Figure 1). These data demonstrate that FKBP5 polymorphisms do not appear to have a simple role in moderating the effects of non–child abuse trauma exposure on PTSD outcomes.
Interaction of FKBP5 Polymorphisms With Child Abuse to Predict PTSD Symptoms
We then regressed PSS scores on FKBP5 genotype, child abuse (3-level variable), and the interaction between genotype and child abuse, adjusting for age and sex. In this analysis, we identified 4 SNPs that showed significant interactions with child abuse after correcting for multiple testing (interaction P < .002)(Figure 1). All 4 associated SNPs are in fairly high linkage disequilibrium, with pair-wiser2 values ranging from 0.53 to 0.87 (Figure 1). The most significant SNP, rs9296158, is located in intron 5 with an interaction P < .0004. Two of the associated SNPs, rs3800373 and rs1360780, have previously been reported to associate with differential glucocorticoid-mediated responses by Binder et al.57 For all 4 significant SNPs, we observed a similar additive mode of interaction, and there appears to be a gene dose-dependent protection from severe child abuse–associated increases in adult PTSD scores (Table 7). In the group of individuals with 2 types of child abuse, individuals homozygous for the protective G allele of rs9296158 had a mean (SEM) PSS score of 13.54 (3.76); in the heterozygous group, the mean PSS score was 21.25 (2.03); and in the group homozygous for the risk allele A, the mean PSS score was 31.11 (5.37). The PSS scores of more than 20 indicate clinical significant PTSD and higher scores indicate more severe PTSD, with a maximum of 51 points that can be reached in that scale.61–63 Interestingly, the genotypes previously associated with a higher number of previous depressive episodes and faster response to antidepressant treatment57 were the genotypes with the highest level of adult PTSD symptoms in the presence of child abuse.
Table 7.
Interaction of FKBP5 Genotype and Level of Child Abuse Predicts PTSD Symptoms in Adults
| PTSD Symptom Score, Mean (SEM) | ||||||
|---|---|---|---|---|---|---|
| FKBP5 Genotypes | No. of Participants | No Child Abuse | No. of Participants | 1 Type of Child Abuse | No. of Participants | 2 Types of Child Abuse |
| rs9296158 | ||||||
| AA | 107 | 7.28 (0.93) | 42 | 16.92 (1.85) | 9 | 31.11 (5.37)a |
| AG | 223 | 7.89 (0.69) | 88 | 12.89 (1.16) | 24 | 21.25 (2.03) |
| GG | 107 | 8.80 (1.08) | 31 | 14.33 (2.28) | 14 | 13.54 (3.76) |
| rs3800373 | ||||||
| CC | 89 | 7.03 (0.98) | 35 | 17.65 (2.04) | 8 | 29.00 (5.60)b |
| AC | 210 | 8.11 (0.74) | 77 | 12.71 (1.24) | 23 | 22.96 (2.35) |
| AA | 137 | 7.97 (0.89) | 49 | 15.17 (1.75) | 16 | 13.75 (3.31) |
| rs1360780 | ||||||
| TT | 81 | 7.17 (1.02) | 36 | 16.82 (2.08) | 7 | 31.00 (6.04)b |
| CT | 222 | 7.97 (0.70) | 77 | 13.12 (1.23) | 26 | 21.28 (2.32) |
| CC | 150 | 8.23 (0.87) | 50 | 14.51 (1.73) | 15 | 14.00 (3.53) |
| rs9470080 | ||||||
| TT | 94 | 6.76 (0.99) | 44 | 16.40 (1.84) | 10 | 31.20 (4.69)b |
| CT | 224 | 8.25 (0.69) | 88 | 13.23 (1.17) | 23 | 20.05 (2.26) |
| CC | 109 | 8.42 (1.07) | 28 | 14.90 (2.39) | 13 | 14.38 (3.97) |
Abbreviation: PTSD, posttraumatic stress disorder.
Interaction P < .001.
Interaction P < .002.
Exploratory/Secondary Genetic Analyses
FKBP5 and Child Abuse Interaction
Given the prior data on FKBP5 and major depression, and the observation that in our sample the 3-level child abuse variable is significantly and positively correlated with our 4-level non–child abuse trauma variable (n = 851, Pearson correlation=0.271; controlling for age and sex, P < .001), we reexamined the above interaction controlling for BDI score as a continuous measure of depression symptoms or the 4-level non–child abuse trauma variable in addition to age and sex. After adjusting for these variables, the interaction terms of child abuse and FKBP5 SNPs remained significant for 2 of the 4 SNPs (rs9296158 and rs3800373; interaction P =.07 for rs1360780 and interaction P = .06 for rs9470080). In addition, we also conducted a regression analysis with BDI as the dependent variable. None of the FKBP5 SNPs showed a significant interaction of genotype and child abuse to predict depression outcome (controlling for age and sex) after controlling for multiple testing. Thus, these data suggest that the FKBP5 genotype may interact with childhood trauma to predict adult PTSD severity, even when controlling for age, sex, non–child abuse trauma level, and severity of depressive symptoms.
Genotype-Dependent Correlations of Child Abuse and PSTD Symptom Severity
We explored the genotype-dependent correlations of child abuse and PSTD symptom severity interaction effect using an ordinal variable of child abuse, which contains 154 points ranging from a CTQ score of 25 to 120. This allowed us to examine correlation analysis without having to group individuals into only 3 child abuse levels, which can lead to small group sizes. There is also importance in validating these effects with a separate, broader, and more continuous measure of child abuse. Thus, we investigated the interaction of the genotypes of the 4-associated FKBP5 SNPs with the CTQ total score to predict PTSD symptom severity. Permutation-based analyses showed an interaction effect with rs9296158 genotype and the continuous environment measure with P = .01 and P = .03 for rs3800373 and P=.07 for rs1360780. To better describe and quantify these differences, we reinvestigated positive correlations of child abuse severity and PSS and BDI scores stratifying by the genotypes of the 4-associated FKBP5 SNPs. Partial correlation (controlling for age and sex) of CTQ total score and PSS scores yielded highly significant correlations of child abuse and PTSD symptom severity in the whole sample (r = 0.347, P = 3.7 × 10−27). However, when stratifying this analysis by the rs9296158 genotypes (Table 7), we observed significantly higher correlation coefficients in the AA vs GG group (z value for difference of correlation coefficient=3.00, P < .01) and the AG vs GG group (z value for difference of correlation coefficient=2.35, P < .05) for correlations between CTQ total score and PSS score. Similar effects were observed with 2 other SNPs, rs1360780 and rs3800373 (Table 8). Interestingly, although BDI scores and the CTQ total scores also showed highly significant positive correlations (R = 0.39, P = 6.9 × 10−33), there were no significant differences of the correlation coefficients among the 3 rs9296158 genotype groups.
Table 8.
FKBP5 SNP Genotype-Dependent Correlations of CTQ Total Score and PSS Scores
| CTQ Correlation With PSS Score Dependent on FKBP5 Genotype | Genotype | ||
|---|---|---|---|
| rs9296158 | |||
| AA | AG | GG | |
| No. of participants | 164 | 360 | 172 |
| Pearson correlation R value | 0.470 | 0.374 | 0.169 |
| Z values for difference | AA/AG = 1.17 | AG/GG = 2.35a | AA/GG = 3.00b |
| rs1360780 | |||
| CC | CT | TT | |
| No. of participants | 238 | 347 | 125 |
| Pearson correlation R value | 0.210 | 0.390 | 0.455 |
| Z values for difference | CC/TT = 2.46a | CT/CC = 2.33a | TT/CT = 0.74 |
| rs3800373 | |||
| AA | AC | CC | |
| No. of participants | 171 | 352 | 156 |
| Pearson correlation R value | 0.248 | 0.350 | 0.435 |
| Z values for difference | AA/CC = 2.59b | AC/AA = 2.30a | CC/AC = .83 |
| rs9470080 | |||
| CC | CT | TT | |
| No. of participants | 222 | 336 | 135 |
| Pearson correlation R value | 0.215 | 0.392 | 0.465 |
| Z values for difference | CC/TT = 1.91 | CT/CC = 1.20 | TT/CT = 1.03 |
Abbreviations: CTQ, Childhood Trauma Questionnaire; PSS, PTSD Symptom Scale; PTSD, posttraumatic stress disorder; SNP, single-nucleotide polymorphism.
P < .05.
P < .01.
Genetic Admixture and Population Stratification
Based on statistical analysis of the ancestry in formative marker genotype data from the subsample, we found that sub-Saharan African ancestry estimates for participants ranged between 41%and100%, with a mean of 83.6%; European ancestry estimate sranged between 0% to 54%, with a mean of 10.9%; Indigenous American ancestry estimates ranged between 0% to 19%, with a mean of 3.3%; and East Asian ancestry estimates ranged from 0% to 33%, with a mean of 2.3%. We found that the degrees of ancestry from these subpopulations were not significantly correlated with total PSS score, level of non–child abuse trauma exposure or presence of child abuse (all Pearson correlation coefficients between 0.01 and −0.04). In addition, using the percentage ancestry estimates for these 4 populations as covariate in the interaction analysis did not alter the observed SNP × environment interactions. This evidence suggests that population stratification is not likely confounding in our analyses.
Effects of CRHR1 SNPs on PTSD Symptom Severity
Because we have recently reported interactions of CRHR1 SNPs with child abuse on adult depression,71 we tested the 3 SNPs with the strongest interaction effects (rs110402, rs242924, and rs7209436) for the effect on PTSD symptom severity. None of the 3 SNPs showed a significant main effect or interaction effect with child abuse or non–child abuse trauma exposure on PTSD symptom severity.
DST, PTSD, and FKBP5 SNP
To assess whether FKBP5 SNPs had a functional consequence on GR sensitivity changes observed in PTSD, we examined the interaction of probable PTSD diagnosis and the 4 FKBP5 SNPs with significant gene × environment interactions on the change in cortisol concentration before and after 0.5-mg dexamethasone. These data were available for 80 individuals and, for any given genotype, we had full genotype, phenotype, and DST data for 75 to 78 individuals. In this sample, the mean (SD) serum concentrations of cortisol on day 1 were 12.08 (5.06) μg/dL (to convert to nmol/L, multiply by 27.588) and following dexamethasone, mean (SD) concentrations were 3.65 (4.38) μg/dL. When we examined the change from baseline cortisol to the dexamethasone-suppressed cortisol concentration using repeated measures analysis of variance, we found significant interactions between risk allele carrier status and PTSD categorical diagnosis (probable diagnosis based on PSS inventory), with repeated measure of cortisol (rs9296158: F1,78 = 8.76, P < .004; rs3800373: F 1,76 = 6.03, P = .02; rs1360780: F1,79 = 3.95, P = .05; and rs9470080 : F 1,77 = 5.32, P = .02) (Figure 2), but no significant main effects. These data suggest that the majority of the patients with the risk alleles with PTSD show enhanced suppression to dexamethasone or enhanced GR sensitivity, which has been reported to be a possible endocrine signature of PTSD. In contrast, those individuals with the putative resilience genotype with PTSD appear to show the opposite effect. These results are supported by permutation-based analyses using percentage suppression as outcome and FKBP5 risk allele carrier status and probable PTSD as predictors.
Figure 2. Glucocorticoid Receptor Sensitivity, PTSD, and FKBP5 SNPs.

PTSD indicates posttraumatic stress disorder; SNPs, single-nucleotide polymorphisms. Mean serum cortisol concentration with 95% confidence interval (CI) is shown in participants who were tested at baseline and after 0.5 mg of dexamethasone (postdexamethasone suppression). The 4 panels represent the mean cortisol concentrations at baseline and postdexamethasone for individuals without probable PTSD (no PTSD) or with probable PTSD stratified by rs3800373, rs9296158, rs1360780, and rs9470080 genotypes. Individuals were categorized as risk allele carriers when they carried the C, A, T, or T alleles of these SNPs, respectively. Carriers of the AA, GG, CC, or CC homozygote genotypes of rs3800373, rs9296158, rs1360780, and rs9470080, respectively, were labeled as carrying the presumed protective genotypes. We found a significant interaction of genotype carrier- and PTSD-status on cortisol suppression (repeated measures analysis of variance: rs3800373, F1,76=6.03, P=.02; rs9296158, F1,78=8.76, P < .004; rs1360780, F1,79=3.95, P =.05; rs9470080, F1,77=5.32, P =.02; but also using permutation-based methods on percentage cortisol suppression). Although the risk alleles seem to be associated with less suppression (ie, glucocorticoid receptor resistance) in the no PTSD group, they are associated with greater suppression of cortisol from baseline to postdexamethasone in the group with PTSD. For rs9296158 GG carrier with no PTSD and rs9470080 CC carrier with no PTSD, the lower bound of the 95% CI had a negative value and was truncated at zero.
COMMENT
Our results indicate that levels of child abuse and non–child abuse trauma each independently predicted the level of adult PTSD symptomatology. Although polymorphisms in FKBP5 did not directly predict the level of PTSD symptoms or interact with the level of non–child abuse trauma to predict PTSD symptoms, SNPs within the FKBP5 locus robustly interacted with the level of child abuse to predict the level of adult PTSD symptoms.
The most novel and important finding of our study was the interaction between FKBP5 polymorphisms and child abuse history to predict the levels of adult PTSD symptoms. The polymorphisms seem to belong to the same bin of SNPs, all in high linkage disequilibrium, which is associated with functional differences in FKBP5 expression and consequent alterations in GR function. Notably, all 4 SNPs showing a significant interaction effect had either been reported to be associated with higher FKBP5 protein levels or a stronger induction of FKBP5 mRNA by cortisol in healthy probands (rs1360780 and rs3800373),57 or were in strong linkage disequilibrium with these SNPs (rs9296158 and rs9470080). The SNP genotypes that were associated with the highest FKBP5 mRNA induction in peripheral blood mononuclear cells by cortisol57 were also the ones that were associated with the highest vulnerability to PTSD symptoms following child abuse. Individuals carrying the other allele seemed to be protected from the development of PTSD symptoms in a gene-dose dependent manner. This is in agreement with the finding of Segman et al59 who showed that trauma-induced increased levels of FKBP5 mRNA expression in peripheral blood mononuclear cells immediately following medical trauma were predictors of the presence of PTSD at 4 months after the trauma.
FKBP5 expression is induced by glucocorticoids as part of an intracellular ultrashort negative feedback loop for GR activity,53 with increased expression of FKBP5 reducing glucocorticoid binding affinity54 and nuclear translocation of the GR,55 resulting in resistance to glucocorticoid activation. Thus, the alleles previously associated with high FKBP5 protein/mRNA expression57 should be associated with GR resistance. This is precisely what we observed in individuals without PTSD symptoms. The healthy carriers of these alleles showed less dexamethasone suppression and thus more GR resistance.
This functional association appears to be switched in patients with PTSD symptoms. These same alleles were associated with a higher dexamethasone suppression ratio and thus increased GR sensitivity, which is associated with PTSD, 43,45 while the protective genotype was associated with relative GR resistance in patients with PTSD symptoms. This environment-dependent reversal of the functional association may be similar to the effects previously reported in patients with depression, where less cortisol response in the dexamethasone-CRH test (an indication of increased GR sensitivity) was observed with the same alleles that had been associated with more FKBP5 protein and thus presumably GR resistance in healthy controls.57
Our study is the first to our knowledge to provide evidence directly supporting a developmental or symptom-dependent difference in the functional consequences of these FKBP5 SNPs on GR sensitivity. We hypothesized that specific FKBP5 alleles may enhance the effects of acutely released cortisol following child abuse on FKBP5 mRNA expression and that abnormal FKBP5 expression leads to maladaptive changes in GR sensitivity. These changes resulted in long-term alterations of HPA axis sensitivity, such as GR hypersensitivity that effect adult response to trauma. Consistent with this notion, alterations of HPA axis responsiveness43,45 have been previously identified as risk factors for PTSD.
PTSD has been suggested to result in part from initial over consolidation of traumatic memories82,83 or, conversely, abnormal extinction of such memories.8,84 Thus, alterations in FKBP5 function could conceivably be involved in abnormal GR-mediated signaling in neurons involved with stress response and memory formation. Polymorphisms within the FKBP5 gene that lead to altered GR responsiveness could promote sensitization of the neural systems involved in stress response and emotional memory processing, thereby placing children who have been abused with specific genetic variants at higher risk for PTSD or PTSD-spectrum symptoms when exposed to other types of traumas. This hypothesis may be supported by the finding that the alleles of rs3800373 and rs1360780, which are associated with higher risk for PTSD symptoms following severe child abuse in our study, were also associated with higher levels of peritraumatic dissociation in children after medical trauma.58 Notably, peritraumatic dissociation has previously been identified as another risk factor for the development of PTSD.10
It is presently not clear if the associated polymorphisms represent the actual functional variants that lead to the differential FKBP5 expression patterns or are in linkage disequilibrium with a so far untyped, potential functional variant. Denser fine-mapping of this region combined with resequencing and in vitro and in vivo functional studies may allow definitive identification of the genetic variants mechanistically responsible for the interactions and associations observed herein. The fact that in our study rs4713916, located in a potential regulatory region 5′ upstream of FKBP5, did not show a significant interaction effect, although in Binder et al52 it had similar association patterns to rs1360780 and rs3800373, is likely due to the different extent of linkage disequilibrium observed among these SNPs in the present sample of black individuals vs the previous sample of ethnic German individuals (linkage disequilibrium was substantially stronger in the German sample).
Depression and PTSD show a high comorbidity,85 so that concurrent depressive symptoms could confound our genetic interaction analysis for PTSD symptom outcome. However, controlling for current depressive symptoms, which were measured with the BDI, did not alter the observed interaction effect on PTSD symptoms and there was no gene × child abuse effect observed when BDI score was used as the outcome. Although the PSS and BDI scores strongly and significantly correlate in our sample (Pearson correlation=0.68, P < .001), our observation of a PTSD symptom-specific effect suggests that interaction of FKBP5 genotypes with child abuse history might influence PTSD symptoms not captured by the BDI, and thereby not shared with depression. Additional evidence suggesting separate gene × environment interactions for depression and PTSD symptomatology also comes from the lack of an interaction effect between SNPs in the CRHR1 gene and child abuse on PTSD symptom severity, despite the fact that these SNPs show strong interaction effects on the symptom severity of adult depression.71
One major limitation of our study is that we cannot present an independent replication, so that at this level of validation, it can only be considered hypothesis generating. The supporting evidence from functional effects of the polymorphisms in the DST serve to strengthen our finding that awaits independent replication. Another limitation is the lack of ancestry informative marker data on the complete sample. We do, however, feel the existing ancestry results among our sample of 280 participants genotyped for the 134 ancestry informative markers help to alleviate the concern of confounding by population stratification. The ancestry estimates in our subsample genotyped for the ancestry informative markers are not correlated with PSS or BDI scores.71 Because confounding due to population stratification only occurs when there is both correlation between outcome and ancestry as well as correlation between SNP genotypes and ancestry, our results are not likely susceptible to bias arising from confounding due to population structure.
Limitations in the phenotype side are that we used retrospective reports of child abuse. It is well-documented that reports of trauma are correlated with PTSD symptoms,86,87 and we have to acknowledge that the retrospective reports of child abuse and also non–child abuse trauma can therefore be biased in favor of an association between these reports and PTSD symptoms. A similar limitation is the lack of examination of the time since the index trauma. Furthermore, because we have used PTSD symptom severity and not diagnosis as an outcome measure and our sample has very high levels of multiple trauma exposure, our results may not be comparable with those obtained in studies with PTSD diagnosis as outcome and studies with less variation in trauma type or fewer overall types of trauma exposure. However, several studies have now indicated that multiple trauma exposure across the lifespan is the rule rather than the exception in samples similar to our study3,88,89 (eg, low income, urban, high percentage of black or Latino participants) and that multiple trauma exposure is related to increased mental and physical health risk.65,90,91 Understanding risk and resilience in response to multiple trauma exposures is of high public health importance.
Our sample was also derived from primarily impoverished, inner-city out-patient primary care clinics and participants were not presenting for treatment for PTSD so that we cannot directly generalize our findings to a clinical or epidemiological setting. Comparability with other studies and populations may be further limited as methods of assessment and definitions of child abuse vary greatly across studies. Finally, the mechanisms accounting for the relationship of child abuse to increased risk for PTSD in adulthood are not well understood. It may be that child abuse directly impacts psychological and biological developmental processes or that child abuse is associated with other variables (child/parental temperament, broader family environment, attachment) that impact psychological and biological development or an interaction of the 2.
CONCLUSIONS
Data reported herein suggest that genetic variation in the FKBP5 gene, which is involved in glucocorticoid signal transduction, may alter sensitization of the stress-response pathway during development, placing those individuals who have had significant child abuse at significant risk for PTSD in the face of other traumatic experiences. These genotypes potentially serve as predictors of both risk and resilience for adult PTSD among survivors of child physical and sexual abuse.
Our genetic results support the hypothesis that the glucocorticoid response system moderates the effects of early life stress on adult PTSD symptoms and that GR hypersensitivity may be important in the pathophysiology of this disorder. These results suggest the possibility that heritable differences in glucocorticoid-mediated neural functioning exacerbate or dampen the effects of child abuse on the stress hormone system, thus altering HPA axis sensitivity and risk for PTSD in adulthood.
Acknowledgments
Funding/Support: This work was primarily supported by grant MH071537 from the National Institute of Mental Health. Support was also received by grants MH069884 (Dr Ressler), MH-42088, and MH-58922 (Dr Nemeroff ) from the National Institute of Mental Health, DA015766 from the National Institute on Drug Abuse (Dr Cubells), Emory and Grady Memorial Hospital General Clinical Research Center, grant M01 RR00039 from the National Institutes of Health National Centers for Research Resources, and the Burroughs Wellcome Fund (Dr Ressler).
Footnotes
Author Contributions: Dr Ressler had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Drs Binder and Bradley contributed equally to this article.
Study concept and design: Binder, Bradley, Epstein, Heim, Schwartz, Cubells, Ressler.
Acquisition of data: Binder, Bradley, Deveau, Mercer, Tang, Gillespie, Heim, Cubells, Ressler.
Analysis and interpretation of data: Binder, Bradley, Liu, Epstein, Mercer, Heim, Nemeroff, Cubells, Ressler.
Drafting of the manuscript: Binder, Bradley, Epstein, Tang, Gillespie, Cubells, Ressler.
Critical revision of the manuscript for important intellectual content: Liu, Epstein, Deveau, Mercer, Heim, Nemeroff, Schwartz, Cubells, Ressler.
Statistical analysis: Binder, Bradley, Liu, Epstein, Tang, Gillespie, Ressler.
Obtained funding: Bradley, Heim, Nemeroff, Cubells, Ressler.
Administrative, technical, or material support: Bradley, Deveau, Mercer, Tang, Gillespie, Heim, Nemeroff, Schwartz, Cubells, Ressler.
Study supervision: Mercer, Nemeroff, Schwartz, Cubells, Ressler.
Financial Disclosures: Dr Binder reported receiving grant support or awards from Pfizer, GlaxoSmithKline, National Alliance in Research in Schizophrenia and Depression (NARSAD), the Doris Duke foundation, and the National Institute of Mental Health (NIMH), and has received speaker’s honoraria from AstraZeneca. Dr Tang reports receiving grant support from NARSAD and speaker’s honoraria from AstraZeneca, Eli Lilly, GlaxoSmithKline, Organon, Pfizer, and Xian-Janssen. Dr Gillespie reports receiving funding from APIRE/Wyeth, NARSAD, and National Institute on Drug Abuse (NIDA). Dr Heim reports receiving financial or grant support from NIMH, Centers for Disease Control and Prevention, NARSAD, the Anxiety Disorders Association of America, Eli Lilly, and Novartis. Dr Nemeroff reports serving on the Scientific Advisory Board for Quintiles, AstraZeneca, Forest Laboratories, Janssen/Ortho-McNeil, American Foundation for Suicide Prevention (AFSP), NARSAD, and PharmaNeuroboost. He holds equity, stock options, or stock in Novadel Pharma, Corcept, CeNeRx, PharmaNeuroboot, and Reevax; is on the board of directors of Novadel Pharma, Mt Cook Pharma, the AFSP, and the George West Mental Health Foundation; and is a grant recipient from the National Institutes of Health. Dr Ressler reports receiving awards, funding support, or both related to other studies from Howard Hughes Medical Institute, Lundbeck, Burroughs Wellcome Foundation, Pfizer, NARSAD, NIMH, and NIDA, and has a consulting agreement with Tikvah Therapeutics for N-methyl-D-aspartate–based therapeutics. No other authors reported any financial disclosures.
Role of the Sponsors: None of the above funding agencies had any role in the design and conduct of the study, in the collection, management, analysis, and interpretation of the data, or in the preparation, review, or approval of the manuscript.
Additional Contributions: Allen Graham, BS, Eboni Johnson, BS, Josh Castleberry, BS, Daniel Crain, BS, Abby Powers, BS, Nineequa Blanding, BS, Daphne Pierre, BS, Rachel Hershenberg, BS (all paid staff from the Grady Trauma Project, Grady Memorial Hospital, Atlanta, Georgia), and Tiina Berg, PhD (Department of Psychiatry, Emory University), provided excellent technical support.
References
- 1.Hoge CW, Castro CA, Messer SC, et al. Combat duty in Iraq and Afghanistan, mental health problems, and barriers to care. N Engl J Med. 2004;351(1):13–22. doi: 10.1056/NEJMoa040603. [DOI] [PubMed] [Google Scholar]
- 2.Keane TM, Marshall AD, Taft CT. Posttraumatic stress disorder: etiology, epidemiology, and treatment outcome. Annu Rev Clin Psychol. 2006;2:161–197. doi: 10.1146/annurev.clinpsy.2.022305.095305. [DOI] [PubMed] [Google Scholar]
- 3.Alim TN, Graves E, Mellman TA, et al. Trauma exposure, posttraumatic stress disorder and depression in an African-American primary care population. J Natl Med Assoc. 2006;98(10):1630–1636. [PMC free article] [PubMed] [Google Scholar]
- 4.Yehuda R, McFarlane AC. Conflict between current knowledge about posttraumatic stress disorder and its original conceptual basis. Am J Psychiatry. 1995;152(12):1705–1713. doi: 10.1176/ajp.152.12.1705. [DOI] [PubMed] [Google Scholar]
- 5.Nemeroff CB, Bremner JD, Foa EB, et al. Posttraumatic stress disorder: a state-of-the-science review. J Psychiatr Res. 2006;40(1):1–21. doi: 10.1016/j.jpsychires.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 6.Yehuda R. Risk and resilience in posttraumatic stress disorder. J Clin Psychiatry. 2004;65(suppl 1):29–36. [PubMed] [Google Scholar]
- 7.Brewin CR, Holmes EA. Psychological theories of posttraumatic stress disorder. Clin Psychol Rev. 2003;23(3):339–376. doi: 10.1016/s0272-7358(03)00033-3. [DOI] [PubMed] [Google Scholar]
- 8.Rothbaum BO, Davis M. Applying learning principles to the treatment of posttrauma reactions. Ann N Y Acad Sci. 2003;1008:112–121. doi: 10.1196/annals.1301.012. [DOI] [PubMed] [Google Scholar]
- 9.Diagnostic and Statistical Manual of Mental Disorders. 4. Washington, DC: American Psychiatric Association; 1994. [Google Scholar]
- 10.Ozer EJ, Best SR, Lipsey TL, Weiss DS. Predictors of posttraumatic stress disorder and symptoms in adults: a meta-analysis. Psychol Bull. 2003;129(1):52–73. doi: 10.1037/0033-2909.129.1.52. [DOI] [PubMed] [Google Scholar]
- 11.Stein MB, Jang KL, Taylor S, et al. Genetic and environmental influences on trauma exposure and posttraumatic stress disorder symptoms: a twin study. Am J Psychiatry. 2002;159(10):1675–1681. doi: 10.1176/appi.ajp.159.10.1675. [DOI] [PubMed] [Google Scholar]
- 12.Broekman BF, Olff M, Boer F. The genetic background to PTSD. Neurosci Biobehav Rev. 2007;31(3):348–362. doi: 10.1016/j.neubiorev.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 13.Koenen KC. Genetics of posttraumatic stress disorder: review and recommendations for future studies. J Trauma Stress. 2007;20(5):737–750. doi: 10.1002/jts.20205. [DOI] [PubMed] [Google Scholar]
- 14.American Psychological Association. Violence and the Family: Report of the American Psychological Association Presidential Task Force on Violence and the Family. Washington, DC: Presidential Task Force on Violence and the Family; 2002. [Google Scholar]
- 15.Gillespie CF, Nemeroff CB. Early life stress and depression. Current Psychiatry. 2005;4:15–30. [Google Scholar]
- 16.Heim C, Nemeroff CB. Neurobiology of early life stress: clinical studies. Semin Clin Neuropsychiatry. 2002;7(2):147–159. doi: 10.1053/scnp.2002.33127. [DOI] [PubMed] [Google Scholar]
- 17.Kaufman J, Plotsky PM, Nemeroff CB, Charney DS. Effects of early adverse experiences on brain structure and function: clinical implications. Biol Psychiatry. 2000;48(8):778–790. doi: 10.1016/s0006-3223(00)00998-7. [DOI] [PubMed] [Google Scholar]
- 18.Bremner JD, Southwick SM, Johnson DR, et al. Childhood physical abuse and combat-related posttraumatic stress disorder in Vietnam veterans. Am J Psychiatry. 1993;150(2):235–239. doi: 10.1176/ajp.150.2.235. [DOI] [PubMed] [Google Scholar]
- 19.Heim C, Nemeroff CB. The role of childhood trauma in the neurobiology of mood and anxiety disorders: preclinical and clinical studies. Biol Psychiatry. 2001;49(12):1023–1039. doi: 10.1016/s0006-3223(01)01157-x. [DOI] [PubMed] [Google Scholar]
- 20.Lang AJ, Laffaye C, Satz LE, et al. Relationships among childhood maltreatment, PTSD, and health in female veterans in primary care. Child Abuse Negl. 2006;30(11):1281–1292. doi: 10.1016/j.chiabu.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 21.Stovall-McClough KC, Cloitre M. Unresolved attachment, PTSD, and dissociation in women with childhood abuse histories. J Consult Clin Psychol. 2006;74(2):219–228. doi: 10.1037/0022-006X.74.2.219. [DOI] [PubMed] [Google Scholar]
- 22.Widom CS. Posttraumatic stress disorder in abused and neglected children grown up. Am J Psychiatry. 1999;156(8):1223–1229. doi: 10.1176/ajp.156.8.1223. [DOI] [PubMed] [Google Scholar]
- 23.Copeland WE, Keeler G, Angold A, Costello EJ. Traumatic events and posttraumatic stress in childhood. Arch Gen Psychiatry. 2007;64(5):577–584. doi: 10.1001/archpsyc.64.5.577. [DOI] [PubMed] [Google Scholar]
- 24.US Department of Health and Human Services. Child Maltreatment 2005. Vol. 16. Washington, DC: US Government Printing Office; 2007. [Google Scholar]
- 25.Dong M, Anda RF, Felitti VJ, et al. The inter relatedness of multiple forms of childhood abuse, neglect, and household dysfunction. Child Abuse Negl. 2004;28(7):771–784. doi: 10.1016/j.chiabu.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 26.Segman RH, Shalev AY. Genetics of posttraumatic stress disorder. CNS Spectr. 2003;8(9):693–698. doi: 10.1017/s1092852900008889. [DOI] [PubMed] [Google Scholar]
- 27.Yehuda R, Halligan SL, Bierer LM. Relationship of parental trauma exposure and PTSD to PTSD, depressive and anxiety disorders in offspring. J Psychiatr Res. 2001;35(5):261–270. doi: 10.1016/s0022-3956(01)00032-2. [DOI] [PubMed] [Google Scholar]
- 28.Yehuda R, Halligan SL, Grossman R. Childhood trauma and risk for PTSD: relationship to intergenerational effects of trauma, parental PTSD, and cortisol excretion. Dev Psychopathol. 2001;13(3):733–753. doi: 10.1017/s0954579401003170. [DOI] [PubMed] [Google Scholar]
- 29.Koenen KC, Lyons MJ, Goldberg J, et al. A high risk twin study of combat-related PTSD comorbidity. Twin Res. 2003;6(3):218–226. doi: 10.1375/136905203765693870. [DOI] [PubMed] [Google Scholar]
- 30.Bachmann AW, Sedgley TL, Jackson RV, et al. Glucocorticoid receptor polymorphisms and posttraumatic stress disorder. Psychoneuroendocrinology. 2005;30(3):297–306. doi: 10.1016/j.psyneuen.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 31.Caspi A, McClay J, Moffitt TE, et al. Role of genotype in the cycle of violence in maltreated children. Science. 2002;297(5582):851–854. doi: 10.1126/science.1072290. [DOI] [PubMed] [Google Scholar]
- 32.Foley DL, Eaves LJ, Wormley B, et al. Childhood adversity, monoamine oxidase a genotype, and risk for conduct disorder. Arch Gen Psychiatry. 2004;61(7):738–744. doi: 10.1001/archpsyc.61.7.738. [DOI] [PubMed] [Google Scholar]
- 33.Widom CS, Brzustowicz LM. MAOA and the “cycle of violence:” childhood abuse and neglect, MAOA genotype, and risk for violent and antisocial behavior. Biol Psychiatry. 2006;60(7):684–689. doi: 10.1016/j.biopsych.2006.03.039. [DOI] [PubMed] [Google Scholar]
- 34.Caspi A, Sugden K, Moffitt TE, et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003;301(5631):386–389. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- 35.Eley TC, Sugden K, Corsico A, et al. Gene-environment interaction analysis of serotonin system markers with adolescent depression. Mol Psychiatry. 2004;9(10):908–915. doi: 10.1038/sj.mp.4001546. [DOI] [PubMed] [Google Scholar]
- 36.Grabe HJ, Lange M, Wolff B, et al. Mental and physical distress is modulated by a polymorphism in the 5-HT transporter gene interacting with social stressors and chronic disease burden. Mol Psychiatry. 2005;10(2):220–224. doi: 10.1038/sj.mp.4001555. [DOI] [PubMed] [Google Scholar]
- 37.Kaufman J, Yang BZ, Douglas-Palumberi H, et al. Social supports and serotonin transporter gene moderate depression in maltreated children. Proc Natl Acad Sci U S A. 2004;101(49):17316–17321. doi: 10.1073/pnas.0404376101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kendler KS, Kuhn JW, Vittum J, et al. The interaction of stressful life events and a serotonin transporter polymorphism in the prediction of episodes of major depression. Arch Gen Psychiatry. 2005;62(5):529–535. doi: 10.1001/archpsyc.62.5.529. [DOI] [PubMed] [Google Scholar]
- 39.Gillespie NA, Whitfield JB, Williams B, et al. The relationship between stressful life events, the serotonin transporter (5-HTTLPR) genotype and major depression. Psychol Med. 2005;35(1):101–111. doi: 10.1017/s0033291704002727. [DOI] [PubMed] [Google Scholar]
- 40.Kaufman J, Yang BZ, Douglas-Palumberi H, et al. Brain-derived neurotrophic factor-5-HTTLPR gene interactions and environmental modifiers of depression in children. Biol Psychiatry. 2006;59(8):673–680. doi: 10.1016/j.biopsych.2005.10.026. [DOI] [PubMed] [Google Scholar]
- 41.Kilpatrick DG, Koenen KC, Ruggiero KJ, et al. The serotonin transporter genotype and social support and moderation of posttraumatic stress disorder and depression in hurricane-exposed adults. Am J Psychiatry. 2007;164(11):1693–1699. doi: 10.1176/appi.ajp.2007.06122007. [DOI] [PubMed] [Google Scholar]
- 42.Yehuda R. Biology of posttraumatic stress disorder. J Clin Psychiatry. 2001;62(suppl 17):41–46. [PubMed] [Google Scholar]
- 43.Yehuda R, Giller EL, Southwick SM, et al. Hypothalamic-pituitary-adrenal dysfunction in posttraumatic stress disorder. Biol Psychiatry. 1991;30(10):1031–1048. doi: 10.1016/0006-3223(91)90123-4. [DOI] [PubMed] [Google Scholar]
- 44.Yehuda R, Golier JA, Halligan SL, Meaney M, Bierer LM. The ACTH response to dexamethasone in PTSD. Am J Psychiatry. 2004;161(8):1397–1403. doi: 10.1176/appi.ajp.161.8.1397. [DOI] [PubMed] [Google Scholar]
- 45.Yehuda R, Golier JA, Yang RK, Tischler L. Enhanced sensitivity to glucocorticoids in peripheral mononuclear leukocytes in posttraumatic stress disorder. Biol Psychiatry. 2004;55(11):1110–1116. doi: 10.1016/j.biopsych.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 46.Watts-English T, Fortson BL, Gibler N, et al. The psychobiology of maltreatment in childhood. J Soc Issues. 2006;62:717–736. [Google Scholar]
- 47.Heim C, Newport DJ, Heit S, et al. Pituitary-adrenal and autonomic responses to stress in women after sexual and physical abuse in childhood. JAMA. 2000;284(5):592–597. doi: 10.1001/jama.284.5.592. [DOI] [PubMed] [Google Scholar]
- 48.Carpenter LL, Tyrka AR, McDougle CJ, et al. Cerebrospinal fluid corticotropin-releasing factor and perceived early-life stress in depressed patients and healthy control subjects. Neuro psycho pharmacology. 2004;29(4):777–784. doi: 10.1038/sj.npp.1300375. [DOI] [PubMed] [Google Scholar]
- 49.Ladd CO, Huot RL, Thrivikraman KV, et al. Long-term behavioral and neuroendocrine adaptations to adverse early experience. Prog Brain Res. 2000;122:81–103. doi: 10.1016/s0079-6123(08)62132-9. [DOI] [PubMed] [Google Scholar]
- 50.Plotsky PM, Thrivikraman KV, Nemeroff CB, et al. Long-term consequences of neonatal rearing on central corticotropin-releasing factor systems in adult male rat offspring. Neuro psycho pharmacology. 2005;30(12):2192–2204. doi: 10.1038/sj.npp.1300769. [DOI] [PubMed] [Google Scholar]
- 51.Schiene-Fischer C, Yu C. Receptor accessory folding helper enzymes: the functional role of peptidyl prolyl cis/trans isomerases. FEBS Lett. 2001;495(1–2):1–6. doi: 10.1016/s0014-5793(01)02326-2. [DOI] [PubMed] [Google Scholar]
- 52.Davies TH, Ning YM, Sanchez ER. A new first step in activation of steroid receptors: hormone-induced switching of FKBP51 and FKBP52 immunophilins. J Biol Chem. 2002;277(7):4597–4600. doi: 10.1074/jbc.C100531200. [DOI] [PubMed] [Google Scholar]
- 53.Vermeer H, Hendriks-Stegeman BI, van der Burg B, et al. Glucocorticoid-induced increase in lymphocytic FKBP51 messenger ribonucleic acid expression. J Clin Endocrinol Metab. 2003;88(1):277–284. doi: 10.1210/jc.2002-020354. [DOI] [PubMed] [Google Scholar]
- 54.Denny WB, Valentine DL, Reynolds PD, et al. Squirrel monkey immunophilin FKBP51 is a potent inhibitor of glucocorticoid receptor binding. Endocrinology. 2000;141(11):4107–4113. doi: 10.1210/endo.141.11.7785. [DOI] [PubMed] [Google Scholar]
- 55.Wochnik GM, Ruegg J, Abel GA, et al. FK506-binding proteins 51 and 52 differentially regulate dynein interaction and nuclear translocation of the glucocorticoid receptor in mammalian cells. J Biol Chem. 2005;280(6):4609–4616. doi: 10.1074/jbc.M407498200. [DOI] [PubMed] [Google Scholar]
- 56.Scammell JG, Denny WB, Valentine DL, Smith DF. Overexpression of the FK506-binding immunophilin FKBP51 is the common cause of glucocorticoid resistance in three New World primates. Gen Comp Endocrinol. 2001;124(2):152–165. doi: 10.1006/gcen.2001.7696. [DOI] [PubMed] [Google Scholar]
- 57.Binder EB, Salyakina D, Lichtner P, et al. Polymorphisms in FKBP5 are associated with increased recurrence of depressive episodes and rapid response to antidepressant treatment. Nat Genet. 2004;36(12):1319–1325. doi: 10.1038/ng1479. [DOI] [PubMed] [Google Scholar]
- 58.Koenen KC, Saxe G, Purcell S, et al. Polymorphisms in FKBP5 are associated with peritraumatic dissociation in medically injured children. Mol Psychiatry. 2005;10(12):1058–1059. doi: 10.1038/sj.mp.4001727. [DOI] [PubMed] [Google Scholar]
- 59.Segman RH, Shefi N, Goltser-Dubner T, et al. Peripheral blood mononuclear cell gene expression profiles identify emergent posttraumatic stress disorder among trauma survivors. Mol Psychiatry. 2005;10(5):500–513. doi: 10.1038/sj.mp.4001636. [DOI] [PubMed] [Google Scholar]
- 60.Newport DJ, Heim C, Bonsall R, et al. Pituitary-adrenal responses to standard and low-dose dexamethasone suppression tests in adult survivors of child abuse. Biol Psychiatry. 2004;55(1):10–20. doi: 10.1016/s0006-3223(03)00692-9. [DOI] [PubMed] [Google Scholar]
- 61.Foa EB, Tolin DF. Comparison of the PTSD Symptom Scale-Interview Version and the Clinician Administered PTSD Scale. J Trauma Stress. 2000;13(2):181–191. doi: 10.1023/A:1007781909213. [DOI] [PubMed] [Google Scholar]
- 62.Coffey SF, Dansky BS, Falsetti SA, et al. Screening for PTSD in a substance abuse sample: psychometric properties of a modified version of the PTSD Symptom Scale Self-Report. J Trauma Stress. 1998;11(2):393–399. doi: 10.1023/A:1024467507565. [DOI] [PubMed] [Google Scholar]
- 63.Falsetti SA, Resnick HS, Resick PA, Kilpatrick D. The Modified PTSD Symptom Scale: a brief self-report measure of posttraumatic stress disorder. Behav Therapist. 1993;16:161–162. [Google Scholar]
- 64.Schwartz AC, Bradley RG, Sexton M, Sherry A, Ressler KJ. Posttraumatic stress disorder among African Americans in an inner city mental health clinic. Psychiatr Serv. 2005;56(2):212–215. doi: 10.1176/appi.ps.56.2.212. [DOI] [PubMed] [Google Scholar]
- 65.Schwartz AC, Bradley R, Penza KM, et al. Pain medication use among patients with posttraumatic stress disorder. Psychosomatics. 2006;47(2):136–142. doi: 10.1176/appi.psy.47.2.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J. An inventory for measuring depression. Arch Gen Psychiatry. 1961;4:561–571. doi: 10.1001/archpsyc.1961.01710120031004. [DOI] [PubMed] [Google Scholar]
- 67.Beck AT, Steer RA, Garbin MG. Psychometric properties of the Beck Depression Inventory: twenty-five years of evaluation. Clin Psychol Rev. 1988;8:77–100. [Google Scholar]
- 68.Anda RF, Felitti VJ, Bremner JD, et al. The enduring effects of abuse and related adverse experiences in childhood: a convergence of evidence from neurobiology and epidemiology. Eur Arch Psychiatry Clin Neurosci. 2006;256(3):174–186. doi: 10.1007/s00406-005-0624-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bernstein DP, Fink L. Childhood Trauma Questionnaire Manual. San Antoinio, TX: Psychological Corporation; 1998. [Google Scholar]
- 70.Bernstein DP, Stein JA, Newcomb MD, et al. Development and validation of a brief screening version of the Childhood Trauma Questionnaire. Child Abuse Negl. 2003;27(2):169–190. doi: 10.1016/s0145-2134(02)00541-0. [DOI] [PubMed] [Google Scholar]
- 71.Bradley RG, Binder EB, Epstein MP, et al. Influence of child abuse on adult depression: moderation by the corticotropin-releasing hormone gene. Arch Gen Psychiatry. 2008;65(2):190–200. doi: 10.1001/archgenpsychiatry.2007.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Livak KJ. Allelic discrimination using fluorogenic probes and the 5′ nuclease assay. Genet Anal. 1999;14(5–6):143–149. doi: 10.1016/s1050-3862(98)00019-9. [DOI] [PubMed] [Google Scholar]
- 73.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 74.Frudakis T, Venkateswarlu K, Thomas MJ, et al. A classifier for the SNP-based inference of ancestry. J Forensic Sci. 2003;48(4):771–782. [PubMed] [Google Scholar]
- 75.Shriver MD, Kittles RA. Genetic ancestry and the search for personalized genetic histories. Nat Rev Genet. 2004;5(8):611–618. doi: 10.1038/nrg1405. [DOI] [PubMed] [Google Scholar]
- 76.Chakraborty R, Weiss KM. Frequencies of complex diseases in hybrid populations. Am J Phys Anthropol. 1986;70(4):489–503. doi: 10.1002/ajpa.1330700408. [DOI] [PubMed] [Google Scholar]
- 77.Rosenberg NA, Pritchard JK, Weber JL, et al. Genetic structure of human populations. Science. 2002;298(5602):2381–2385. doi: 10.1126/science.1078311. [DOI] [PubMed] [Google Scholar]
- 78.Shriver MD, Mei R, Parra EJ, et al. Large-scale SNP analysis reveals clustered and continuous patterns of human genetic variation. Hum Genomics. 2005;2(2):81–89. doi: 10.1186/1479-7364-2-2-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Epstein MP, Satten GA. Inference on haplotype effects in case-control studies using unphased genotype data. Am J Hum Genet. 2003;73(6):1316–1329. doi: 10.1086/380204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhao JH, Curtis D, Sham PC. Model-free analysis and permutation tests for allelic associations. Hum Hered. 2000;50(2):133–139. doi: 10.1159/000022901. [DOI] [PubMed] [Google Scholar]
- 81.Blalock H. Social Statistics. New York, NY: McGraw-Hill; 1972. pp. 406–407. [Google Scholar]
- 82.Pitman RK, Delahanty DL. Conceptually driven pharmacologic approaches to acute trauma. CNS Spectr. 2005;10(2):99–106. doi: 10.1017/s109285290001943x. [DOI] [PubMed] [Google Scholar]
- 83.Davidson JR, Stein DJ, Shalev AY, Yehuda R. Posttraumatic stress disorder: acquisition, recognition, course, and treatment. J Neuropsychiatry Clin Neurosci. 2004;16(2):135–147. doi: 10.1176/jnp.16.2.135. [DOI] [PubMed] [Google Scholar]
- 84.Gillespie CF, Ressler KJ. Emotional learning and glutamate: translational perspectives. CNS Spectr. 2005;10(10):831–839. doi: 10.1017/s1092852900010439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kessler RC, Sonnega A, Bromet E, Hughes M, Nelson CB. Posttraumatic stress disorder in the National Comorbidity Survey. Arch Gen Psychiatry. 1995;52(12):1048–1060. doi: 10.1001/archpsyc.1995.03950240066012. [DOI] [PubMed] [Google Scholar]
- 86.Southwick SM, Morgan CA, III, Nicolaou AL, Charney DS. Consistency of memory for combat-related traumatic events in veterans of Operation Desert Storm. Am J Psychiatry. 1997;154(2):173–177. doi: 10.1176/ajp.154.2.173. [DOI] [PubMed] [Google Scholar]
- 87.Roemer L, Litz BT, Orsillo SM, Ehlich PJ, Friedman MJ. Increases in retrospective accounts of warzone exposure over time: the role of PTSD symptom severity. J Trauma Stress. 1998;11(3):597–605. doi: 10.1023/A:1024469116047. [DOI] [PubMed] [Google Scholar]
- 88.Liebschutz J, Saitz R, Brower V, et al. PTSD in urban primary care: high prevalence and low physician recognition. J Gen Intern Med. 2007;22(6):719–726. doi: 10.1007/s11606-007-0161-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Schilling EA, Aseltine RH, Jr, Gore S. Adverse childhood experiences and mental health in young adults. BMC Public Health. 2007;7:30. doi: 10.1186/1471-2458-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Green BL, Goodman LA, Krupnick JL, et al. Outcomes of single versus multiple trauma exposure in a screening sample. J Trauma Stress. 2000;13(2):271–286. doi: 10.1023/A:1007758711939. [DOI] [PubMed] [Google Scholar]
- 91.Breslau N, Chilcoat HD, Kessler RC, Davis GC. Previous exposure to trauma and PTSD effects of subsequent trauma: results from the Detroit Area Survey of Trauma. Am J Psychiatry. 1999;156(6):902–907. doi: 10.1176/ajp.156.6.902. [DOI] [PubMed] [Google Scholar]