

Abstract
Treatment of emotional disorders involves the promotion of extinction processes, which are defined as the learned reduction of fear. The molecular mechanisms underlying extinction have only begun to be elucidated. By employing genetic and pharmacological approaches in mice, we show here that extinction requires downregulation of Rac-1 and cyclin-dependent kinase 5 (Cdk5), and upregulation of p21 activated kinase-1 (PAK-1) activity. This is physiologically achieved by a Rac-1–dependent relocation of the Cdk5 activator p35 from the membrane to the cytosol and dissociation of p35 from PAK-1. Moreover, our data suggest that Cdk5/p35 activity prevents extinction in part by inhibition of PAK-1 activity in a Rac-1–dependent manner. We propose that extinction of contextual fear is regulated by counteracting components of a molecular pathway involving Rac-1, Cdk5 and PAK-1. Our data suggest that this pathway could provide a suitable target for therapeutic treatment of emotional disorders.
The pathogenesis of emotional disorders, post-traumatic stress disorders in particular, often involves associative learning that links anxiogenic stimuli to certain life experiences1–4. These disorders severely affect the lives of patients and are an increasing burden on our societies1. Treatment of such disorders generally involves the promotion of extinction processes, which are defined as the reduction of an aversively-motivated behavior2. Therefore, understanding the molecular mechanisms underlying extinction may help to develop therapeutic strategies for emotional disorders.
A well-established procedure for investigating extinction in rodents is Pavlovian fear conditioning. In this procedure, a single exposure of rodents to a novel context followed by an electric foot shock elicits the acquisition of conditioned fear. On the basis of associative learning, the animals show an inborn aversive freezing behavior upon re-exposure to the conditioned context. This form of contextual fear conditioning is hippocampus dependent and leads to a long-lasting fear memory5. During extinction, animals are repeatedly re-exposed to the conditioned context without receiving the foot shock again (extinction trial), which eventually results in the decline of the aversive freezing behavior6.
At present, the molecular mechanisms underlying extinction are not well understood. A number of studies have shown that some, but not all, components of the molecular machinery that is required for the initial encoding of fear memories also regulate extinction7–10. Recent studies indicate that synaptic remodeling, for example, mediated by actin re-arrangement, may be important for extinction3,6. Actin dynamics are intimately involved in synaptic plasticity, synapse formation and neuronal morphology6,11,12. Notably, Cdk5 has been described as an important regulator of synaptic function and actin dynamics13–16.
Cdk5 is a serine/threonine protein-kinase that is critical for the development of the central nervous system17. The related neuron-specific proteins p35 and p39 are required and sufficient to activate Cdk5 on direct binding14. Recently, there has been emerging evidence for a role of Cdk5/p35 in the adult brain. Cdk5 phosphorylates a number of synaptic proteins and regulates dendritic spine morphogenesis, synaptic plasticity and learning18–26. Cdk5 regulates actin dynamics via its upstream regulator Rac-1 and downstream target PAK-1, which have both also been implicated in synaptic plasticity15,27,28. Whether a Cdk5 regulated pathway is implicated with extinction of learned fear has not been addressed previously.
In this study, we show that inhibition of Cdk5 in the hippocampus facilitates extinction of learned contextual fear. Conversely, extinction was severely impaired when Cdk5 activity was upregulated. During physiological extinction, Cdk5 activity was downregulated by a reduced membrane association of p35 via the small GTPase Rac-1. Furthermore, our data suggests that inhibition of PAK-1 activity by Cdk5 impedes extinction.
RESULTS
Inhibition of Cdk5 activity facilitates extinction
To test whether Cdk5 activity has a role in extinction of learned fear, we implanted mice with microcannula into the dorsal hippocampus (intrahippocampal; Supplementary Fig. 1 online) and trained in the contextual fear-conditioning procedure. Fear conditioning consisted of a 3-min context exposure followed by a single electric foot shock (0.7 mA, constant current, 2 s). Subsequently, all mice were subjected to a daily extinction trial on 6 consecutive d (E1–E6). Each extinction trial consisted of a 3 min re-exposure to the conditioned context without presenting the foot shock again. It was previously shown that in this extinction procedure, mice usually show a substantial reduction of aversive freezing behavior in 4–6 extinction trials that are performed on consecutive days6.
Immediately after the exposure to E1–E3, mice were injected (intrahippocampal) with either vehicle or three different concentrations of the Cdk5 inhibitor butyrolactone I. Injection of 50 ng of butyrolactone 1 significantly reduced freezing behavior on E2 and E3 when compared with the vehicle group (P < 0.05, Fig. 1a). A reminder shock procedure6 was able to reinstate freezing behavior, suggesting that the original fear memory was not entirely erased (Supplementary Fig. 2 online). In agreement with previous data, the recovery of the fear response was rarely complete and may indicate that unlearning and new learning may coexist during extinction29,30. The injection of lower concentrations of butyrolactone 1 (25 or 12.5 ng) did not reduce hippocampal Cdk5 activity, as measured by histone H1 phosphorylation (Supplementary Fig. 2), and had no significant effect on the reduction of freezing behavior (P = 0.3781, Fig. 1b,c). Notably, injection of roscovitine (intrahippocampal, 50 ng), another well-established Cdk5 inhibitor, into the dorsal hippocampus immediately after E1–E3 also reduced freezing behavior when compared with a vehicle group (Supplementary Fig. 2). Consistent with the data presented in Figure 1a, a single injection of butyrolactone I (50 ng) immediately after E1 significantly diminished freezing behavior in the subsequent extinction trials when compared with the vehicle group (P < 0.0001, Fig. 1d). Similar results were obtained when mice were injected 1 h after E1 (Fig. 1e). In contrast, when butyrolactone I (50 ng) was administered 3 h after E1, freezing behavior was indistinguishable between butyrolactone I– and vehicle-injected mice (Fig. 1f). We estimated that the employed concentrations of butyrolactone I and roscovitine permit a specific inhibition of Cdk5. However, higher levels of those drugs may also affect Erk-1/2 kinases31. Notably, injection of butyrolactone I (50 ng) into the hippocampus did not affect the MAP kinase signaling pathway, which is important for extinction of contextual fear9,32 (Supplementary Fig. 2). Furthermore, intrahippocampal injection of butyrolactone I did not affect extinction of amygdala-dependent cued fear, demonstrating that Cdk5 inhibition in the hippocampus has a specific effect on contextual fear. Moreover, freezing behavior was not generally affected. (Supplementary Fig. 2). In addition, we found that intrahippocampal injection of butyrolactone I 15 min before E1 or E2 did not affect freezing behavior during the extinction trial, suggesting that Cdk5 activity affects the consolidation of extinction in our procedure (Supplementary Fig. 2).
Figure 1.
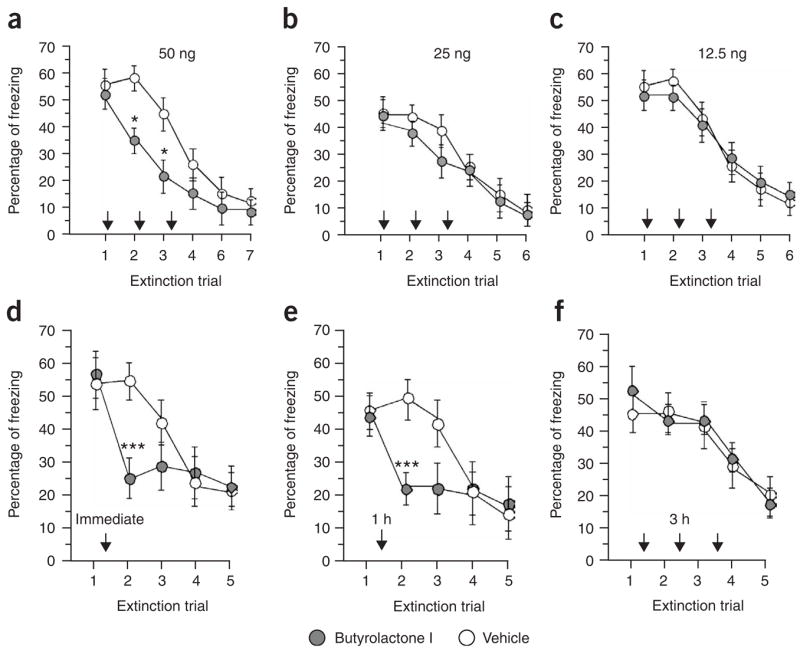
Inhibition of hippocampal Cdk5 activity facilitates extinction. (a) Mice (ten per group) were subjected to our extinction procedure and injected (intrahippocampal) with butyrolactone I (50 ng) immediately after E1–E3. When compared with the vehicle group, butyrolactone I–injected mice showed reduced freezing behavior on E2 and E3, indicating facilitated extinction. (b) The experiment was carried out as described in a, but a lower concentration (25 ng) of butyrolactone I was injected after E1–E3. Extinction was similar in vehicle and butyrolactone I–injected mice. (c) Similar results were obtained when mice were administered 12.5 ng of butyrolactone I. (d) Consistent with the data shown in a, a single injection of 50 ng of butyrolactone immediately after E1 facilitated extinction, as indicated by a significant reduction of freezing behavior on E2 when compared with a vehicle group. The reduced freezing behavior persisted throughout extinction trials in the absence of further butyrolactone I injection. (e) Mice were subjected to our extinction procedure and injected with butyrolactone I (50 ng) 1 h after E1. This facilitated extinction, as indicated by a significant reduction of freezing behavior on E2 when compared with a vehicle group. (f) Intrahippocampal injection of butyrolactone I 3 h after E1 had no significant effect on extinction when compared with the vehicle group. *P < 0.05 versus vehicle group, n = 9–10 mice per group. Error bars represent s.e.m.
In summary, these data show that inhibition of hippocampal Cdk5 facilitates extinction of aversive freezing behavior. Furthermore, our findings suggest that Cdk5 activity is critically involved in regulating extinction within an interval of 1 h after exposure to the extinction trial.
Increased Cdk5 activity impairs extinction
We next investigated the effect of increased Cdk5 activity on extinction. To this end we employed the CK-p25 Tg transgenic mice, in which forebrain-specific expression of the potent Cdk5 activator p25 can be switched on and off by a doxycycline diet (Supplementary Fig. 3 online)19. We previously showed that expression of p25 for 1–2 weeks increased Cdk5 activity in the hippocampus without causing any neuronal loss19. Here we subjected CK-p25 Tg and control mice to contextual fear conditioning. p25 expression was induced 24 h later for 1 week (Fig. 2a,b), which led to increased hippocampal Cdk5 activity (Fig. 2c). At this time point mice were subjected to six consecutive extinction trials (Fig. 2a). In contrast to control mice, no reduction of freezing behavior was observed in CK-p25 Tg mice (Fig. 2d). Next, doxycycline was re-introduced to the diet, leading to the repression of p25 expression (Fig. 2b). Consistently, hippocampal Cdk5 activity declined to baseline levels when measured 1 week later (Fig. 2c; see 1 week On 1 week Off group). Subsequently, all mice were exposed to five additional extinction trials. Notably, the 1 week ON 1 week OFF CK-p25 Tg showed a significant reduction in freezing behavior (P < 0.0001, Fig. 2d). Thus, increased Cdk5 activity prevented extinction, which can be reversed on downregulation of Cdk5 activity to baseline levels. These observations suggest that Cdk5 activity negatively regulates extinction.
Figure 2.
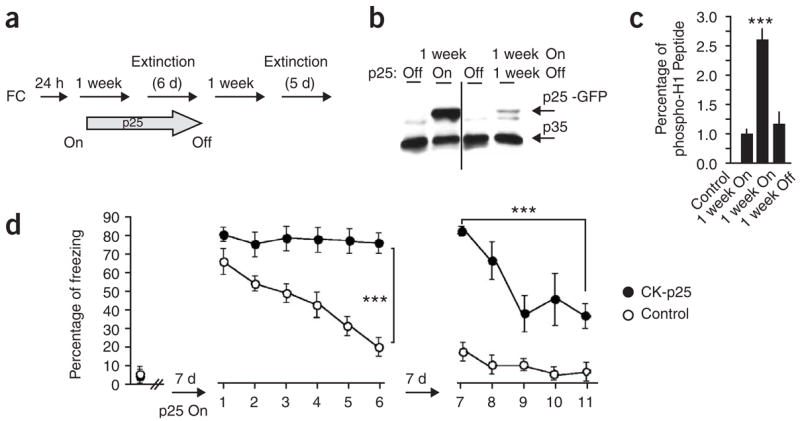
Increased Cdk5 activity in CK-p25 Tg mice impairs extinction. (a) Experimental design. CK-p25 Tg (n = 9) and control mice (n = 9) were trained by contextual fear conditioning, and p25 expression was induced 24 h later for 1 week. Afterwards, all mice were exposed to six extinction trials before repression of p25 production in CK-p25 Tg by reintroduction of doxycycline to the diet. After 1 week, all mice were subjected to another five extinction trials on consecutive days. (b) Immunoblot analysis of hippocampal lysates from CK-p25 Tg and control mice. Antibody to p35 (C-19) was employed to detected endogenous p25 and p25-GFP in CK-p25 Tg mice 1 week after p25 induction and 1 week after p25 repression, respectively. (c) Cdk5 kinase activity was analyzed by p35 (C-19) immunoprecipitation assays using histone H1 as an in vitro substrate. Cdk5 activity was significantly increased after 1 week of induction, and decreased to baseline levels after 1 week of p25 repression. (d) CK-p25 Tg and control mice were subjected to the experimental procedure described in a. As long as p25 was expressed in CK-p25 Tg, leading to increased Cdk5 activity, the extinction of freezing behavior was abolished. When p25 production was repressed and Cdk5 activity was reduced to baseline levels, extinction occurred in the same mice. ***P < 0.0001, n = 3–5 for biochemical analysis. Error bars represent s.e.m.
Extinction depletes Cdk5 activity from the membranes
To gain further insight into the mechanisms by which Cdk5 regulates extinction, we measured Cdk5 and p35 levels during extinction. To this end, we compared E1 versus E3, because in our procedure the molecular mechanisms activated after E1 (mice showed no extinction during subsequent E2) versus E3 (mice showed extinction during subsequent E4) might be different. Because we previously observed that Cdk5 activity was required for extinction within 1 h after an extinction trial (see Fig. 1d–f), we focused the molecular analysis on the time point 0.5 h after an extinction trial.
Immunoblot analysis of total-protein lysates prepared from the hippocampus 0.5 h after E1, E3 or from naive mice revealed no significant difference in p35 and Cdk5 levels (P = 0.2982, Supplementary Fig. 4 online). We next examined the subcellular distribution of Cdk5 and p35 during extinction. Hippocampal lysates were prepared 0.5 h after E1 and E3 and from naive animals, and were separated into membrane and nonmembrane fractions (Supplementary Fig. 4). Notably, we found that membrane-associated p35 levels were significantly reduced after E3 when compared with E1 (P < 0.05, Fig. 3a).
Figure 3.

Membrane association of Cdk5 and p35 during extinction is regulated by Rac-1. (a) Membrane and cytosolic fractions were prepared from the hippocampus of mice 0.5 h after exposure to E1, E3 or from naive animals. We observed a significant reduction of membrane-associated p35 that was accompanied by increased cytosolic p35 levels after E3. Synaptophysin (SVP) served as a loading control for the membrane fraction, whereas GAPDH was employed to verify equal loading in the cytosolic fraction. (b) Membrane lysates prepared from mice 0.5 h after E3 and from naive mice were used in a Rac-1 kinase assay. Rac-1 activity was significantly decreased after E3. (c) Hippocampal cytosolic and membrane fractions were prepared on E3 0.5 h after mice were injected (ICV) with either vehicle or Rac-1 inhibitor. Immunoblot analysis showed a significant reduction of membrane-associated Cdk5/p35 when compared with vehicle-injected mice. (d) Consistent application of Rac-1 inhibitor facilitated extinction. Mice (10 per group) were subjected to our extinction procedure and injected (ICV) with Rac-1 inhibitor (NSC23760; 10 μg μl−1) immediately after E1–E3. When compared with the vehicle group, Rac-1 inhibitor injected mice showed reduced freezing, indicating facilitated extinction. n = 3–5 for biochemical analysis. ***P < 0.0001 versus naive/vehicle group, *P < 0.05 versus naive/vehicle group. Error bars represent s.e.m.
Notably, when mice were exposed to E1, and hippocampal protein lysates were prepared 2 d later without any further extinction trial, Cdk5 and p35 protein levels and distribution were not affected (Supplementary Fig. 5), suggesting that membrane depletion of p35 is specific to extinction. Taken together, these data show that the reduced freezing behavior during extinction correlates with a reduction of membrane-associated p35 levels.
Rac-1 regulates the localization of Cdk5/p35 kinase and extinction
The small GTPase Rac-1 was shown to regulate the localization and activity of Cdk5/p35 kinase during neurite outgrowth33. This prompted us to investigate the involvement of Rac-1 in Cdk5-mediated extinction. Rac-1 is only active in its GTP-bound form (GTP–Rac-1)34. By employing a Rac-1–activity assay (Upstate) we found that Rac-1 activity was markedly reduced in membrane fraction after E3 when compared with the naive group (Fig. 3b). Total-protein levels of Rac-1 were not significantly different between naive and E3 (P = 0.2561, Supplementary Fig. 4), suggesting that membrane-associated Rac-1 activity decreased after E1. The correlation between reduction of membrane-associated Rac-1 activity and p35 levels after E3 prompted us to test the possibility of a direct relationship of Rac-1 activity and p35 localization during extinction.
We implanted microcannula into the lateral ventricles of mice that were subsequently trained by contextual fear conditioning and exposed to our extinction procedure. Immediately after E1–E3, mice were injected with a Rac-1 inhibitor35(NSC23760, 10 μg μl−1) or vehicle. Hippocampal membrane and cytosolic fractions were prepared 0.5 h after E3 and analyzed by immunoblot. When compared to the lysates prepared from the vehicle-injected group, mice that were administered with the Rac-1 inhibitor showed a marked redistribution of membrane-associated p35 and Cdk5 to the cytosol (Fig. 3c). This result suggests that Rac-1 activity regulates the distribution of p35/Cdk5 kinase during extinction.
Next we wanted to determine the effect of Rac-1 on extinction directly. Microcannula were implanted into the lateral ventricles of mice that were subsequently trained by contextual fear conditioning and exposed to our extinction procedure. Immediately after E1–E3, mice were injected with a Rac-1 inhibitor (NSC23760, 10 μg μl−1) or vehicle. Notably, mice injected with the Rac-1 inhibitor showed facilitated extinction as indicated by a significant reduction of freezing behavior when compared with the vehicle group (P < 0.0001, Fig. 3d). Similar results were obtained by intrahippocampal injections (Supplementary Fig. 6 online). Thus, although it is difficult to directly compare the effects of butyrolactone I and Rac-I inhibitors on the dynamics of extinction in absolute terms, Rac-1 activity, similar to Cdk5, appears to inhibit extinction.
A Rac-1–Cdk5–PAK-1 pathway regulates extinction
The Cdk5/p35 kinase was previously shown to regulate PAK-1 in a Rac-1–dependent manner33,36. As PAK-1 is implicated in the dynamics of actin cytoskeleton, learning and synaptic remodeling in the adult brain28, we sought to examine whether PAK-1 has a role in Cdk5-mediated extinction.
First, we measured PAK-1 phosphorylation at threonine 212 during extinction, as this site is phosphorylated by the p35/Cdk5 kinase (ref. 33). We found that PAK-1T212 levels were markedly reduced in the E3 membrane fraction when compared with naive mice or E1 (Fig. 4a). These data are consistent with decreased membrane-associated p35 levels observed after E3 (see Fig. 3a) and further support the view that Cdk5 activity declines during extinction. We also analyzed the activity of PAK-1 using a phospho-T423 antibody to PAK-1 (ref. 37) in hippocampal lysates prepared from naive mice and from mice 0.5 h after E1 or E3 by immunoblotting. We detected a significant increase in membrane-associated PAK-1Thr423 levels after E3, when compared with E1 (P < 0.05, Fig. 4b). In the cytosolic fraction, PAK-1Thr423 levels were downregulated after E1 when compared with naive mice, and increased after E3 when compared with E1 (Fig. 4b). These data suggest that the activity of PAK-1 is upregulated during extinction.
Figure 4.

Inhibition of Rac-1 facilitates extinction. (a) Membrane and cytosolic fractions were prepared from the hippocampus of mice 0.5 h after exposure to E1, E3 or from naive animals and analyzed for PAK-1 by immunoblotting. Levels of PAK-1Thr212 were significantly decreased in the membrane fraction after E3. (b) The same lysates as in a were used to analyze the levels of pPAK-1T423, a non-Cdk5 phosphorylation site that indicates active PAK-1. pPAK-1T423 levels were significantly increased after E3 in the membrane fraction. In the cytosolic fraction, pPAK-1T423 levels decreased after E1 when compared with naive mice, but increased back to baseline levels after E3. (c) Hippocampal cytosolic and membrane fractions were prepared on E3 0.5h after mice were injected (ICV) with either vehicle or Rac-1 inhibitor. Immunoblot analysis showed a significant reduction of membrane-associated p35/Cdk5, pPAK-1T212 and PAK-1 levels. (d) The same lysates as described in c were used to immunoprecipitate PAK-1. A hemagglutinin antibody served as a control for nonspecific binding of proteins to the protein A–agarose beads. The precipitates were immunoblotted with antibodies to PAK-1 and p35. There was a substantial reduction in the amount of p35 that co-immunoprecipitated with PAK-1 in lysates from mice that were administered with Rac-1 inhibitor. Error bars represent s.e.m.
Next, we set out to determine if the regulation of PAK-1 activity during extinction depended on the activities of Rac-1 and the Cdk5/p35 kinase. To this end, we analyzed lysates prepared from mice that were injected with a Rac-1 inhibitor or vehicle (as described in Fig. 3c) by immunoblot. When compared with the vehicle-injected group, mice that were administered with the Rac-1 inhibitor showed a substantial reduction of membrane-associated PAK-1 and PAK-1T212 levels (Fig. 4c). Consistently, PAK-1 levels increased in the cytosol of mice treated with Rac-1 inhibitor. However, we observed no corresponding increase in p-PAK-1T212 levels in the cytosol. In fact, cytosolic PAK-1T212 levels were also decreased (Fig. 4c). On co-immunoprecipitation of PAK-1 and p35 from cytosolic fractions, we found that Rac-1 inhibitor treatment resulted in a reduced amount of p35 being co-immunoprecipitated with PAK-1 when compared with the vehicle group (Fig. 4d). This result suggests that cytosolic p35 is sequestered from PAK-1 on Rac-1 inhibition during extinction. In summary, these data suggest that Rac-1 activity regulates PAK-1 during extinction.
To investigate the impact of Cdk5/p35 kinase activity on PAK-1 function during extinction, we implanted mice with microcannula in the lateral ventricles and injected them with 50 ng of butyrolactone I immediately after E1. Because a single injection of butyrolactone I after E1 was sufficient to facilitate extinction (see Fig. 1d), we prepared hippocampal lysates 0.5 h after E1 and analyzed them by immunoblotting using PAK-1 antibodies (P < 0.05, Fig. 5a). Butyrolactone I injection significantly reduced Cdk5-dependent phosphorylation of PAK-1, as PAK-1T212 levels in both the membrane and cytosolic fractions were decreased (Fig. 5b,c). In contrast, PAK-1T423 levels increased in lysates obtained from butyrolactone I–injected mice (Fig. 5b,c). Moreover, similar to treatment with Rac-1 inhibitor, inhibition of Cdk5 caused a redistribution of total PAK-1 from the membrane to the cytosol (Fig. 5d). These data provide evidence that Rac-1 and Cdk5/p35 kinase regulate the activity of PAK-1 during extinction.
Figure 5.
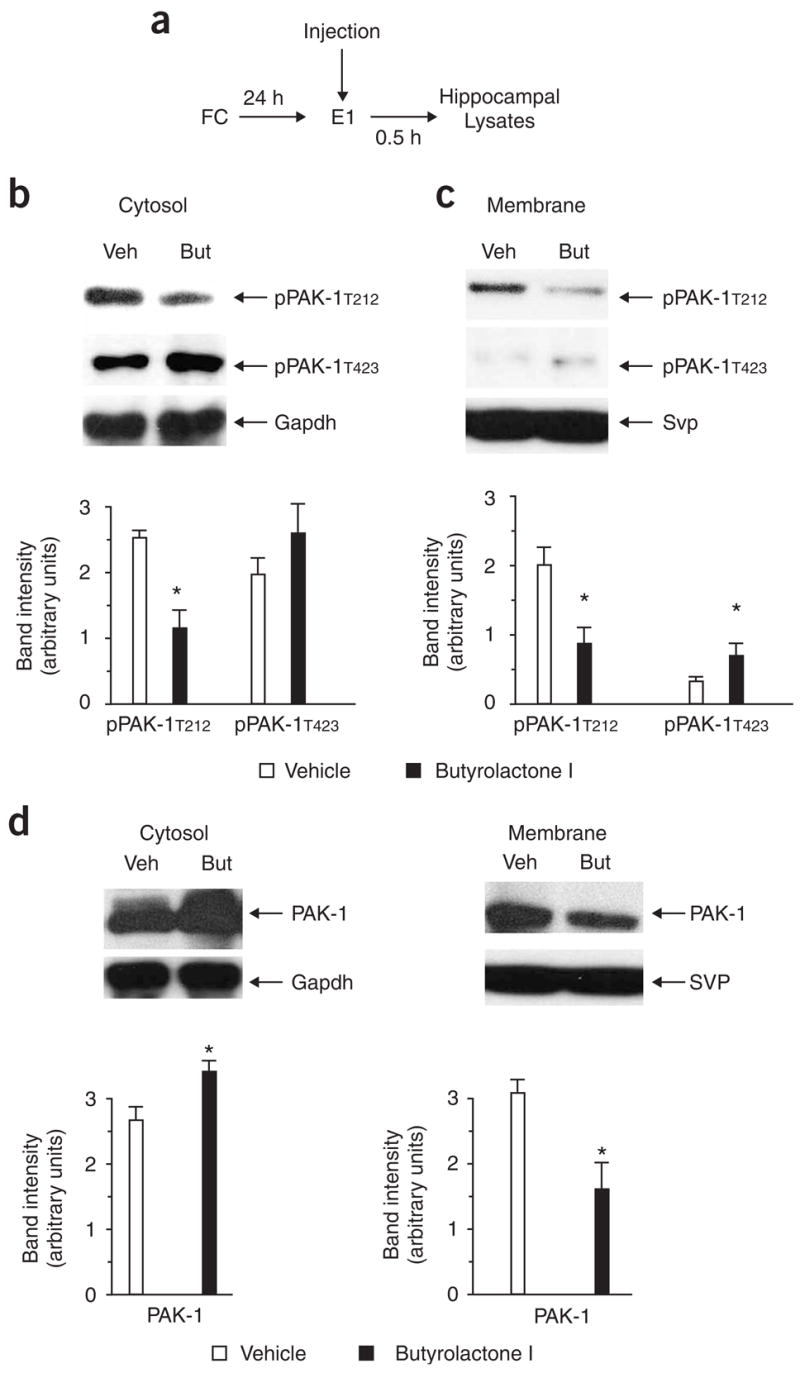
Cdk5 affects PAK-1 activity during extinction. (a) Experimental design. Microcannula were implanted into the hippocampus of mice that were subjected to contextual fear conditioning. The Cdk5 inhibitor butyrolactone I (50 ng) was injected immediately after exposure to E1 and hippocampal cytosolic and membrane fractions were prepared 0.5 h later. (b) Mice injected with butyrolactone I (But) showed significantly reduced levels of PAK-1T212 in the cytosolic fraction. (c) Similarly, PAK-1T212 levels were reduced in the membrane fraction. Conversely, levels of pPAK-1T423 were significantly increased. (d) Notably, butyrolactone-injected mice also showed a significant redistribution of PAK-1 from the membrane to the cytosol when compared with vehicle-injected mice. Error bars represent s.e.m.
PAK-1 promotes extinction
To further determine a role for PAK-1 activity during extinction, we introduced a herpes virus expressing green fluorescent protein (GFP) and a dominant-negative PAK-1 (HSV–dnPAK-1) into the dorsal hippocampus via a microcannula. It was previously shown that the dnPAK-1 protein substantially inhibited Pak-1 activity in vivo28. It is important that mice first normally acquire fear memories, and that any molecular manipulation is only applied after the extinction trial. To this end, we first subjected mice to contextual fear conditioning. After 24 h, HSV–dnPAK-1 or HSV-GFP (control group) was injected into the hippocampus. Because we found in pilot experiments that expression of these constructs (as measured by GFP signal) was first detectable 1 d after injection (Fig. 6), all mice were subjected to extinction trials the next day (Fig. 6a). Notably, we found that mice injected with HSV–dnPAK-1 showed a significant impairment in extinction when compared with the control group (P < 0.05, Fig. 6c). These data suggest that inhibition of PAK-1 activity impairs extinction.
Figure 6.
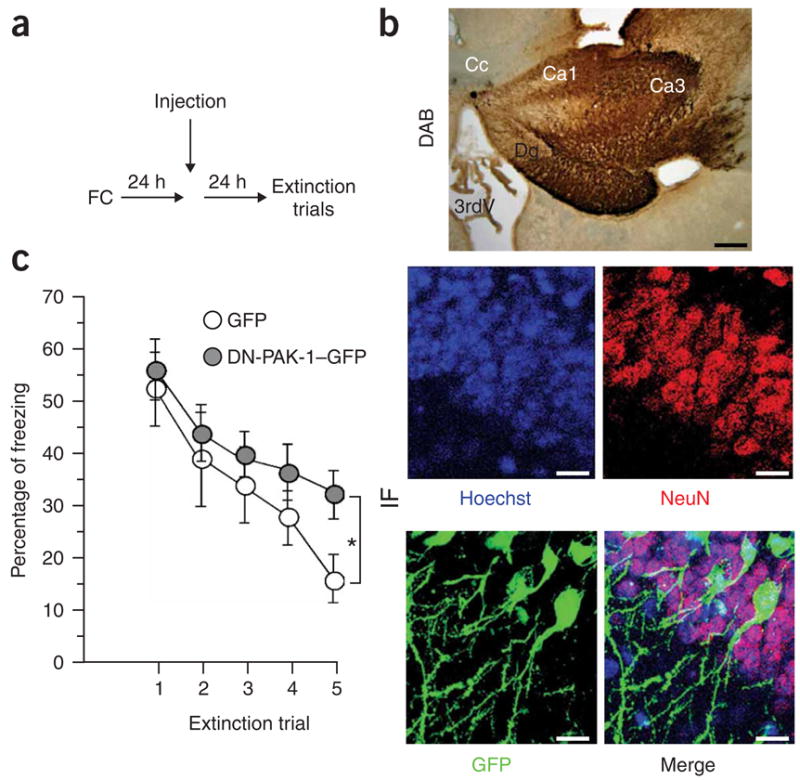
PAK-1 activity promotes extinction. (a) Experimental design. Mice were implanted with microcannula in the dorsal hippocampus and subjected to contextual fear conditioning. After 24 h, mice were injected with either HSV-GFP or HSV-GFP– dnPAK-1 virus into the dorsal hippocampus. As of the next day, all mice were subjected to our extinction procedure. (b) Herpes virus expressing GFP and a dnPAK-1 construct or only GFP were injected into the dorsal hippocampus of mice. Upper, DAB staining for GFP of mice injected with dnPAK-1–GFP. Lower, high-magnification pictures of the hippocampal region showing staining for Hoechst, NeuN and GFP. The transfection efficiency was about 30%. Analysis was carried out 48 h after injection. (c) Mice (n = 10 per group) expressing dnPAK-1 in the hippocampus show a significantly impaired reduction of freezing behavior indicating impaired extinction. Error bars represent s.e.m.
DISCUSSION
In this study, we show that pharmacological inhibition of hippocampal Cdk5 activity by butyrolactone I or roscovitine facilitates extinction in the contextual fear-conditioning procedure. Conversely, increased Cdk5 activity in CK-p25 Tg mice severely impaired extinction, as indicated by the persistence of aversive freezing behavior. Therefore, our data suggest that although Cdk5 activity seems to be important for the initial encoding of the contextual fear memory trace18, it subsequently impairs contextual fear extinction. Notably, similar observations have been made for cAMP signaling. To this end, cAMP is required for the acquisition of fear memory38, but mice that overexpress the type I adenylyl cyclase Adcy1 in the forebrain and that have elevated hippocampal cAMP levels show slower extinction of fear39.
Besides the hippocampus, the prefrontal cortex and the amygdala have been implicated with the extinction of cued and contextual fear4,40–42. The possible role of Cdk5 activity in those brain areas during extinction is a question that remains unanswered.
To elucidate the molecular mechanism by which Cdk5 activity regulates extinction, we took advantage of the employed experimental extinction procedure. As such, after a single fear-conditioning training mice showed freezing behavior on E1–E3. When retested on E4, however, freezing was usually substantially decreased. We therefore speculated that the key molecular processes triggered after E1 (mice showed freezing on subsequent E2) and E3 (mice showed reduced freezing on subsequent E4) are different.
Notably, we observed a redistribution of membrane-associated hippocampal p35 levels to the cytosol after E3. Depletion of p35 from the membrane correlated with reduced phosphorylation of the Cdk5 substrate PAK-1, indicating that extinction leads to reduced membrane-associated Cdk5 activity. Our data suggests that membrane depletion of p35 is a mechanism for locally reducing Cdk5 activity, and thereby allowing extinction to occur.
Previous reports showed that the small GTPase Rac-1 binds p35, thereby affecting the subcellular distribution of Cdk5 activity15,36. Moreover, Rac-1 activity has been implicated in synaptic plasticity27. Therefore, we investigated the role of Rac-1 during Cdk5-dependent extinction. Notably, we found that extinction led to a reduction of membrane-associated GTP-bound Rac-1 after E3 without affecting the total amount of Rac-1 protein, suggesting a reduction in Rac-1 activity. The fact that reductions of membrane-associated Rac-1 and Cdk5 activity after E3 occurred simultaneously indicates that Rac-1 and Cdk5 might be part of the same signaling pathway during extinction. In support of this notion, inhibition of Rac-1 activity reduced the levels of membrane-associated p35 protein, thereby recapitulating the cellular changes observed after E3 when freezing behavior declined during physiological extinction. Notably, when Rac-1 activity was inhibited during extinction, we observed a redistribution of both p35 and Cdk5 from the membrane to the cytosol, whereas only p35 was affected during physiological extinction. As our data indicate that Rac-1 activity regulates extinction via Cdk5/p35 kinase, it is plausible that pharmacological inhibition of Rac-1 causes more marked changes on the molecular level than the alterations that occur during extinction under physiological conditions. Taken together, our data suggest that Rac-1 and Cdk5/p35 kinase are part of a distinctive signaling pathway that prevents extinction under physiological conditions.
Cdk5 directly phosphorylates PAK-1 on threonine 212 and inhibits PAK1 activity. Our observations that PAK-1T212 levels decreased during extinction suggest that PAK-1 activity might be upregulated. Consistent with this assumption, we found that phosphorylation of membrane-associated PAK-1 at the non-Cdk5 site threonine 423 was increased after E3. The autophosphorylation of threonine 423 of PAK-1 liberates the Pak-1 inhibitory domain and is a marker for active PAK-1 (refs. 28,37). Cytosolic PAK-1T423 levels decreased after E1, but increased back to baseline after E3. These findings suggest that the reduction of aversive freezing behavior correlates with increased PAK-1 activity.
Notably, we found that inhibition of Rac-1 and Cdk5 activity during extinction reduced PAK-1T212 levels, providing direct evidence that Rac-1 and Cdk5 regulate PAK-1 through threonine 212 phosphorylation during extinction. Also, inhibition of Cdk5 led to increased PAK-1T423 levels. These data suggest that Cdk5 activity regulates PAK-1 during extinction.
Inhibition of Rac-1 and Cdk5 also induced a redistribution of PAK-1 from the membrane to the cytosol. This is consistent with previous findings demonstrating that Cdk5 activity affects the subcellular localization of PAK-1 in neuronal growth cones36. However, despite increased cytosolic p35 and Cdk5 levels, there was no corresponding increase in the phosphorylation of PAK-1 on threonine 212 in the cytosol. It was actually reduced. We speculate that this could be due to the fact that p35 is sequestered from PAK-1 on redistribution to the cytosol, because the p35–PAK-1 interaction, independent of phosphorylation, has been implicated with inhibition PAK-1 activity36. Indeed, we found that less p35 co-immunoprecipitated with PAK-1 in hippocampal lysates prepared from mice injected with the Rac-1 inhibitor when compared with the vehicle group.
In summary, our data indicate that during extinction membrane depletion of Cdk5 activity and dissociation of p35 from PAK-1 in the cytosol removes the inhibitory tone on PAK-1 activity. This eventually results in an increase of mainly cytosolic PAK-1 activity. It is likely that PAK-1 activity during extinction is regulated by additional mechanisms, such as protein phosphatases. In fact, protein phosphatase 1 has been implicated in extinction processes43. We provide direct evidence that PAK-1 activity is required for extinction, as inhibition of PAK-1 by viral-mediated hippocampal expression of a dominant-negative PAK-1 mutant substantially impaired extinction. As such, the effect of PAK-1 inhibition on extinction is opposite to that of Rac-1 and Cdk5 inhibition. This finding supports the idea that RAC-1, Cdk5/p35 kinase and PAK-1 are counteracting components of a hippocampal signaling pathway that regulates extinction of contextual fear. When taken together, our data delineate a molecular pathway whereby extinction requires the downregulation of Rac-1 and Cdk5, and the upregulation of PAK-1 activity. Notably, Rac-1, Cdk5/p35 kinase and PAK-1 have all been implicated in the regulation of the actin cytoskeleton15.
Remodeling of synaptic circuits via actin dynamics is an important molecular mechanism during the acquisition and extinction of fear. For example, actin dynamics regulate spine morphology, synaptogenesis12,44 and hippocampal long-term potentiation11. Moreover, a dynamic actin cytoskeleton is required for extinction6. In fact, recent data suggest that remodeling of synaptic circuits via actin dynamics is an important molecular mechanism during extinction3,6. PAK-1 phosphorylates LIM-kinase45, which in turn regulates the actin-binding protein cofilin46. This suggests the intriguing possibility that a Rac-1–Cdk5/p35–PAK-1 pathway affects the dynamics of the actin cytoskeleton, thereby affecting neuronal morphology. Consistent with this assumption, it should be noted that increased Cdk5 activity has been implicated with the formation and strengthening of new synapses19, whereas decreased Cdk5 activity is associated with a reduced number of dendritic spines19,47. The possibility arises that the observed redistribution of Cdk5 activity during extinction allows for synaptic remodeling via actin dynamics. Cdk5 activity likely regulates extinction via multiple mechanisms. For example, deletion of Cdk5 in the hippocampus was recently shown to facilitate the extinction of contextual fear via NMDA receptor–mediated synaptic transmission26.
Our findings provide evidence that, in addition to mechanisms that promote extinction, strategies that impair extinction have evolved as molecular counter players. In fact, the Rac-1–Cdk5–PAK-1 signaling pathway described here may provide an example whereby pathways that impair and promote extinction are mechanistically linked. From an evolutionary point of view, mechanisms that impair extinction seem reasonable, as rapid extinction of fear after exposure to threatening situations, such as places where a predator attacked, could be disadvantageous for survival. In fact, in contrast to other experimental procedures, where extinction sometimes occurs in a single trial48, in the fear-conditioning procedure employed in this study, extinction only occurs after 4–6 extinction trials. This is consistent with the fact that patients suffering from emotional disorders such as phobia usually require multiple exposure sessions before the aversively motivated behavior is extinguished4. As such, molecular mechanisms that impair extinction, such as Rac-1 and Cdk5 activity, could be promising drug targets for emotional disorders.
METHODS
Animals
Mice (Balb/c, 12 weeks old) were housed under standard conditions with access to food and water ad libitum. All mice were purchased from Taconic. As this is a collaborative study, we should mention that behavior testing was carried out in such a way that the mice were either housed in a separated compartment in the testing room, or different rooms were used for housing and testing. In the latter case, the mice were brought to the testing room before the procedure. Although it appears that the dynamics of extinction behavior are somewhat variable depending on the employed behavior facility, the trends remained the same in different experiments. All experiments were approved by the Institutional Animal Care and Use Committees of Harvard University, Massachusetts Institute of Technology and Northwestern University.
Extinction of contextual freezing behavior
Extinction and reminder shock procedures were carried out as described previously6. In brief, mice were exposed to contextual fear conditioning (3 min context followed by at mild electric foot shock, 2 s, 0.7 mA). In some experiments, a tone (30 s, 10 kHz, 75 dB sound pressure level) was presented for 30 s before the shock. Extinction was carried out on consecutive days. An extinction trial consisted of a 3 min re-exposure to the conditioning context. In our procedure, substantial reduction of freezing was observed during E4. However, we found that this time course strongly depended on the experimental setting.
Surgery and injections
Custom-made microcannula were purchased from PlasticOne. The gauges of the guide and injection cannula were 26 and 28, respectively. To insert cannula into the dorsal hippocampus, we used the coordinates anterior-posterior −1.5 mm, lateral 1 mm, depth 2 mm as described previously18. To insert cannula intracerebroventricular (ICV), we used the coordinates AP +0.5 mm, lateral 1 mm, depth 2 mm. Butyrolactone I and roscovitine were dissolved and injected as described previously18. Rac-1 inhibitor was dissolved in artificial cerebrospinal fluid and delivered bilaterally (0.5 μl per side) over a 1-min period. The correct localization of the cannula was verified at the end of an experiment by methylene blue injection. The cistronic herpes virus expressing GFP and dnPAK-1 or GFP alone was injected into the hippocampus bilaterally (0.5 μl per side) over a 2-min period. dnPAK-1 was a gift from S. Tonegawa (Massachusetts Institute of Technology), and consisted of the DNA encoding amino acid 83–149 of PAK-1. It has been previously demonstrated that this fragment acts as a dominant-negative protein toward Pak-1 and impairs PAK-1 activity in vitro and in vivo28.
Immunoblot analysis
Immunoblot analysis was performed as described previously 19. The antibody detecting pPAK-1Thr212 was a gift from M. Nikolic (Imperial College London) and used in a 1:250 dilution. Antibody to PAK-1 was from Santa Cruz. All other antibodies were used in a 1:1,000 dilution: pPAK-1Thr423 (Cell Signaling), synaptophysin (Sigma), GAPDH and Rac-1 (Upstate), Cdk5 (J3, Santa Cruz) and p35 (C-19, Santa Cruz).
Immunohistochemistry
Immunohistochemical analyses were carried out as described previously19. Antibodies were used in a 1:1,000 concentration: GFP (Molecular Probes) and NeuN (Chemicon).
Statistical analysis
The data were analyzed by unpaired Student’s t-test. We employed one-way ANOVAs, followed by post hoc Scheffe’s test to compare means from several groups at the same time. Data are presented as s.e.m. A detailed description of the statistics employed in this study can be found in ref. 49.
Supplementary Material
Note: Supplementary information is available on the Nature Neuroscience website.
Acknowledgments
We thank B. Samuels for reading the manuscript and for critical discussion, all members of the Tsai lab for helpful advice, M. Nikolic for the pPAK-1T212 antibody and S. Tonegawa for the dominant-negative PAK-1 construct. L.-H.T. is an investigator of the Howard Hughes Medical Institute. This work was partially supported by a US National Institutes of Health grant (NS051874) to L.-H.T. This work was also partially supported by a US National Institute of Mental Health grant MH073669 to J.R., and a Humboldt/Deutsche Forschungsgemeinschaft fellowship to F.S. and A.F., respectively, and by funds from the European Neuroscience Institute Goettingen to A.F. The European Neuroscience Institute is jointly funded by the Medical School University Goettingen and the Max Planck Society.
Footnotes
AUTHOR CONTRIBUTIONS
The studies were conceived and designed by F.S., A.F., J.R. and L.-H.T. F.S., A.F., X.W., C.S., R.N. and J.R. contributed to the experiments in this work. The paper was written by A.F. and L.-H.T.
COMPETING INTERESTS STATEMENT
The authors declare no competing financial interests.
References
- 1.Myers KM, Davis M. Behavioral and neural analysis of extinction. Neuron. 2002;36:567–584. doi: 10.1016/s0896-6273(02)01064-4. [DOI] [PubMed] [Google Scholar]
- 2.Davis M, Ressler K, Rothbaum BO, Richardson R. Effects of D-cycloserine on extinction: translation from preclinical to clinical work. Biol Psychiatry. 2006;60:369–375. doi: 10.1016/j.biopsych.2006.03.084. [DOI] [PubMed] [Google Scholar]
- 3.Lattal KM, Radulovic J, Lukowiak K. Extinction: does it or doesn’t it? The requirement of altered gene activity and new protein synthesis. Biol Psychiatry. 2006;60:344–351. doi: 10.1016/j.biopsych.2006.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sotres-Bayon F, Cain CK, LeDoux JE. Brain mechanisms of fear extinction: historical perspectives on the contribution of prefrontal cortex. Biol Psychiatry. 2006;60:329–336. doi: 10.1016/j.biopsych.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 5.Kim JJ, Fanselow MS. Modality-specific retrograde amnesia of fear. Science. 1992;256:675–677. doi: 10.1126/science.1585183. [DOI] [PubMed] [Google Scholar]
- 6.Fischer A, Sananbenesi F, Schrick C, Spiess J, Radulovic J. Distinct roles of hippocampal de novo protein synthesis and actin rearrangement in extinction of contextual fear. J Neurosci. 2004;24:1962–1966. doi: 10.1523/JNEUROSCI.5112-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Falls WA, Miserendino MJ, Davis M. Extinction of fear-potentiated startle: blockade by infusion of an NMDA antagonist into the amygdala. J Neurosci. 1992;12:854–863. doi: 10.1523/JNEUROSCI.12-03-00854.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chhatwal JP, Stanek-Rattiner L, Davis M, Ressler KJ. Amygdala BDNF signaling is required for consolidation but not encoding of extinction. Nat Neurosci. 2006;9:870–872. doi: 10.1038/nn1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Szapiro G, Vianna MR, McGaugh JL, Medina JHII. The role of NMDA glutamate receptors, PKA, MAPK and CAMKII in the hippocampus in extinction of conditioned fear. Hippocampus. 2003;13:53–58. doi: 10.1002/hipo.10043. [DOI] [PubMed] [Google Scholar]
- 10.Marsicano G, et al. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- 11.Fukazawa Y, et al. Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron. 2003;38:447–460. doi: 10.1016/s0896-6273(03)00206-x. [DOI] [PubMed] [Google Scholar]
- 12.Matus A. Actin-based plasticity in dendritic spines. Science. 2000;290:754–758. doi: 10.1126/science.290.5492.754. [DOI] [PubMed] [Google Scholar]
- 13.Fischer A, Sananbenesi F, Spiess J, Radulovic J. Cdk5 in the adult non-demented brain. Curr Drug Targets CNS Neurol Disord. 2003;2:375–381. doi: 10.2174/1568007033482706. [DOI] [PubMed] [Google Scholar]
- 14.Dhavan R, Tsai LH. A decade of CDK5. Nat Rev Mol Cell Biol. 2001;2:749–759. doi: 10.1038/35096019. [DOI] [PubMed] [Google Scholar]
- 15.Nikolic M. The role of Rho GTPases and associated kinases in regulating neurite outgrowth. Int J Biochem Cell Biol. 2002;34:731–745. doi: 10.1016/s1357-2725(01)00167-4. [DOI] [PubMed] [Google Scholar]
- 16.Cheung ZH, Fu AK, Ip NY. Synaptic roles of Cdk5: implications in higher cognitive functions and neurodegenerative diseases. Neuron. 2006;50:13–18. doi: 10.1016/j.neuron.2006.02.024. [DOI] [PubMed] [Google Scholar]
- 17.Ohshima T, et al. Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Natl Acad Sci USA. 1996;93:11173–11178. doi: 10.1073/pnas.93.20.11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fischer A, Sananbenesi F, Schrick C, Spiess J, Radulovic J. Cyclin-dependent kinase 5 is required for associative learning. J Neurosci. 2002;22:3700–3707. doi: 10.1523/JNEUROSCI.22-09-03700.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fischer A, Sananbenesi F, Pang PT, Lu B, Tsai LH. Opposing roles of transient and prolonged expression of p25 in synaptic plasticity and hippocampus-dependent memory. Neuron. 2005;48:825–838. doi: 10.1016/j.neuron.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 20.Angelo M, Plattner F, Irvine EE, Giese KP. Improved reversal learning and altered fear conditioning in transgenic mice with regionally restricted p25 expression. Eur J Neurosci. 2003;18:423–431. doi: 10.1046/j.1460-9568.2003.02746.x. [DOI] [PubMed] [Google Scholar]
- 21.Li BS, et al. Regulation of NMDA receptors by cyclin-dependent kinase-5. Proc Natl Acad Sci USA. 2001;98:12742–12747. doi: 10.1073/pnas.211428098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tan TC, et al. Cdk5 is essential for synaptic vesicle endocytosis. Nat Cell Biol. 2003;5:701–710. doi: 10.1038/ncb1020. [DOI] [PubMed] [Google Scholar]
- 23.Tomizawa K, et al. Cophosphorylation of amphiphysin I and dynamin I by Cdk5 regulates clathrin-mediated endocytosis of synaptic vesicles. J Cell Biol. 2003;163:813–824. doi: 10.1083/jcb.200308110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim Y, et al. Phosphorylation of WAVE1 regulates actin polymerization and dendritic spine morphology. Nature. 2006;442:814–817. doi: 10.1038/nature04976. [DOI] [PubMed] [Google Scholar]
- 25.Lee SY, Wenk MR, Kim Y, Nairn AC, De Camilli P. Regulation of synpatojanin 1 by cyclin-dependent kinase 5 at synapses. Proc Natl Acad Sci USA. 2004;101:546–551. doi: 10.1073/pnas.0307813100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hawasli AH, et al. Cyclin-dependent kinase 5 governs learning and synaptic plasticity via control of NMDAR degradation. Nat Neurosci. doi: 10.1038/nn1914. advance online publication 27 May 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakayama AY, Harms MB, Luo L. Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J Neurosci. 2000;20:5329–5338. doi: 10.1523/JNEUROSCI.20-14-05329.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hayashi ML, et al. Altered cortical synaptic morphology and impaired memory consolidation in forebrain- specific dominant-negative PAK transgenic mice. Neuron. 2004;42:773–787. doi: 10.1016/j.neuron.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 29.Delamater AR. Experimental extinction in Pavlovian conditioning: behavioural and neuroscience perspectives. Q J Exp Psychol B. 2004;57:97–132. doi: 10.1080/02724990344000097. [DOI] [PubMed] [Google Scholar]
- 30.Myers KM, Davis M. Mechanisms of fear extinction. Mol Psychiatry. 2007;12:120–150. doi: 10.1038/sj.mp.4001939. [DOI] [PubMed] [Google Scholar]
- 31.Kitagawa M, et al. Butyrolactone I, a selective inhibitor of cdk2 and cdc2 kinase. Oncogene. 1993;8:2425–2432. [PubMed] [Google Scholar]
- 32.Fischer A, et al. Hippocampal Mek/Erk signaling mediates extinction of contextual freezing behavior. Neurobiol Learn Mem. 2007;87:149–158. doi: 10.1016/j.nlm.2006.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nikolic M, Chou MM, Lu W, Mayer BJ, Tsai LH. The p35/Cdk5 kinase is a neuron-specific Rac effector that inhibits Pak1 activity. Nature. 1998;395:194–198. doi: 10.1038/26034. [DOI] [PubMed] [Google Scholar]
- 34.Zhang QG, et al. Akt inhibits MLK3/JNK3 signaling by inactivating Rac1: a protective mechanism against ischemic brain injury. J Neurochem. 2006;98:1886–1898. doi: 10.1111/j.1471-4159.2006.04020.x. [DOI] [PubMed] [Google Scholar]
- 35.Desire L, et al. RAC1 inhibition targets amyloid precursor protein processing by gamma-secretase and decreases Abeta production in vitro and in vivo. J Biol Chem. 2005;280:37516–37525. doi: 10.1074/jbc.M507913200. [DOI] [PubMed] [Google Scholar]
- 36.Rashid T, Banerjee M, Nikolic M. Phosphorylation of Pak1 by the p35/Cdk5 kinase affects neuronal morphology. J Biol Chem. 2001;276:49043–49052. doi: 10.1074/jbc.M105599200. [DOI] [PubMed] [Google Scholar]
- 37.Lei M, et al. Structure of PAK1 in an autoinhibited conformation reveals a multistage activation switch. Cell. 2000;102:387–397. doi: 10.1016/s0092-8674(00)00043-x. [DOI] [PubMed] [Google Scholar]
- 38.Wong ST, et al. Calcium-stimulated adenylyl cyclase activity is critical for hippocampus-dependent long-term memory and late phase LTP. Neuron. 1999;23:787–798. doi: 10.1016/s0896-6273(01)80036-2. [DOI] [PubMed] [Google Scholar]
- 39.Wang H, Ferguson GD, Pineda VV, Cundiff PE, Storm DR. Overexpression of type-1 adenylyl cyclase in mouse forebrain enhances recognition memory and LTP. Nat Neurosci. 2004;7:635–642. doi: 10.1038/nn1248. [DOI] [PubMed] [Google Scholar]
- 40.Burgos-Robles A, Vidal-Gonzalez I, Santini E, Quirk GJ. Consolidation of fear extinction requires NMDA receptor–dependent bursting in the ventromedial prefrontal cortex. Neuron. 2007;53:871–880. doi: 10.1016/j.neuron.2007.02.021. [DOI] [PubMed] [Google Scholar]
- 41.Vianna MR, Coitinho AS, Izquierdo I. Role of the hippocampus and amygdala in the extinction of fear-motivated learning. Curr Neurovasc Res. 2004;1:55–60. doi: 10.2174/1567202043480170. [DOI] [PubMed] [Google Scholar]
- 42.Berlau DJ, McGaugh JL. Enhancement of extinction memory consolidation: the role of the noradrenergic and GABAergic systems within the basolateral amygdala. Neurobiol Learn Mem. 2006;86:123–132. doi: 10.1016/j.nlm.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 43.Lin CH, et al. Identification of calcineurin as a key signal in the extinction of fear memory. J Neurosci. 2003;23:1574–1579. doi: 10.1523/JNEUROSCI.23-05-01574.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bonhoeffer T, Yuste R. Spine motility. Phenomenology, mechanisms and function. Neuron. 2002;35:1019–1027. doi: 10.1016/s0896-6273(02)00906-6. [DOI] [PubMed] [Google Scholar]
- 45.Edwards DC, Sanders LC, Bokoch GM, Gill GN. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat Cell Biol. 1999;1:253–259. doi: 10.1038/12963. [DOI] [PubMed] [Google Scholar]
- 46.Arber S, et al. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 1998;393:805–809. doi: 10.1038/31729. [DOI] [PubMed] [Google Scholar]
- 47.Fu WY, et al. Cdk5 regulates EphA4-mediated dendritic spine retraction through an ephexin1-dependent mechanism. Nat Neurosci. 2007;10:67–76. doi: 10.1038/nn1811. [DOI] [PubMed] [Google Scholar]
- 48.Berman DE, Dudai Y. Memory extinction, learning anew and learning the new: dissociations in the molecular machinery of learning in cortex. Science. 2001;291:2417–2419. doi: 10.1126/science.1058165. [DOI] [PubMed] [Google Scholar]
- 49.Howell DC. In: Statistical Methods for Psychology. Crokett C, editor. Duxbury Thompson Learning; Duxbury: 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Note: Supplementary information is available on the Nature Neuroscience website.