

Abstract
Brain energy metabolism is increased during postnatal development and diminished in neurodegenerative diseases linked to senescence. The objective of this study was to determine if these conditions could involve postnatal or senescence-related shifts in activity or expression of dihydrolipoamide dehydrogenase (DLDH), a key mitochondrial oxidoreductase. Rats ranging from 10 to 60 days of age were used in studies of postnatal development, whereas rats aged 5 or 30 months were used in the aging studies. The expression of DLDH was determined by Western blot analysis using anti-DLDH antibodies and DLDH diaphorase activity was measured by an in-gel activity staining method using nitroblue tetrazolium (NBT)/NADH. Activity of DLDH dehydrogenase was measured as NAD+ oxidation of dihydrolipoamide. When these measures were considered in separate groups of 10-, 20-, 30-, or 60-day-old rats, all three showed an increase between 10 and 20 days of age. However, dehydrogenase activity of DLDH showed a further, progressive increase from 20 days to adulthood, in the absence of any further change in DLDH expression or diaphorase activity. No age-related decline in DLDH activity or expression was evident over the period from 5 to 30 months of age. Moreover, aging did not render DLDH more susceptible to oxidative inactivation by mitochondria-generated reactive oxygen species (ROS). Taken together, results of the present study indicate that (1) brain DLDH expression and activity undergo independent postnatal maturational increases; (2) Senescence does not confer any detectable change in the activity of DLDH or its susceptibility to inactivation by mitochondrial oxidative stress.
Keywords: aging, brain, dihydrolipoamide dehydrogenase, mitochondria, oxidative stress, postnatal development, reactive oxygen species
1. Introduction
Mammalian mitochondrial dihydrolipoamide dehydrogenase (DLDH, EC 1.8.1.4) is a flavin-dependent, pyridine dinucleotide oxidoreductase. It is the third component of pyruvate dehydrogenase complex, α-ketoglutarate dehydrogenase complex, and branched chain keto acid dehydrogenase complex (Patel and Roche, 1990, Williams, 1992). DLDH is also the L protein in the glycine cleavage system (Faure, et al., 2000, Neuburger, et al., 2000). Structurally, the active form of DLDH is a stable homodimer, with each monomer possessing a non-covalently but tightly bound FAD molecule, a transiently bound NAD+ or NADH molecule, and a redox active center containing two cysteine residues (Cys-45 and Cys-50 in both human and rat) that are directly engaged in thiol-disulfide exchange reactions during catalysis (Brautigam, et al., 2005, Ciszak, et al., 2006, Thorpe and Williams, 1976, Williams, 1992). In vivo, DLDH oxidizes dihydrolipoamide that is covalently linked to acyltransferase using NAD+ as the electron acceptor, leading to the release of NADH (Patel and Roche, 1990, Vettakkorumakankav and Patel, 1996, Williams, 1992). This reaction is usually referred to as the forward reaction of DLDH, as opposed to the reverse reaction, in which DLDH catalyzes the oxidation of NADH using lipoamide as the electron acceptor.
DLDH exhibits a number of additional functional capacities. In vitro, DLDH can act as a diaphorase (Massey, et al., 1960) that is capable of transferring electrons from NADH to electron acceptors such as cytochrome c (Igamberdiev, et al., 2004) and ubiquinone (Olsson, et al., 1999, Xia, et al., 2001), and to electron-accepting dyes such as 2,6-dichlorophenolindophenol (DCPIP) (Patel, et al., 1995) and nitroblue tetrazolium (NBT) (Scouten and McManus, 1971, Sokatch, et al., 1981). While DLDH itself may be a source of reactive oxygen species (Bando and Aki, 1991, Gazaryan, et al., 2002, Sreider, et al., 1990, Tahara, et al., 2007), it is also capable of scavenging nitric oxide (Igamberdiev, et al., 2004) and can serve as an antioxidant by protecting other proteins against oxidative inactivation by 4-hydroxyl-2-nonenal (Korotchkina, et al., 2001). Moreover, DLDH can also act as a proteolytic enzyme when the stability of its homodimer is altered (Babady, et al., 2007).
It is noteworthy that decreases in the activity of the DLDH-associated complexes α-ketoglutarate dehydrogenase and pyruvate dehydrogenase, in brain, represent a common element in several age-associated neurodegenerative diseases, including Alzheimer’s and Parkinson’s diseases (Gibson, et al., 2000, Sullivan and Brown, 2005). Studies of adult DLDH-deficient mice have suggested that a partial decrease of DLDH, which is sufficient to diminish activity of its associated enzyme complexes (Johnson, et al., 1997), results in an elevated level of susceptibility to chemical neurotoxicity (Klivenyi, et al., 2004). Moreover, variations in the DLD gene have been linked to Alzheimer’s disease (Brown, et al., 2004, Brown, et al., 2007).
Whereas a loss of DLDH activity can be linked to neurodegenerative disease, the effect of aging, per se, on the function of DLDH has not been investigated. Thus, one purpose of the current study was to determine whether or not DLDH of brain mitochondria exhibits an age-related decline in its function, or an increase in its susceptibility to oxidative inactivation. It is well established that oxidative stress is associated with aging and brain dysfunction (Navarro and Boveris, 2007), and a significant contributing factor may be the oxidative inactivation of selected proteins (Smith, et al., 1992). For example, it has been previously reported that upon brain aging, both complex I and complex IV activities decrease in accordance with an increase in mitochondrial oxidative stress (Navarro and Boveris, 2004, Navarro and Boveris, 2007).
A second purpose of the current study was to determine the extent to which maturational changes in DLDH could be involved in postnatal ontogeny of the rodent brain. The rat brain is functionally and bioenergetically immature at birth. During the normal development of the rat brain, ketone bodies and glucose are used as respiratory substrates, whereas the energy requirements of the adult rat brain are almost exclusively dependent on glucose oxidation (Malloch, et al., 1986). Such a transition in fuel utilization is accompanied by two general characteristics that occur in mitochondria. First, mitochondria isolated from the brain of newborn rats have a poor capacity of oxidative phosphorylation (Land, et al., 1977). Second, as energy demand and oxygen consumption increase postnatally, the activities of many mitochondrial enzymes involved in energy metabolism are gradually increased (Kalous, et al., 2001). These enzymes include adenine nucleotide translocase (Schonfeld and Bohnensack, 1995), creatine kinase (Holtzman, et al., 1993, Schonfeld and Reiser, 2007), malic enzyme (Bukato, et al., 1992), the pyruvate dehydrogenase complex (Malloch, et al., 1986), the electron transport chain complexes (Almeida, et al., 1995, Bates, et al., 1994), Krebs cycle enzymes (Clark, et al., 1981, Leong and Clark, 1984), glutamate dehydrogenase (Leong and Clark, 1984), as well as antioxidant enzymes such as superoxide dismutase, catalase and glutathione peroxidase (Mavelli, et al., 1982). To our knowledge, however, changes in rat brain DLDH expression and activity during postnatal development have not been reported.
Development-related studies were conducted in rats ranging from 10 to 60 days of age, while aging-related studies were conducted in rats when they were 5 or 30 months of age. In these age groups we studied (i) DLDH expression measured by Western blot immunostaining using anti-DLDH antibodies (ii) DLDH diaphorase activity measured by an in-gel activity staining method using NBT/NADH (Yan, et al., 2007) and (iii) DLDH dehydrogenase activity measured spectrophotometrically using dihydrolipoamide as the substrate in the presence of NAD+ (Patel and Hong, 1998, Patel, et al., 1995). Results of the present study suggest that (i) DLDH undergoes postnatal maturation during brain development, and (ii) DLDH does not lose its activity or gain susceptibility to oxidative stress during the normal brain aging process.
2. Experimental procedures
2.1. Animal and chemicals
Pregnant female Sprague-Dawley rats and adult male Sprague-Dawley rats were obtained from Harlan (Indianapolis, IN). In postnatal development studies, pups of either sex were used, while in the aging studies, only male rats were used. All chemicals were purchased from Sigma (St. Louis, MO) unless otherwise stated. Dihydrolipoamide was prepared using sodium borohydride reduction of lipoamide as previously described (Patel and Hong, 1998, Patel, et al., 1995).
2.2. Preparation of mitochondria
The isolation of mitochondria from whole brain was carried out as previously outlined (Sims, 1993) with modifications (Yan, et al., 2007). Brains were removed rapidly and homogenized in 15 ml of ice-cold, mitochondrial isolation buffer containing 0.32 M sucrose, 1 mM EDTA and 10 mM Tris-HCl, pH 7.1. The homogenate was centrifuged at 1,330 g for 10 min and the supernatant was saved. The pellet was resuspended in 0.5 (7.5 ml) volume of the original isolation buffer and centrifuged again under the same conditions. The two supernatants were combined and centrifuged further at 21,200 g for 10 min. The resulting pellet was resuspended in 12% Percoll solution that was prepared in mitochondrial isolation buffer and centrifuged at 6,900 g for 10 min. The resulting supernatant was then carefully removed by vacuum. The obtained soft pellet was resuspended in 10 ml of the mitochondrial isolation buffer and centrifuged again at 6,900 g for 10 min. Preparation of mitochondria from liver, heart and kidney was carried out as previously described (Navarro, et al., 2004, Yan, et al., 2002, Yan, et al., 2005). All of the mitochondrial pellets obtained after centrifugation were either used immediately or frozen at −80°C until analysis. All the protein concentrations were determined by bicinchoninic acid assay (Smith, et al., 1985) using a kit obtained from Pierce (Rockford, IL).
2.3. Preparation of mitochondrial extracts
Whole mitochondrial extracts as the source of DLDH analyses were prepared as previously described (Patel and Hong, 1998, Patel, et al., 1995). Briefly, mitochondrial pellet (either fresh or frozen) was resuspended at a protein concentration of approximately 0.5 mg/ml in a hypotonic buffer containing 20 mM potassium phosphate (pH 7.4), 1% Triton X-100, 2 mM EDTA, 2 mM EGTA and protease inhibitors. The suspension was sonicated in a Branson Sonifier 150, four times for 30 s at 1-min intervals. The sonicated sample was kept on ice for 30 min and clarified by centrifugation at 20,000 g for 30 min. The clear DLDH-containing supernatant was then used for mitochondrial DLDH assays. Where indicated, mitochondrial extracts were passed through PD-10 columns (Amersham Bioscience) to remove small molecular weight molecules such as NADH according to the instructions given by the manufacturer.
2.4. Western blot detection and densitometric quantification of DLDH expression
For detection and densitometric quantification of DLDH expression, mitochondrial extracts were resolved by SDS-PAGE (10% w/v) followed by electrophoretic gel transfer to Hybond-C membranes with a Mini-Trans-Blot electrophoretic transfer cell (Bio-Rad, Richmond, CA) according to the method described by Towbin et al. (Towbin, et al., 1979) with slight modifications (Yan and Sohal, 1998). Transference was carried out at 100 V (constant voltage) for 1 h in a buffer containing 25 mM Tris, 192 mM glycine, and 10% methanol (v/v), pH 8.3. Western blot detection of DLDH using anti-DLDH antibodies was performed as previously described (Yan, et al., 1998). The blots were incubated with 50 ml of 5% nonfat dried milk (w/v) for 1 h, followed by three washes, 10 min each, with Tris-buffered saline that contained 0.1% Tween-20 (TBST). Blots were then incubated over night at 4°C with anti-DLDH antibodies purchased from US Biological (Swampscott, MA) (diluted 1:25,000 in TBST containing 0.2% BSA). The primary antibody was removed and the blots were washed three times, 10 min each, with TBST. The blots were then incubated in horse-radish peroxidase-conjugated goat anti-rabbit IgG (diluted 1:50,000 in TBST containing 0.2% BSA) for 3 h at room temperature. After washing the blots with TBST three times (10 min each), the DLDH protein band was visualized with an enhanced chemiluminescence kit obtained from Amersham Bioscience. All gel and immunoblot images were scanned by an EPSON PERFECTION 1670 scanner and densitometric quantifications were performed using Scion Image software (version 4.0.3).
2.5. Spectrophotometric measurement of DLDH dehydrogenase activity
DLDH dehydrogenase activity was measured in the forward reaction or in the reverse reaction where indicated. In the forward reaction, the activity was measured by DLDH catalyzed oxidation of dihydrolipoamide at the expense of NAD+ (Patel and Hong, 1998, Patel, et al., 1995). The final volume of reaction was 1 ml, and the mixture contained 100 mM potassium phosphate, pH 8.0, 1.5 mM EDTA, 0.6 mg/ml BSA, 3.0 mM dihydrolipoamide and 3.0 mM NAD+. In the reverse reaction, DLDH dehydrogenase activity was measured by reduction of lipoamide at the expense of NADH. The final volume of reaction was also 1 ml, and the mixture contained 100 mM potassium phosphate, pH 6.3, 1.5 mM EDTA, 0.6 mg/ml BSA, 0.6 mM lipoamide, 0.1 mM NAD+ and 0.1 mM NADH. For both the forward and the reverse reactions, a solution containing all the assay components except mitochondrial extracts was used as the blank. The reaction was initiated by the addition of appropriate amount of mitochondrial extracts (10–20 μg/ml assay solution) and the change in absorbance at 340 nm was followed at room temperature. An extinction coefficient of 6.22 mM−1 cm−1 for NADH was used for the calculation of enzyme activity (Patel and Hong, 1998, Patel, et al., 1995). One unit of dehydrogenase activity was defined as 1 μmol of NAD+ reduced or 1 μmol of NADH oxidized per min.
2.6. In-gel DLDH diaphorase activity staining
In-gel staining of DLDH diaphorase activity by NBT/NADH was performed using blue native polyacrylamide gel electrophoresis (BN-PAGE) as recently described (Yan, et al., 2007). Essentially, a non-gradient blue native gel (9%, w/v) was performed. The gel buffer contained 500 mM aminocaproic acid and 50 mM Bis-Tris, pH 7.0. The cathode buffer contained 50 mM tricine, 15 mM Bis-Tris, pH 7.0, and 0.02% Serva blue G-250, whereas the anode buffer contained 50 mM Bis-Tris, pH 7.0. The sample buffer was 50 mM aminocaproic acid (final concentration) containing 0.3% Serva blue G-250 (w/v, final concentration). Following sample loading (typically, 20 μg protein/lane), the gel was run at 150 V until the front line had entered into one-third of the gel where the cathode buffer was replaced by the one that did not have Serva blue G-250 (50 mM Tricine, 15 mM Bis-Tris, pH 7.0). Gel running was then continued at 200 V until complete. The gel was incubated in 50 mM potassium phosphate buffer (pH 7.0) containing 0.2 mg/ml NBT and 0.1 mg/ml NADH. When an appropriate color of DLDH band appeared, the staining solution was discarded and the gel was scanned immediately for gel documentation.
2.7. Spectrophotometric measurement of DLDH diaphorase activity
Where indicated, the diaphorase activity of DLDH was also measured by DLDH catalyzed reduction of 2,6-dichlorophenolindophenol (DCPIP) at the expense of NADH (Patel, et al., 1995). The final volume of reaction was 1 ml, and the mixture contained 1.5 mM EDTA, 0.6 mg/ml BSA, 40μM DCPIP, and 200 μM NADH. The reaction was initiated by the addition of an appropriate amount of mitochondrial extract (10–20 μg/ml assay solution) and the decrease in absorbance at 600 nm was followed at room temperature. An extinction coefficient of 21 mM−1 cm−1 for the oxidized form of DCPIP was used for the calculation of diaphorase activity (Patel, et al., 1995).
2.8. In vitro DLDH oxidative inactivation by mitochondria-generated ROS
DLDH oxidative inactivation in isolated brain mitochondria was performed by supplementing mitochondria with respiratory substrates in the presence of Antimycin A, a condition that is known to enhance mitochondrial ROS generation (Turrens, 1997, Turrens, et al., 1985). Mitochondrial incubation was carried out as previously described (Schonfeld and Reiser, 2006). Essentially, mitochondria (0.25 mg/ml) were incubated at 25°C for 60 min in incubation buffer (110 mM mannitol, 10 mM KH2PO4, 60 mM Tris, 60 mM KCl, and 0.5 mM EGTA, pH 7.4) in the presence of 50 μM Antimycin A. The mixture was then supplemented with pyruvate plus malate (5 mM each) or succinate alone (10 mM) as mitochondrial respiratory substrates. At the end of the incubation, mitochondria were pelleted by centrifugation at 8,000 g for 10 min, and mitochondrial extracts were then prepared as described above.
2.9. Data analysis
All experiments were performed independently at least three times using different animals. Results are expressed as means ± SEM. Statistical analysis was performed with Welch’s t test using Instat software (Graphad Software, San Diego, CA). A probability value less than 0.05 (p < 0.05) was considered significant.
3. Results
3.1. Changes in DLDH expression and activity during postnatal development in the rat brain
Table 1 shows the brain weights at the different ages examined. As expected, the brain weight increased during postnatal development. However, there was no further increase beyond 60 days of age (data not shown). Comparison of DLDH expression, diaphorase activity and dehydrogenase activity was made between 10-, 20-, 30-, and 60-day-old rats. Fig. 1A shows DLDH expression as determined by anti-DLDH Western blots. A quantitative result derived from this figure shows that there was approximately a 30% increase in DLDH expression between 10 and 20 days of age, whereas there was no further increase during the period from 20 to 60 days (Fig. 1B). Similarly, the diaphorase activity, determined by in-gel NBT/NADH staining using blue native PAGE (Fig. 2A), showed a 46% increase between 10 and 20 days of age, and there was also no further increase between 20 and 60 days (Fig. 2B). DLDH dehydrogenase activity, however, increased progressively over the period from 10 to 60 days of age (Fig. 3). The incremental increase was: 63% between 10 and 20 days, 30% between 20 and 30 days, and 25% between 30 and 60 days of age.
Fig. 1.

Comparison of DLDH expression at different postnatal ages. A shows Western blot detection of DLDH expression; B shows the relative quantitative result derived from A. Values, expressed as percentage of 10 days of age, are mean ± SEM of three independent experiments. *p < 0.05 (all vs.10-day-old); Welch’s t test.
Fig. 2.

Comparison of DLDH diaphorase activity determined by in-gel activity staining. DLDH was resolved by blue native PAGE followed by diaphorase activity staining using the NBT/NADH system as described in the text. A shows the gel image of activity staining; B shows quantification of relative diaphorase activity derived by scanning the band intensity in A. Values, expressed as percentage of 10 days of age, are mean ± SEM of three independent experiments. *p < 0.05 (all vs. 10-day-old); Welch’s t test.
Fig. 3.
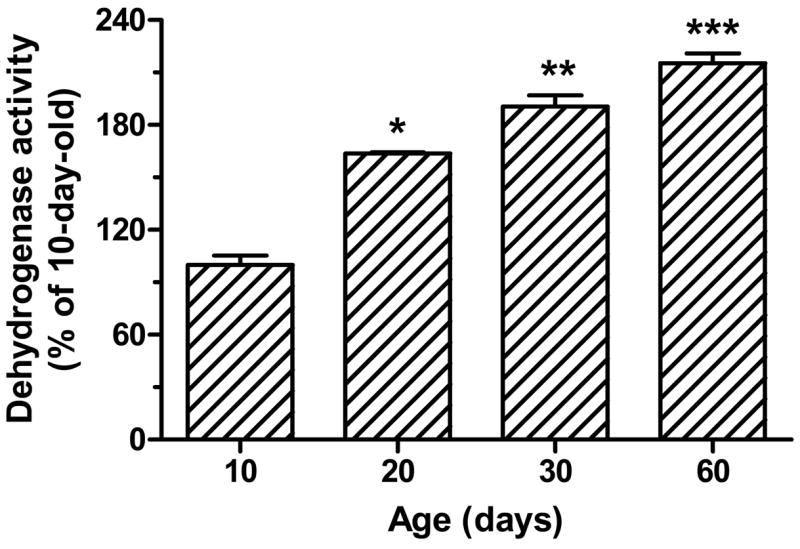
Comparison of DLDH dehydrogenase activity measured spectrophotometrically. The spectrophotometric measurement was performed by monitoring the increase of NADH absorbance at 340 nm upon DLDH-mediated, NAD+-dependent oxidation of dihydrolipoamide (the forward reaction). A solution containing all assay components except mitochondrial sample was used as the blank. The dehydrogenase activity value was derived from the ratio between the enzyme unit and the corresponding protein’s content that was determined densitometrically as shown in Fig. 1B. Data, expressed as percentage of the values for 10 days of age, are mean ± SEM of three animals for each age group. *p < 0.05 (20-day vs. 10-day), **p < 0.05 (30-day vs. 20-day), and ***p < 0.05 (60-day vs. 30-day) all indicate statistical significance (Welch’s t test).
3.2. Changes in postnatal DLDH activity in heart, liver and kidney
For the purpose of comparing DLDH in brain and peripheral tissues, we also investigated changes in DLDH diaphorase and dehydrogenase activities in heart, liver and kidney using the same approach as described above for brain DLDH. The results in Fig. 4 show DLDH diaphorase and dehydrogenase activity in these tissues as determined in 10-, 20-, 30-, or 60-day-old rats. Between 10 and 60 days of age, no increase in DLDH diaphorase activity could be detected in any of the three tissues. In both heart and kidney, a small, albeit significant increase in DLDH dehydrogenase activity was observed only for the period between 30 and 60 days of age. In liver, an increase in DLDH dehydrogenase activity of similar small magnitude was observed between 10 and 20 days of age, with no further increase evident thereafter.
Fig. 4.

Changes in DLDH diaphorase activity and dehydrogenase activity during postnatal development in heart, kidney and liver. DLDH protein content was determined as described for Fig. 1. DLDH diaphorase activity was measured by the activity staining method described for Fig 2. DLDH dehydrogenase activity was measured spectrophotometrically by following the increase of NADH absorbance at 340 nm in the presence of dihydrolipoamide. Both diaphorase activity and dehydrogenase activity were normalized based on the corresponding DLDH expression and are expressed as a percentage of the values for 10-day-old pups. *p < 0.05 (Welch’s t test) for 60 vs. 30 days of age for heart and kidney and for 20 vs. 10 days for liver.
3.3. Changes in DLDH expression and activity during brain aging
We further evaluated changes in DLDH expression, diaphorase activity and dehydrogenase activity during the aging process using male rats aged 5 or 30 months. Although the objective of this particular experiment was to focus on senescence-related changes in DLDH, 20-day-old pups were also included in this experiment for comparative purposes. The results are summarized in Fig. 5. A densitometric quantification of the immunostaining shown in Fig. 5A did not indicate a significant difference in DLDH expression among the three age groups examined (Fig. 5B). Similarly, DLDH diaphorase activity, determined by in-gel NBT/NADH staining (Fig. 5C), did not show any change when quantified densitometrically (Fig. 5D). Furthermore, there was no detectable decrease in DLDH dehydrogenase activity between 5 and 30 months of age (Fig. 5E). As expected, there was a significant increase in dehydrogenase activity between 20 days and 5 months of age that was in accordance with the results obtained in the developmental studies (Fig. 3).
Fig. 5.
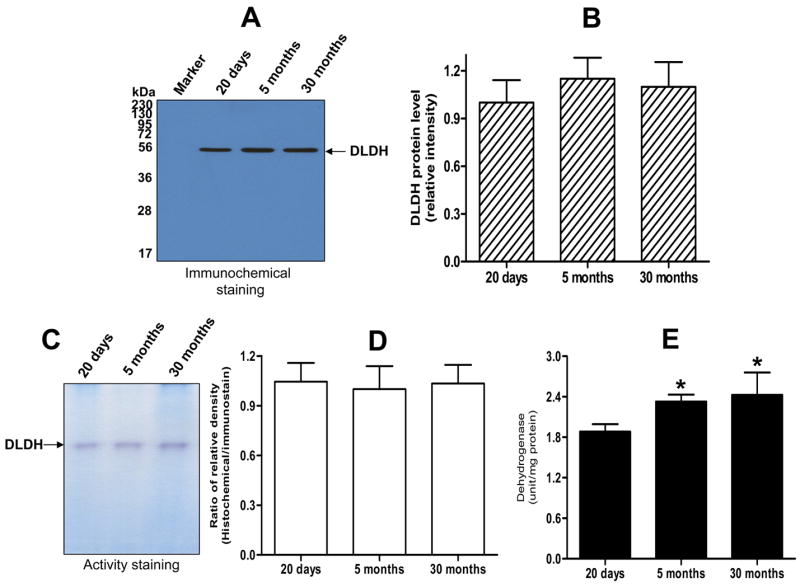
Comparison of DLDH expression, diaphorase activity and dehydrogenase activity between 20-day-old, 5-month-old, and 30-month-old rats. A shows the anti-DLDH Western blot images; B shows quantitative results of DLDH expression derived from A; C shows in-gel activity staining of diaphorase activity; D shows quantitative results of DLDH diaphorase activity derived from C; E shows DLDH dehydrogenase activity determined spectrophotometrically. *p < 0.05 (both ages vs. 20-day-old); Welch’s t test. All values are mean ± SEM of three independent experiments.
3.4. Comparison of DLDH activity following gel filtration of mitochondrial extracts
Given the fact that DLDH dehydrogenase activity can be inhibited by an excessive amount of NADH via a feedback inhibitory mechanism (Wilkinson and Williams, 1981), we then addressed the possibility that the relatively lower DLDH dehydrogenase activity observed at 20 days of age (Fig. 5E) is due to NADH inhibition. In this study, enzyme activity was measured after the mitochondrial extracts had been passed through PD-10 columns, so that NADH would be removed from the preparations. As there was no difference in DLDH dehydrogenase activity between 5 and 30 months of age (Fig. 5E), this comparison of enzyme activity involved samples from 20-day-old and 5-month-old rats. Moreover, DLDH dehydrogenase activity was assayed in both the forward and the reverse reactions in this study, along with a spectrophotometric measurement of DLDH diaphorase activity using DCPIP as the artificial electron acceptor. The results shown in Fig. 6 indicate that after gel filtration of mitochondrial extracts, DLDH dehydrogenase activity at 20 days of age was still significantly lower than at 5 months of age, and the magnitude of the difference was the same in both the forward and the reverse reactions. This result ruled out the possibility that the lower DLDH dehydrogenase activity observed in pups was due to NADH inhibition. No difference in diaphorase activity was evident between 20 days and 5 months of age, a finding that was in accordance with the result obtained by the in-gel NBT/NADH staining method (Fig. 5D).
Fig. 6.
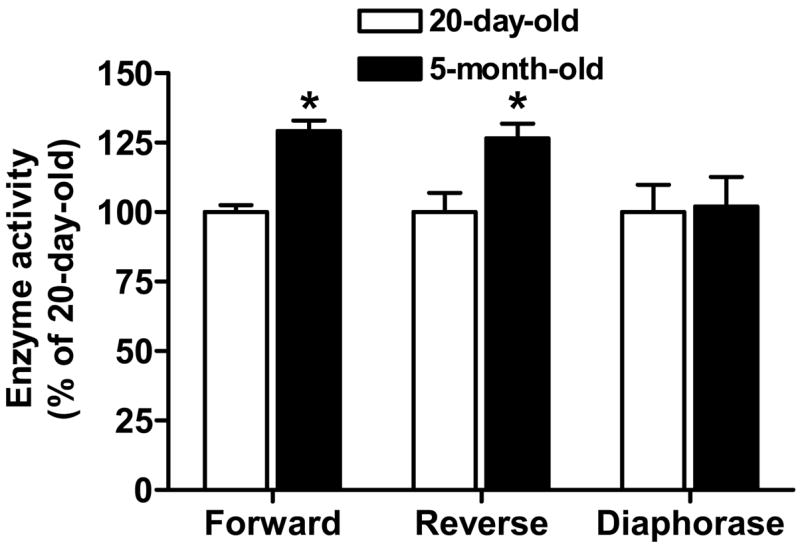
Measurements of DLDH activities following treatment of mitochondrial extracts by PD-10 columns. Dehydrogenase activities in both the forward and the reverse reactions were determined, along with determination of diaphorase activity. Values, expressed as percentage of the values for 20-day-olds, are mean ± SEM of three independent experiments. *p < 0.05; Welch’s t test.
3.5. In vitro DLDH oxidative inactivation by mitochondria-generated ROS
The finding that brain DLDH dehydrogenase activity did not decline between 5 and 30 months of age (Fig. 5E) indicates that DLDH does not undergo a significant aging-related oxidative inactivation, but did not necessarily rule out the possibility of an age-dependent increase in susceptibility to inactivation by mitochondria-generated ROS. Therefore, we investigated the status of in vitro DLDH inactivation by mitochondria-generated ROS in the different age groups. The results shown in Fig. 7 indicate that DLDH did indeed lose activity under the experimental conditions, but the percentage loss was virtually the same for the three age groups examined. Therefore, DLDH oxidative inactivation in vitro was age-independent, indicating that the susceptibility of DLDH to ROS attack does not change during brain development and aging. In addition, the percentage loss in DLDH dehydrogenase activity in each age group was independent of mitochondrial respiratory substrates in the presence of Antimycin A.
Fig. 7.
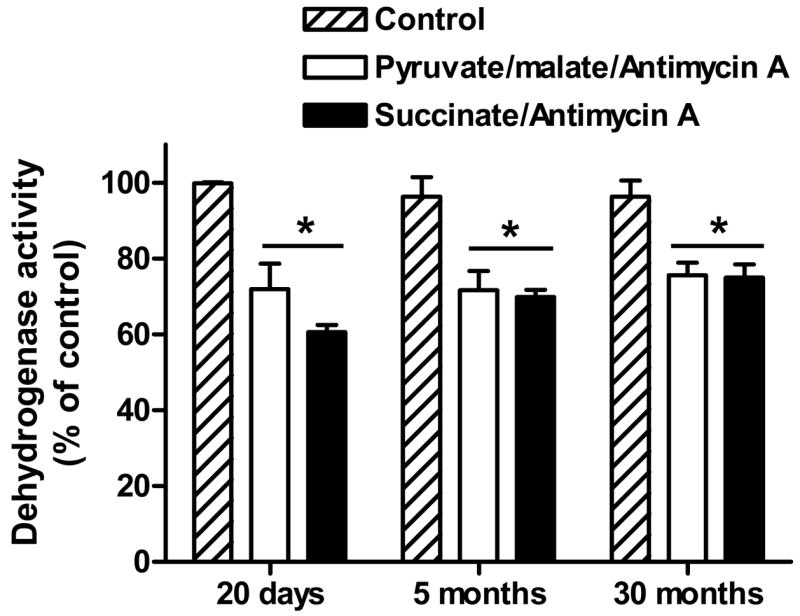
In vitro DLDH oxidative inactivation by mitochondria-generated ROS. Mitochondrial respiration buffer contained 110 mM mannitol, 10 mM KH2PO4, 60 mM Tris, 60 mM KCl, 0.5 mM EGTA, pH 7.4. Mitochondria (0.25 mg/ml) were incubated in the presence of Antimycin A (50μM) and the indicated respiratory substrates (pyruvate/malate: 5 mM/5 mM; succinate: 10 mM). Control contained neither Antimycin A nor substrates. DLDH dehydrogenase activity was measured spectrophotometrically as described in Figure legend 4. Values, expressed as percentage of control in each group, are mean ± SEM of three independent experiments. * p < 0.05 for comparison between the experimental groups and the control within each age group.
4. Discussion
The main findings of the present study are: (i) DLDH dehydrogenase activity in brain tissue increases progressively from 10 days of age into adulthood, whereas DLDH expression and diaphorase activity show little change after 20 days; (ii) Neither the activity nor the expression of DLDH showed significant senescence-related change and; (iii) Aging did not result in greater susceptibility of DLDH to oxidative inactivation by mitochondria-generated ROS (Fig. 7).
Data collected in the present study indicate that postnatal maturation of DLDH occurs in distinguishable stages. In an initial stage, ending between postnatal days 10 and 20, diaphorase and dehydrogenase activity of DLDH both increase in accordance with an augmentation of DLDH expression. Given that postnatal increases in brain mitochondrial mass are nearly complete by 20 days of age (Dahl and Samson, 1959, Land, et al., 1977), our results for DLDH expression over the period from 10 to 20 days postnatally (Fig 1B) are in accordance with the overall growth of brain mitochondrial mass during postnatal development. The increase in DLDH expression during this period would thus fully account for the observed increase in diaphorase and dehydrogenase activity, and these effects may be driven by a postnatal increase in brain energy demand. Studies of caloric restriction in adult rodents suggest that DLDH expression is indeed responsive to shifts in energy demand (Poon, et al., 2006). Moreover, a development-associated increase in DLDH expression caused by an increased energy demand has been previously reported in a study of pea seedling development (Luethy, et al., 2001).
During a second phase, beginning after postnatal day 20 and continuing into adulthood, dehydrogenase activity shows an additional, progressive, age-related increase in the absence of any detectable change in DLDH expression or diaphorase activity. The observed increase in DLDH dehydrogenase activity against an unchanged DLDH expression during this stage could be due to an increased catalytic efficiency of the enzyme in association with an increased energy demand during brain development. That DLDH diaphorase activity is not similarly changed during this stage could be attributed to three factors: (i) there is no change in DLDH expression; (ii) there are no defined binding sites on DLDH for non-lipoyl electron acceptors in the presence of NADH; and (iii) DLDH catalyzed diaphorase reactions are achieved by a four-electron-reduced enzyme (Argyrou, et al., 2003), while DLDH catalyzed dehydrogenase reactions are accomplished by a two-electron-reduced enzyme (Argyrou, et al., 2003).
The pattern of postnatal DLDH maturation in the brain differed markedly from that of peripheral organs, including heart, kidney and liver. Over the period from 10 to 60 days of age, there was a progressive, nearly 2-fold, age-associated increase in brain DLDH dehydrogenase activity (Fig. 3), whereas in heart, liver and kidney, the increases were much smaller (between 20 and 30%) and did not occur incrementally as a function of age. The more prominent increases in DLDH dehydrogenase activity in brain, when compared with the peripheral organs, may reflect unique energy requirements associated with the relatively greater amount and complexity of postnatal maturation occurring within the brain. These changes include development of microneurons, cell migration, axonal and dendritic growth, synapse proliferation, and myelination (Bates, et al., 1994, Kalous, et al., 2001).
It is established that an elevated level of protein oxidation is associated with aging (Berlett and Stadtman, 1997, Smith, et al., 1992). For example, mitochondrial aconitase (Yan, et al., 1997, Yarian, et al., 2006) and adenine nucleotide translocase (Yan and Sohal, 1998) can lose their functional activities due to age-related oxidative damage. Interestingly, DLDH, as a redox enzyme, does not incur age-related oxidative inactivation in the rat brain. This is in contrast to in vitro studies where DLDH can be inactivated by mitochondria-generated ROS (Fig. 7 of this study), by metal-catalyzed oxidation (Gutierrez-Correa and Stoppani, 1995), as well as by the myeloperoxidase system (Gutierrez-Correa and Stoppani, 1999). Our observation that DLDH did not lose activity and did not become more susceptible to oxidative inactivation during aging not only indicate that DLDH is stable during brain aging, but also confirm the notion that protein oxidation during aging is a highly selective process (Das, et al., 2001, Jana, et al., 2002, Prokai, et al., 2007, Toroser, et al., 2007, Yan, et al., 1997, Yan, et al., 2000, Yan and Sohal, 1998). Additionally, given that mitochondrial ROS generation capacity does not change during postnatal brain development (Schonfeld and Reiser, 2007), it is reasonable to think that the lower DLDH dehydrogenase activity observed in rat pups is not a consequence of oxidative stress, but rather an adaptive component of postnatal development reflective of changes in energy demand.
It should be pointed out that under certain in vivo oxidative stress conditions, DLDH has been found to be oxidatively modified. For instance, DLDH was found to be modified by 4-hydroxynonenal (HNE) in the ventilatory muscles of rats challenged by lipopolysaccharide (Hussain, et al., 2006), and found to be nitrosylated in rat liver (Foster and Stamler, 2004) and in Mycobacterium tuberculosis (Rhee, et al., 2005). Because DLDH activity was not measured in these studies, it is not yet known whether or not the HNE modification or nitrosylation had any significant inhibitory effects. In the case of the HNE modification, a loss of DLDH activity would not necessarily be an outcome, as such modifications do not always lead to loss of function (Moreau, et al., 2003). In the case of DLDH nitrosylation, it may be more probable that DLDH activity is compromised. When isolated mitochondria were incubated with nitric oxide donors, DLDH exhibited a substantial loss of activity (Yan, et al., 2007), indicating that, in vivo, DLDH nitrosylation may have a deleterious effect on DLDH activity.
Because the current study was based on mitochondria from samples of whole brain, the current studies do not rule out the additional possibility that activity of DLDH, or its associated complexes, is altered selectively within different regions of the brain. Indeed, the degree of age-associated oxidative stress/damage differs markedly among different regions of the brain (Dubey, et al., 1996, Forster, et al., 1996, Rebrin, et al., 2007). Furthermore, studies of α-ketoglutarate dehydrogenase complex activity in neurodegenerative diseases have also suggested a dependence on brain region (Klivenyi, et al., 2004).
Finally, it should be pointed out that a comparison of amino acid sequences (retrieved from Protein Database Bank) between rat and human DLDH reveals a 93% homology, and all the key amino acid residues involved in catalysis such as the redox active center (Cys-45, Cys-50) in one monomer and the essential histidine glutamic acid pair (His-452, Glu-457) in the other monomer are conserved (Brautigam, et al., 2005, Ciszak, et al., 2006, Jentoft, et al., 1992). Therefore, rat brain would be a good model for studying DLDH dysfunctions that might be implicated in human brain development and neurodegenerative disorders.
Table I.
Increase in rat brain weight during development
| Age (days) | Brain weights (gram) |
|---|---|
| 10 | 0.80 ± 0.06 |
| 20 | 1.17 ± 0.03 |
| 30 | 1.37 ± 0.03 |
| 60 | 1.77 ± 0.03 |
Data are expressed as mean ± SEM, n = 3.
Acknowledgments
The authors thank Drs. Renqi Huang and Zhenglan Chen for assistance. This work was supported in part by the National Institutes of Health (Grant: PO1 AG022550).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Almeida A, Brooks KJ, Sammut I, Keelan J, Davey GP, Clark JB, Bates TE. Postnatal development of the complexes of the electron transport chain in synaptic mitochondria from rat brain. Dev Neurosci. 1995;17:212–218. doi: 10.1159/000111289. [DOI] [PubMed] [Google Scholar]
- Argyrou A, Sun G, Palfey BA, Blanchard JS. Catalysis of diaphorase reactions by Mycobacterium tuberculosis lipoamide dehydrogenase occurs at the EH4 level. Biochemistry. 2003;42:2218–2228. doi: 10.1021/bi020654f. [DOI] [PubMed] [Google Scholar]
- Babady NE, Pang YP, Elpeleg O, Isaya G. Cryptic proteolytic activity of dihydrolipoamide dehydrogenase. Proc Natl Acad Sci U S A. 2007;104:6158–6163. doi: 10.1073/pnas.0610618104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bando Y, Aki K. Mechanisms of generation of oxygen radicals and reductive mobilization of ferritin iron by lipoamide dehydrogenase. J Biochem (Tokyo) 1991;109:450–454. doi: 10.1093/oxfordjournals.jbchem.a123402. [DOI] [PubMed] [Google Scholar]
- Bates TE, Almeida A, Heales SJ, Clark JB. Postnatal development of the complexes of the electron transport chain in isolated rat brain mitochondria. Dev Neurosci. 1994;16:321–327. doi: 10.1159/000112126. [DOI] [PubMed] [Google Scholar]
- Berlett BS, Stadtman ER. Protein oxidation in aging, disease, and oxidative stress. J Biol Chem. 1997;272:20313–20316. doi: 10.1074/jbc.272.33.20313. [DOI] [PubMed] [Google Scholar]
- Brautigam CA, Chuang JL, Tomchick DR, Machius M, Chuang DT. Crystal structure of human dihydrolipoamide dehydrogenase: NAD+/NADH binding and the structural basis of disease-causing mutations. J Mol Biol. 2005;350:543–552. doi: 10.1016/j.jmb.2005.05.014. [DOI] [PubMed] [Google Scholar]
- Brown AM, Gordon D, Lee H, Caudy M, Hardy J, Haroutunian V, Blass JP. Association of the dihydrolipoamide dehydrogenase gene with Alzheimer’s disease in an Ashkenazi Jewish population. Am J Med Genet B Neuropsychiatr Genet. 2004;131:60–66. doi: 10.1002/ajmg.b.30008. [DOI] [PubMed] [Google Scholar]
- Brown AM, Gordon D, Lee H, Vrieze FW, Cellini E, Bagnoli S, Nacmias B, Sorbi S, Hardy J, Blass JP. Testing for linkage and association across the dihydrolipoyl dehydrogenase gene region with Alzheimer’s disease in three sample populations. Neurochem Res. 2007;32:857–869. doi: 10.1007/s11064-006-9235-3. [DOI] [PubMed] [Google Scholar]
- Bukato G, Kochan Z, Swierczynski J. Changes of malic enzyme activity in the developing rat brain are due to both the increase of mitochondrial protein content and the increase of specific activity. Int J Biochem. 1992;24:267–273. doi: 10.1016/0020-711x(92)90257-2. [DOI] [PubMed] [Google Scholar]
- Ciszak EM, Makal A, Hong YS, Vettaikkorumakankauv AK, Korotchkina LG, Patel MS. How dihydrolipoamide dehydrogenase-binding protein binds dihydrolipoamide dehydrogenase in the human pyruvate dehydrogenase complex. J Biol Chem. 2006;281:648–655. doi: 10.1074/jbc.M507850200. [DOI] [PubMed] [Google Scholar]
- Clark JB, Leong SF, Munday LA. The postnatal development of brain metabolism. Biochem Soc Trans. 1981;9:372–373. doi: 10.1042/bst0090372. [DOI] [PubMed] [Google Scholar]
- Dahl DR, Samson FE., Jr Metabolism of rat brain mitochondria during postnatal development. Am J Physiol. 1959;196:470–472. doi: 10.1152/ajplegacy.1959.196.2.470. [DOI] [PubMed] [Google Scholar]
- Das N, Levine RL, Orr WC, Sohal RS. Selectivity of protein oxidative damage during aging in Drosophila melanogaster. Biochem J. 2001;360:209–216. doi: 10.1042/0264-6021:3600209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey A, Forster MJ, Lal H, Sohal RS. Effect of age and caloric intake on protein oxidation in different brain regions and on behavioral functions of the mouse. Arch Biochem Biophys. 1996;333:189–197. doi: 10.1006/abbi.1996.0380. [DOI] [PubMed] [Google Scholar]
- Faure M, Bourguignon J, Neuburger M, MacHerel D, Sieker L, Ober R, Kahn R, Cohen-Addad C, Douce R. Interaction between the lipoamide-containing H-protein and the lipoamide dehydrogenase (L-protein) of the glycine decarboxylase multienzyme system 2. Crystal structures of H- and L-proteins. Eur J Biochem. 2000;267:2890–2898. doi: 10.1046/j.1432-1033.2000.01330.x. [DOI] [PubMed] [Google Scholar]
- Forster MJ, Dubey A, Dawson KM, Stutts WA, Lal H, Sohal RS. Age-related losses of cognitive function and motor skills in mice are associated with oxidative protein damage in the brain. Proc Natl Acad Sci U S A. 1996;93:4765–4769. doi: 10.1073/pnas.93.10.4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster MW, Stamler JS. New insights into protein S-nitrosylation. Mitochondria as a model system. J Biol Chem. 2004;279:25891–25897. doi: 10.1074/jbc.M313853200. [DOI] [PubMed] [Google Scholar]
- Gazaryan IG, Krasnikov BF, Ashby GA, Thorneley RN, Kristal BS, Brown AM. Zinc is a potent inhibitor of thiol oxidoreductase activity and stimulates reactive oxygen species production by lipoamide dehydrogenase. J Biol Chem. 2002;277:10064–10072. doi: 10.1074/jbc.M108264200. [DOI] [PubMed] [Google Scholar]
- Gibson GE, Park LC, Sheu KF, Blass JP, Calingasan NY. The alpha-ketoglutarate dehydrogenase complex in neurodegeneration. Neurochem Int. 2000;36:97–112. doi: 10.1016/s0197-0186(99)00114-x. [DOI] [PubMed] [Google Scholar]
- Gutierrez-Correa J, Stoppani AO. Inactivation of heart dihydrolipoamide dehydrogenase by copper Fenton systems. Effect of thiol compounds and metal chelators. Free Radic Res. 1995;22:239–250. doi: 10.3109/10715769509147543. [DOI] [PubMed] [Google Scholar]
- Gutierrez-Correa J, Stoppani AO. Inactivation of myocardial dihydrolipoamide dehydrogenase by myeloperoxidase systems: effect of halides, nitrite and thiol compounds. Free Radic Res. 1999;30:105–117. doi: 10.1080/10715769900300111. [DOI] [PubMed] [Google Scholar]
- Holtzman D, Tsuji M, Wallimann T, Hemmer W. Functional maturation of creatine kinase in rat brain. Dev Neurosci. 1993;15:261–270. doi: 10.1159/000111343. [DOI] [PubMed] [Google Scholar]
- Hussain SN, Matar G, Barreiro E, Florian M, Divangahi M, Vassilakopoulos T. Modifications of proteins by 4-hydroxy-2-nonenal in the ventilatory muscles of rats. Am J Physiol Lung Cell Mol Physiol. 2006;290:L996–1003. doi: 10.1152/ajplung.00337.2005. [DOI] [PubMed] [Google Scholar]
- Igamberdiev AU, Bykova NV, Ens W, Hill RD. Dihydrolipoamide dehydrogenase from porcine heart catalyzes NADH-dependent scavenging of nitric oxide. FEBS Lett. 2004;568:146–150. doi: 10.1016/j.febslet.2004.05.024. [DOI] [PubMed] [Google Scholar]
- Jana CK, Das N, Sohal RS. Specificity of age-related carbonylation of plasma proteins in the mouse and rat. Arch Biochem Biophys. 2002;397:433–439. doi: 10.1006/abbi.2001.2690. [DOI] [PubMed] [Google Scholar]
- Jentoft JE, Shoham M, Hurst D, Patel MS. A structural model for human dihydrolipoamide dehydrogenase. Proteins. 1992;14:88–101. doi: 10.1002/prot.340140110. [DOI] [PubMed] [Google Scholar]
- Johnson MT, Yang HS, Magnuson T, Patel MS. Targeted disruption of the murine dihydrolipoamide dehydrogenase gene (Dld) results in perigastrulation lethality. Proc Natl Acad Sci U S A. 1997;94:14512–14517. doi: 10.1073/pnas.94.26.14512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalous M, Rauchova H, Drahota Z. Postnatal development of energy metabolism in the rat brain. Physiol Res. 2001;50:315–319. [PubMed] [Google Scholar]
- Klivenyi P, Starkov AA, Calingasan NY, Gardian G, Browne SE, Yang L, Bubber P, Gibson GE, Patel MS, Beal MF. Mice deficient in dihydrolipoamide dehydrogenase show increased vulnerability to MPTP, malonate and 3-nitropropionic acid neurotoxicity. J Neurochem. 2004;88:1352–1360. doi: 10.1046/j.1471-4159.2003.02263.x. [DOI] [PubMed] [Google Scholar]
- Korotchkina LG, Yang H, Tirosh O, Packer L, Patel MS. Protection by thiols of the mitochondrial complexes from 4-hydroxy-2-nonenal. Free Radic Biol Med. 2001;30:992–999. doi: 10.1016/s0891-5849(01)00491-9. [DOI] [PubMed] [Google Scholar]
- Land JM, Booth RF, Berger R, Clark JB. Development of mitochondrial energy metabolism in rat brain. Biochem J. 1977;164:339–348. doi: 10.1042/bj1640339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leong SF, Clark JB. Regional development of glutamate dehydrogenase in the rat brain. J Neurochem. 1984;43:106–111. doi: 10.1111/j.1471-4159.1984.tb06684.x. [DOI] [PubMed] [Google Scholar]
- Leong SF, Clark JB. Regional enzyme development in rat brain. Enzymes of energy metabolism. Biochem J. 1984;218:139–145. doi: 10.1042/bj2180139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luethy MH, Gemel J, Johnston ML, Mooney BP, Miernyk JA, Randall DD. Developmental expression of the mitochondrial pyruvate dehydrogenase complex in pea (Pisum sativum) seedlings. Physiol Plant. 2001;112:559–566. doi: 10.1034/j.1399-3054.2001.1120414.x. [DOI] [PubMed] [Google Scholar]
- Malloch GD, Munday LA, Olson MS, Clark JB. Comparative development of the pyruvate dehydrogenase complex and citrate synthase in rat brain mitochondria. Biochem J. 1986;238:729–736. doi: 10.1042/bj2380729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey V, Gibson QH, Veeger C. Intermediates in the catalytic action of lipoyl dehydrogenase (diaphorase) Biochem J. 1960;77:341–351. doi: 10.1042/bj0770341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavelli I, Rigo A, Federico R, Ciriolo MR, Rotilio G. Superoxide dismutase, glutathione peroxidase and catalase in developing rat brain. Biochem J. 1982;204:535–540. doi: 10.1042/bj2040535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau R, Heath SH, Doneanu CE, Lindsay JG, Hagen TM. Age-related increase in 4-hydroxynonenal adduction to rat heart alpha-ketoglutarate dehydrogenase does not cause loss of its catalytic activity. Antioxid Redox Signal. 2003;5:517–527. doi: 10.1089/152308603770310167. [DOI] [PubMed] [Google Scholar]
- Navarro A, Boveris A. Rat brain and liver mitochondria develop oxidative stress and lose enzymatic activities on aging. Am J Physiol Regul Integr Comp Physiol. 2004;287:R1244–1249. doi: 10.1152/ajpregu.00226.2004. [DOI] [PubMed] [Google Scholar]
- Navarro A, Boveris A. Brain mitochondrial dysfunction in aging: conditions that improve survival, neurological performance and mitochondrial function. Front Biosci. 2007;12:1154–1163. doi: 10.2741/2133. [DOI] [PubMed] [Google Scholar]
- Navarro A, Boveris A. The mitochondrial energy transduction system and the aging process. Am J Physiol Cell Physiol. 2007;292:C670–C686. doi: 10.1152/ajpcell.00213.2006. [DOI] [PubMed] [Google Scholar]
- Navarro A, Gomez C, Lopez-Cepero JM, Boveris A. Beneficial effects of moderate exercise on mice aging: survival, behavior, oxidative stress, and mitochondrial electron transfer. Am J Physiol Regul Integr Comp Physiol. 2004;286:R505–R511. doi: 10.1152/ajpregu.00208.2003. [DOI] [PubMed] [Google Scholar]
- Neuburger M, Polidori AM, Pietre E, Faure M, Jourdain A, Bourguignon J, Pucci B, Douce R. Interaction between the lipoamide-containing H-protein and the lipoamide dehydrogenase (L-protein) of the glycine decarboxylase multienzyme system. 1 Biochemical studies. Eur J Biochem. 2000;267:2882–2889. doi: 10.1046/j.1432-1327.2000.01301.x. [DOI] [PubMed] [Google Scholar]
- Olsson JM, Xia L, Eriksson LC, Bjornstedt M. Ubiquinone is reduced by lipoamide dehydrogenase and this reaction is potently stimulated by zinc. FEBS Lett. 1999;448:190–192. doi: 10.1016/s0014-5793(99)00363-4. [DOI] [PubMed] [Google Scholar]
- Patel MS, Hong YS. Lipoic acid as an antioxidant: the role of dihydrolipoamide dehydrogenase. In: Armstrong D, editor. Free Radical and Antioxidant Protocols. Humana Press; Totowa, NJ: 1998. pp. 337–346. [DOI] [PubMed] [Google Scholar]
- Patel MS, Roche TE. Molecular biology and biochemistry of pyruvate dehydrogenase complexes. FASEB J. 1990;4:3224–3233. doi: 10.1096/fasebj.4.14.2227213. [DOI] [PubMed] [Google Scholar]
- Patel MS, Vettakkorumakankav NN, Liu TC. Dihydrolipoamide dehydrogenase: activity assays. Methods Enzymol. 1995;252:186–195. doi: 10.1016/0076-6879(95)52022-8. [DOI] [PubMed] [Google Scholar]
- Poon HF, Shepherd HM, Reed TT, Calabrese V, Stella AM, Pennisi G, Cai J, Pierce WM, Klein JB, Butterfield DA. Proteomics analysis provides insight into caloric restriction mediated oxidation and expression of brain proteins associated with age-related impaired cellular processes: Mitochondrial dysfunction, glutamate dysregulation and impaired protein synthesis. Neurobiol Aging. 2006;27:1020–1034. doi: 10.1016/j.neurobiolaging.2005.05.014. [DOI] [PubMed] [Google Scholar]
- Prokai L, Yan LJ, Vera-Serrano JL, Stevens SM, Forster MJ. Mass spectrometry-based survey of age-associated protein carbonylation in rat brain mitochondria. J Mass Spectrom. 2007;42:1583–1589. doi: 10.1002/jms.1345. [DOI] [PubMed] [Google Scholar]
- Rebrin I, Forster MJ, Sohal RS. Effects of age and caloric intake on glutathione redox state in different brain regions of C57BL/6 and DBA/2 mice. Brain Res. 2007;1127:10–18. doi: 10.1016/j.brainres.2006.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee KY, Erdjument-Bromage H, Tempst P, Nathan CF. S-nitroso proteome of Mycobacterium tuberculosis: Enzymes of intermediary metabolism and antioxidant defense. Proc Natl Acad Sci U S A. 2005;102:467–472. doi: 10.1073/pnas.0406133102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonfeld P, Bohnensack R. Developmental changes of the adenine nucleotide translocation in rat brain. Biochim Biophys Acta. 1995;1232:75–80. doi: 10.1016/0005-2728(95)00114-9. [DOI] [PubMed] [Google Scholar]
- Schonfeld P, Reiser G. Rotenone-like action of the branched-chain phytanic acid induces oxidative stress in mitochondria. J Biol Chem. 2006;281:7136–7142. doi: 10.1074/jbc.M513198200. [DOI] [PubMed] [Google Scholar]
- Schonfeld P, Reiser G. Ca(2+) Storage Capacity of Rat Brain Mitochondria Declines During the Postnatal Development Without Change in ROS Production Capacity. Antioxid Redox Signal. 2007;9:191–199. doi: 10.1089/ars.2007.9.191. [DOI] [PubMed] [Google Scholar]
- Scouten WH, McManus IR. Microbial lipoamide dehydrogenase. Purification and some characteristics of the enzyme derived from selected microorganisms. Biochim Biophys Acta. 1971;227:248–263. doi: 10.1016/0005-2744(71)90058-1. [DOI] [PubMed] [Google Scholar]
- Sims NR. Methods in Toxicology: Mitochondrial Dysfunction. Academic Press; San Diego: 1993. pp. 51–59. [Google Scholar]
- Smith CD, Carney JM, Tatsumo T, Stadtman ER, Floyd RA, Markesbery WR. Protein oxidation in aging brain. Ann N Y Acad Sci. 1992;663:110–119. doi: 10.1111/j.1749-6632.1992.tb38654.x. [DOI] [PubMed] [Google Scholar]
- Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- Sokatch JR, McCully V, Gebrosky J, Sokatch DJ. Isolation of a specific lipoamide dehydrogenase for a branched-chain keto acid dehydrogenase from Pseudomonas putida. J Bacteriol. 1981;148:639–646. doi: 10.1128/jb.148.2.639-646.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreider CM, Grinblat L, Stoppani AO. Catalysis of nitrofuran redox-cycling and superoxide anion production by heart lipoamide dehydrogenase. Biochem Pharmacol. 1990;40:1849–1857. doi: 10.1016/0006-2952(90)90366-s. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Brown MR. Mitochondrial aging and dysfunction in Alzheimer’s disease. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:407–410. doi: 10.1016/j.pnpbp.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Tahara EB, Barros MH, Oliveira GA, Netto LE, Kowaltowski AJ. Dihydrolipoyl dehydrogenase as a source of reactive oxygen species inhibited by caloric restriction and involved in Saccharomyces cerevisiae aging. Faseb J. 2007;21:274–283. doi: 10.1096/fj.06-6686com. [DOI] [PubMed] [Google Scholar]
- Thorpe C, Williams CH., Jr Differential reactivity of the two active site cysteine residues generated on reduction of pig heart lipoamide dehydrogenase. J Biol Chem. 1976;251:3553–3557. [PubMed] [Google Scholar]
- Toroser D, Orr WC, Sohal RS. Carbonylation of mitochondrial proteins in Drosophila melanogaster during aging. Biochem Biophys Res Commun. 2007;363:418–424. doi: 10.1016/j.bbrc.2007.08.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrens JF. Superoxide production by the mitochondrial respiratory chain. Biosci Rep. 1997;17:3–8. doi: 10.1023/a:1027374931887. [DOI] [PubMed] [Google Scholar]
- Turrens JF, Alexandre A, Lehninger AL. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys. 1985;237:408–414. doi: 10.1016/0003-9861(85)90293-0. [DOI] [PubMed] [Google Scholar]
- Vettakkorumakankav NN, Patel MS. Dihydrolipoamide dehydrogenase: structural and mechanistic aspects. Indian J Biochem Biophys. 1996;33:168–176. [PubMed] [Google Scholar]
- Wilkinson KD, Williams CH., Jr NADH inhibition and NAD activation of Escherichia coli lipoamide dehydrogenase catalyzing the NADH-lipoamide reaction. J Biol Chem. 1981;256:2307–2314. [PubMed] [Google Scholar]
- Williams CHJ. Lipoamide dehydrogenase, glutathione reductase, thioredoxin reductase, and mercuric ion reductase-a family of flavoenzyme transhydrogenases. In: Muller F, editor. Chemistry and Biochemistry of Flavoenzymes. III. CRC Press; Boca Raton: 1992. pp. 121–212. [Google Scholar]
- Xia L, Bjornstedt M, Nordman T, Eriksson LC, Olsson JM. Reduction of ubiquinone by lipoamide dehydrogenase. An antioxidant regenerating pathway. Eur J Biochem. 2001;268:1486–1490. doi: 10.1046/j.1432-1327.2001.02013.x. [DOI] [PubMed] [Google Scholar]
- Yan LJ, Christians ES, Liu L, Xiao X, Sohal RS, Benjamin IJ. Mouse heat shock transcription factor 1 deficiency alters cardiac redox homeostasis and increases mitochondrial oxidative damage. Embo J. 2002;21:5164–5172. doi: 10.1093/emboj/cdf528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan LJ, Levine RL, Sohal RS. Oxidative damage during aging targets mitochondrial aconitase. Proc Natl Acad Sci USA. 1997;94:11168–11172. doi: 10.1073/pnas.94.21.11168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan LJ, Levine RL, Sohal RS. Effects of aging and hyperoxia on oxidative damage to cytochrome c in the housefly, Musca domestica. Free Radic Biol Med. 2000;29:90–97. doi: 10.1016/s0891-5849(00)00323-3. [DOI] [PubMed] [Google Scholar]
- Yan LJ, Orr WC, Sohal RS. Identification of oxidized proteins based on sodium dodecyl sulfate-polyacrylamide gel electrophoresis, immunochemical detection, isoelectric focusing, and microsequencing. Anal Biochem. 1998;263:67–71. doi: 10.1006/abio.1998.2799. [DOI] [PubMed] [Google Scholar]
- Yan LJ, Rajasekaran NS, Sathyanarayanan S, Benjamin IJ. Mouse HSF1 disruption perturbs redox state and increases mitochondrial oxidative stress in kidney. Antioxid Redox Signal. 2005;7:465–471. doi: 10.1089/ars.2005.7.465. [DOI] [PubMed] [Google Scholar]
- Yan LJ, Sohal RS. Mitochondrial adenine nucleotide translocase is modified oxidatively during aging. Proc Natl Acad Sci USA. 1998;95:12896–12901. doi: 10.1073/pnas.95.22.12896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan LJ, Yang SH, Shu H, Prokai L, Forster MJ. Histochemical staining and quantification of dihydrolipoamide dehydrogenase diaphorase activity using blue native PAGE. Electrophoresis. 2007;28:1036–1045. doi: 10.1002/elps.200600574. [DOI] [PubMed] [Google Scholar]
- Yarian CS, Toroser D, Sohal RS. Aconitase is the main functional target of aging in the citric acid cycle of kidney mitochondria from mice. Mech Ageing Dev. 2006;127:79–84. doi: 10.1016/j.mad.2005.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]