


Abstract
The tumor suppressor p53 is a sequence-specific transcription factor, which regulates the expression of target genes involved in different stress responses. To understand p53's essential transcriptional functions, unbiased analysis of its DNA-binding repertoire is pivotal. In a genome-wide tiling ChIP-on-chip approach, we have identified and characterized 1546 binding sites of p53 upon Actinomycin D treatment. Among those binding sites were known as well as novel p53 target sites, which included regulatory regions of potentially novel transcripts. Using this collection of genome-wide binding sites, a new high-confidence algorithm was developed, p53scan, to identify the p53 consensus-binding motif. Strikingly, this motif was present in the majority of all bound sequences with 83% of all binding sites containing the motif. In the surrounding sequences of the binding sites, several motifs for potential regulatory cobinders were identified. Finally, we show that the majority of the genome-wide p53 target sites can also be bound by overexpressed p63 and p73 in vivo, suggesting that they can possibly play an important role at p53 binding sites. This emphasizes the possible interplay of p53 and its family members in the context of target gene binding. Our study greatly expands the known, experimentally validated p53 binding site repertoire and serves as a valuable knowledgebase for future research.
INTRODUCTION
The tumor suppressor gene p53 is the most frequently mutated gene in human cancers (1). It can be activated by a large number of stress signals. The p53 protein is able to function as a sequence-specific transcription factor (2) and it regulates the expression of target genes involved in growth arrest, apoptosis, DNA repair, senescence, differentiation and other responses (3). Substantial evidence indicates that the transcriptional functions of p53 are necessary for p53-mediated tumor suppression (4), although it has also been reported that p53 can induce apoptosis without a functional transactivation domain (5).
The tumor suppressor p53 in a sequence-dependent manner to a so-called p53 consensus-binding motif. This motif is found in many identified binding sites of, mostly upregulated, p53 target genes and consists of two copies of the palindromic consensus half-site RRRCWWGYYY separated by a spacer of 0–13 bp, in which R =purine, W = A or T and Y = pyrimidine (6). The p53 binding ability and its transcriptional activity might be influenced by the sequence of the two half-sites as well as their mutual orientation (7,8). Up to now it was thought that this p53 response element is mostly found within a few thousand base pairs of the transcriptional start site (TSS) (4). In addition, binding sites which differ from the classical p53 binding motif have been reported (9,10). It has been suggested that the deviations from the consensus sequence hint at the possibility that DNA topology also determines p53 binding (11) and that even the DNA structure might totally replace the consensus sequence (12). However, these findings are largely based on single target genes; a genome-wide analysis of binding sequences for common motifs will be very informative.
The transcriptional activity of p53 can be regulated by posttranslational modifications (13,14) as well as transcriptional cofactors and p53-binding proteins (4,15). p300 acts as an p53-dependent coactivator for p53 target genes by acetylating p53 (16,17) and it binds to various corecruited factors that enhance the p53 response (18). Two of those have recently been identified, JMY and Strap, and both factors are required for p53 activity (19,20). Recent studies have also provided evidence that the selection of p53 target genes can be modulated by p53 interacting proteins. The ASPP (apoptosis-stimulating protein of p53) proteins have been shown to interact with p53 and to specifically modulate p53-induced apoptosis but not cell cycle arrest (21). Interestingly, the hCAS (human cellular apoptosis susceptibility) protein has been reported to be part of a distinct macromolecular complex of p53 at specific subsets of p53 target genes, e.g. Pig3 and p53AIP1 and its knockdown attenuates the p53-dependent apoptosis (22). Other transcription factors that might be involved in the target gene selection of p53 are the Brn3 family of POU domain transcription factors (23), the YB1 protein (24), NF-κB and IKKα (25,26) as well as the hematopoietic zinc-finger cofactor (HZF) protein (27). In addition to transcriptional factors that influence the p53-target gene binding, there is also evidence that the p53 family members p63 and p73 can contribute to the p53-recruitment at specific target genes. Both p63 and p73 were reported to be required for the p53 binding to the p53 response elements of the target genes Perp, Bax and Noxa, but not to those of p21 or Mdm2 (28). A ‘priming model’ was suggested, in which p63 and p73 can bind to a specific chromatin-embedded response element not accessible for p53, and subsequently modify the context of the response element in such a way that it becomes available for p53 binding (29). So far, a systematic analysis of the capability of p63 and p73 to play a role in vivo at p53-target sites has not been performed.
Many p53 target genes are currently known, e.g. identified with microarray expression profiling (30,31), and at the moment it is intensively studied how p53 determines which target genes to activate or repress in a certain stress response (4,32). In addition to the experimentally identified p53 target genes, there are also computationally predicted binding sites (33,34). These predictions do not necessarily reflect the actual target sites bound in vivo by p53. For the selection of functional binding sites, the involvement of other cellular factors, chromatin accessibility, DNA sequences surrounding the potential binding site and DNA topology have to be taken into consideration, in addition to the consensus-binding sequence itself. These factors can not yet be accurately modeled. It is estimated that there are between 300 and 3000 binding sites for p53 in the human genome, based on studies from Hoh et al. (34), ChIP-on-chip (chromatin immunoprecipitated DNA hybridized on DNA arrays) data extrapolated from chromosome 21–22 (35) and ChIP-on-chip data derived from ENCODE regions (36). Since there are only about 180 experimentally confirmed target genes (30), and 542 high-probability binding sites (37), it is expected that there are still many unidentified binding sites and target genes, notwithstanding several studies that have reported binding sites for p53 (35–42). Furthermore, it remains to be seen how a comprehensive set of p53-DNA binding sites in vivo can be used to give more insight into the different transcriptional functions of p53.
Here, we report a genome-wide ChIP-on-chip study of p53 employing high-resolution tiling arrays with an average probe spacing of 100 bp. We have identified 1546 high-confidence sites and performed extensive analysis of the in vivo binding sites with respect to their sequence as well as surroundings and nearby genes. We report the development of a new publicly available algorithm, p53scan, to identify the p53 consensus binding motif with high specificity. The motif is present in 83% of all the p53-bound sequences and in almost all highly enriched binding sites. Potential novel functions of p53 derived from the global binding sites were investigated and validated. To obtain a more complete picture of the in vivo bound target genes of p53, we have also performed ChIP-on-chip analyses with two of its family members, p63 and p73. We show that a large fraction of these newly identified binding sites for p53 could also be bound by p63 and p73 in vivo.
MATERIALS AND METHODS
Cell culture and drug treatment
The human osteosarcoma cell lines U2OS expressing endogenous wild-type p53, and Saos-2, which are p53 null (43), were maintained in Dulbecco's modified Eagle medium supplemented with 10% fetal calf serum at 37°C. The Tet-on inducible expression system (BD Biosciences, Breda, The Netherlands) was used in Saos-2 cells to generate cell lines that conditionally express FLp53, TAp63α or TAp73α. cDNA of the gene of interest was cloned into the pTRE vector and cotransfected with the pZoneXN, which has a puromycin selection marker, into Saos-2 cells. Transfections were performed by the calcium phosphate precipitation method. Stable clones were selected with 1μg/ml puromycin (Sigma, Zwijndrecht, The Netherlands). To induce the expression of FLp53, TAp63α or TAp73α, 2 μg/ml doxycyclin (Sigma), a Tetracyclin homologue, was added to the medium. The inducible Saos-2 cell lines were first induced with doxycyclin for 24 h and then treated with 5 nM Actinomycin D (Sigma) for another 24 h. The U2OS cells were treated with 5 nM Actinomycin D for 24 h.
Cell cycle analysis
Cells were induced and treated as described above. The cells were fixed with 96% ethanol and stained with propidium iodide (Sigma). DNA content was analyzed by flow cytometry (Becton Dickinson FACScan) and analyzed using CellQuest Pro software.
Immunoblotting
To assess protein levels, proteins from whole-cell extracts were harvested, lyzed and separated by SDS–PAGE and analyzed by western blotting with α-p53 (DO1, BD PharMingen, Breda, The Netherlands).
Transactivation assays
The selected binding sites were amplified from genomic U2OS DNA and cloned behind the luciferase gene into the pGL3-promoter vector (Invitrogen, Breda, The Netherlands), which contains a luciferase reporter gene behind a SV40 promoter. U2OS cells were transiently transfected with pGL3 constructs and a pRL-TK reporter (Promega, Leiden, The Netherlands), constitutively expressing Renilla as a normalization control, by calcium phosphate transfection. Cells were lyzed and luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega).
RNA isolation and RT–PCR
Total RNA was extracted using the RNeasy Mini kit according to protocol (Qiagen, Venlo, The Netherlands). For cDNA synthesis, reverse transcription was performed with 1 μg of the total RNA, oligodT anchor primers, dNTPS, DTT, buffer and Superscript Retrotranscriptase (Invitrogen). cDNA was analyzed by qPCR using a MyIQ machine (Biorad, Veenendaal, The Netherlands). Primers used for real-time PCR are available upon request.
ChIP and ChIP-on-chip
Chromatin immunoprecipitation (ChIP) was essentially performed as described by Denissov et al. (44). The cells were sonicated using a Bioruptor sonicator (Diagenode, Liege, Belgium) for 15 min at high power, 30 s ON, 30 s OFF. Antibody incubation with chromatin from U2OS cells treated with Actinomycin D was performed overnight at 4°C with 2 μg of DO1 antibody (BD PharMingen). For ChIP experiments in Saos-2 inducible cell lines DO1 (BD PharMingen), 4A4 (Abcam, Cambridge, UK) and BL906 (Abcam) were used for p53, p63 and p73, respectively. Real-time PCR was performed using the SYBR Green mix (Biorad) with the MyIQ machine (Biorad). Primers used for real-time PCR are available upon request. To produce more material for a ChIP-on-chip, the total DNA and ChIP DNA needed to be amplified. For genome-wide hybridization, the material was amplified using LM-PCR amplification (45). The T7-based amplification procedure (46) was used for the hybridizations on the dedicated arrays. The total DNA and ChIP DNA were hybridized to whole genome tiling arrays (HG17Tiling Set) or custom designed microarrays manufactured by NimbleGen Systems, Inc., Madison, WI, USA. Raw data for all microarray hybridizations are available at ArrayExpress (http://www.ebi.ac.uk/arrayexpress) under accession E-TABM-442.
Custom microarray design
Peak detection (see below) was performed on the genome-wide dataset and all probes within the positive regions recognized by the peak detection procedure, extended equally up- and down-stream to a total of 2 kb, were spotted on a custom design array (NimbleGen Systems), hereafter referred to as dedicated design. All the probes from a continuous region of chromosome 21 (from 28 692 406 to 41 270 931) on the hg17 array were included in the dedicated design to provide a baseline for normalization purposes. This region is hereafter referred to as tilepath.
Data normalization
The probe sequences from both the whole genome and the dedicated design were compared to the human genomic sequence with BLAT (47). Probes with 10 or more matches were discarded for use in the subsequent analysis. The raw probe ratios were normalized within arrays using Tukey's biweight. For all hybridizations performed on the dedicated array, the ratios were normalized against the tilepath region.
Peak detection
The microarray data were analyzed using three different peak detection programs to identify putative targets with a high degree of confidence. Default parameters were used except where noted. The proprietary program provided by NimbleGen was run with a 1% false positive rate. For Tilemap (48), hybridization length was set to 50 and maximal gap size to 100. All probes with posterior probability of at least 0.9 were defined as peaks. For Mpeak (49), the maximum gap was set to 300, and a minimum log2 ChIP/Total ratio of at least 2.5 SD was used as a threshold. All positive regions or peaks <1 kb in length were extended equally up- and down-stream to cover 1 kb. Per biological replicate all regions determined to be positive by one of the programs were combined. Finally, a peak was defined as a binding target if positive regions shared any overlap in each biological replicate.
Mapping binding sites to genes
Target locations were mapped to NCBI 36 coordinates using the Batch Coordinate Conversion (liftOver) utility provided by the UCSC Genome Bioinformatics group. Gene locations of all genes were downloaded from Ensembl [release 43, February 2007 (50)]. To map a target to a gene, the distance from the middle of the target to the Ensembl gene start was used.
Annotation of genes
For annotation of genes, only target genes with a binding site within 5 kb of the gene were used. Overrepresented Gene Ontology (GO) (51) categories within annotation of the target genes were determined with Ontologizer using the parent–child method, which takes into account the parent–child relationships of the GO hierarchy (52). The P-values were adjusted using Westfall–Young single-step multiple testing correction and a corrected P-value threshold of 0.05 was used as a cut-off for reporting significant matches. Genes were annotated with KEGG pathways (53) using Fatigo+ (54). Overrepresented pathways were determined according to the hypergeometric distribution with a P-value threshold of 0.05.
The p53scan algorithm and motif analysis
Sequences of equal length were selected for all targets by determining the probe with the highest mean ratio value within each peak and selecting a 500 bp region centered on this probe. All probes within regions on the slide of at least 10 consecutive probes, or 1 kb, with a maximum mean log2 ChIP/Total ratio of 0.5 were selected as background sequences. These sequences were divided in 500 bp regions to create sequences of the same length as the target sequences.
To determine the optimal positional weight matrix (PWM) for p53scan, the de novo motif discovery program MDmodule (55) was applied to half of the 500 bp target sequences, 773 in total. Sequences were ordered based on the ChIP/Total ratio of the highest probe in the peak. MDmodule was run with a width of 20 and the number of top sequences to look for motifs was set to 200. Default parameters were used for all other options. The MDmodule output was subsequently converted to a PWM.
In p53scan, the score of a subsequence × of length L is calculated as follows:
where fi,b is the fraction of each nucleotide at position i, g is 0.25 and z is 0.01.
To incorporate a variable spacer length, the two half-sites are scanned separately and the scores for each half-site are combined. Cutoffs for spacer lengths greater than 0 were determined by scanning 10 times as many random nonbound sequences and choosing the threshold that result in no hits. This enables p53scan to take high-scoring motifs with spacer lengths other than 0 into account without drastically changing the false positive rate (FPR).
To test the performance of the algorithm and to compare it to other available algorithms, the 773 sequences not used for training were compared to a background set of five times as many random nonbound sequences. Three different metrics were used for comparison:
The area under the receiver operator curve (ROC AUC), which reflects the balance between the false positive rate and the true positive rate.
The mean normalized conditional probability (MNCP) as described by Clarke and Granek (56).
The maximum F-measure or weighted harmonic mean of precision and recall.
 |
The process of randomly selecting training, test and background sequences and subsequent performance comparison was repeated in 10 independent runs. In all cases the performance was comparable. The best scoring PWM was kept and implemented. All 1546 targets were compared to five times as many randomly selected noncoding sequences of equal length to produce Figure 4B. Subsequently, p53scan was used to analyze the complete set of target sequences with a score cutoff of 4.393 for spacer length 0 resulting in the highest F-measure, corresponding to an estimated FPR of ∼7%.
Figure 4.

Development of p53scan, a p53-motif finding algorithm. (A) p53 motif identified de novo with MD-module, visualized using WebLogo. (B) ROC curve comparing p53scan, p53MH and Match. The FPR used to characterize the p53-binding sites (∼7%) is marked. (C) Distribution of spacer length in the p53 consensus sites based on the p53scan algorithm.
For de novo motif prediction of possible cofactor motifs, the motif discovery program MDmodule was applied to the 500 bp target sequences. The same procedure as described for the p53scan PWM was followed. The number of top sequences to look for motifs was set to 100, all widths from 6 to 16 were considered, and the number of motifs to report was set to 10. To calculate the significance of the discovered motifs, the number of sequences with at least one motif instance with 0.8 × maximum possible score was determined in both the sample and the background sequences using TAMO (Tools for Analysis of Motifs) (57). For each motif a P-value was calculated using the hypergeometric distribution. The corresponding significance value was calculated as Significance 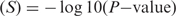 .
.
As selection criteria, we used a significance cut-off of 1.3 corresponding to a P-value of 0.05 with a minimum of occurrence of at least 10 times. All significantly enriched motifs were clustered using a k-medoids clustering algorithm as described in (58). Clustered and aligned motifs were averaged to produce consensus motifs. The significance of the resulting motifs was determined as described in the previous paragraph.
To analyze target sequences for known motifs, the sequences were scanned with all the position weight matrices from the TRANSFAC database (public release version 6.0) (59) and further analyzed as described in the previous de novo motif prediction section. Similar motifs were grouped and averaged.
RESULTS
Genome-wide identification of p53-binding sites using ChIP-on-chip
In order to detect in vivo binding sites for p53 on a genome-wide scale, we applied the ChIP-on-chip approach to endogenous p53 expressing U2OS osteosarcoma cells. In unstressed cells, endogenous p53 is maintained at low levels. To activate p53, cells were treated with 5 nM Actinomycin D for 24 h (60). Upon Actinomycin D treatment, p53 is stabilized and growth arrest is induced (Figure 1A and B). ChIP was performed upon this treatment. To assess the specificity of our ChIP-results, p53 binding to exon 2 of the myoglobin gene was determined as background signal and enrichment was calculated as fold binding over background signal (Figure 1C). For the p21-promoter, an enrichment of p53 binding of almost 600-fold was attained showing that the immunoprecipitation was highly specific. For the global binding site analysis, the enriched (ChIP) sample and the nonenriched (Total) DNA sample were amplified, differentially labeled and cohybridized to 38 DNA arrays covering the whole human genome (repeat masked) with a probe spacing of 100 bp and a probe length of 50 bp (NimbleGen Systems, Inc.).
Figure 1.

Genome-wide identification of p53-binding sites using ChIP-on-chip. (A) Representative cell cycle profile of U2OS cells untreated or treated for 24 h with 5 nM Actinomycin D. (B) Western blot showing p53 expression levels of U2OS cells, untreated or treated for 24 h with 5 nM Actinomycin D. (C) ChIP enrichment (fold over negative control, myoglobin) of p53 at the p21 promoter and the intronic binding site of GADD45A. (D) ChIP-on-chip profile of p53 binding to chromosome 6 visualized using Signalmap (NimbleGen Inc.). Shown are the log2 ratio of ChIP/Total signal derived from the genome-wide tiling data and a zoomed-in view of binding to the p21 promoter for all three biological replicates. Genes are represented by thick horizontal bars (plus-strand positive, minus-strand negative). (E) Overlap of the ChIP-on-chip derived p53-binding sites with PET5+ data from Wei et al.
We called putative p53-binding sites combining three different peak extraction algorithms to maximize the number of potential peaks. The genomic loci of the combined peaks were combined to generate a so-called dedicated array (61), which was used to hybridize two further biological replicate experiments (Figure 1D). If a peak was identified in all three biological replicates, it was considered as a high-confidence p53-binding site. This way, we identified in total 1546 high-confidence binding sites for p53. Verification of the identified binding sites was performed by quantitative PCR with three independent ChIP experiments. In total, 50 potential binding sites were randomly selected and tested. This resulted in a confirmation of 48 out of 50 sites. We conclude that we identified 1546 genome-wide p53-binding sites with a false positive rate of ∼4%.
Since several studies, using different ChIP-based techniques, have identified binding sites for p53 in various cell systems (35–38,62), we compared these to the collection of our binding sites (Table 1). The overlap between our data set and the PET5 cluster in the ChIP-PET data set for p53 by Wei et al. (37) was 69%, even though different cell lines and treatments were used (Figure 1E). The extensive overlap with the highest ranked targets of the ChIP-PET data (69%) and lower overlap (17%) in the low ranked targets with ChIP-PET data is in concordance with the study of Euskirchen et al. (63), where they compared ChIP-sequencing with ChIP-on-chip under the same conditions for STAT1 and also found the most overlap in the highest ranked regions. From the genes that were identified by a ChIP-based screen in yeast (41), 50% are found in our study. Thus, even when using different cellular systems and physiological conditions as well as various ChIP-based techniques, half or more of the p53-binding sites appear to be overlapping with our global ChIP-on-chip approach, showing we created a high-confidence p53-binding dataset.
Table 1.
Overlap with published data sets
| Published study | Total number of binding sites | Overlap with p53 of this study (1546 total) | |
|---|---|---|---|
| Yang et al. (62) | 5807 | 383 | |
| Wei et al. (37) | PET2+ | 1773 | 301 |
| PET2+ with p53PET motif | 542 | 262 | |
| PET3+ | 327 | 170 | |
| PET4+ | 169 | 111 | |
| PET5+ | 106 | 73 | |
| Krieg et al. (38) | Low | 113 | 3 |
| Mid | 34 | 1 | |
| High | 8 | 1 | |
| Cawley et al. (35) | 48 | 15 | |
| Kaneshiro et al. (36) | 37 | 16 | |
| Hearnes et al. (41) | 38 | 26 | |
Characterization of identified p53-binding sites
To annotate the identified p53 binding sites, their locations were analyzed with respect to annotated Ensembl genes. We found that 21% of all p53-binding sites mapped to TSS flanking regions (5 kb upstream, first exon and intron), 28% were within a gene (excluding first exon and intron), 3% within 5 kb downstream, 16% within 5–25 kb upstream or downstream and 32% in intergenic regions (Figure 2A). We compared the frequency of p53 binding in specific genomic regions to the distribution of these genomic regions over the whole genome (using Ensembl gene annotations) and found that p53-binding sites are significantly enriched in TSS flanking regions and within 5–25 kb upstream or downstream of a gene (P = 1.49E-009 and 0.0094, respectively) (Figure 2A).
Figure 2.

Characterization of identified p53 binding sites. (A) Distribution of the p53-binding site location relative to Ensembl genes (upper panel) compared to the genome-wide distribution (lower panel). Locations of binding sites are divided in TSS flanking region (5 kb upstream of TSS + first exon + first intron), intragenic region (all introns and exons except first), 5 kb downstream (5 kb downstream of last exon), 5–25 kb up- or down-stream or intergenic regions (everything else). The asterisk represents significant enrichment. (B) Expression change of genes which have a p53 binding site in the TSS flanking region. The expression change is shown after 24 h of 5 nM Actinomycin D treatment in fold over untreated U2OS cells. Error bars represent standard deviation of three independent experiments. (C) Transactivation assay of intronic (yellow) and intergenic (blue) p53-binding sites. The relative luciferase activity is plotted in fold over empty vector. Error bars represent standard deviations of three independent biological replicates. (D) Binding profile and expression change of four ESTs bound by p53. In the upper panel, the ChIP-on-chip data is visualized. In the lower left panel, these binding sites are confirmed by targeted ChIP for p53. Shown is the enrichment in fold over negative control (myoglobin). In the lower right panel, the expression change of the EST is shown after 24 h of 5 nM Actinomycin D treatment in fold over untreated U2OS cells. Error bars represent standard deviation of three independent experiments.
To study if binding of p53 in TSS flanking regions can regulate the transcription of the corresponding gene products, we randomly selected 11 genes in this group to test their changes of expression upon p53 activation (Figure 2B). Four of these genes were indeed more than 2-fold upregulated, two were >2-fold downregulated, and five did not change >2-fold. Thus, upon p53 binding in TSS flanking regions, genes can get activated or repressed by p53, which is in accordance with the described function of p53 as transcriptional activator and repressor.
While the biological function of p53 binding to promoter regions is well established, we wanted to study the functional potential of intronic as well as intergenic p53 binding. We first tested nine of the intronic and intergenic binding sites (randomly chosen) in transactivation assays cloning them into a pGL3-promoter-luciferase vector, which can be used to test enhancer functions. U2OS cells were transiently transfected with these luciferase constructs and treated with Actinomycin D to activate p53. For each of the selected binding sites, the luciferase activity increased two to nine times compared to the pGL3prom control vector (Figure 2C). This indicates that p53-binding sites in introns and intergenic regions can play a role as transcriptional enhancers.
To test whether the p53 binding in intergenic regions could also regulate the transcription of novel gene products, we mapped the intergenic binding sites to human expressed sequence tags (ESTs). This revealed that 67% of intergenic p53-binding sites are located within 5 kb up- or down-stream of an EST. This is a significant enrichment (P = 1.50E-4) compared to the proportion of the complete genome that falls within this category. From these binding sites close to an EST, we chose four sites for further analysis (Figure 2D, upper panel). We validated binding of p53 to these sites in targeted ChIP experiments (Figure 2D, lower left panel) and tested changes of expression of these novel transcripts upon p53 activation. The four chosen transcripts showed a 2- to 15-fold induction (Figure 2D, lower right panel) upon p53 activation. This indicates that many novel, currently unannotated transcripts may be regulated by p53.
Functional annotation of the p53-binding sites
Since we found well-known p53 target genes involved in pathways such as DNA repair (GADD45A, DDB2), cell cycle regulation (MDM2, p21) and apoptosis (BAX, DR5) (Table S1), we wanted to analyze the possible biological roles of all the p53-binding sites in our dataset. When we grouped our p53 targets, which have a binding site within 5 kb, according to function in GO using Ontologizer (52,64), we found several new groups of target genes that have not been linked to p53 function before or that expand p53's functions such as the phosphorus and biopolymer metabolism group (Figure 3A). Metabolic changes occur in many cancers and recently p53 has been linked to changes in metabolism (65). The fact that we find metabolism-related genes significantly enriched in our binding site list could well indicate that p53 plays an even wider role in metabolic changes besides the so far described function of p53 in glucose metabolism and oxidative stress (66).
Figure 3.

Functional annotation of identified p53-binding sites. (A) All Ensembl genes within 5 kb of an identified p53-binding site were annotated according to the Gene Ontology (GO) using Ontologizer. Significantly enriched groups (P < 0.05) are indicated with an asterisk. Shown is a selection of GO categories. (B) All Ensembl genes within 5 kb of an identified p53-binding site were annotated according to Kyoto Encyclopedia of Genes and Genomes (KEGG) using FatiGO+. Significantly enriched pathways (P < 0.05) are indicated with an asterisk. Shown are the five pathways with the highest number of p53-bound genes. (C) Binding profile of p53 target genes involved in the axon guidance pathway. In the upper panel the ChIP-on-chip data is visualized. In the lower panel these binding sites are confirmed by targeted ChIP for p53. Shown is the enrichment in fold over negative control (myoglobin).
To study the involvement of our identified p53-target genes in entire biological pathways, we classified them according to the Kyoto Encyclopaedia of Genes and Genomes (KEGG) using FatiGO+ (54). Axon guidance and calcium-signaling pathways (Figure 3B) are significantly overrepresented in our dataset, suggesting a hitherto undescribed role for p53 in these biological processes. To study the axon guidance target gene group further, we randomly chose three genes from this group for our analysis: SEMA3C, SEMA6A and SEMA3A (Figure 3C, upper panel). We validated the ChIP-on-chip data (Figure 3C, upper panel) by targeted ChIP and found a significant enrichment of p53 binding upon Actinomycin D treatment of U2OS cells to all tested sites (Figure 3C, lower panel). Thus, it remains to be elucidated which role the target genes from the axon guidance group could play during the p53-mediated stress response.
Development of p53scan, a novel p53-motif finding algorithm
The p53 DNA binding site has been characterized and is consistently described as two copies of the half-site RRRCWWGYYY separated by a spacer of 0–13 bp, where R = purine, W = A or T and Y = pyrimidine. Although this may be the most optimal sequence for p53 binding, only 52 out of the 1546 binding targets in this study contain a perfect match to this sequence. Thus, the p53 motif shows a high degree of degeneracy, which could create the versatility of different p53-mediated stress responses in vivo. Different approaches for identifying the degenerate p53-consensus binding motif have been described. Most are based on a PWM scoring method although recently an algorithm based on experimentally measured binding affinity was shown to give interesting results (33).
An accurate PWM which correctly reflects the binding preference of a transcription factor is a crucial parameter for identifying binding sites with PWM algorithms. The different PWMs that have been described for p53 until now were constructed based on only 17 (TRANSFAC) to 39 (37) binding sequences. Consequently, these PWMs only reflect the information present in those few sequences. Therefore, not surprisingly, using p53MH (which uses a PWM based on 37 sequences) with a cutoff of 90 as suggested by the authors (34), we find that only 33% of our high-confidence binding sequences contain a motif. Having identified a set of genome-wide binding sites, we wondered if we could use the information encompassed in a wide variety of p53-binding sites to develop a more sensitive algorithm. We randomly selected 773 sequences (one half of our identified binding targets) and ordered these based on the ChIP/Total ratio of the highest probe in the peak. We used the de novo motif prediction program MDmodule (55) on the ordered sequences to predict the p53 motif (Figure 4A). The PWM was constructed from these results (matrix shown in Table S2) and combined with a scoring scheme adapted from the p53MH model. This approach enabled us to greatly increase the amount of binding sequences with an identified p53 motif up to 83% (FPR ∼7%).
The performance of our algorithm, p53scan, was benchmarked on the binding sites identified in this study (the ones not used for training) and the human p53-binding sites identified previously by ChIP in combination with paired end tag sequencing [ChIP-PET 3+ (37)]. We compared the performance to p53MH and to the Match algorithm (67) with the p53 PWM from TRANSFAC, using three different metrics: ROC AUC, MNCP (56) and the harmonic mean of precision and recall (F-measure). The training and benchmarking process was repeated in ten independent runs and the average results are shown in Table 2. We implemented the best performing PWM in p53scan, and compared the performance on all binding targets to p53MH and Match (Figure 4B). We also compared p53scan to the ChIP-PET algorithm described by Wei et al. (37) using the ChIP-PET 3+ sequences. The authors identified 72% of these sequences as having a motif using p53PET with an estimated FPR of 0.68%. Using p53scan on their sequence set with the same estimated FPR, we find a p53-binding motif in 82% of the binding sites. These results clearly show that p53scan can identify more p53 motifs in the evaluated sequence sets than the currently described algorithms, without lowering specificity.
Table 2.
The performance of p53scan
| p53 ChIP-on-chip | p53 ChIP-PET 3+ | |||||
|---|---|---|---|---|---|---|
| MNCP | AUC | F-measure | MNCP | AUC | F-measure | |
| p53scan | 8.67 | 0.90 | 0.86 | 3.97 | 0.93 | 0.91 |
| p53MH | 5.62 | 0.83 | 0.76 | 3.37 | 0.86 | 0.81 |
| Match | 7.35 | 0.82 | 0.82 | 3.49 | 0.87 | 0.81 |
The possibility of a spacer within the p53 motif deserves special consideration. If all spacer lengths from 1 to 13 are considered without specifying additional constraints the false positive rate of a PWM algorithm like p53scan greatly increases, due to the greater number of possible motifs that is evaluated. As can be seen in Figure 4C most of our p53-binding motifs actually do not have a spacer between the two half sites. Therefore by default p53scan employs a strict score threshold for all spacer lengths other than 0. The score threshold for each individual spacer length was selected by scanning a background sequence set (random nonbound sequences of equal length) for each spacer length and selecting the cutoff that resulted in no hits. Thus, we have developed a highly specific and inclusive algorithm to identify p53-binding motifs, which is freely available at: http://www.ncmls.nl/bioinfo/p53scan.
Characterization of the p53-binding motif
The p53scan cutoff resulting in the highest F-measure (FPR ∼7%, as marked in Figure 4B) represents a balance between retrieving false positives and missing false negatives. We have used this setting to analyze and further characterize the bound target sites identified in this study. We found the p53 motif in 1281 out of 1546 (83%) binding sites. We determined the location of the consensus motif with respect to the ChIP-on-chip data and found it to be located mainly in the centre of the peaks (Figure 5A). To study whether there is a correlation between the ChIP/Total binding ratio of p53 and the occurrence of the p53-binding motif, we ranked the identified p53-binding sites according to their relative binding enrichment (log2 ChIP/Total) and divided them into three subgroups of low, medium and high ratios. The percentage of binding sites, which contain the p53-binding motif increased slightly with the binding ratio of the peaks. In the high-enrichment subgroup (468 sites with a log2 ChIP/Total ratio of at least 3.17), almost 90% of the p53-binding sites contain a p53-binding motif (Figure 5B).
Figure 5.

Characterization of the p53-binding motif. (A) Histogram of the distance of the p53 consensus site to the probe with the highest mean ratio within a binding site, based on the p53scan algorithm. (B) Percentage of binding sites containing a motif relative to binding ratio. The identified p53-binding sites were ranked according to their binding ratio (log2 ChIP/Total) and divided into three groups ranging from low to high ratios. Shown for each group is the percentage of sequences containing a motif, based on the p53scan algorithm. (C) Average binding ratio (log2 of ChIP/Total) relative to the number of mismatches to the p53 consensus site visualized in a boxplot. Data based on p53scan results.
Next, we analyzed the binding enrichment as measured by ChIP/Total signal ratio in relation to the exact composition of the consensus site. We averaged the binding ratio as a function of the number of mismatches to the consensus motif (Figure 5C). This shows that there is a correlation between enrichment in vivo as measured by ChIP-on-chip and the nucleotide composition of the p53-binding motif.
Since 52 (∼11%) of the sequences in the high subgroup, consisting of the most highly enriched targets (Figure 5B), do not contain a p53-binding motif as identified by p53scan, we tried to further characterize the motifs in these sequences. When we expand the peak area to 1.5 kb, p53scan can find a binding motif in 43 out of the 52 sequences, using settings that result in only 15 motifs found in 1500 random coding sequences of equal length (FPR ∼1%). In these cases, the actual calling of the peak area could have been imprecise, most likely due to the binding site being located in repeat-masked areas. Remarkably, when we take these matches into account, in total 98% of the sequences in the high subgroup contain a p53 motif.
Transcription factor binding motifs in the vicinity of the identified p53-binding sites
Corecruited DNA-binding factors (18) have been invoked to play a role in the flexible response of p53 to various stress signals. We therefore analyzed the vicinity of the p53 sites (500 bp centered on the peak of the p53 binding) for the potential presence of other known transcription factor binding sites, using TAMO (57) with the TRANSFAC database (59). Predicted binding sites of eight different motifs of transcription factors were found to be significantly overrepresented in the surrounding sequences of the p53-binding sites (Figure 6). Among those were potential binding sites for Krüppel-like factors (KLF), Sp1/Sp3, the group of basic helix–loop–helix (bHLH) proteins, AP1, AP2, MZF1, CP2 and ETS2. Many of these factors that we have found to be statistically enriched in our genome-wide collection of p53-binding sites have been experimentally shown to influence p53-dependent transcriptional activity for single target genes. The most overrepresented motif in our dataset are the motifs for KLF; it has been suggested that KLF 4 is a mediator of p53 in controlling progression of the cell cycle following DNA damage (68). Interestingly, p53 has been reported to require cooperation of Sp1 or a Sp1-like factor for the transcriptional activation of the human BAX promoter (69) as well as for p21 (70). To find out if the potentially cobinding transcription factors might influence p53-transcriptional activity towards a specific direction of the response pathway, we looked at the different subsets of the p53-binding sites containing a specific motif. We analyzed the potential biological significance of the genes, which are within 5 kb of these binding sites using GO annotations, as described above. Three significantly enriched GO categories were found for the cobinding factors: developmental, metabolic and cell–cell signaling pathways (Table 3). In all three pathways p53 has been reported to play a role. Our data thus supports the notion that the response pathways of p53 might be influenced by the identified potential cobinding transcription factors.
Figure 6.

Overrepresented transcription factor binding motifs in the vicinity of identified p53-binding sites. Known transcription factors motifs identified in the 500 bp region centered around p53-binding sites based on TRANSFAC 6 database. Statistical significance (P < 0.05) was calculated using TAMO. Motifs were visualized using WebLogo.
Table 3.
Enriched TRANSFAC motifs involved in a biological function
| TRANSFAC | GO term | GO description | P-value |
|---|---|---|---|
| MZF1 | GO:0007275 | Multicellular organismal development | 0.03 |
| bHLH | GO:0007275 | Multicellular organismal development | 0.04 |
| bHLH | GO:0019219 | Regulation of nucleobase, nucleoside, nucleotide and nucleic acid metabolic process | 0.04 |
| CP2 | GO:0007267 | Cell–cell signaling | 0.04 |
| AP1 | GO:0007275 | Multicellular organismal development | 0.05 |
p63 and p73 binding to p53 targets
The p53 family members p63 and p73 have been reported to contribute to the p53 stress response in certain tissues in vivo (28,71). They might play a role in the regulation of transcriptional activities of p53 as well as potential cobinding transcription factors as evidenced by their influence on p53's ability to bind to various apoptotic promoters in vivo (28,71). Furthermore, Yang et al. (62) studied the genome wide binding of p63 and identified a p63-specific motif. Since this motif strongly resembles the motif to which p53 binds, we were interested if and to what extend p63, and the other p53 family member p73, can bind to our identified p53 global binding sites. Because of possible cell-type specific differences in the transcriptional response pathways of the p53-family, a cellular system was needed that would allow a direct comparison between p53, p73 and p63. We generated Saos-2 cell lines expressing TAp63α, TAp73α or p53 under a tetracycline-inducible promoter. We performed ChIP-on-chip analysis for p63 and p73 as well as overexpressed p53 on the dedicated array. Comparing the binding sites identified for endogenous p53 in U2OS cells to those identified for exogenous p53 in the Saos-2 p53 cell line, we found that 1112 of the endogenous binding sites (72%) are bound by exogenous p53 as well. Very interestingly, if we compare these 1112 p53 exogenous-binding sites, which overlap with the binding sites occupied by wt endogenous p53, 72% of those p53-binding sites could also be bound by p73 and/or p63 in vivo (Figure 7). With the majority of the p53-binding sites also being bound by p73 and p63, there seems to be good evidence that p63 and p73 could play an important role in the p53 transcriptional response pathways.
Figure 7.

p63 and p73 binding to p53 targets. (A) Overlap of p53-binding sites also bound by p63 and p73 in Saos2 inducible cell lines as determined by ChIP-on-chip on the dedicated p53 array. (B) p53 motif identified with p53scan, visualized using WebLogo for p53-binding sites also bound by p63 or p73 (upper panel) and binding sites only bound by p53 (lower panel).
To investigate whether binding sites that can be bound only by p53 or also by p73 and p63 show sequence differences, we compared the p53 motif based on p53scan in shared binding sites (Figure 7B, upper panel) and binding sites which are not bound by p63 and p73 (Figure 7B lower panel). These motifs very much resemble each other, independently of whether they are bound by p53 only or by all three family members. Accordingly, p63 and p73 are actually able to bind sequences containing this p53-binding motif on a global scale. The p53-binding motif was identified in 86% of the shared binding sites and in 76% of the binding sites for p53 only. It remains to be elucidated which other parameters of a p53-binding site determine whether it can be bound by p53 only or also by the family members. It has been shown in vitro that five specific bases in the p53 consensus sequence are important for stable binding of p73 to DNA (72). These specific nucleotides are present in 9.6% of the motifs found by p53scan in the shared binding sites, and in 10.8% of the motifs in p53-only binding sites. Therefore, according to our observations this specific characteristic of the p53 consensus motif cannot explain the difference between p53 only and shared binding site of our genome-wide in vivo binding data.
Besides differences in the p53 motif, we also analyzed the p53 only versus the shared binding sites in respect to their genomic location, GO annotation, and the potential presence of other known transcription factor binding sites, as described above, but could not observe significant differences (data not shown). Thus, we found that the DNA binding characteristics of the p53-binding sites which were bound by its family members closely resemble those that were bound by p53 only.
DISCUSSION
Genome-wide identification of p53-binding sites
To characterize the transcriptional mechanisms of the p53-mediated stress response, we analyzed p53 binding to chromatin on a genome-wide scale using the ChIP-on-chip approach and identified 1546 high-confidence binding sites.
While these binding sites were significantly enriched in TSS flanking regions, encompassing possible promoters, a large fraction was located in intragenic or intergenic regions, as also observed in other studies (37,41). We and others have provided evidence for functionality of these intergenic binding sites. In our reporter assays, we could show that the intergenic and intragenic p53-binding sites can function as enhancers. Likely, the interaction between enhancer and target gene is mediated via loop formation, as shown for example for the Hoxd gene cluster and the β-globin locus (73). Furthermore, the intergenic-binding sites could be involved in regulation of nonprotein coding genes as well as other novel transcripts, as has been suggested by smaller scale ChIP-on-chip analyses (35). In our study, we discovered that unannotated transcripts located near intergenic p53-binding sites can be upregulated upon p53 activation.
Motifs in the p53-binding site
We have developed a new algorithm, p53scan, which incorporates the motif derived de novo from the genome-wide binding sites identified in this study as shown in Figure 4A. Although this motif resembles the different versions of the p53-binding motif described up to now, it more accurately reflects the in vivo binding preference of p53, as this new motif is based on information from hundreds of binding sites. Indeed, comparisons of p53scan to other publicly available algorithms, including p53MH, which has been most widely used, show that it produces markedly better results in the metrics that were tested in this study. In addition, we compared p53scan to the algorithm developed by Wei et al. (37), called p53PET model, with the sequences identified in their study as input. We found more sequences containing a motif with p53scan than with p53PET model (82% versus 72%) at the same specificity level. This confirms that the sensitivity of p53scan is not limited to the binding sites of our ChIP-on-chip dataset, but that it can also be used for future analysis of other binding data. As a publicly accessible, intuitive and above all sensitive and specific algorithm, p53scan is a useful addition to the available tools that will help characterize the widely diverse binding preference of p53.
When analyzing our p53-binding sites with p53scan, 83% of the identified binding sites contained a motif that is reminiscent of the p53-binding motif. The predominant motif has no spacer in between the two half-sites, although there is a small fraction with a spacer of one nucleotide. This is in agreement with the genome-wide spacer distribution found previously (37). In the most highly enriched group of target sites, nearly all bound sequences contain our p53-consensus motif. This suggests that almost all highly enriched p53-binding sites are bound in a direct sequence-specific manner dependent on the consensus motif. Of all the identified binding sites, in 17% p53scan cannot detect the p53 consensus motif. Although previous studies have also found p53-binding sites without a detectable p53-consensus motif (36,37), we find less of those binding sites in our study using the more inclusive identification of the motif by p53scan. The fact that no p53 motif can be identified in a small subset of binding sites can have several reasons: either p53 is also able to bind purely on the basis of DNA topology independent of the sequence (11), or it might bind to a different motif like microsatellites for the PIG3 target gene (9). The remaining sites without a common motif could of course also be bound due to indirect binding of p53 to chromatin.
By grouping the binding sites according to their ChIP/Total ratio, we found a positive correlation with both the percentage of binding sites containing a p53-binding motif, as well as the degree of similarity of the identified motifs with the p53 consensus motif. Thus, the more the binding sequence resembled the p53 consensus motif, the higher the ChIP/Total ratio. This is in accordance with the structural data of DNA-bound p53 that showed that protein–DNA interfaces vary as a function of the specific base sequence of the DNA (74). From this structural data, it was also concluded that the differential binding affinity is correlated with sequence-specific variations, which have a direct influence on the protein-DNA contact geometry.
The binding of p53 to DNA occurs in the context of other transcription factors and cofactors. It has been shown for individual target genes that other transcriptional activators or repressors can act together with p53 and can have differential effects on the transcription of target genes. Therefore, we analyzed common cis-elements among the genome-wide set of p53-binding sites. We find potential SP1-binding sequences to be highly enriched in the vicinity of p53-binding sites in our global approach; p53 has been reported to require the cooperation of Sp1 or a Sp1-like factor for transcriptional activation of the human BAX and p21 promoter (69,75). For bHLH motifs, which we found to be enriched in our set of target sites, it is known that the p53 promoter itself contains a functional consensus sequence for bHLH proteins. In the murine p53 promoter, this element has been shown to be required for full promoter activity (20). The fact that we find bHLH motifs enriched in the vicinity of p53-binding sites, shows that these factors might be involved in a positive feedforward regulation of p53 pathways. Thus, our findings extend the analysis of the transcriptional environment of single targets to a larger subset of target genes derived from a global screen. In the future, we will need to elucidate what biological consequences might result from certain combinatorial interplay between p53 and other cobinding factors. Therefore, an interesting challenge will lie in the elucidation of which transcription factor complexes can be found at which p53-target genes and whether certain biological responses appear to be dependent on the macro-molecular transcription factor complexes at p53-binding sites. A first interesting approach to purify macromolecular complexes at different subsets of p53-target genes was done by Tanaka et al. (22) by fractionating cross-linked p53-associated chromatin and identifying the human cellular apoptosis susceptibility protein (hCAS/CSE1L) in the one fraction of a subset of p53 target promoters, including PIG3, in a p53-autonomous manner. Thus, it remains to be seen whether also for other cellular response pathways specific combinations of cobinding factors can be isolated at p53-target genes.
Global binding of p73 and p63 to p53-binding sites
For the first time, we investigated on a global scale to which extent p53-binding sites can be occupied by p73 and p63 upon a specific stress signal in vivo. We found that 72% of the p53-binding sites can also be bound by p73 and/or p63. Some groups have postulated differences in binding motif for the p53 family members, but this is mostly based on individual targets or in vitro derived data. Lokshin et al. (72) showed the importance of five bases in the p53 consensus sequence for stable binding of in vitro p73 to DNA. In our global binding site set, we cannot differentiate between the sets of shared and p53-only binding sites on the basis of this sequence difference. The fraction of motifs containing these five bases was comparable in both sets. We could also not identify a significant difference between the motif for p53 in shared binding sides or sites exclusively bound by p53. The fact that we did not find specific motif variation, is in agreement with the genome-wide screen for p63-binding sites by Yang et al. (62) and in vitro studies from Perez et al. (76), which showed with a SELEX approach that p63 binds principally to the p53-consensus motif, preferentially to a slightly more degenerate form of it. Therefore, we conclude that p63 and p73 can bind to p53-binding sites on a large scale, which may imply that the stress response is mediated in part by either competitive or cooperative binding of p53 family members to target genes. Alternatively, this could hint at the possibility that p63 and p73 are capable of taking over part of the function of p53 if needed.
This study provides a global set of high-confidence p53-binding sites, which greatly expands the known, experimentally validated p53 binding repertoire and gives a global insight into their characteristics. These data can serve as a valuable knowledgebase for further research, in which new functional studies will help to further clarify the complex role of p53 and its family members.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank Dr K. Vousden for p53-constructs; Dr H. van Bokhoven for p63-constructs and Drs Shinji Takagi and G. Melino for p73-constructs. We thank Colin Logie, Gert Jan Veenstra and Willem Welboren for helpful discussions and critical reading of the article. Funding for this work was provided by Dutch Cancer Foundation (KWF) (KUN 2003-2926 to L.S., S.D. and M.K., KUN 2005-3347 to L.S.); Netherlands Organisation for Scientific Research (NWO) (Vidi 846.05.002 to S.J.vH., S.J.J.B. and M.L.); EPIgenetic TReatment Of Neoplastic disease (EPITRON) (LSH-2004-2.2.0-2 to MAvD); X-TRA-NET (LSHG-CT-2005-018882 to M.A.vD.). Funding to pay the Open Access publication charges for this article was provided by KWF and NWO.
Conflict of interest statement. None declared.
REFERENCES
- 1.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 2.Kern SE, Kinzler KW, Bruskin A, Jarosz D, Friedman P, Prives C, Vogelstein B. Identification of p53 as a sequence-specific DNA-binding protein. Science. 1991;252:1708–1711. doi: 10.1126/science.2047879. [DOI] [PubMed] [Google Scholar]
- 3.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 4.Laptenko O, Prives C. Transcriptional regulation by p53: one protein, many possibilities. Cell Death Differ. 2006;13:951–961. doi: 10.1038/sj.cdd.4401916. [DOI] [PubMed] [Google Scholar]
- 5.Erster S, Moll UM. Stress-induced p53 runs a transcription-independent death program. Biochem. Biophys. Res. Commun. 2005;331:843–850. doi: 10.1016/j.bbrc.2005.03.187. [DOI] [PubMed] [Google Scholar]
- 6.el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Definition of a consensus binding site for p53. Nat Genet. 1992;1:45–49. doi: 10.1038/ng0492-45. [DOI] [PubMed] [Google Scholar]
- 7.Zhao R, Gish K, Murphy M, Yin Y, Notterman D, Hoffman WH, Tom E, Mack DH, Levine AJ. Analysis of p53-regulated gene expression patterns using oligonucleotide arrays. Genes Dev. 2000;14:981–993. [PMC free article] [PubMed] [Google Scholar]
- 8.Inga A, Storici F, Darden TA, Resnick MA. Differential transactivation by the p53 transcription factor is highly dependent on p53 level and promoter target sequence. Mol. Cell. Biol. 2002;22:8612–8625. doi: 10.1128/MCB.22.24.8612-8625.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Contente A, Dittmer A, Koch MC, Roth J, Dobbelstein M. A polymorphic microsatellite that mediates induction of PIG3 by p53. Nat. Genet. 2002;30:315–320. doi: 10.1038/ng836. [DOI] [PubMed] [Google Scholar]
- 10.Ho JS, Ma W, Mao DY, Benchimol S. p53-Dependent transcriptional repression of c-myc is required for G1 cell cycle arrest. Mol. Cell. Biol. 2005;25:7423–7431. doi: 10.1128/MCB.25.17.7423-7431.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gohler T, Reimann M, Cherny D, Walter K, Warnecke G, Kim E, Deppert W. Specific interaction of p53 with target binding sites is determined by DNA conformation and is regulated by the C-terminal domain. J. Biol. Chem. 2002;277:41192–41203. doi: 10.1074/jbc.M202344200. [DOI] [PubMed] [Google Scholar]
- 12.Jett SD, Cherny DI, Subramaniam V, Jovin TM. Scanning force microscopy of the complexes of p53 core domain with supercoiled DNA. J. Mol. Biol. 2000;299:585–592. doi: 10.1006/jmbi.2000.3759. [DOI] [PubMed] [Google Scholar]
- 13.Bode AM, Dong Z. Post-translational modification of p53 in tumorigenesis. Nat. Rev. 2004;4:793–805. doi: 10.1038/nrc1455. [DOI] [PubMed] [Google Scholar]
- 14.Brooks CL, Gu W. Ubiquitination, phosphorylation and acetylation: the molecular basis for p53 regulation. Curr. Opin. Cell. Biol. 2003;15:164–171. doi: 10.1016/s0955-0674(03)00003-6. [DOI] [PubMed] [Google Scholar]
- 15.Aylon Y, Oren M. Living with p53, dying of p53. Cell. 2007;130:597–600. doi: 10.1016/j.cell.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 16.Avantaggiati ML, Ogryzko V, Gardner K, Giordano A, Levine AS, Kelly K. Recruitment of p300/CBP in p53-dependent signal pathways. Cell. 1997;89:1175–1184. doi: 10.1016/s0092-8674(00)80304-9. [DOI] [PubMed] [Google Scholar]
- 17.Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 18.Coutts AS, La Thangue NB. The p53 response: emerging levels of co-factor complexity. Biochem. Biophys. Res. Commun. 2005;331:778–785. doi: 10.1016/j.bbrc.2005.03.150. [DOI] [PubMed] [Google Scholar]
- 19.Shikama N, Lee CW, France S, Delavaine L, Lyon J, Krstic-Demonacos M, La Thangue NB. A novel cofactor for p300 that regulates the p53 response. Mol. Cell. 1999;4:365–376. doi: 10.1016/s1097-2765(00)80338-x. [DOI] [PubMed] [Google Scholar]
- 20.Demonacos C, Krstic-Demonacos M, La Thangue NB. A TPR motif cofactor contributes to p300 activity in the p53 response. Mol. Cell. 2001;8:71–84. doi: 10.1016/s1097-2765(01)00277-5. [DOI] [PubMed] [Google Scholar]
- 21.Sullivan A, Lu X. ASPP: a new family of oncogenes and tumour suppressor genes. Br. J. Cancer. 2007;96:196–200. doi: 10.1038/sj.bjc.6603525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tanaka T, Ohkubo S, Tatsuno I, Prives C. hCAS/CSE1L associates with chromatin and regulates expression of select p53 target genes. Cell. 2007;130:638–650. doi: 10.1016/j.cell.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 23.Budhram-Mahadeo VS, Bowen S, Lee S, Perez-Sanchez C, Ensor E, Morris PJ, Latchman DS. Brn-3b enhances the pro-apoptotic effects of p53 but not its induction of cell cycle arrest by cooperating in trans-activation of bax expression. Nucleic Acids Res. 2006;34:6640–6652. doi: 10.1093/nar/gkl878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Homer C, Knight DA, Hananeia L, Sheard P, Risk J, Lasham A, Royds JA, Braithwaite AW. Y-box factor YB1 controls p53 apoptotic function. Oncogene. 2005;24:8314–8325. doi: 10.1038/sj.onc.1208998. [DOI] [PubMed] [Google Scholar]
- 25.Huang W-C, Ju T-K, Hung M-C, Chen C-C. Phosphorylation of CBP by IKK[alpha] promotes cell growth by switching the binding preference of CBP from p53 to NF-[kappa]B. Mol. Cell. 2007;26:75–87. doi: 10.1016/j.molcel.2007.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schumm K, Rocha S, Caamano J, Perkins ND. Regulation of p53 tumour suppressor target gene expression by the p52 NF-kappaB subunit. EMBO J. 2006;25:4820–4832. doi: 10.1038/sj.emboj.7601343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Das S, Raj L, Zhao B, Kimura Y, Bernstein A, Aaronson SA, Lee SW. Hzf determines cell survival upon genotoxic stress by modulating p53 transactivation. Cell. 2007;130:624–637. doi: 10.1016/j.cell.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flores ER, Tsai KY, Crowley D, Sengupta S, Yang A, McKeon F, Jacks T. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature. 2002;416:560–564. doi: 10.1038/416560a. [DOI] [PubMed] [Google Scholar]
- 29.Espinosa JM. Mechanisms of regulatory diversity within the p53 transcriptional network. Oncogene. 2008 doi: 10.1038/onc.2008.37. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sbisa E, Catalano D, Grillo G, Licciulli F, Turi A, Liuni S, Pesole G, De Grassi A, Caratozzolo MF, D’Erchia AM, et al. p53FamTaG: a database resource of human p53, p63 and p73 direct target genes combining in silico prediction and microarray data. BMC Bioinform. 2007;8(Suppl 1):S20. doi: 10.1186/1471-2105-8-S1-S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.el-Deiry WS. Regulation of p53 downstream genes. Semin. Cancer Biol. 1998;8:345–357. doi: 10.1006/scbi.1998.0097. [DOI] [PubMed] [Google Scholar]
- 32.Horn HF, Vousden KH. Coping with stress: multiple ways to activate p53. Oncogene. 2007;26:1306–1316. doi: 10.1038/sj.onc.1210263. [DOI] [PubMed] [Google Scholar]
- 33.Veprintsev DB, Fersht AR. Algorithm for prediction of tumour suppressor p53 affinity for binding sites in DNA. Nucleic Acids Res. 2008;36:1589–1598. doi: 10.1093/nar/gkm1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoh J, Jin S, Parrado T, Edington J, Levine AJ, Ott J. The p53MH algorithm and its application in detecting p53-responsive genes. Proc. Natl Acad. Sci. USA. 2002;99:8467–8472. doi: 10.1073/pnas.132268899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cawley S, Bekiranov S, Ng HH, Kapranov P, Sekinger EA, Kampa D, Piccolboni A, Sementchenko V, Cheng J, Williams AJ, et al. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell. 2004;116:499–509. doi: 10.1016/s0092-8674(04)00127-8. [DOI] [PubMed] [Google Scholar]
- 36.Kaneshiro K, Tsutsumi S, Tsuji S, Shirahige K, Aburatani H. An integrated map of p53-binding sites and histone modification in the human ENCODE regions. Genomics. 2006;89:178–188. doi: 10.1016/j.ygeno.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 37.Wei CL, Wu Q, Vega VB, Chiu KP, Ng P, Zhang T, Shahab A, Yong HC, Fu Y, Weng Z, et al. A global map of p53 transcription-factor binding sites in the human genome. Cell. 2006;124:207–219. doi: 10.1016/j.cell.2005.10.043. [DOI] [PubMed] [Google Scholar]
- 38.Krieg AJ, Hammond EM, Giaccia AJ. Functional analysis of p53 binding under differential stresses. Mol. Cell. Biol. 2006;26:7030–7045. doi: 10.1128/MCB.00322-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ceribelli M, Alcalay M, Vigano MA, Mantovani R. Repression of new p53 targets revealed by ChIP on chip experiments. Cell Cycle. 2006;5:1102–1110. doi: 10.4161/cc.5.10.2777. [DOI] [PubMed] [Google Scholar]
- 40.Jen KY, Cheung VG. Identification of novel p53 target genes in ionizing radiation response. Cancer Res. 2005;65:7666–7673. doi: 10.1158/0008-5472.CAN-05-1039. [DOI] [PubMed] [Google Scholar]
- 41.Hearnes JM, Mays DJ, Schavolt KL, Tang L, Jiang X, Pietenpol JA. Chromatin immunoprecipitation-based screen to identify functional genomic binding sites for sequence-specific transactivators. Mol. Cell. Biol. 2005;25:10148–10158. doi: 10.1128/MCB.25.22.10148-10158.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zeng J, Yan J, Wang T, Mosbrook-Davis D, Dolan KT, Christensen R, Stormo GD, Haussler D, Lathrop RH, Brachmann RK, et al. Genome wide screens in yeast to identify potential binding sites and target genes of DNA-binding proteins. Nucleic Acids Res. 2008;36:e8. doi: 10.1093/nar/gkm1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kubbutat MH, Ludwig RL, Ashcroft M, Vousden KH. Regulation of Mdm2-directed degradation by the C terminus of p53. Mol. Cell. Biol. 1998;18:5690–5698. doi: 10.1128/mcb.18.10.5690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Denissov S, van Driel M, Voit R, Hekkelman M, Hulsen T, Hernandez N, Grummt I, Wehrens R, Stunnenberg H. Identification of novel functional TBP-binding sites and general factor repertoires. EMBO J. 2007;26:944–954. doi: 10.1038/sj.emboj.7601550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ren B, Robert F, Wyrick JJ, Aparicio O, Jennings EG, Simon I, Zeitlinger J, Schreiber J, Hannett N, Kanin E, et al. Genome-wide location and function of DNA binding proteins. Science. 2000;290:2306–2309. doi: 10.1126/science.290.5500.2306. [DOI] [PubMed] [Google Scholar]
- 46.Liu CL, Schreiber S, Bernstein B. Development and validation of a T7 based linear amplification for genomic DNA. BMC Genomics. 2003;4:19. doi: 10.1186/1471-2164-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kent WJ. BLAT–the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ji H, Wong WH. TileMap: create chromosomal map of tiling array hybridizations. Bioinformatics. 2005;21:3629–3636. doi: 10.1093/bioinformatics/bti593. [DOI] [PubMed] [Google Scholar]
- 49.Zheng M, Barrera LO, Ren B, Wu YN. ChIP-chip: data, model, and analysis. Biometrics. 2007;63:787–796. doi: 10.1111/j.1541-0420.2007.00768.x. [DOI] [PubMed] [Google Scholar]
- 50.Hubbard TJ, Aken BL, Beal K, Ballester B, Caccamo M, Chen Y, Clarke L, Coates G, Cunningham F, Cutts T, et al. Ensembl 2007. Nucleic Acids Res. 2007;35:D610–D617. doi: 10.1093/nar/gkl996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Gen. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grossmann S, Bauer S, Robinson PN, Vingron M. Improved detection of overrepresentation of Gene-Ontology annotations with parent child analysis. Bioinformatics. 2007;23:3024–3031. doi: 10.1093/bioinformatics/btm440. [DOI] [PubMed] [Google Scholar]
- 53.Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, Kawashima S, Katayama T, Araki M, Hirakawa M. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 2006;34:D354–D357. doi: 10.1093/nar/gkj102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Al-Shahrour F, Diaz-Uriarte R, Dopazo J. FatiGO: a web tool for finding significant associations of Gene Ontology terms with groups of genes. Bioinformatics. 2004;20:578–580. doi: 10.1093/bioinformatics/btg455. [DOI] [PubMed] [Google Scholar]
- 55.Liu XS, Brutlag DL, Liu JS. An algorithm for finding protein-DNA binding sites with applications to chromatin-immunoprecipitation microarray experiments. Nat. Biotechnol. 2002;20:835–839. doi: 10.1038/nbt717. [DOI] [PubMed] [Google Scholar]
- 56.Clarke ND, Granek JA. Rank order metrics for quantifying the association of sequence features with gene regulation. Bioinformatics. 2003;19:212–218. doi: 10.1093/bioinformatics/19.2.212. [DOI] [PubMed] [Google Scholar]
- 57.Gordon DB, Nekludova L, McCallum S, Fraenkel E. TAMO: a flexible, object-oriented framework for analyzing transcriptional regulation using DNA-sequence motifs. Bioinformatics. 2005;21:3164–3165. doi: 10.1093/bioinformatics/bti481. [DOI] [PubMed] [Google Scholar]
- 58.Harbison CT, Gordon DB, Lee TI, Rinaldi NJ, Macisaac KD, Danford TW, Hannett NM, Tagne JB, Reynolds DB, Yoo J, et al. Transcriptional regulatory code of a eukaryotic genome. Nature. 2004;431:99–104. doi: 10.1038/nature02800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Matys V, Fricke E, Geffers R, Gossling E, Haubrock M, Hehl R, Hornischer K, Karas D, Kel AE, Kel-Margoulis OV, et al. TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 2003;31:374–378. doi: 10.1093/nar/gkg108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ashcroft M, Taya Y, Vousden KH. Stress signals utilize multiple pathways to stabilize p53. Mol. Cell. Biol. 2000;20:3224–3233. doi: 10.1128/mcb.20.9.3224-3233.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim TH, Barrera LO, Zheng M, Qu C, Singer MA, Richmond TA, Wu Y, Green RD, Ren B. A high-resolution map of active promoters in the human genome. Nature. 2005;436:876–880. doi: 10.1038/nature03877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang A, Zhu Z, Kapranov P, McKeon F, Church GM, Gingeras TR, Struhl K. Relationships between p63 binding, DNA sequence, transcription activity, and biological function in human cells. Mol. Cell. 2006;24:593–602. doi: 10.1016/j.molcel.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 63.Euskirchen GM, Rozowsky JS, Wei CL, Lee WH, Zhang ZD, Hartman S, Emanuelsson O, Stolc V, Weissman S, Gerstein MB, et al. Mapping of transcription factor binding regions in mammalian cells by ChIP: comparison of array- and sequencing-based technologies. Genome Res. 2007;17:898–909. doi: 10.1101/gr.5583007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Robinson PN, Wollstein A, Bohme U, Beattie B. Ontologizing gene-expression microarray data: characterizing clusters with Gene Ontology. Bioinformatics. 2004;20:979–981. doi: 10.1093/bioinformatics/bth040. [DOI] [PubMed] [Google Scholar]
- 65.Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–120. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 66.Bensaad K, Vousden KH. p53: new roles in metabolism. Trends Cell. Biol. 2007;17:286–291. doi: 10.1016/j.tcb.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 67.Kel AE, Gossling E, Reuter I, Cheremushkin E, Kel-Margoulis OV, Wingender E. MATCH: a tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res. 2003;31:3576–3579. doi: 10.1093/nar/gkg585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yoon HS, Chen X, Yang VW. Kruppel-like factor 4 mediates p53-dependent G1/S cell cycle arrest in response to DNA damage. J. Biol. Chem. 2003;278:2101–2105. doi: 10.1074/jbc.M211027200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thornborrow EC, Manfredi JJ. The tumor suppressor protein p53 requires a cofactor to activate transcriptionally the human BAX promoter. J. Biol. Chem. 2001;276:15598–15608. doi: 10.1074/jbc.M011643200. [DOI] [PubMed] [Google Scholar]
- 70.Koutsodontis G, Tentes I, Papakosta P, Moustakas A, Kardassis D. Sp1 plays a critical role in the transcriptional activation of the human cyclin-dependent kinase inhibitor p21(WAF1/Cip1) gene by the p53 tumor suppressor protein. J. Biol. Chem. 2001;276:29116–29125. doi: 10.1074/jbc.M104130200. [DOI] [PubMed] [Google Scholar]
- 71.Flores ER, Sengupta S, Miller JB, Newman JJ, Bronson R, Crowley D, Yang A, McKeon F, Jacks T. Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell. 2005;7:363–373. doi: 10.1016/j.ccr.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 72.Lokshin M, Li Y, Gaiddon C, Prives C. p53 and p73 display common and distinct requirements for sequence specific binding to DNA. Nucleic Acids Res. 2007;35:340–352. doi: 10.1093/nar/gkl1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li Q, Barkess G, Qian H. Chromatin looping and the probability of transcription. Trends Genet. 2006;22:197–202. doi: 10.1016/j.tig.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 74.Kitayner M, Rozenberg H, Kessler N, Rabinovich D, Shaulov L, Haran TE, Shakked Z. Structural basis of DNA recognition by p53 tetramers. Mol. Cell. 2006;22:741–753. doi: 10.1016/j.molcel.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 75.Koutsodontis G, Vasilaki E, Chou WC, Papakosta P, Kardassis D. Physical and functional interactions between members of the tumour suppressor p53 and the Sp families of transcription factors: importance for the regulation of genes involved in cell-cycle arrest and apoptosis. Biochem. J. 2005;389:443–455. doi: 10.1042/BJ20041980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Perez CA, Ott J, Mays DJ, Pietenpol JA. p63 consensus DNA-binding site: identification, analysis and application into a p63MH algorithm. Oncogene. 2007;26:7363–7370. doi: 10.1038/sj.onc.1210561. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.