

Abstract
Fra-1 as an integral part of AP-1 (Jun/Fos) drives transcriptional programs involved in several physiologic and pathologic processes. It is also critical for tumor cell motility and metastasis. We have previously shown that two critical elements of Fra-1 promoter, the upstream TPA response element (TRE) and the SRE are necessary for its induction in response to phorbol esters in human pulmonary epithelial cell lines. Here, we have investigated the roles of various MAP kinases in regulating Fra-1 expression in response to TPA. Using pharmacologic and genetic tools, we demonstrate a prominent role for ERK1/2, but not JNK1/2 and p38, signaling in the TPA-induced activation of specific transcription factors that bind to the AP1 site and the SRE. Inhibition of ERK1/2 pathway suppresses Elk1 activation, and c-Jun and Fra-2 recruitment to the promoter.
Keywords: Lung cancer cells, MAP kinases, ERK1/2 kinases, AP-1, c-Jun
INTRODUCTION
Members of the AP-1 (Jun and Fos) family regulate the expression of genes involved in cell proliferation, differentiation, inflammation, host defenses and malignant transformation [1]. Among the Fos (c-Fos, Fra-1, Fra-2 and Fos-B) subfamily of proteins, a prominent role for Fra-1 in regulating tumor progression and metastasis has been well established in a variety of cancer cells [2; 3]. In steady state, the expression of Fra-1 in lung tissues is low. However, upon exposure to tumor-promoting phorbol esters, mitogens, and pro-inflammatory cytokines these cells express a very high level of Fra-1 [4; 5]. In addition, we and others have shown induction of Fra-1 expression in the lung tissue/cell types by pro-carcinogenic and pro-oxidant environmental stimuli, such as cigarette smoke [6; 7], diesel exhaust particles [8], and asbestos [9]. Although the exact roles of Fra-1 in malignant and non-malignant lung diseases are not clearly established in vivo, some cell culture studies have shown that this transcription factor regulates asbestos-induced malignant mesothelioma [9; 10]. We have recently shown that ectopic Fra-1 expression in a human type II-like adenocarcinoma cell line (A549) induces fibroblastoid characteristics and greatly enhances the cell motility and invasion in vitro, and tumor metastasis in vivo [11].
Indeed, a number of independent studies showed that Fra-1 regulates the motility and invasive behavior of various tumorigenic cells by altering gene expression [12]. For example, ectopic Fra-1 has been shown to induce the expression matrix mettaloproteinases-1 and -9, and urokinase-type plasminogen activator receptor, through TPA response elements (TREs) [13]. Consistent with these results, we have found that ectopic expression of Fra-1 in A549 cells upregulates gene expression associated with metastasis and tumor progression (unpublished data). Although studies by us and others [2; 3] have further reinforced the significance of Fra-1 in epithelial cancer cell progression, the exact mechanisms that result in its induction have not been understood.
Our studies demonstrated that the TPA –induced binding of c-Jun to a TRE located at −318 bp of the Fra-1 promoter is critical for its transcriptional induction in human lung epithelial cells [4; 5]. The temporally later induction of Fra-1 than c-Fos was mainly due to the lack a functional serum response element (SRE) in the proximal promoter region [14]. However, we demonstrated that human Fra-1, like c-Fos, contains a functional SRE and the flanking ATF site (located at position −276/−237) which is constitutively occupied by the member of ternary complex factor (TCF) such as Elk1, SRF, and CREB/ATF-1. Mutations within the SRE and ATF sites or −318 TRE abolished the promoter activity suggesting that protein interactions occurring at these elements are critical for TPA stimulated Fra-1 transcription [4; 5]. Here, we have further investigated the impact of MAP kinase signaling on the TPA induced recruitment of transacting factors to the Fra-1 promoter. We report that ERK1/2 signaling, but not JNK1/2 and p38 pathway, is critical for this process. We demonstrate that ERK1/2 signaling, besides TCF activation, modulates the binding of AP-1 proteins (c-Jun and Fra-2) to the promoter in response to TPA stimulation.
EXPERIMENTAL PROCEDURES
Cell culture and reagents
Human lung adenocarcinoma cell line, A549, was grown in RPMI 1640 medium supplemented with 5% fetal bovine serum and antibiotics. Mouse embryonic fibroblasts (MEFs) lacking the erk1 gene (erk1−/−) [15] and isogenic wildtype (erk1 +/+) cells (kindly provided by J Pouyssegur, France) were cultured as previously described [16]. c-Jun (SC-45X), Fra-1(SC-605X), JNK1 (SC-474), ERK2 (SC-154), Elk1 (SC-355), and phospho-Elk1 (ser 383, SC-8406) antibodies were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). The JNK (T183/Y185), c-Jun (Ser73) and ERK (T202/Y204) antibodies were purchased from Cell Signaling Technology, Inc. (Danvers, MA). Pharmacologic inhibitors PD98059, UO126, SP600125, and SB203580 were obtained from EMD Chemicals, Inc. (NJ, USA). Unless indicated otherwise, they were used at 10 µM concentration (SB203580 at 20 µM) in most experiments.
Gene expression analyses
For immunoblot analyses, total protein (30–40 µg) isolated from cells was separated on a 10% SDS-PAGE, transferred to PVDF membranes (Millipore, Inc) and probed with specific antibodies. For Northern blot analysis, 32P-labeled cDNAs specific for Fra-1 and 28S RNA were used as probes and Fra-1 mRNA expression was quantified as described previously [4]. Quantitative real time PCR (qRT-PCR) analysis was performed using TaqMan® assays specific for human and mouse Fra-1 (Applied Biosystems, CA). The threshold cycle (Ct) values of sample (n=3) for each gene were normalized to that of β-actin. The relative value for the vehicle-treated (control) cells was set as one arbitrary unit (AU).
Reporter assays
Cells were transfected with the human Fra1 promoter (−379 to +32 bp )placed upstream of a luciferase reporter gene, (hereafter notated as 379-Luc, for details see [4]) along with the Renilla luciferase (pRL-TK) vector (Promega Corp, Madison, WI). After overnight incubation, cells were serum-starved for 24 h and then treated with either vehicle (DMSO) or TPA (50 ng/ml) for 5 h. Luciferase activity was quantified as described previously [4].
Electrophoretic mobility shift assay (EMSA)
Nuclear extracts (2–3 µg) from control and TPA-treated cells were incubated with a 32P-labeled DNA probe that bore the −318 TRE of human Fra-1 promoter and EMSA was carried out [4]. In some experiments, nuclear extracts were incubated with 2 µg of specific antibodies or non-immune IgG on ice for 2 h prior to their use in EMSA to demonstrate the presence of specific proteins in the complexes.
Chromatin immunoprecipitation (ChIP) assays
Cells (~ 1×107) were treated with TPA for 60 min, and nucleo-protein complexes were cross linked with formaldehyde, soluble chromatin was prepared using a commercially available kit (Upstate Biotechnology Inc., NY) and incubated with specific antibodies. DNA recovered from the immunoprecipitated (IP) products was used as a template for PCR reactions with Fra-1 promoter-specific primers as detailed elsewhere [5].
Statistical Analysis
All assays were performed with triplicates in each experiment, and data from several experiments was used for plotting. Data are represented as mean ± S.D. Statistical significance of the differences between the control and the corresponding treated samples was determined Student’s t-test.
RESULTS
The ERK1/2 pathway is essential for TPA-induced Fra-1 expression
To determine the role of MAP kinases in regulating Fra-1 expression, we first determined the activation of JNK1/2, ERK1/2, and p38 kinase in response to TPA using phospho-specific antibodies in immunoblot analyses (Fig. 1A). As anticipated, TPA robustly stimulated ERK1/2 phosphorylation, which persisted up to 120 min (top). In contrast to this, JNK1/2 was maximally activated by 30min, which returned to basal line by 120 min. The activation of p38 was barely detected under these conditions. We next analyzed the impact of MAP kinase inhibition on TPA stimulated Fra-1 mRNA expression using Northern analyses. As shown in Fig. 1B, UO126 (ERK1/2 pathway inhibitor) and SP600125 (JNK-inhibitor) strongly blocked the TPA-induced expression of Fra-1. The effect of UO126 on TPA-induced Fra-1 mRNA expression was further verified using qRT-PCR (Fig. 1C). TPA strongly stimulated Fra-1expression (bar 2), but this stimulation was suppressed in the presence of UO126 (bar 4). Thus, ERK1/2 pathway appears to be critical for TPA induced Fra-1 expression.
Figure 1. Effect of MAP kinase inhibition on TPA induced Fra-1expression.
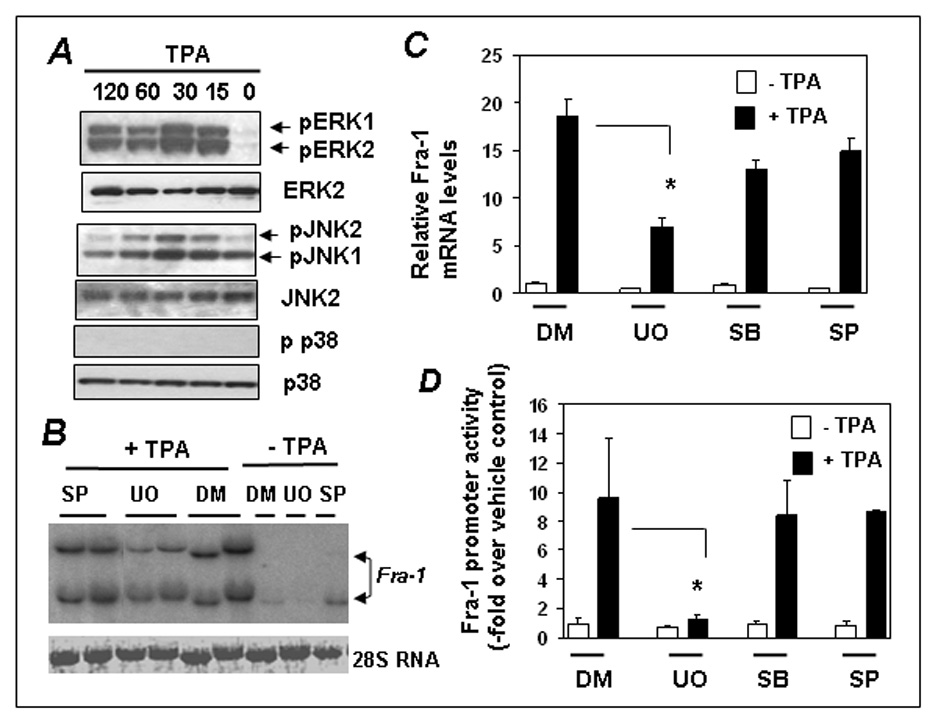
(A) Cells were stimulated with TPA for 0–120 min, cell extracts were isolated, and immunoblotted and JNK1/2, ERK1/2 and p38 kinase activation was analyzed by phosphospecific or native antibodies. (B) Cells were incubated with UO126 (UO) or SP600125 (SP6) for 30 min and then treated with DMSO (DM) or TPA for 3 h. Total RNA was isolated and Fra-1 expression was analyzed by Northern blot analysis. A representative blot of two independent experiments is shown. (C) Cells were incubated with UO126 (UO), SB203580 (SB), or SP600125 (SP6) inhibitors as in panel B for 30 min and then treated with TPA for 90 min, and Fra-1 mRNA expression was analyzed by real-time RT-PCR. Bars represent the mean ± SD (n = 3). * = p<0.001 (D) Cells were transfected with 379-Luc along with the pRL-TK. Cells were serum-starved for 24 h and then stimulated without or with TPA for 5 h. The promoter activity of the 379-Luc under basal condition (control) was set to one. Data represent the mean ± SD (n = 3). * = p<0.03.
We have previously shown that a fragment bearing human −379 to +32 bp of Fra-1 promoter contains required cis-elements for TPA inducible expression [4]. To assess the importance of ERK1/2 signaling on this promoter, we transfected cells with the 379-Luc and the TPA-stimulated luciferase activity was analyzed in the presence or absence of kinase inhibitors. The TPA induced luciferase activity, was blunted only in the presence of UO126; but not SP600125 or SB203580 (Fig. 1D). Therefore, ERK1/2 signaling appears to be important for the TPA induced expression of Fra-1.
The effect of MAP kinase inhibition on TPA induced AP-1 protein binding to the −318 TRE
We have previously shown that Elk1 and ATF/CREB proteins are constitutively bound to the human Fra-1 promoter at a putative SRE and TPA treatment did not markedly affect it [5]. In contrast, we observed an enhanced recruitment of c-Jun to the −318 TRE of Fra-1 promoter following TPA stimulation [5]. Therefore, we next examined the effects of various MAP kinases on AP-1 protein binding to the −318 TRE (Fig. 2). In previous studies we have clearly established the specificity of AP-1 complex binding to this site [4; 5]. As expected, we noticed the formation of a specific AP-1 complex formed with this −318 TRE, which was induced further upon TPA treatment (Fig. 2A, compared lanes 1 and 2 to lanes 6 and 7). MAP kinase inhibition did not significantly affect the AP-1 complex formed under steady state. However, UO126 (lane 8) significantly decreased TPA-induced AP-1 complex formation in contrast to DMSO-treatment (lanes 6 and 7). The p38 (lane 9) or JNK1/2 (lane 10) inhibitors had no such inhibitory effect. This result suggests that ERK pathway regulates TPA-induced AP-1 binding to the −318 TRE.
Figure 2. The effects of MAP kinase inhibition on Jun protein binding to the −318 TRE.
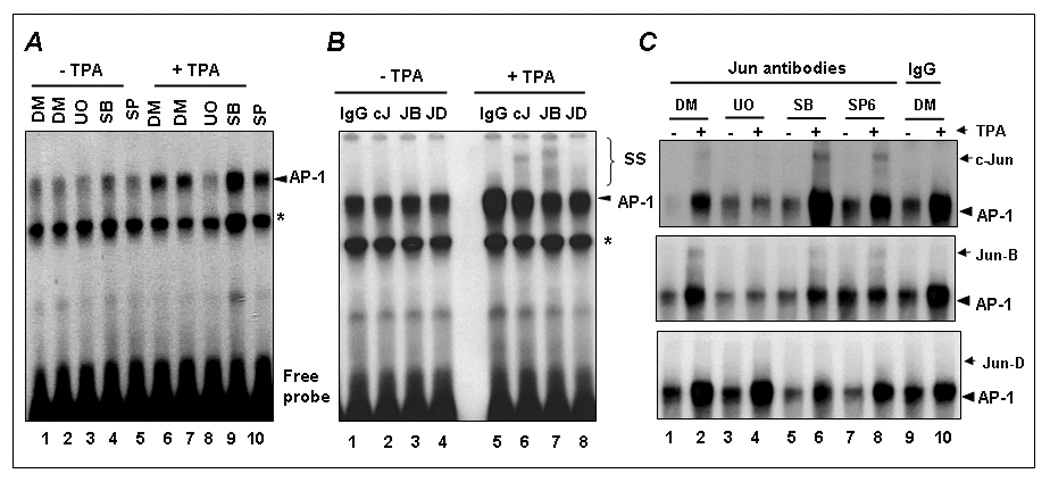
(A) The effect of MAP kinase inhibitors on AP-1 binding to the −318 TRE. Cells were incubated with and without UO126 (UO), SB203580 (SB), or SP600125 (SP) for 30 min and then stimulated with TPA (+) for 60 min. DMSO was used as vehicle control. Nuclear extracts were isolated and EMSA analysis was performed using the labeled −318 TRE as a probe. Arrow indicates the position of AP-1 complex. * indicates the position of non-specific complex. (B) EMSA supershift analysis of the complexes formed at the −318 TRE was performed by incubating nuclear extracts with the indicated Jun antibodies. IgG antibody was used as negative control. The position of AP-1 and supershifted (SS) complexes is indicated. (C) The effects of UO126 (UO), SB203580 (SB), or SP600125 (SP) inhibitors on c-Jun (top), Jun-B (middle), and Jun-D (bottom) binding to the −318 TRE is shown.
We next used antibodies specific for c-Jun, Jun-B and Jun-D in EMSA-complex supershift analyses to determine whether ERK1/2 pathway affects the binding of specific member of the c-Jun family to the −318 TRE. Prior to TPA-treatment, these proteins were not detected in the AP1 complex (Fig. 2B, lanes 2–4). However, anti-c-Jun (lane 6) and anti-Jun-B (lane 7) antibodies supershifted AP-1 protein complex formed in response to TPA treatment. Under these conditions, Jun-D binding (lane 8) was barely detected. As shown in Fig. 2C, inhibition of ERK pathway completely blocked the binding of c-Jun (top panel) and Jun-B (middle panel) to the TRE (lane 4); however, the p38 (lane 6) or JNK1/2 (lane 8) inhibitors had no effect on either c-Jun or Jun-B binding to the −318 TRE. These results suggest that the ERK pathway regulates the binding of c-Jun and Jun-B to the −318 TRE following TPA stimulation.
Inhibition of ERK1/2 blocks TPA-induced recruitment of c-Jun but not Jun-B to the Fra-1 promoter in vivo
EMSA results suggest the ERK1/2 pathway regulates the recruitment of c-Jun and Jun-B with the −318 TRE. To verify whether c-Jun and Jun-B recruitment to the promoter is similarly affected in vivo, we performed a ChIP assay with the human Fra-1 promoter specific primers spanning the −318 TRE (see schematic Fig. 3A). As shown in Fig. 3B, TPA strongly induced the recruitment of c-Jun and Jun-B to the Fra-1 promoter (compare lane 1 to lanes 2 and 3). However, in the presence of UO126 c-Jun binding to the promoter was suppressed (lanes 5 and 6). In contrast to EMSA results, UO126 did not affect Jun-B binding to the endogenous promoter. We have previously shown that Fra-2 binds to the Fra-1 promoter following TPA stimulation both in vitro and in vivo [4]. Therefore, we have examined whether ERK1/2 signaling also controls Fra-2 binding to the endogenous Fra-1 promoter. Consistent with the previous results, TPA stimulated the binding of Fra-2 to the endogenous Fra-1 promoter, which was inhibited by UO126. We examined the effect of ERK inhibition on Elk1 activation following TPA stimulation by western blot analysis. We chose not to perform ChIP assays as Ekl1 constitutively bind to the Fra-1 promoter and this binding was altered upon mitogenic stimulation. Following serum starvation overnight cells were treated with vehicle or UO126 and then stimulated TPA for 30 min. Cell lysates were prepared and probed with phosphospecific Elk1 antibodies by immunoblot analysis. As shown in Fig. 3C, TPA strongly induced Elk1 phosphorylation (lane 1), but such activation was blocked by ERK1/2 inhibitor UO126 (lane 4).
Figure 3. The effects of MAP kinase inhibition on Jun protein binding to the −318 TRE.
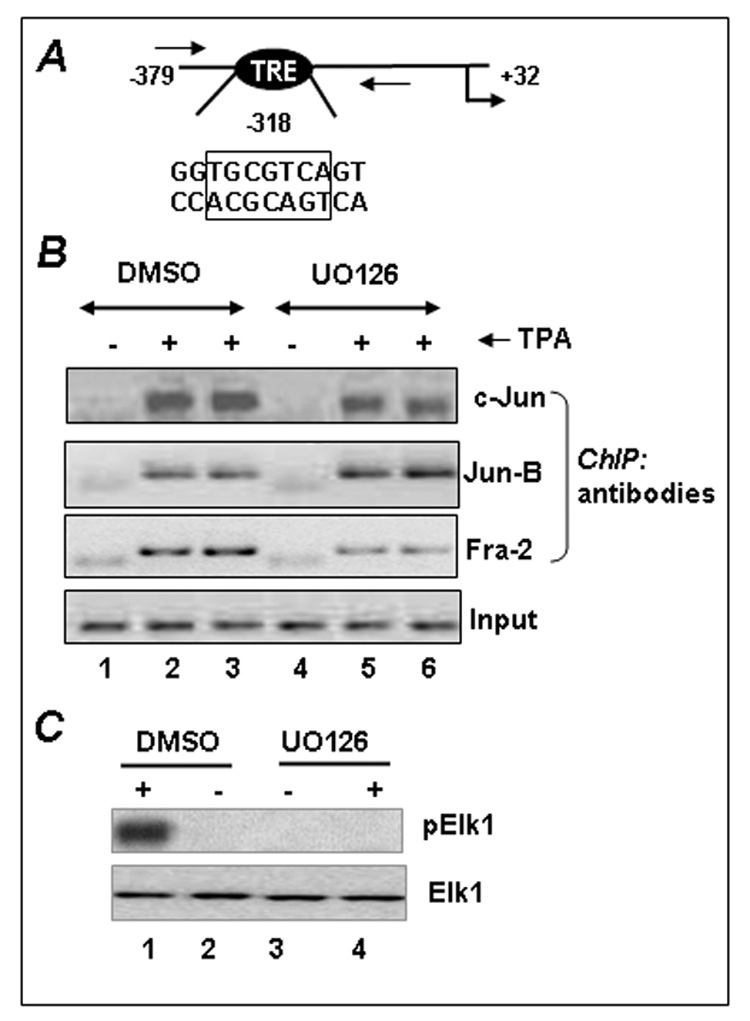
(A) The position and sequence of the functional TRE of human Fra-1 promoter located at position −318 is shown. Forward and reverse arrows indicate the position of the primers used in ChIP assays (see Ref 2 for more details). (B) The effect of ERK inhibition on the recruitment of c-Jun to the Fra-1 promoter. A549 cells were serum-starved for 24 h and incubated with UO126 or DMSO for 30 min prior to treatment without (−) or with TPA (+). After a 60-min incubation, chromatin protein-DNA complexes were cross-linked and ChIP assays were performed using c-Jun and Fra-2 antibodies and non-immune IgG (data not shown). Experiments were repeated at least twice to obtain reproducible results. (C) Immunoblot analysis of Elk1 activation by TPA in the presence and absence of U0126. A549 cells were serum-starved for 12 h and then stimulated with TPA for 30 min, and Elk1 activation was analyzed by Western blot analysis using phopshospecific (p) and native Elk1 antibodies.
ERK1 kinase regulates Fra-1 induction by TPA
To firmly establish the roles of the ERK1 and ERK2 kinases in the regulation of Fra-1 induction by TPA, we have utilized MEFs lacking the erk1 gene (Erk1 −/−). The extent of Fra-1 induction by TPA in these cells was compared to that of wildtype (Erk1 +/+) cells. First, we examined TPA-stimulated ERK1/2 activation by immunoblot analysis using phosphospecific antibodies. As shown in Fig. 4A, TPA strongly stimulated ERK1/2 phosphorylation in Erk1 +/+ cells (compare lanes 1 and 2). As anticipated, only ERK2 was activated in Erk1 −/− cells (lanes 4–6). We next performed a Northern blot analysis to determine the effect of Erk1-deficiency on TPA-stimulated Fra-1 expression, TPA strongly stimulated Fra-1 mRNA expression in erk1/2 +/+ cells (lanes 9–11), and UO126 blocked it (lanes 14 and 15). TPA-stimulated expression of Fra-1 in Erk1 +/+ cells (lanes 9–11) and it was inhibited by UO126. However, Fra-1 induction was remarkably weaker Erk1 −/− cells (lanes 1–3) than the Erk1 +/+ cells (lanes 9–11). These results were confirmed using a qRT-PCR analysis of the Fra-1 transcripts (data not shown). These results collectively support a prominent role for ERK1 kinase in regulating the TPA induced expression of Fra-1.
Figure 4. Erk1 signaling is critical for TPA-stimulated Fra-1 induction.
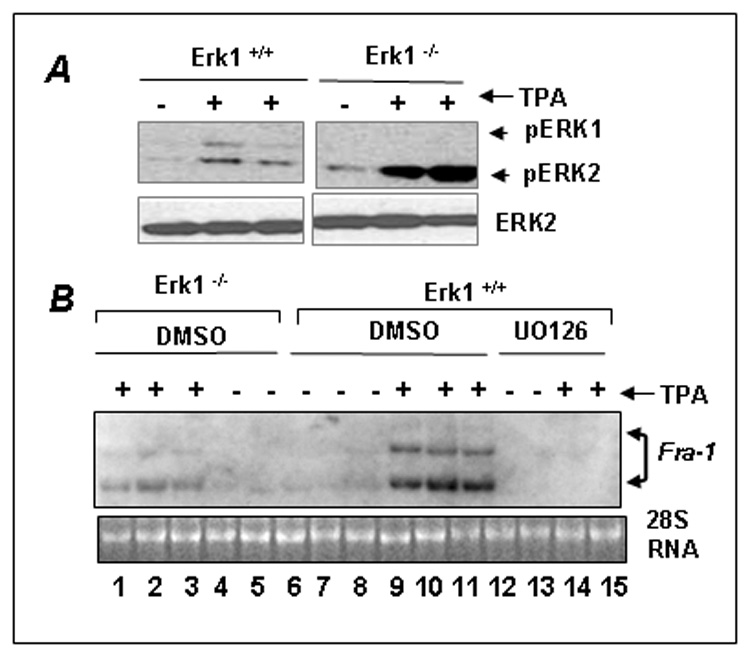
(A) A Western blot showing the activation of ERK1 and ERK2 kinases by TPA in the wildtype (Erk1+/+) and erk1- deficient (Erk1−/−) MEFs. The MEFs were serum-starved for 2 h and then stimulated with TPA for 15 min, and ERK activation was analyzed by Western blot analysis. (B) The effect of erk1-deficiency on TPA-stimulated Fra-1 expression. Erk1+/+ and Erk1−/− cells were serum starved for 2 h and then stimulated with vehicle DMSO (−) or with (+) TPA for 3 h. To determine, the effects of ERK1/2 inhibition on Fra-1 expression, Erk1+/+ cells were incubated with DMSO or UO126 for 30 min prior to TPA stimulation. Northern blot was performed using Fra-1 cDNA as a probe. 28S RNA stained with ethidium bromide is shown as loading control.
DISCUSSION
In the present study, we have demonstrated that ERK signaling plays an important role in regulating tumor promoter TPA stimulated Fra-1 transcription in human lung epithelial cells. Inhibition of either JNK1/2 or p38 signaling failed to suppress TPA-stimulated Fra-1 expression. Moreover, using MEFS lacking erk1 gene, we have provided genetic evidence for a prominent role played by erk1 signaling in regulating the induction of Fra-1. Importantly, in the present study we have demonstrated that inhibition of ERK pathway blocks the recruitment of c-Jun and Fra-2 transcription factors at the endogenous Fra-1 promoter following TPA-stimulation. We have previously shown using ChIP assays that Elk1, SRF, and CREB/ATF proteins, the well established targets of ERK signaling, are constitutively bound to a critical SRE of the Fra-1 promoter in pulmonary epithelial cells, and treatment with either TPA or cigarette smoke did not alter their DNA binding patterns [5; 6] suggesting that ERK1/2 probably stimulates phosphorylation of these factors after engagement with the promoter. Indeed, we found that Inhibition of the ERK pathway suppresses TPA-induced Elk1 phosphorylation in A549 cells (Fig. 3C). Thus, it seems that the ERK1/2 pathway controls Fra-1 transcription both by regulating the activation of promoter bound Elk1 and facilitating an enhanced recruitment of c-Jun and Fra-2 to the TRE.
c-Jun is a major target of JNK signaling. The requirement of c-Jun but not its upstream activator JNK1/2 pathway in TPA-stimulated Fra-1 transcription strongly suggests that in the absence of JNK1/2 activation, and ERK1/2 signaling can drive the Fra-1 transcription by regulating the activation of AP-1 and TCF factors. Previously, we have shown that JNK pathway minimally, if any, contributes to TNFα- stimulated Fra-1 transcription in A549 cells, despite a high level of JNK activation by this pro-inflammatory cytokine. In the present study, we have also shown that TPA strongly stimulates JNK1/2 phosphorylation, but inhibition of this pathway had a minimal effect on TPA-stimulated Fra-1 mRNA expression and promoter activity (Fig. 1). These results suggest that ERK1/2 pathway, but not JNK1/2, drives Fra-1 transcription in human lung cell lines. We have shown that mutations in either −318 TRE or in the SRE cripples the mitogen-inducibility of the human Fra-1 promoter [5]. The diminished levels of c-Jun binding at the Fra-1 promoter in the absence of ERK1/2 signaling suggest that this MAP kinase pathway also plays a key role in the recruitment of c-Jun at the Fra-1 promoter in response to TPA. Intriguingly, TPA has been shown to stimulate c-Jun phosphorylation via ERK1/2 signaling, in the absence of JNK1/2 signaling [17].
We have also provided genetic evidence that ERK1-mediated signaling is critical for TPA-stimulated Fra-1 transcription. Despite the elevated levels of ERK2 activation in response to TPA in the erk1−/− cells, the magnitude of Fra-1 induction by TPA is strikingly lower, as compared to wildtype cells (Fig. 4). This result suggests that ERK1 and ERK2 distinctly regulate gene transcription depending on promoter context. In support of this notion, erk1-deficient mice display decreased levels of TPA-promoted incidence and progression of skin cancer and mouse keraticocytes lacking erk1 proliferate poorly, as compared to their wildtype counter parts [18]. Conversely, it has been shown that ERK2 but not ERK1 promotes Ras-induced fibroblast cell transformation and tumor formation in nude mice [19]. However, our current findings that ERK1 deficiency dampens tumor promoter induced Fra-1 expression and that overexpression of Fra-1 greatly enhances malignant potential of lung cancer cells [11] collectively support a potential role for ERK1/Fra-1 mediated transcriptional program in lung cancer cell progression and invasion. Further studies using Fra-1 and Erk1/2 deficient genetic mouse models are warranted to firmly establish this concept in vivo.
In conclusion, the present study demonstrates that ERK1/2 signaling plays a prominent role in regulating tumor promoter induced Fra-1 proto-oncogene induction in human lung cancer cells. Our results with erk1-deficient cells highlight a prominent role for ERK1 signaling in mediating this process. Furthermore, we have dissected the mechanisms by which ERK signaling regulates the Fra-1 induction and it seems that this appears to be regulated at least at two levels: 1) through Elk1 activation, and 2) the binding of c-Jun and Fra-2 to the TRE.
Acknowledgments
We thank Jacques Pouyssegur (Center A. Lacassagne, Nice, France) for providing us with erk1-deficient and isogenic wildtype cells used in this study. These studies are supported by National Institutes of Health Grants ES11863 and HL66109 and (to SPR) and to CA87282 and CA105005 to DVK. We apologize for not citing several references in the article section, due to space limitation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer. 2003;3:859–868. doi: 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- 2.Young MR, Colburn NH. Fra-1 a target for cancer prevention or intervention. Gene. 2006;379:1–11. doi: 10.1016/j.gene.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 3.Giancotti V. Breast cancer markers. Cancer Lett. 2006;243:145–159. doi: 10.1016/j.canlet.2006.01.035. [DOI] [PubMed] [Google Scholar]
- 4.Adiseshaiah P, Papaiahgari SR, Vuong H, Kalvakolanu DV, Reddy SP. Multiple cis-Elements Mediate the Transcriptional Activation of Human fra-1 by 12-O-Tetradecanoylphorbol-13-acetate in Bronchial Epithelial Cells. J Biol Chem. 2003;278:47423–47433. doi: 10.1074/jbc.M303505200. [DOI] [PubMed] [Google Scholar]
- 5.Adiseshaiah P, Peddakama S, Zhang Q, Kalvakolanu DV, Reddy SP. Mitogen regulated induction of FRA-1 proto-oncogene is controlled by the transcription factors binding to both serum and TPA response elements. Oncogene. 2005;24:4193–4220. doi: 10.1038/sj.onc.1208583. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Q, Adiseshaiah P, Reddy SP. Matrix metalloproteinase/epidermal growth factor receptor/mitogen-activated protein kinase signaling regulate fra-1 induction by cigarette smoke in lung epithelial cells. Am J Respir Cell Mol Biol. 2005;32:72–81. doi: 10.1165/rcmb.2004-0198OC. [DOI] [PubMed] [Google Scholar]
- 7.Hirama N, Shibata Y, Otake K, Machiya J, Wada T, Inoue S, Abe S, Takabatake N, Sata M, Kubota I. Increased surfactant protein-D and foamy macrophages in smoking-induced mouse emphysema. Respirology. 2007;12:191–201. doi: 10.1111/j.1440-1843.2006.01009.x. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Q, Kleeberger SR, Reddy SP. DEP-induced fra-1 expression correlates with a distinct activation of AP-1-dependent gene transcription in the lung. Am J Physiol Lung Cell Mol Physiol. 2004;286:L427–L436. doi: 10.1152/ajplung.00221.2003. [DOI] [PubMed] [Google Scholar]
- 9.Ramos-Nino ME, Timblin CR, Mossman BT. Mesothelial cell transformation requires increased AP-1 binding activity and ERK-dependent Fra-1 expression. Cancer Res. 2002;62:6065–6069. [PubMed] [Google Scholar]
- 10.Ramos-Nino ME, Scapoli L, Martinelli M, Land S, Mossman BT. Microarray Analysis and RNA Silencing Link fra-1 to cd44 and c-met Expression in Mesothelioma. Cancer Res. 2003;63:3539–3545. [PubMed] [Google Scholar]
- 11.Adiseshaiah P, Lindner DJ, Kalvakolanu DV, Reddy SP. FRA-1 proto-oncogene induces lung epithelial cell invasion and anchorage-independent growth in vitro, but is insufficient to promote tumor growth in vivo. Cancer Res. 2007;67:6204–6211. doi: 10.1158/0008-5472.CAN-06-4687. [DOI] [PubMed] [Google Scholar]
- 12.Guo W, Giancotti FG. Integrin signalling during tumour progression. Nat Rev Mol Cell Biol. 2004;5:816–826. doi: 10.1038/nrm1490. [DOI] [PubMed] [Google Scholar]
- 13.Tulchinsky E. Fos family members: regulation, structure and role in oncogenic transformation. Histol Histopathol. 2000;15:921–928. doi: 10.14670/HH-15.921. [DOI] [PubMed] [Google Scholar]
- 14.Tsuchiya H, Fujii M, Niki T, Tokuhara M, Matsui M, Seiki M. Human T-cell leukemia virus type 1 Tax activates transcription of the human fra-1 gene through multiple cis elements responsive to transmembrane signals. J Virol. 1993;67:7001–7007. doi: 10.1128/jvi.67.12.7001-7007.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pages G, Guerin S, Grall D, Bonino F, Smith A, Anjuere F, Auberger P, Pouyssegur J. Defective thymocyte maturation in p44 MAP kinase (Erk 1) knockout mice. Science. 1999;286:1374–1377. doi: 10.1126/science.286.5443.1374. [DOI] [PubMed] [Google Scholar]
- 16.Roy SK, Hu J, Meng Q, Xia Y, Shapiro PS, Reddy SP, Platanias LC, Lindner DJ, Johnson PF, Pritchard C, Pages G, Pouyssegur J, Kalvakolanu DV. MEKK1 plays a critical role in activating the transcription factor C/EBP-beta-dependent gene expression in response to IFN-gamma. Proc Natl Acad Sci U S A. 2002;99:7945–7950. doi: 10.1073/pnas.122075799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morton S, Davis RJ, McLaren A, Cohen P. A reinvestigation of the multisite phosphorylation of the transcription factor c-Jun. Embo J. 2003;22:3876–3886. doi: 10.1093/emboj/cdg388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bourcier C, Jacquel A, Hess J, Peyrottes I, Angel P, Hofman P, Auberger P, Pouyssegur J, Pages G. p44 mitogen-activated protein kinase (extracellular signal-regulated kinase 1)-dependent signaling contributes to epithelial skin carcinogenesis. Cancer Res. 2006;66:2700–2707. doi: 10.1158/0008-5472.CAN-05-3129. [DOI] [PubMed] [Google Scholar]
- 19.Vantaggiato C, Formentini I, Bondanza A, Bonini C, Naldini L, Brambilla R. ERK1 and ERK2 mitogen-activated protein kinases affect Ras-dependent cell signaling differentially. J Biol. 2006;5:14. doi: 10.1186/jbiol38. [DOI] [PMC free article] [PubMed] [Google Scholar]