

Summary
Cryptosporidium parvum is an important human pathogen and potential bio-terrorism agent. No vaccines exist against C. parvum, the drugs currently approved to treat cryptosporidiosis are ineffective, and drug discovery is challenging because the parasite cannot be maintained continuously in cell culture. Mining the sequence of the C. parvum genome has revealed that the only route to guanine nucleotides is via inosine-5′-monophosphate dehydrogenase (IMPDH). Moreover, phylogenetic analysis suggests that the IMPDH gene was obtained from bacteria by lateral gene transfer. Here we exploit the unexpected evolutionary divergence of parasite and host enzymes by designing a high throughput screen to target the most diverged portion of the IMPDH active site. We have identified four parasite-selective IMPDH inhibitors that display antiparasitic activity with greater potency than paromomycin, the current “gold standard” for anticryptosporidial activity.
Introduction
The “vicious cycle of diarrhea and malnutrition” in developing countries could be broken with the advent of effective chemotherapy against Cryptosporidium parvum (Berkman et al., 2002; Huang et al., 2004; Huang and White, 2006). C. parvum is also an important pathogen in the developed world, where AIDS patients are at risk of severe infection (Carey et al., 2004; Fayer, 2004). The parasite produces spore-like oocysts that are resistant to common methods of water treatment, so Cryptosporidium also poses a credible bioterrorism threat (DuPont et al., 1995). The tools to respond to such an incident are woefully inadequate: no vaccines or effective drug treatments are currently available. The damage would be substantial: the economic cost of the 1993 Milwaukee outbreak, where ~400,000 individuals contracted disease, totaled $31.7 million in medical costs and another $64.6 million in productivity losses (Corso et al., 2003). Independent of such bio-terrorism scenarios, effective drugs are urgently needed for the management of cryptosporidiosis in AIDS patients and epidemic outbreaks.
The search for drugs to treat cryptosporidiosis has been almost futile. Compounds such as spiramycin, clarithromycin, paromomycin and nitazoxanide display modest activity in model systems, but limited efficacy in clinical trials with immunocompetent patients and poor efficacy in immunocompromised patients (Abubakar et al., 2007; Mead, 2002). Commonly used antiparasitic drugs fail against C. parvum, which is not surprising given that the C. parvum genome has undergone massive gene loss and horizontal transfer when compared with related apicomplexan parasites such as Plasmodium and Toxoplasma (Abrahamsen et al., 2004; Striepen et al., 2004; Templeton et al., 2004; Xu et al., 2004). C. parvum cannot be maintained in continuous cell culture and genetic tools do not exist, so the validation of new drug targets is thwarted by a dearth of information about parasite metabolism. Nevertheless, the C. parvum genome has revealed the presence of a very streamlined purine salvage pathway that relies on the uptake of adenosine (Abrahamsen et al., 2004; Striepen et al., 2004; Xu et al., 2004). The only route to guanine nucleotides is via IMPDH, which catalyzes the conversion of IMP to XMP with the concomitant reduction of NAD+ (Fig. 1A). Phylogenetic analysis suggests that C. parvum IMPDH was obtained from a bacterial source by lateral gene transfer (Striepen et al., 2004; Striepen et al., 2002), and C. parvum IMPDH is only ~39% identical to the human isozymes IMPDH1 and IMPDH2. While the IMP site is conserved, the NAD site is highly diverged and several inhibitors, most notably mycophenolic acid, bind selectively to the NAD site of human IMPDHs (Ratcliffe, 2006). Thus the NAD site is the most promising target for parasite-selective inhibitors (Fig. 1B).
Fig. 1. Mechanism and structure of IMPDH.
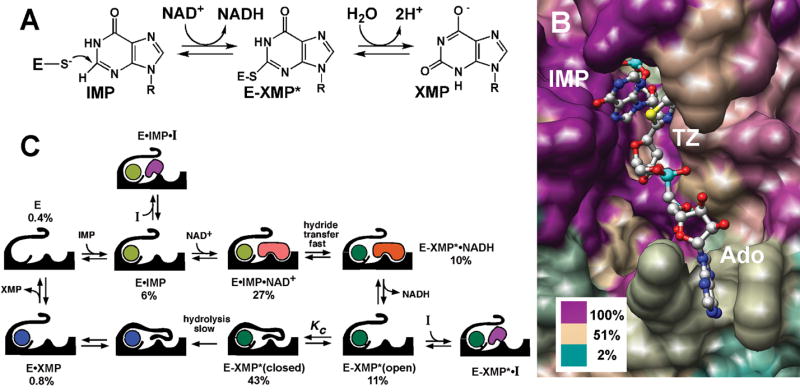
(A) The IMPDH reaction. (B) The active site, rendered by sequence conservation. The structure of E•IMP•tiazofurin adenine dinucleotide complex of IMPDH from Tritrichomonas foetus (PDB accession code 1LRT). Image was produced using the UCSF Chimera package (Pettersen et al., 2004). Percentage of sequence identity is colored as shown using the alignment from (Striepen et al., 2002). TZ, tiazofurin; Ado, adenosine. (C) The kinetic mechanism of IMPDH, showing the distribution of enzyme under the conditions of the HTS (250 μM IMP and 500 μM NAD+), determined as described in Supplemental Material. Not shown: E•NAD+ ≤ 0.7%, E-XMP*•NAD+ = 1%.
Here we devise a screen to target the NAD site of C. parvum IMPDH and identify ten parasite-selective inhibitors with values of IC50 ranging from 0.1–20 μM. The best inhibitors display antiparasitic activity in a cell culture model of infection. These compounds are the first parasite-specific IMPDH inhibitors and the first target-based antibiotics for C. parvum.
Results and Discussion
High throughput screening
We devised a HTS to identify inhibitors that target the NAD+ site of C. parvum IMPDH, taking advantage of a detailed knowledge of the kinetic mechanism which allows us to calculate the distribution of enzyme-substrate complexes at various substrate concentrations (Fig. 1C, Table S1) (Digits and Hedstrom, 1999; Riera et al.; Umejiego et al., 2004). We chose high IMP concentrations (250 μM), so that IMP binds first. Under these conditions, only ~0.4% of the enzyme is in the E state and less than 0.7% will be present as E•NAD+, so the IMP site is virtually inaccessible to inhibitors; only compounds with low nanomolar affinities for the IMP site would be identified in this screen. NAD+ binds second and hydride transfer is rapid to form the covalent intermediate E-XMP* and NADH. NADH then departs and a mobile flap folds into the vacant site, forming the closed conformation required for the hydrolysis of E-XMP* (Hedstrom and Gan, 2006). We chose an NAD+ concentration (500 μM) high enough to generate a robust signal in the HTS, but low enough that significant fractions of the E•IMP and E-XMP*open complexes are present (7 and 11%, respectively). Therefore HTS should yield micromolar inhibitors that bind to the highly diverged NAD site. The HTS protocol is summarized in Table S2 and the results for a typical plate are shown in Fig. S1.
This screen identified 134 compounds that inhibited C. parvum IMPDH by at least 45% (z scores ≤ −10; hit rate of 0.3%). Eighteen of these compounds inhibited C. parvum IMPDH in the secondary screen, and eleven of these 18 compounds did not inhibit human IMPDH2. Authentic samples of these compounds were purchased or synthesized, and compound structure and purity were confirmed by NMR and mass spectroscopy (see Supporting Information for details on synthesis). One of the authentic samples did not inhibit C. parvum IMPDH. The remaining 10 compounds were characterized further (Compounds A-K; Fig. 2).
Fig. 2. Characterization of the parasite-selective inhibitors.

note: there is no “I”. IC50 values are reported for enzyme inhibition. Conditions are described in Materials and Methods. a. ≤ 30% inhibition is observed at 50 μM inhibitor except as noted; b. ≤ 20% inhibition is observed at 50 μM inhibitor; c. 40% inhibition at 25 μM; d. 45% inhibition at 25 μM. Inhibition of C. parvum growth was assessed in a cell culture model using an ELISA assay or by real-time PCR (denoted with superscript RT), as described in Materials and Methods. Cytotoxicity was assessed by measuring the release of LDH using the CytoTox assay (Promega). Cytostatic effects were evaluated with the LIVE/DEAD assay (Molecular Probes). (−) denotes no cytostatic effect; (+) denotes slightly cytostatic (<20%).
Characterization of the principal hits
The values of IC50 for these compounds range from 0.13–19 μM, with only compound K in excess of 10 μM. In all cases, the inhibition data is well described by a simple binding function (Eq. S1), which indicates that the compounds are reversible inhibitors (Fig. 3A). Selectivity ranges from ≥9-fold higher affinity for the parasite enzyme in the worst case (J) to more than 400-fold (G) (Fig. 2).
Fig. 3. Inhibitor characterization.

(A) IC50 determination for compound F. (B) The mechanism of inhibition for compound F versus IMP, [F] =
 0.0 μM; ▼ 0.5 μM; ▲ 1.0 μM; ■ 2.5μM; ◆ 5.0 μM. (C) The mechanism of inhibition for compound F versus NAD+, [F] = ● 0.0 μM; ◆ 0.5 μM; ▼ 1.0 μM; ▲ 2.5μM; ■ 5.0 μM. (D) Dual inhibitor experiments for compound F versus tiazofurin, [tiazofurin] = ● 0.0 mM; ▼ 2.0 mM; ■ 4.0 mM; ◆ 6.0 mM; ▲ 8.0 mM. (E) Dual inhibitor experiments for compound F versus ADP, [ADP] = ● 0.0 mM; ▼ 5.0 mM; ■ 10.0 mM; ◆ 15.0 mM; ▲ 20.0 mM. (F) Dual inhibitor experiment for compound H versus ADP, [ADP] = ● 0.0 mM; ▼ 5.0 mM; ■ 10.0 mM; ◆ 15.0 mM; ▲ 20.0 mM. Conditions as described in Materials and Methods.
0.0 μM; ▼ 0.5 μM; ▲ 1.0 μM; ■ 2.5μM; ◆ 5.0 μM. (C) The mechanism of inhibition for compound F versus NAD+, [F] = ● 0.0 μM; ◆ 0.5 μM; ▼ 1.0 μM; ▲ 2.5μM; ■ 5.0 μM. (D) Dual inhibitor experiments for compound F versus tiazofurin, [tiazofurin] = ● 0.0 mM; ▼ 2.0 mM; ■ 4.0 mM; ◆ 6.0 mM; ▲ 8.0 mM. (E) Dual inhibitor experiments for compound F versus ADP, [ADP] = ● 0.0 mM; ▼ 5.0 mM; ■ 10.0 mM; ◆ 15.0 mM; ▲ 20.0 mM. (F) Dual inhibitor experiment for compound H versus ADP, [ADP] = ● 0.0 mM; ▼ 5.0 mM; ■ 10.0 mM; ◆ 15.0 mM; ▲ 20.0 mM. Conditions as described in Materials and Methods.
The mechanism of inhibition
All of the compounds are uncompetitive inhibitors with respect to IMP and noncompetitive (mixed) with respect to NAD+ (Fig. 3B, C), indicating that the inhibitors bind to both E•IMP and E-XMP*open as designed. Most of the compounds display similar affinities for both complexes, but D and J display a preference for E•IMP (Table 1). To further localize the site of inhibitor binding within the NAD site, we analyzed how the compounds interact with tiazofurin, which binds in the nicotinamide subsite (Hedstrom et al., 1990). Typical dual inhibitor experiments are shown in Fig. 3D–. If two inhibitors are mutually exclusive, a parallel line pattern is observed (Fig. 3D), and α the interaction constant, is infinity (∞). The binding of tiazofurin is mutually exclusive with all of the inhibitors, indicating that all bind in the nicotinamide subsite (G is too potent and K is too weak to permit this analysis). However, none of the inhibitors block ADP binding (Fig. 3E, F; Table 1), which is somewhat surprising since the adenosine subsite is the most different from the host enzyme (Fig. 1B). Compounds B, C, D, H and J interact either synergistically or independently with ADP (α ≤ 1), which indicates that the binding site for these inhibitors does not extend into the ADP subsite. In contrast, the binding of A, E and F antagonizes ADP binding (α ≥ 1), indicating that the binding site for these compounds impinges on the ADP site, either directly, or by inducing a conformation that decreases the affinity for ADP. Compounds A, E and F are not significantly larger than the other inhibitors, which suggests that these compounds have an alternative binding mode. Thus the screen has identified two classes of parasite-selective inhibitors.
Table 1. Mechanism of inhibition.
Nomenclature of Cleland (Cleland, 1963), where noncompetitive (mixed) inhibition is described by two inhibition constants, Kis and Kii. In the present case, Kis and Kii describe the affinity of the inhibitor for the E•IMP and E-XMP* complexes, respectively. UC, uncompetitive; NC, noncompetitive. ME, mutually exclusive; I, independent; S, synergistic; A, antagonistic.
| Inhibitor | vs IMP (UC) Kii (μM) | vs NAD (NC) | Dual Inhibitor Analysis α | ||
|---|---|---|---|---|---|
| E•IMP Kis (μM) | E-XMP* Kii (μM) | Tiazofurin | ADP | ||
| A | 5.6 ± 0.5 | 3 ± 2 | 4 ± 2 | ∞ (ME) | 1.3 (I) |
| B | 2.7 ± 0.2 | 1.7 ± 0.6 | 2.5 ± 1.2 | ∞ (ME) | 0.6 (S) |
| C | 1.5 ± 0.1 | 1.5 ± 0.7 | 1.4 ± 0.2 | ∞ (ME) | 0.3 (S) |
| D | 4.5 ± 0.4 | 1.8 ± 0.5 | 7 ± 4 | ∞ (ME) | 0.7 (S) |
| E | 1.0 ± 0.1 | 4.1 ± 2.0 | 1.5 ± 0.3 | ∞ (ME) | 2.7 (A) |
| F | 3.7 ± 0.4 | 1.4 ± 0.2 | 2.7 ± 0.6 | ∞ (ME) | 2.0 (A) |
| G | 0.17 ± 0.01 | 0.14 ± 0.04 | 0.20 ± 0.05 | n.d. | n.d. |
| H | 0.61 ± 0.02 | 1.3 ± 0.4 | 1.1 ± 0.4 | ∞ (ME) | 0.8 (I) |
| J | 11.4 ± 0.3 | 4.1 ± 0.2 | 15 ± 3 | ∞ (ME) | 0.6 (S) |
Antiparasitic activity in cell culture model of parasite growth
We next assessed if the inhibitors have antiparasitic activity. C. parvum is an obligate intracellular parasite that cannot be maintained in continuous cell culture. However, sporozoites readily infect a variety of epithelial cells, undergoing the first two asexual replication cycles. Even though the lifecycle is incomplete, these in vitro infections are sufficient for an initial evaluation of antiparasitic activity (Arrowood, 2002; Upton, 1997). Since parasite and host cell metabolism are intertwined, we first assessed the cytotoxic and cytostatic effects on human ileocecal adenocarcinoma epithelial HCT-8 cells, a widely used host cell model. Only compound B displayed significant cytotoxic effects in an LDH release assay; G and H are mildly cytotoxic while A, J and K are slightly cytostatic (Fig. 2 and Fig. 4A).
Fig. 4. Activity of the principle hits in a cell culture model of Cryptosporidium infection.
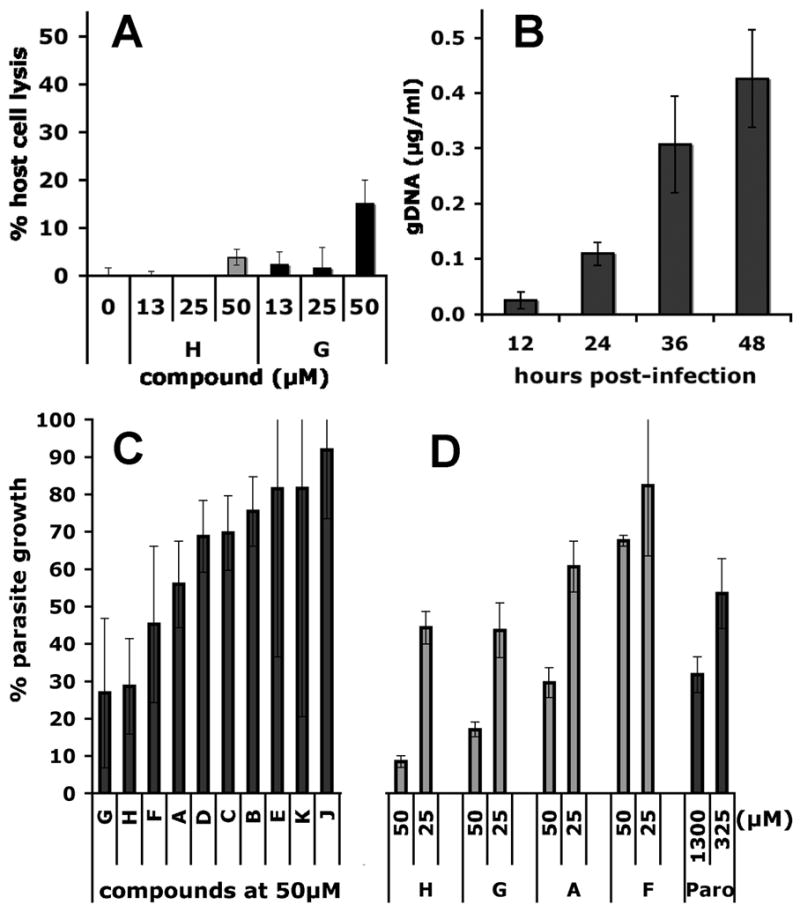
(A) Representative cytoxocity assay showing the cytolytic effects of compounds G and H monitored by the release of lactate dehydrogenase (LDH). (B) Parasite proliferation as measured by RT-PCR over 48 hours, the period over which the anti-parasitic effects of compounds A-K were determined. (C) VVL-ELISA C. parvum growth assay; (D) RT-PCR C. parvum growth assay. Assays as described in Materials and Methods.
HCT-8 cells were infected with oocysts of C. parvum Iowa isolate and parasites proliferated ~15-fold over 48 h as measured by quantitative real time PCR (Fig. 4B). Parasites were also quantified using a biotin-conjugated VVL lectin that specifically recognizes sporozoites and intracellular stages, but not the outer oocyst wall (see Materials and Methods) (Gut and Nelson, 1999b; Hashim et al., 2004; Hashim et al., 2006; Winter et al., 2000). Paromomycin inhibited parasite growth with a value of EC50 = 120 μM, validating the assay (literature values of EC50 = 65–130 μM (Perkins et al., 1998; Woods et al., 1995; You et al., 1996)) (Fig. S2). Compounds A, F, G and H display significant dose-dependent antiparasitic activity in this assay (Fig. 2 and Fig. 4C). We confirmed the antiparasitic activity of A, F, G and H using an RT-PCR assay that measures the abundance of parasite ribosomal RNA genes (Fig. 4D) (Cai et al., 2005). No correlation exists between the antiparasitic activity of the compounds and cytotoxic/cytostatic effects, so the antiparasitic activities must result from direct effects on the parasite. These observations suggest that at least four compounds (A, F, G and H) enter the parasite, which is a very important finding given that failure of drug uptake has been frequently invoked to explain the resistance of Cryptosporidium to antiparasitic drugs (Griffiths et al., 1998; Mead, 2002).
Significance
C. parvum is an important pathogen of both the developed and developing world and a potential bio-warfare agent. This protozoan parasite has eluded drug treatment and no vaccines exist. Eukaryotic pathogens present a particularly challenging problem for drug design because the targets generally bear a close resemblance to host proteins. Surprisingly, the purine salvage pathway of C. parvum relies on an IMPDH likely obtained from a prokaryote through horizontal gene transfer, and thus is very different from the host counterpart (Striepen et al., 2004; Striepen et al., 2002). We designed a HTS to target the highly diverged NAD site by exploiting our detailed understanding of the kinetic mechanism of C. parvum IMPDH. This screen identified ten parasite-selective inhibitors, all of which bind in the NAD site as designed. Two different classes of interactions are observed: five compounds bind only in the nicotinamide portion of the NAD site while three extend from the nicotinamide site into the adenosine subsite. Four compounds display antiparasitic activity in a cell culture model of infection, further validating the choice of IMPDH as target against cryptosporidiosis. Importantly, the new inhibitors are already more potent than paromomycin, the current gold standard for anti-cryptosporidial activity.
Experimental Procedures
Materials
The compound collections were provided by the National Screening Laboratory for the Regional Centers of Excellence in BioDefense and Emerging Infectious Disease (NSRB) at Harvard Medical School. The initial screen included 44,522 compounds from the following libraries: ActiMol TimTec 1, Bionet 2, ChemDiv 2, ChemDiv 3, Maybridge 4, NINDS custom collection, SpecPlus collection, BIOMOL ICCB known bioactives, ICBG 1 fungal extracts and Starr Foundation extracts 1. Compounds D, E, F, G, H, and L were purchased from ChemDiv Inc. (San Diego, CA), K from TimTec Inc. (Newark, DE) and J from Asinex Ltd. (Moscow). Compounds A, B, and C were synthesized as described in the Supporting Material. C. parvum oocysts of the Iowa strain were a kind gift from Dr. Mead (Emory University). VVL-biotin was purchased from Vector Labs (Burlingame, CA).
Enzyme Purification and assays
Recombinant human IMPDH1 and IMPDH2 and C. parvum were expressed in guaB strains of Escherchia coli (which lack endogenous IMPDH) and purified as described previously (Farazi et al., 1997; Mortimer and Hedstrom, 2005; Umejiego et al., 2004). Assays were routinely performed in 50 mM Tris-HCl, pH 8.0, 100 mM KCl, 3 mM EDTA and 1 mM dithiothreitol (assay buffer) at 25 °C. The production of NADH was monitored by following changes in absorbance or fluorescence.
Primary Screen
Table S2 describes the assay protocol. Assays were performed in duplicate in 384 well, clear flat bottom polystyrene non-binding surface microplates (Corning 3640). Inhibitors (100 nl) were added to 30 μl of enzyme solution. An initial absorbance measurement at 340 nm was obtained using an Envision plate reader, and the reaction was initiated by the addition of IMP and NAD (40 μl) to give final assay conditions: 250 μM IMP, 500 μM NAD and 70 nM C. parvum IMPDH in assay buffer. The reaction proceeded for ~3 hrs, then was quenched by the addition of GMP (10 μl, final concentration 25 mM). The absorbance at 340 nm was measured, and the change in absorbance was determined by subtracting the initial value. Positive (12–25 mM GMP) and negative (no inhibitor) controls were included on each plate. This screen identified 134 compounds that displayed >45% inhibition with z scores ≤−10.
Inhibitor characterization
The values of Ki with respect to NAD+ were determined using a saturating IMP concentration (150 μM; Km = 29 μM; (Umejiego et al., 2004)) and varied NAD+ concentrations. The values of Ki with respect to IMP were determined by using fixed NAD+ concentration (500 μM; Km = 150 μM (Umejiego et al., 2004)). The mechanism of inhibition was determined by the fit to the appropriate equation for uncompetitive or noncompetitive inhibition (Cleland, 1963). Dual inhibition experiments with tiazofurin and ADP were performed with constant concentrations of IMP (250 μM) and NAD+ (250 μM). Data were fit an equation describing multiple inhibition (Equation 1).
| (Eq. 1) |
where I and J are the inhibitors; v0 is the initial velocity in the absence of inhibitors; α is the interaction constant and Ki and Kj are the inhibition constants of I and J respectively. Values of α > 8000 were assigned as ∞. Detailed protocols are provided in the Supporting Material.
Cell culture model of C. parvum infection
The human ileocecal adenocarcinoma epithelial cell line, HCT-8, was used to support C. parvum infection in vitro. Oocysts were treated with 10 mM HCl to facilitate excystation (Gut and Nelson, 1999a) and applied to a HCT-8 monolayer. Parasites were measured with biotin-conjugated VVL (Vector Labs) in a cell-based ELISA adapted to a 96 well format (Gut and Nelson, 1999b; Moriarty et al., 2005), or by RT-PCR as described by Cai et al. (Cai et al., 2005). Detailed protocols are provided in the Supporting Material.
Effects of the compounds on the host cells
The cytotoxic and cytostatic effects of compounds A-K on HCT-8 cells were determined using the CytoTox96 assay (Promega) and the LIVE/DEAD assay (Molecular Probes).
Supplementary Material
Acknowledgments
This work was supported by NIH RO1 AI055268 (B.S.). Facilities and compound libraries for high-throughput screening were made available through the National Screening Laboratory for the Regional Centers of Excellence in BioDefense and Emerging Infectious Disease (NSRB/NERCE). We thank members of the NSRB and ICCB-Longwood (Harvard Medical School) for their ongoing assistance. Molecular graphics images were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR-01081).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abrahamsen MS, Templeton TJ, Enomoto S, Abrahante JE, Zhu G, Lancto CA, Deng M, Liu C, Widmer G, Tzipori S, et al. Complete genome sequence of the apicomplexan, Cryptosporidium parvum. Science. 2004;304:441–5. doi: 10.1126/science.1094786. [DOI] [PubMed] [Google Scholar]
- Abubakar I, Aliyu SH, Arumugam C, Usman NK, Hunter PR. Treatment of cryptosporidiosis in immunocompromised individuals: Systematic review and meta-analysis. Br J Clin Pharmacol. 2007;63:387–393. doi: 10.1111/j.1365-2125.2007.02873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrowood MJ. In vitro cultivation of Cryptosporidium species. Clin Microbiol Rev. 2002;15:390–400. doi: 10.1128/CMR.15.3.390-400.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkman DS, Lescano AG, Gilman RH, Lopez SL, Black MM. Effects of stunting, diarrhoeal disease, and parasitic infection during infancy on cognition in late childhood: A follow-up study. Lancet. 2002;359:564–571. doi: 10.1016/S0140-6736(02)07744-9. [DOI] [PubMed] [Google Scholar]
- Cai X, Woods KM, Upton SJ, Zhu G. Application of quantitative real-time reverse transcription-pcr in assessing drug efficacy against the intracellular pathogen cryptosporidium parvum in vitro. Antimicrob Agents Chemother. 2005;49:4437–4442. doi: 10.1128/AAC.49.11.4437-4442.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey CM, Lee H, Trevors JT. Biology, persistence and detection of Cryptosporidium parvum and Cryptosporidium hominis oocyst. Water Res. 2004;38:818–862. doi: 10.1016/j.watres.2003.10.012. [DOI] [PubMed] [Google Scholar]
- Cleland WW. The kinetics of enzyme-catalyzed reactions with two or more substrates or products. Ii Inhibition: Nomenclature and theory. Biochim Biophys Acta. 1963;67:173–187. doi: 10.1016/0006-3002(63)91815-8. [DOI] [PubMed] [Google Scholar]
- Corso PS, Kramer MH, Blair KA, Addiss DG, Davis JP, Haddix AC. Cost of illness in the 1993 waterborne Cryptosporidium outbreak, Milwaukee, Wisconsin. Emerg Infect Dis. 2003;9:426–431. doi: 10.3201/eid0904.020417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digits JA, Hedstrom L. Kinetic mechanism of Tritrichomonas foetus inosine-5′-monophosphate dehydrogenase. Biochemistry. 1999;38:2295–2306. doi: 10.1021/bi982305k. [DOI] [PubMed] [Google Scholar]
- DuPont HL, Chappell CL, Sterling CR, Okhuysen PC, Rose JB, Jakubowski W. The infectivity of Cryptosporidium parvum in healthy volunteers. N Engl J Med. 1995;332:855–859. doi: 10.1056/NEJM199503303321304. [DOI] [PubMed] [Google Scholar]
- Farazi T, Leichman J, Harris T, Cahoon M, Hedstrom L. Isolation and characterization of mycophenolic acid resistant mutants of inosine 5′-monophosphate dehydrogenase. J Biol Chem. 1997;272:961–965. doi: 10.1074/jbc.272.2.961. [DOI] [PubMed] [Google Scholar]
- Fayer R. Cryptosporidium: A water-borne zoonotic parasite. Veterinary Parasitology. 2004;126:37–56. doi: 10.1016/j.vetpar.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Griffiths JK, Balakrishnan R, Widmer G, Tzipori S. Paromomycin and geneticin inhibit intracellular cryptosporidium parvum without trafficking through the host cell cytoplasm: Implications for drug delivery. Infect Immun. 1998;66:3874–3883. doi: 10.1128/iai.66.8.3874-3883.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gut J, Nelson RG. Cryptosporidium parvum: Lectins mediate irreversible inhibition of sporozoite infectivity in vitro. J Eukaryot Microbiol. 1999a;46:48S–49S. [PubMed] [Google Scholar]
- Gut J, Nelson RG. Cryptosporidium parvum: Synchronized excystation in vitro and evaluation of sporozoite infectivity with a new lectin-based assay. J Eukaryot Microbiol. 1999b;46:56S–57S. [PubMed] [Google Scholar]
- Hashim A, Clyne M, Mulcahy G, Akiyoshi D, Chalmers R, Bourke B. Host cell tropism underlies species restriction of human and bovine Cryptosporidium parvum genotypes. Infect Immun. 2004;72:6125–6131. doi: 10.1128/IAI.72.10.6125-6131.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashim A, Mulcahy G, Bourke B, Clyne M. Interaction of Cryptosporidium hominis and Cryptosporidium parvum with primary human and bovine intestinal cells. Infect Immun. 2006;74:99–107. doi: 10.1128/IAI.74.1.99-107.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedstrom L, Cheung K, Wang CC. A novel mechanism of mycophenolic acid resistance in the protozoan parasite Tritrichomonas foetus. Biochem Pharm. 1990;39:151–160. doi: 10.1016/0006-2952(90)90659-9. [DOI] [PubMed] [Google Scholar]
- Hedstrom L, Gan L. IMP dehydrogenase: Structural schizophrenia and an unusual base. Curr Opin Chem Biol. 2006;10:520–525. doi: 10.1016/j.cbpa.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Huang DB, Chappell C, Okhuysen PC. Cryptosporidiosis in children. Semin Pediatr Infect Dis. 2004;15:253–259. doi: 10.1053/j.spid.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Huang DB, White AC. An updated review on Cryptosporidium and Giardia. Gastroenterol Clin North Am. 2006;35:291–314. viii. doi: 10.1016/j.gtc.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Mead JR. Cryptosporidiosis and the challenges of chemotherapy. Drug Resist Updat. 2002;5:47–57. doi: 10.1016/s1368-7646(02)00011-0. [DOI] [PubMed] [Google Scholar]
- Moriarty EM, Duffy G, McEvoy JM, Caccio S, Sheridan JJ, McDowell D, Blair IS. The effect of thermal treatments on the viability and infectivity of Cryptosporidium parvum on beef surfaces. J Appl Microbiol. 2005;98:618–623. doi: 10.1111/j.1365-2672.2004.02498.x. [DOI] [PubMed] [Google Scholar]
- Mortimer SE, Hedstrom L. Autosomal dominant retinitis pigmentosa mutations in inosine 5′-monophosphate dehydrogenase type I disrupt nucleic acid binding. Biochem J. 2005;390:41–47. doi: 10.1042/BJ20042051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins ME, Wu TW, Le Blancq SM. Cyclosporin analogs inhibit in vitro growth of Cryptosporidium parvum. Antimicrob Agents Chemother. 1998;42:843–848. doi: 10.1128/aac.42.4.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera- a visualization system for exploratory research and analysis. J Comp Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Ratcliffe AJ. Inosine 5′-monophosphate dehydrogenase inhibitors for the treatment of autoimmune diseases. Curr Opin Drug Discov Devel. 2006;9:595–605. [PubMed] [Google Scholar]
- Riera TV, Wang W, Josephine H, Hedstrom L. manuscript in preparation. [Google Scholar]
- Striepen B, Pruijssers AJ, Huang J, Li C, Gubbels MJ, Umejiego NN, Hedstrom L, Kissinger JC. Gene transfer in the evolution of parasite nucleotide biosynthesis. Proc Natl Acad Sci USA. 2004;101:3154–3159. doi: 10.1073/pnas.0304686101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striepen B, White MW, Li C, Guerini MN, Malik SB, Logsdon JM, Jr, Liu C, Abrahamsen MS. Genetic complementation in apicomplexan parasites. Proc Natl Acad Sci USA. 2002;99:6304–6309. doi: 10.1073/pnas.092525699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Templeton TJ, Iyer LM, Anantharaman V, Enomoto S, Abrahante JE, Subramanian GM, Hoffman SL, Abrahamsen MS, Aravind L. Comparative analysis of apicomplexa and genomic diversity in eukaryotes. Genome Res. 2004;14:1686–1695. doi: 10.1101/gr.2615304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umejiego NN, Li C, Riera T, Hedstrom L, Striepen B. Cryptosporidium parvum IMP dehydrogenase: Identification of functional, structural and dynamic properties that can be exploited for drug design. J Biol Chem. 2004;279:40320–40327. doi: 10.1074/jbc.M407121200. [DOI] [PubMed] [Google Scholar]
- Upton SJ. In vitro cultivation. In: Fayer R, editor. Cryptosporidium and cryptosporidiosis. CRC Press; Boca Raton: 1997. pp. 181–207. [Google Scholar]
- Winter G, Gooley AA, Williams KL, Slade MB. Characterization of a major sporozoite surface glycoprotein of Cryptosporidum parvum. Funct Integr Genomics. 2000;1:207–217. doi: 10.1007/s101420000028. [DOI] [PubMed] [Google Scholar]
- Woods KM, Nesterenko MV, Upton SJ. Development of a microtitre elisa to quantify development of Cryptosporidium parvum in vitro. FEMS Microbiol Lett. 1995;128:89–94. doi: 10.1111/j.1574-6968.1995.tb07505.x. [DOI] [PubMed] [Google Scholar]
- Xu P, Widmer G, Wang Y, Ozaki LS, Alves JM, Serrano MG, Puiu D, Manque P, Akiyoshi D, Mackey AJ, et al. The genome of Cryptosporidium hominis. Nature. 2004;431:1107–1112. doi: 10.1038/nature02977. [DOI] [PubMed] [Google Scholar]
- You X, Arrowood MJ, Lejkowski M, Xie L, Schinazi RF, Mead JR. A chemiluminescence immunoassay for evaluation of Cryptosporidium parvum growth in vitro. FEMS Microbiol Lett. 1996;136:251–256. doi: 10.1111/j.1574-6968.1996.tb08057.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.