

Abstract
Imaging oxygen in 3D with submicron spatial resolution can be made possible by combining phosphorescence quenching technique with multiphoton laser scanning microscopy. Because Pt and Pd porphyrin-based phosphorescent dyes, traditionally used as phosphors in biological oxygen measurements, exhibit extremely low two-photon absorption (2PA) cross-sections, we designed a nanosensor for oxygen, in which a 2P absorbing antenna is coupled to a metalloporphyrin core via intramolecular energy transfer (ET) with the purpose of amplifying the 2PA induced phosphorescence of the metalloporphyrin. The central component of the device is a polyfunctionalized Pt porphyrin, whose triplet state emission at ambient temperatures is strong, occurs in the near infrared and is sensitive to O2. The 2PA chromophores are chosen in such a way that their absorption is maximal in the near infrared (NIR) window of tissue (e.g., 700−900 nm), while their fluorescence is overlapped with the absorption band(s) of the core metalloporphyrin, ensuring an efficient antenna-core resonance ET. The metalloporphyrin-antenna construct is embedded inside the protecting dendritic jacket, which isolates the core from interactions with biological macromolecules, controls diffusion of oxygen and makes the entire sensor water-soluble. Several Pt porphyrin-coumarin based sensors were synthesized and their photophyics studied to evaluate the proposed design.
Introduction
Imaging tissue oxygen in vivo presents a challenging and important problem in modern physiology and medicine. Oxygen is a key metabolite, and tissue hypoxia is a critical parameter with respect to various tissue pathologies, such as retinal diseases,1 brain abnormalities2 and cancer.3 Nevertheless, imaging technologies for mapping tissue oxygenation4 (e.g., NMR/EPR,5 PET,6 near infrared tomographic techniques,7 etc)8 are yet to be adequately developed. One method, superior in its ability to directly detect oxygen in tissue, is based on oxygen dependent quenching of phosphorescence.9 When a phosphorescent dye is dissolved in the blood and excited using appropriate illumination, its phosphorescence lifetime and intensity become robust indicators of oxygen concentration in the environment. Phosphorescence quenching is exquisitely sensitive and selective to oxygen, possesses excellent temporal resolution and can be implemented for high-resolution hypoxia imaging in 2D.10 In addition, a variant of the technique employing NIR absorbing phosphors11 is currently being developed into a 3D near infrared tomographic modality.12 However, like all “diffuse” NIR methods,13 3D phosphorescence imaging suffers from intrinsically low spatial resolution, which is due to the strong scattering of photons in tissues. For a number of biologically important applications, an oxygen imaging method, capable of submicron spatial resolution when looking at the areas below the surface of the tissue, would be extremely desirable.
A way to develop a high-resolution oxygen imaging modality would be to combine phosphorescence lifetime imaging10 with Two-Photon (or, more generally, Multiphoton) Laser Scanning Microscopy (2P LSM).14 2P LSM is based on the two-photon absorption (2PA) phenomenon,15 which occurs with high efficiency only at extremely high local instantaneous intensities of the excitation light, i.e., in the focus of an ultrafast pulsed laser beam. Raster-scanning such a beam in the axial plane at a selected depth (usually no more than 500 μm) within the object permits its effective 3D optical sectioning, provided that the object contains a 2P absorbing luminescent dye. 2PA is typically initiated by lasers operating in the NIR region of the spectrum, where absorption by natural tissue chromophores is diminished, and light can penetrate deeper into the tissue. On the other hand, photodamage, associated with high laser power,16 is confined in the case of 2PA LSM to the immediate vicinity of the focal plane, while being kept minimal along the excitation path, unlike in conventional linear optical methods.
A difficulty in using 2P LSM in combination with phosphorescence quenching is that the phosphors for biological oxygen measurements are typically based on Pt or Pd porphyrins,9 whose 2PA cross-sections are very low,17 i.e., in the order of just a few GM units.18 While in principle phosphorescence of metalloporphyrins can be induced via 2P excitation,19 for practical applications 2PA cross-sections of biological probes must be considerably higher.
In recent years, much attention has been given to nonlinear absorption of tetrapyrroles, because of their potential usefulness for 2P PDT.20 It has been shown that the 2PA cross-section of the basic tetrapyrrole macrocycle can be increased up to hundreds of GM units by an appropriate substitution and/or due to the resonant one-photon enhancement of 2PA.21 In addition, recent studies of porphyrin oligomers with acetylenic bridges revealed that these materials can exhibit extremely high 2PA cross-sections (up to 8000 GM).22 Unfortunately, no data is available yet on the triplet state emission of Pt and Pd complexes of porphyrin oligomers, and it is not clear how modification of the porphyrin electronic system, leading to an increase in 2PA, would affect its' triplet state properties.
An alternative approach to amplify apparent 2PA cross-sections of metalloporphyrins, while leaving their electronic systems intact, would be to couple them to 2PA antenna-chromophores, so that the 2P excitation energy is channeled via the energy transfer (ET). The theoretical and experimental literature on the ET in porphyrin based systems is very extensive, reflecting its' analogy with processes in the natural photosynthesis. The most relevant to the present design, however, are the reports on ET in dendrimers,23,24 porphyrin-cored dendrimers,25 dendritic systems in which energy is transferred to other triplet state emitters26 and in which the ET is coupled to the multiphoton light harvesting.27,28 The dendritic configuration provides an opportunity to pack many 2PA chromophores in a small volume, resulting in an increase in the effective molecular 2PA cross-section.29 Moreover, it has been shown that the 2PA enhancement in multibranched molecules is not only due to the increase in the local number density, but also due to cooperative effects of the branches.30 An extensive theoretical treatment of nonlinear ET in dendrimers was recently published, where several mechanisms of energy migration upon absorption of two or more photons in multichromophoric systems were discussed.31
Herein, we describe a design of the phosphorescent oxygen nanosensor, in which 2P light harvesting and intramolecular ET are succeeded by the intersystem crossing within the acceptor chromophore (i.e., metalloporphyrin) to induce its' phosphorescence, sensitive to oxygen. To protect the phosphorescent emitter from interactions with macromolecules in the medium, to control oxygen diffusion to the core and to solubilize the whole molecule, the functional elements of the device are encapsulated inside the dendrimer. The role of the dendrimer in this construct is deemed to mimic that of a protein matrix, into which the active sites of a protein are embedded and isolated from the environment.
To probe the proposed design we synthesized several model compounds, all adducts of coumarins and Pt tetraarylporphyrins, performed their linear and 2P photophysical characterization and measured their oxygen quenching parameters. By demonstrating enhancement of the metalloporphyrin phosphorescence via intramolecular ET from the 2P antenna dyes and obtaining Stern-Volmer oxygen quenching plots upon 2P excitation we show the adequacy of the proposed design for oxygen measurements by phosphorescence quenching using 2P LSM. The obtained results provide guidelines for the future development of amplified 2PA sensors for biological applications.
Results and Discussion
Design of the Sensor
A schematic drawing of the 2PA oxygen nanosensor together with its' Jablonski diagram and a wavelength scale, depicting the order of all relevant transitions, are shown in Figure 1.
Figure 1.
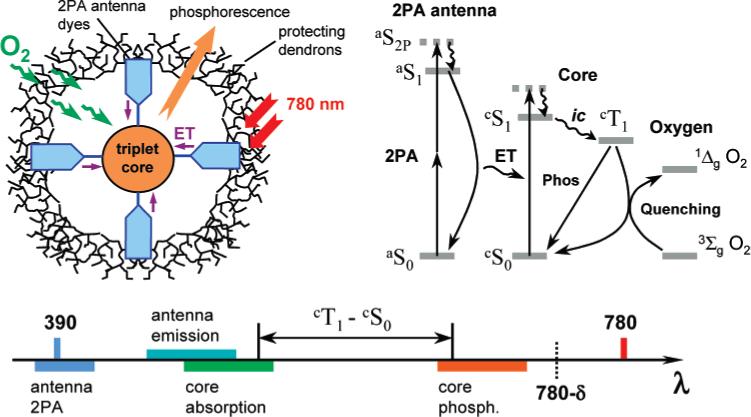
Scheme of the 2P phosphorescent oxygen sensor, its' Jablonski diagram and related wavelength scale.
Ti:Sapphire lasers, routinely used in multiphoton experiments, produce radiation at around 780 nm, which excites 2P transitions at twice the frequency, i.e., near 390 nm. After the 2P states of the antenna (aS2P) are populated and have internally converted into the lowest excited singlet state aS1, the excess energy is transferred to the core of the molecular device, presumably via the Förster type ET.32 The Förster mechanism assumes that the fluorescence (aS1 → aS0) of the donor (2P antenna) overlaps with an absorption band cSn ← cS0 (n = 1,2...) of the acceptor (core). Therefore, the core must possess linear absorption band-(s) somewhere in the region extending to the red from 390 nm. Exact positions of these bands are defined by the Stokes shift of the fluorescence of the antenna relative to its' absorption at 390 nm. The ET from the antenna to the core results either in the direct population of its' lowest singlet excited state (cS1), or cS1 is populated via internal conversion from the higher states cSn (n > 1). Finally, the cS1 state is depopulated via the intersystem crossing (ic) to yield the triplet state cT1, which, in turn, releases its' energy by either emitting phosphorescence or by passing it onto a quencher molecule, e.g., oxygen. The latter is consequently promoted to its' excited state, which in the case of oxygen is 1ΔgO2, and the energy is further consumed either chemically, or emitted as radiation or released to the ‘energy sink’ as heat.
If the cS1 state of the core is populated by the ET directly, the intersystem crossing cS1 → cT1 already results in a large red shift of the core phosphorescence relative to the original 2P absorption band of the antenna (e.g., at 390 nm). This shift becomes even larger if the cS1 state is populated via the cSn → cS1 internal conversion process. Overall, the whole energy cascade can result in a large separation of the final emission (phosphorescence of the core) from the primary 2P absorption of the antenna. Consequently, phosphorescence may occur close to the frequency of the laser. Therefore, the components of the device must be chosen in such a way that the whole energy cascade would fall within the wavelength range [λlaser/2; λlaser–δ], where δ is a shift from the peak of the laser spectrum, that is large enough to allow the discrimination of the phosphorescence. Of course, the phosphorescence may also occur in the region below the energy of the laser, i.e., above the wavelength (λlaser+δ), but very few such phosphors are known.
Another set of restrictions, imposed on the components of the device, concerns its' actual performance in the oxygen sensing application. It is convenient to define a gain coefficient γ, relating the intensity of emission from the molecular device D with a 2P antenna (I(D)) to that of the “naked” core C (I(C)), measured under the same conditions and normalized by the corresponding molar concentrations
| (1) |
The coefficient γ should be maximized in order that the smallest possible energies can be used in applications.
In general, the intensity of emission from the device (D) depicted in Figure 1 is proportional to the product of the 2PA cross-section of the antenna (σ2), the quantum yield of the energy transfer (ϕET) and the quantum yield of the core phosphorescence (ϕp)
| (2) |
where k is the proportionality constant, depending on the experimental conditions. It is reasonable to expect that neither the ET nor the emission from the triplet state cT1 depend on the mechanism of generating the excited states. Thus, coefficient γ, after being normalized by the values of the ET and emission quantum yields, should relate the effective 2PA cross section of the whole construct D (σ2(D)), to that of the core C (σ2(C)). It is useful, however, to distinguish between the measured gain γ and the “expected” gain γe, calculated assuming that the 2PA cross-section of the complex molecule is the sum of the 2PA cross sections of its' components (σ2(sum)) and the values of ϕET and ϕp are the same as in independent linear experiments (ϕET 1P and ϕp 1P)
| (3) |
Comparing the values of γ and γe gives an idea of how the energy is distributed within the sensor molecule D upon 2P excitation, without a priori assuming that neither the ET nor the triplet formation are affected by the nature of the excitation (2P vs 1P). Both parameters γ and γe are wavelength dependent, but in our experiments only one laser frequency (λex=780 nm) was used, so we omit the dependence on λ from the definitions 1−3.
Other critical parameters with respect to oxygen sensing are the phosphorescence lifetime of the sensor (τ0) and its' phosphorescence oxygen quenching constant kq. Phosphorescence or, more generally, luminescence quenching by oxygen in solution is a diffusion-controlled process, which typically follows the linear Stern–Volmer relationship in the range of physiologically relevant oxygen concentrations
| (4) |
where τ is the phosphorescence lifetime, τ0 is the phosphorescence lifetime in the absence of oxygen, pO2 is oxygen pressure and kq is the bimolecular rate constant. This form of the Stern–Volmer equation assumes that oxygen concentration in solution follows Henry law, and thus the quenching constant kq accounts for both the diffusion coefficient of oxygen in the immediate vicinity of the luminescent chromophore as well as for the oxygen solubility in the environment. Since the latter is typically unknown, it is useful to use the pressure rather that the concentration form of the Stern–Volmer equation.
The probability of collisional quenching by oxygen increases with an increase in the excited state lifetime, making longer decaying probes more sensitive to small changes in oxygen concentration. Consequently, phosphorescent chromophores, characterized by long lifetimes (tens to hundreds of microseconds), are preferred over generally brighter but much faster decaying (nanoseconds) fluorescent chromophores, even though the probability of the fluorescence quenching by oxygen may be as much as 9 times higher than that of the phosphorescence quenching, based on spin-statistical considerations.33
Model Pt Porphyrin-(tetra-Coumarin) Adducts
We selected Pt tetraarylporphyrin to be the phosphorescent core of the device, serving as the terminal acceptor of the excitation energy. Phosphorescence of Pt porphyrins at ambient temperatures is strong,34 occurs in the near infrared and is sensitive to O2.

For example, Pt meso-tetra(4-methoxycarbonylphenyl)porphyrin (PtTMCPP) shows phosphorescence with λmax = 668 nm, quantum yield ϕp = 7.5%, lifetime τ0 = 30.3 μs in the absence of oxygen and an oxygen quenching constant kq = 13 755 mmHg1−s−1 in DMF at 23 °C. The value of σ2 of PtTMCPP at 780 nm, determined by the relative emission method,35 is about 1.7 GM, which, as expected based on its' symmetry, is very low.
Förster-type ET onto the B-band (Soret) of a porphyrin has advantages because of its very high extinction coefficient, e.g., for PtTMCPP: λmax(B) = 403 nm, ε(B) = 249 650 M−1cm−1. Unfortunately, appropriately functionalized 2PA dyes, characterized by high σ2 values and possessing emission bands overlapping with porphyrin absorptions36 were unavailable to us. Such dyes should be considered in the future for construction of optimized 2P oxygen sensors. However, the present goal was to evaluate the feasibility of the entire scheme and elucidate its' potential difficulties. Consequently, we turned our attention to commercial dyes, for which σ2 values have already been reported, and which are routinely used in biomedical and other applications.37
Although coumarins are rather moderate 2P absorbers, their 2PA cross-sections (tens of GM units)37 are still higher than those of Pt and Pd porphyrins, and the ET from coumarins in dendrimers is well documented in the literature,23c–e in dendrimer-like porphyrin-based structures in particular.25f,38 In addition, ET from coumarins to other types of luminescent cores, i.e., Ru2+(bpy) complexes, has also been described.26b,c Attachment of four coumarin carboxylic acid (CCA) moieties to Pt meso-tetra(4-carboxyphenyl)porphyrin (PtTCPP) via ethylenediamine (EDA) linkers (Scheme 1) gave us the first model compound 3–a close relative of the recently reported tetracarboxyphenylporphyrin-CCA adduct with piperazine linkers.25f The photophysical data for compound 1 and all the other compounds discussed in the text are summarized in Table 1.
Scheme 1.

Synthesis of Pt Porphyrin-coumarin Adducts with EDA Linkers
Table 1.
Photophysical Data for the Compounds Described in the Textj
| no. | λabs (log ∊), nm (M−1cm−1) | λemis (λexcit), emis. typea | ϕ,b % | ϕET | σ2,d GM | Expected gain,e γe | Actual gain,e γ | kq, (mmHg1−s−1) | τ0, (μs) |
|---|---|---|---|---|---|---|---|---|---|
| PtTMCPP | 539 (3.77) | ||||||||
| 509 (4.41) | 668 (509), p | 7.5 (p) | (1)c | 1.7 | 1e | 1e | 13,755 ± 55 | 30.3 | |
| 403 (5.40) | 661 (510), pg | 11.0 (p)g | 42.1g | ||||||
| PtOBCPP | 540 (3.72) | ||||||||
| 511 (4.31) | 668 (511), p | 10.3 (p) | (1)c | 1.4 | 1g | 1g | 12,669 ± 266 | 48.8 | |
| 403 (5.34) | |||||||||
| 2 | 433 (4.58) | 489 (433), f | 67.5 (f) | 16 | |||||
| 3 | 539 (3.68) | ||||||||
| 509 (4.39) | 668 (509), p | 7.7 (p) | 0.98 | 31.2 | |||||
| 403 (5.28) | |||||||||
| 293 (4.63) | |||||||||
| 4 | 540 (3.78) | ||||||||
| 510 (4.43) | 667 (510), p | 6.5 (p) | 0.85 | (65.7) | 28.5f | 4.8−7.9f | 13,159 ± 200 | 27.6 | |
| 403 (5.40) | 489 (460), f | 4.6 (f) | |||||||
| 5 | 585 (3.27) | 746 (585) | |||||||
| 541 (3.76) | |||||||||
| 510 (4.19) | 668 (510), p | 0.65 (p) | 0.82 | (129.7) | 4.8g | 2.1g | 6.1 | ||
| 402 (5.32) | 491 (460), f | 6.3 (f) | |||||||
| 7 | 590 (2.61) | 752 (590) | |||||||
| 540 (3.73) | |||||||||
| 510 (4.43) | 668 (510), p | 1.4 (p) | 0.75 | (65.7) | 5.6f | 1.6f | 10,132 ± 117 | 18.6 | |
| 403 (5.46) | 492 (460), f | 8.7 (f) | |||||||
| 8 | 587 (3.47) | ||||||||
| 540 (3.94) | |||||||||
| 514 (4.49) | 672 (514), p | 6.1 (p) | 0.83 | (65.7) | 26.1f | 3.7f | 287.3 ± 0.9h,j | 45.1h,j | |
| 403 (5.55) | 491 (460), f | 2.5 (f) | |||||||
| 12 | 590 (3.18) | ||||||||
| 540 (3.95) | 669 (511), p | 2.3 (p) | 0.78 | (65.7) | 9.2f | 2.2f | 5,657 ± 226 | 20.1 | |
| 511 (4.61) | 485 (460), f | 1.8 (f) | |||||||
| 403 (5.59) | 668 (510), pg | 6.6 (p)g | 0.81 | 38.1g | |||||
| 13 | 590 (3.59) | ||||||||
| 540 (4.03) | |||||||||
| 515 (4.56) | 673 (515), ph | 1.4 (p)h | 0.76h | (65.7) | 5.5f | 1.9f | 332.6 ± 10.1h | 37.4h | |
| 403 (5.49) | 494 (460), fh | 1.3 (f)h |
f-antenna fluorescence; p-core phosphorescence.
Emission quantum yield.
The ET quantum yield for the core molecules themselves is assumed to be 1 (shown in parentheses).
Ti:Sapphire laser, 780 nm, 110 fs; σ2 value is shown in parentheses when is estimated as the sum of the 2PA cross-sections of the components; 1 GM = 10−50 cm4 s photon−1 molecule−1.
λex=780 nm.
Relative to PtTMCPP.
Relative to PtOBCPP.
Phosphate buffer, pH=7.4; deoxygenated using glucose/glucose oxidase/catalase system.
i In deoxygenated toluene.
All measurements were performed in deoxygenated DMF at r.t., unless noted otherwise. (Definitions of all parameters are given in the text.)
The absorption spectrum of 3 in DMF (Figure 2) was nearly identical to the superposition of its components, i.e., CCA and PtTMCPP, indicating no interaction between them in the ground state. Linear experiments revealed that the excitation energy delivered to coumarins in 3 (λex = 290 nm) was quantitatively transferred to the Pt porphyrin core and emitted as phosphorescence (λmax = 667 nm), proving functionality of this part of the energy pathway in the construct.
Figure 2.
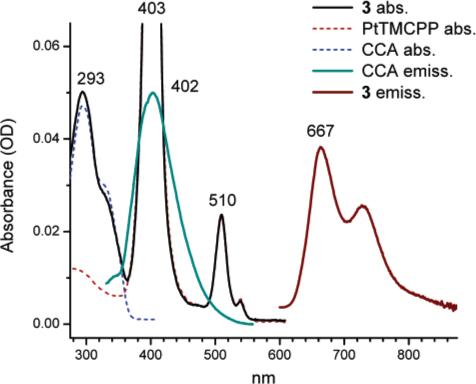
Absorption and emission (linear excitation) spectra of 3 and reference compounds in DMF. For emission: (1) samples were deoxygenated; (2) ordinate values are given in arbitrary units.
The ϕp and τ0 values of 3 were found to be almost the same as for PtTMCPP, used as a standard. The ET efficiency, determined by comparing the excitation and absorption spectra of 3, normalized by the maximum of the Pt porphyrin phosphorescence, was about 98%. This quite high yield of the ET was not surprising because of the excellent overlap of the coumarin emission (λmax = 402 nm) with the B-band of PtTMCPP and a close distance between the chromophores in 3 (rav = 13.6 Å according to the MM2 simulations). The Förster distance R0 for PtTMCPP–CCA pair was estimated to be 72.2 Å.
The 2PA induced phosphorescence of 3 was, however, very weak. The linear absorption band of CCA is positioned at around 290 nm, and since coumarin is not a centrosymmetrical molecule, its 2P transitions are likely to be close to its regular linear absorption bands. Since the latter lie higher than the twice-frequency of 780 nm laser (390 nm), used in our experiments, the 2PA cross-section of CCA at 780 nm was low and too few excitation photons were captured. In addition, the phosphorescent band, centered at 667 nm was located on the descending wing of the Rayleigh peak from the laser. While shorter excitation wavelength (<700 nm) would most likely increase the 2PA in 3, it would simultaneously shift the laser spectrum closer to the phosphorescence (Figure 1). Besides, for systems in which the spin—orbit coupling is large, as in Pt porphyrins (ϕic ≈ 1),39 triplet states can be, in principle, attained via direct T1 → S0 transitions,40 which would become increasingly more probable as the laser frequency approaches the phosphorescent absorption band. Linear absorption would present an unwanted source of background signal.
The necessity to shift the 2P absorption to the red, i.e., to about 2 × 390 nm, suggested that the Q-band of PtTMCPP (λmax(Q) = 510 nm, ε(Q) = 25 800 M−1cm−1) be used in the energy transfer step. One such dye is Coumarin-343 (C343, Scheme 1), whose emission (λmax = 489 nm) overlaps significantly with the Q-band of PtTMCPP. C343 has been used in ET studies in dendritic systems.23c—e It fluoresces with quantum yield ϕf = 75% in DMF and its' σ2 value is about 25 GM.37 The ϕf and σ2 values for C343 with the EDA linker (2) were found to be 67.5% and 16 GM, respectively. The linear absorption band of C343 (λmax = 433 nm) overlaps with the B-band of PtTMCPP, but its' red shoulder extends into the gap between the B and Q-bands of the porphyrin. For example, excitation at 460 nm, would almost exclusively excite the C343 fragment in a Pt porphyrin-C343 adduct.
Four C343 moieties could be attached to PtTCPP via EDA linkers using essentially the same chemistry as in the synthesis of 3, taking advantage of structural similarities between CCA and C343 molecules (Scheme 1).
The absorption spectrum of the resulting adduct 4 closely resembled that of the 1:4 mixture of PtTMCPP and 2 (Figure 3), but the red shoulder of the porphyrin Soret band, which is due to the absorption of C343, was slightly attenuated. This difference indicates a small interaction between the chromophores. This interaction could also be responsible for the small decrease in the phosphorescence quantum yield of 4 (ϕp = 6.5%), compared to that of PtTMCPP (ϕp = 7.5%). The ET efficiency in 4 was found to be also slightly lower than in 3, i.e., about 85%, reflecting the lower extinction coefficient of the Q-band of PtTMCPP and a smaller absorption/emission overlap between the donor and acceptor in the molecule. Consequently, the residual fluorescence of C343 in 4 was still noticeable, i.e., ϕf = 3.6% (λex = 460 nm). On the other hand, the increase in the phosphorescence of the Pt porphyrin in 4, relative to that of the 4:1 mixture of 2 (Scheme 1) and PtTMCPP, was about 4.5 times. This increase is quite close to the expected enhancement factor of 4.3, calculated as the product of the ratio of the molar extinctions ε(4)/ε(PtTMCPP) at λex (5.8 at 460 nm), the ratio of the phosphorescence quantum yields ϕp(4)/ϕp(PtTMCPP) (0.87) and the ET quantum yield ϕET(4) (0.95).
Figure 3.
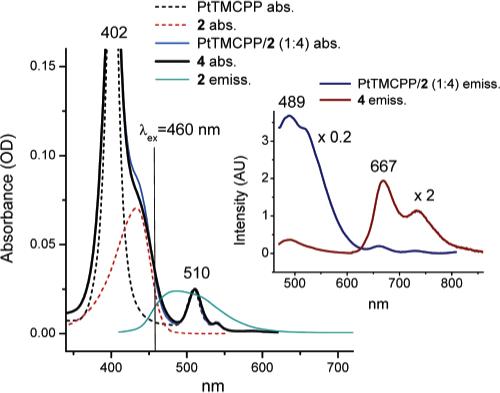
Absorption and emission spectra of 4 and of the reference compounds in DMF upon linear excitation (λex = 460 nm). Emission of 2 is also shown (main graph) to demonstrate the overlap with the Q-band of PtTMCPP (λmax = 510 nm). Emission spectra of 4 (multiplied by 2) and of the 1:4 mixture of PtTMCPP and 2 (divided by 5) are shown in the inset. Spectra are normalized by the absorption at 510 nm. Samples were deoxygenated by Ar bubbling.
The key result for this work, however, was the observed large amplification of the phosphorescent signal upon 2P excitation, which presented the main proof of the principle for the proposed scheme. When the excitation was carrier out at 780 nm, using an amplified Ti:Sapphire laser (110 fs pulse width, 1 kHz repetition rate), the increase in the phosphorescence of 4, compared to that of PtTMCPP, was about 8-fold (Figure 4, B).
Figure 4.
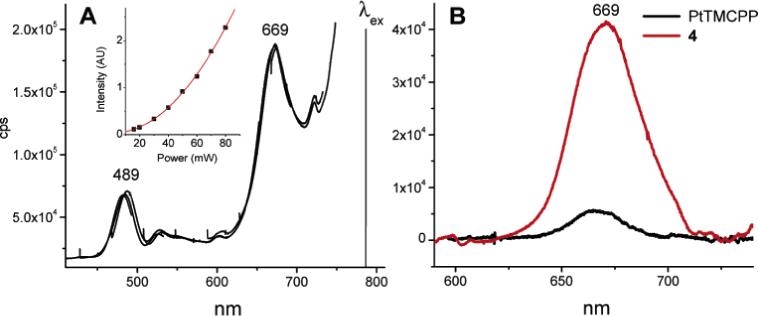
Emission spectra of 4 in deoxygenated DMF upon 2P excitation (λex = 780 nm, 110 fs, 1 kHz). A) Uncorrected spectrum (raw data), showing the wing of the laser Rayleigh peak. The inset shows the dependence of the emission amplitude on the excitation intensity (the solid line is a quadratic function). B) Spectra of 4 and PtTMCPP (reference) after the baseline subtraction.
The emission from PtTMCPP and 4 was confirmed to have a quadratic dependence on the excitation power (Figure 4, A(inset)), proving its' origin in the 2P process.41 The relative intensities of the fluorescence of C343 (λmax = 489 nm) and the phosphorescence of the Pt porphyrin (λmax = 667 nm) were similar to those in the linear experiments.
The observed increase in the phosphorescence of 4, compared with that of the core (PtTMCPP), i.e., γ = 7.8, was significantly lower than the expected value γe = 28.5 (see Table 1). Following eq 3, γe was calculated assuming that the 2PA cross-section of 4 is the sum of the cross-sections of the core porphyrin (1.7 GM) and the four antenna fragments 2 (64 GM), i.e., 65.7 GM; and that the ET efficiency (ϕET = 0.85) and the emission quantum yields (ϕp(4) = 0.065, ϕp(PtTMCPP) = 0.075) upon 2P excitation of 4 were the same as determined in independent linear experiments. As mentioned earlier, the accuracy of the determination of the phosphorescence intensities in the steady state experiments was limited due to the difficulties in detecting signals in the proximity of the laser spectrum. Therefore, we additionally measured the phosphorescence intensities of 4 and of PtTMCPP upon 2P excitation using a pulsed time-resolved setup (see Experimental Section for details). However, the value of the gain factor (γ = 4.8−4.9) turned out to be even lower than in the steady-state experiments. Several possibilities were envisioned to explain this result, e.g., the 2PA cross-section σ2 of 4 was considerably below expected (Table 1); the ET and/or the phosphorescence in 4 were affected by the ultrafast excitation. The large Stokes shift of the C343 fluorescence (2645 cm−1 in DMF) makes the self-quenching between C343 fragments in 4 an unlikely cause of the diminished emission from the core.42 Besides, such self-quenching should, in principle, be the same for 1P and 2P excitation processes.
The ET efficiencies in molecules with multiple antenna dyes (e.g., dendritic molecules) may be reduced by “annihilation” of two or more excitations.43 However, the probability of two excitations occurring in the same molecule of 4 was estimated to be very low, i.e., only about 10−5. This value was calculated assuming that the 2PA cross section of each coumarin in 4 was 20 GM and taking into account that the concentration of molecules was about 10−4 M (i.e., 0.6 × 1017 molecule cm−3) and that the beam was focused into a spot of about 3 mm in diameter, resulting in the photon flux of about 5 × 1028 photon s−1 cm−1.
The triplet state of the Pt porphyrin also should not have been affected or destroyed by the laser, which would result in a lower phosphorescence quantum yield. Indeed, the 110 fs excitation pulses in our experiments were comparable or faster than the intersystem system crossing, and they were separated by 1 ms periods, corresponding to more than 10 triplet lifetimes (τ0(4) = 27.6 μs). Therefore, the Pt porphyrin triplet population had ample time to decay naturally prior to the arrival of the next excitation pulse.
It is interesting to mention a similar experiment, reported recently by Dichtel and co-workers,28 in which a free-base porphyrin was modified with several powerful 2PA antenna dyes. The ET efficiency in their system, as judged from the emission/absorption overlap obtained from the published graph, was quite high. Nevertheless, the increase in the porphyrin fluorescence (estimated from the published graph) upon 2P excitation was also much lower than expected from the ratio of the combined σ2's of the 2P antenna chromophores (several thousands GM units) to that of the core porphyrin. However, in their case the lower apparent emission may have been a result of the diminished fluorescence quantum yield of the adduct, compared to that of the core porphyrin.
More comprehensive photophysical studies will be required to understand the reason for the discrepancy between the expected value of γe and the experimentally obtained gain γ in our case and in similar model systems. However, if confirmed on the fundamental level this discrepancy might limit the applicability of dendritic 2PA antennas in combination with porphyrin-emitters.
Model Pt Porphyrin-(octa-Coumarin) Adduct
The next step in our experiments was to establish whether an increase in the number of antenna chromophores would result in further amplification of the signal from the Pt porphyrin. With that in mind, we synthesized construct 5 (Scheme 1), in which the number of C343 fragments was doubled compared to that in 4. Compound 5 is based on Pt octabutoxycarbonylporphyrin (PtOBCPP), whose phosphorescence quantum yield (ϕp = 10.3%) is slightly higher than that of PtTMCPP. 5 was synthesized by implementing the same reaction sequence as in the synthesis of 3 and 4, but employing Pt octacarboxyphenylporphyrin (PtOCPP) as the core (Scheme 1). Extremely low solubility of 5 made handling of this material very difficult, but was sufficient for performing the photophysical measurements.
As expected, the absorption spectrum of 5 (Figure 5) very closely resembled the superposition of its' functional components, however, a small extra band could be observed at 587 nm. The emission spectrum of 5 also contained bands from both C343 and Pt porphyrin, but the longer wavelength shoulder of the phosphorescence signal was extended further to the red, practically forming a separate peak at λmax = 743 nm. The excitation spectrum of 5 at λemis = 743 nm matched the absorption spectrum, except that the band at 587 nm could be seen more distinctly. The small absorption at 587 nm and its' emission counterpart at 743 nm could be due to a ground-state interaction between the Pt porphyrin and one of the coumarins in the molecule.
Figure 5.
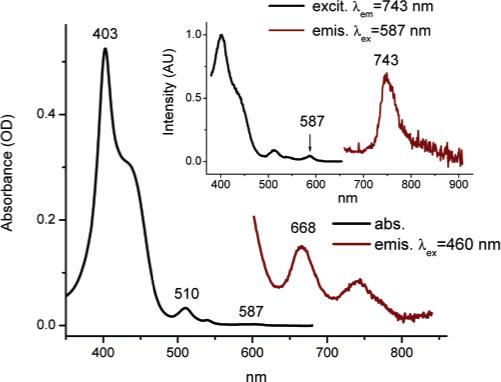
Linear absorption, emission and excitation spectra of 5 in deoxygenated DMF.
The possibility of formation of an intramolecular complex was further supported by the phosphorescence measurements. It appeared that compound 5 possessed an extremely low phosphorescence quantum yield (ϕp = 0.65%), i.e., more than 15 times lower than that of the parent porphyrin. Such weak phosphorescence immediately placed 5 out of consideration for practical oxygen sensing, but at the same time was rather interesting by itself. Since nothing in the structure of the PtOCPP macrocycle was likely to change upon appending C343-EDA fragments (e.g., no nonplanar perturbations, leading to decrease in emission),44 quenching of the phosphorescence in 5 was most probably associated with an interaction between the core and the peripheral C343 dyes. It is quite likely that such an interaction could facilitate charge transfer (CT) between Pt porphyrin in its' excited triplet state and one of the antenna coumarins. Charge transfer upon energy transfer in dendritic systems has been documented in the literature,45 and in our case, it could be additionally favored by a very long lifetime of the triplet state Pt porphyrin (tens of microseconds) as well as by the presence of electron-donor amino groups in C343 molecules. Meta-orientation of the anchor groups in meso-aryl rings in the porphyrin, to which EDA-C343 fragments are linked, also could have played a role in facilitating the interaction between the peripheral groups and the core. C343 molecules, fixed at the ends of the flexible linkers, could encounter close face-to-face contacts with the porphyrin, resulting in direct orbital overlaps and an efficient CT. In 4, on the other hand, para-orientation of the EDA linkers made such interactions much less probable.
The 1H NMR data supported the hypothesis regarding the proximity of the C343 fragments to the Pt porphyrin in 5 (see Supporting Information, Table 1). The resonances corresponding to C434 in 5 are 0.18 to 0.53 ppm upfield shifted (0.3 ppm average) compared to the resonances of Coumarin-343 itself, with aromatic protons being shifted the most. At the same time, the corresponding 1H resonances in 4 are shifted on average only by about 0.05 ppm relative to those of Coumarin-343, suggesting a much weaker interaction between C343 and PtP moieties.
An alternative cause of the phosphorescence quenching in 5 could be the triplet—triplet ET32 between the Pt porphyrin and a C343 molecule. Triplet—triplet ET between the core and the peripheral groups has also been described for dendritic systems.46 In our experiments, however, we could not detect any deviations from pure Pt porphyrin phosphorescence (i.e., extra bands due to the C343 phosphorescence) when 5 was excited directly at the Pt porphyrin Q-band at 510 nm. Nevertheless, the possibility of the triplet—triplet ET cannot be completely ruled out, as it is still possible that the triplet of C343 emission strongly overlaps with Pt porphyrin phosphorescence and/or the internal conversion is the predominant pathway of its' deactivation. We could not locate the experimental value for the C343 triplet state energy in the literature. The computational data,47 however, suggest the values of 25 205 and 16 581 cm−1 respectively for S1 ← S0 and T1 ← S0 transitions in the C343 molecule. Given the experimental fluorescence maximum of C343 at 489 nm and considering proportional shift of the C343 phosphorescence relative to the computationally predicted value (603 nm), the triplet emission of C343 would be expected at about 740 nm. Obviously, even if there was a weak signal from the C343 triplet, we could easily miss it due to the overlap with Pt porphyrin emission.
In addition to negligible phosphorescence, the ET in 5 was also not very high, i.e., ϕET = 0.82, and the amplification of the emission upon 2P excitation (γ = 2.1) was significantly below the expected value (γe = 4.8), making 5 a very poor overall performer. However, an important realization that came from the experiments with this compound was that the CT (and/or triplet—triplet ET) can in principle present a significant obstacle for the design depicted in Figure 1. Indeed, all known efficient 2PA dyes are easily polarizable molecules, comprising of π-electron donor and π-electron acceptor groups,48 which are likely to be active in CT processes. Therefore, CT either from the triplet state porphyrin to the antenna, or backward, may always compete with the porphyrin phosphorescence. Accordingly, a very special caution should be exercised to prevent the CT in the sensor molecule. Either the 2P dyes incapable of the electron exchange with the triplet state porphyrin will have to be used, or, more realistically, the 2P dyes will have to be placed at such a distance from the core, that the CT efficiency will be minimal, while the ET efficiency will be still very high. Such distance-tuning should be possible if the antenna-core pair is chosen to have a large Förster R0 distance. Moving the antenna out of the core, but leaving it within the R0, would lower the CT rate, falling exponentially with the distance, while maintaining a high rate of the ET.
Stern—Volmer Quenching of 2PA Induced Phosphorescence
Since compound 4 turned out to be the brightest phosphor among all the studied adducts, we chose it as a model to demonstrate oxygen sensing by phosphorescence lifetime and to measure its' lifetime-based Stern—Volmer plot upon 2P excitation. The Stern—Volmer oxygen quenching constant of 4 in DMF at 23.5 °C, determined using linear excitation and a standard lifetime/oxygen titration setup,49 was 13 159 ± 200 mmHg−1s−1, which is consistent with values obtained typically for phosphorescent porphyrins in DMF solutions at ambient temperatures.49 To retrieve the kq value for the same compound upon 2P excitation, the detection system of a time-domain phosphorometer was synchronized with the laser pulses, so that the emission could be registered for about 500 μs following the flash. The resulting phosphorescence decays (Figure 6A), generated upon 2P excitation, were analyzed by fitting to single exponentials, giving a 2P Stern—Volmer plot (Figure 6B).
Figure 6.
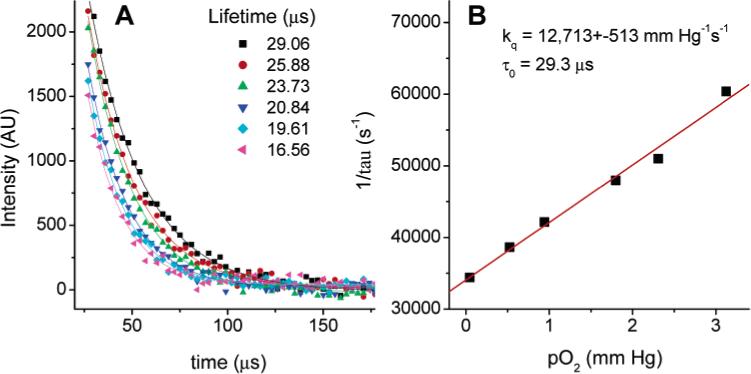
Phosphorescence decays of 4 in DMF upon 2P excitation (λex = 780 nm, 110 fs, 1 kHz) at different oxygen concentrations (A) and the corresponding Stern—Volmer plot (B).
As expected, the value of the constant kq, determined from the 2P experiment, basically matched that obtained in a regular titration, i.e., 12 713 ± mmHg−1s−1. This result proves the applicability of the method for oxygen measurements and shows that the last part of the energy pathway depicted in Figure 1 remained unaffected by the nature of excitation.
Dendritically Protected Pt Porphyrin—Coumarin Adducts
Oxygen sensing in biological tissues assumes that the phosphorescent dyes are soluble in water, inert toward biological objects in the blood (e.g. bio-macromolecules, cell and blood vessel surfaces) and, at the same time, that the oxygen quenching constants (kq, eq 3) of the phosphors are not excessively high, so that some phosphorescence still can be detected at ambient oxygen pressures. On the other hand, simple metalloporphyrins modified with hydrophilic groups usually exhibit extremely high oxygen quenching constants in aqueous solutions (e.g., 3000−4000 mmHg−1s−1),49 they aggregate and, when in the blood, bind to bio-membranes, proteins and other macromolecules. To protect phosphorescent chromophores and make them suitable for biological applications, it has been proposed to encapsulate porphyrins inside dendrimers.11a,49,50 Dendritic encapsulation,51 of porphyrins in particular,25,49,50,52 is known to be an effective means for tuning thermodynamic properties of the dendrimer cores52b,e,m and building kinetic barriers for small molecules.49,50,52a,c,f
While tuning the oxygen quenching constants is the most important functional role of dendrimers in oxygen nanosensors, for such large complex molecules as 4 and 5 dendritic encapsulation was expected to first of all play a simple solubilizing role. Both the central core porphyrin and the antenna dyes are very hydrophobic molecules, requiring many external hydrophilic groups on the dendrimer to prevent their aggregation and provide enough solubility in water. Newkome-type poly-(ester amide) dendrimers53 have been shown to be extremely effective in solubilizing porphyrins in aqueous solutions and preventing their aggregation.49,52b,e,j,m We chose to complement standard Newkome dendrons with arylglycine fragments,54 which have been recently found effective in hydrophobic shielding of the porphyrin cores. A prototype of the water soluble Pt porphyrin-based dendritic oxygen sensor with 2P antenna was assembled using a mixed divergent/convergent synthesis shown in Scheme 2.
Scheme 2.

Synthesis of Dendritic Pt Porphyrin-C343 Adducts
At first, PtTCPP was modified with four lys(cBz)(OtBu) fragments, resulting in the orthogonally protected compound 6. Deprotection of amino groups in 6 by Pd/C catalyzed hydrogenolysis, followed by coupling of four C343 fragments, gave tetra-Boc-protected compound 7, which was converted into tetra-acid 8 upon treatment with TFA. Only acid-cleavable protecting groups (e.g., Boc) could be used in these syntheses, as C343 fragments are very unstable under basic conditions as well as under conditions of the hydrogenolysis. Consequently, we used a method, developed recently by Cardona and Cawley,55 to synthesize tBu esters of Newkome-type dendrons 9, which were further coupled to arylglycine diacid 1054 to give cBz-protected mixed dendron 11. After deprotection, 11a was coupled to 8, yielding Pt porphyrin dendrimer 12 with four C343 antenna dyes, attached to the ends of the lysine linkers. Cleaving tBu esters in 12 by TFA gave the final water-soluble dendrimer 13. All the compounds preceding the dendrimer 12 in Scheme 2 were unambiguously characterized by standard methods. However, so far we failed to obtain an adequate MALDI spectrum of 12, despite trying numerous supporting matrixes, salts, and changing conditions. Apparently, ionization of this compound is extremely difficult, which is in tune with our previous experience of work with arylglycine porphyrin-dendrimers.54 Nevertheless, based on the NMR and GPC analysis (see Supporting Information) we concluded that porphyrin dendrimer 12 had adequate purity for subsequent photophysical characterization. In addition, a broad peak centered close to the theoretically predicted mass was seen in the MALDI spectrum of compound 13 (see below), obtained from 12 by hydrolysis.
The first compound in Scheme 2, suitable for 2P experiments, i.e., adduct 7, is different from the compound 4 by the length of the linkers, i.e., six vs two CH2 groups. This increase in the linker length and, perhaps, the linker flexibility reduced the ET efficiency in 7 (ϕET = 0.75) and, more importantly, its' phosphorescence (ϕp = 1.44%) was nearly five times weaker than that of the parent PtTMCPP. Again, we suggest that the most probable cause for such attenuation is the charge transfer, made possible by longer more flexible lysine linkers, facilitating direct contacts between the coumarines and the porphyrin. The absorption and the emission spectra of 7 support this hypothesis, as they contain extra bands reminiscent of those in 5. And again, the gain factor measured for 7 in 2P experiments was quite low (γ = 1.6), constituting only a fraction of the expected value γe = 5.6.
As expected, cleavage of tBu ester groups in 7 did not achieve the desired water solubility. The absorption spectra of 8 in aqueous solutions revealed its' strong aggregation. However, upon binding to bovine serum albumin (BSA), which is known to be an excellent solubilizing agent for porphyrinic compounds, the aggregation could be entirely avoided.9a Interestingly, the overall performance of 8 in water in the presence of BSA turned out to be much higher than that of its' esterified analogue 7 in DMF. The phosphorescence quantum yield of 8 went up to 6.1% and the ET efficiency increased as well (ϕET = 0.83). It is likely that BSA provided a more restrictive environment to the C343 moieties in the BSA-8 complex, rigidifying the whole structure, limiting nonradiative deactivation pathways and, perhaps, prohibiting contacts between C343 molecules and Pt porphyrin. As a result, the gain factor upon 2P excitation for 8 was found to be considerably higher (γ = 3.7) than that for 7. Although far from being optimal, complex 8 with BSA presents a model for a fully synthetic hydrophilic sensor. The role of BSA in this complex is perhaps what we deem to be the role of the dendrimer, i.e., solubilization, fixation of the antenna fragments at a distance from the core and, of course, attenuation of the oxygen quenching constant. The latter determined for the complex 8-BSA was 287 mmHg−1s−1, which is almost 13 times lower than the quenching constant of the reference PtOCPP in aqueous buffer (kq = 4239 mmHg−1s−1).
Modification of 8 with four dendritic branches lead to a well soluble dendritic Pt porphyrin dendrimer 12. Its' phosphorescence quantum yield was somewhat higher than that of the nondendritic precursor 7 (ϕp = 2.3%) and so was the gain coefficient γ = 2.2. A useful feature of compound 12, compared to virtually all other compounds described, was its' much improved solubility in organic solvents. The possibility of dissolve 12 in solvents less polar than DMF enabled us to test solvent effects on the phosphorescence quantum yield of this compound. More polar solvents would likely to stabilize the CT state, resulting in a more pronounced phosphorescence quenching, should this quenching involve the CT processes. Indeed, in toluene the phosphorescence quantum yield of 12 was decreased by 1.7 times (ϕp(12) = 6.6%), compared to that of PtTMCPP (ϕp(PtTMCPP) = 11%); whereas in DMF the phosphorescence of 12 dropped by as much as 3.3 times, suggesting that the charge transfer was at least in part responsible for the static quenching.
The most dramatic change upon attachment of the dendrons occurred with the oxygen quenching constant, which for 12 in DMF was almost one-half that of 7 (kq = 10 132 mmHg−1s−1), i.e., kq = 5657 (mmHg−1s−1). This attenuation demonstrates how dendrimer can control oxygen diffusion to the core.
Not surprisingly, a much stronger attenuation was achieved in the case of water-soluble dendrimer 13 (Figure 7). Its' oxygen quenching constant (kq = 333 mmHg−1s−1) was, in fact, almost as low as that of the 8-BSA complex, indicating an efficient protection of the phosphorescent core by the dendritic branches. The possibility was considered that some of this almost “too good” protection could be explained by partial aggregation of 13, notwithstanding its' 24 peripheral carboxylates. However, the addition of BSA or any other conventional aggregate-solubilizing agent, such as Tween 80, did not affect broadening of the absorption spectrum. It was, therefore, reasonable to conclude that the hydrophobic folding of the dendrons and of the lysine-C343 groups, was the primary cause of the oxygen diffusion attenuation.
Figure 7.
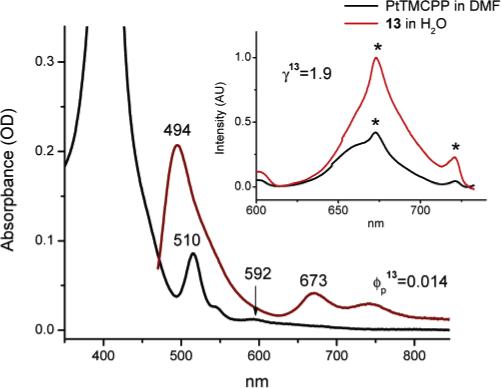
Linear absorption and emission spectra of 13 in deoxygenated aqueous buffer solution (pH = 7.4) (main graph). Phosphorescence of 13 upon 2P excitation (λex = 780 nm, 110 fs, 1 kHz) and the reference spectrum of PtTMCPP in deoxygenated DMF. Spectra are normalized by concentrations. (Asterisks show the “artifact” peaks occurring due to the high excitation power).
As seen from Table 1 and Figure 7 (inset) the overall performance of the construct 13 in 2P experiments was not impressive (γ = 1.9), mainly due to its' very low phosphorescence quantum yield (ϕp = 1.4%). The absorption spectrum of 13 contained a detectable band at 592 nm, manifesting an interaction between the chromophores in the ground state, probably responsible for the static quenching. Nevertheless, all the main features of the design were implemented and proven functional in compound 13. Indeed, this compound possesses a phosphorescent oxygen sensitive core, 2P antenna dyes and solubilizing/protecting dendrons, and, therefore, it can be considered a functional prototype of the protected 2PA sensor for biological oxygen measurements.
Conclusion
The foregoing experiments demonstrate that a water soluble 2PA sensor suitable for oxygen measurements using 2P LSM can be constructed by combining in one molecule a phosphorescent emitter with an array of 2P absorbing chromophores and encapsulating the whole system inside a protecting dendrimer with hydrophilic periphery. Several important guidelines for the future design and optimization, which follow from this work, are summarized below.
First of all, the gain in the emission from the triplet state core does not seem to be directly proportional to the increase in the 2PA cross-section of the antenna dyes. This notion, although consistently supported by the experiments described in this work, will require verification on other systems. It is possible that the discrepancy between the expected and the observed gain in emission is related to the ET processes initiated by 2P excitation.
Second, CT from the 2P antenna to the triplet state core, or backward, can play a significant role in quenching the core phosphorescence. Since 2PA dyes are usually easily polarizable molecules, they can stabilize charges and thus be active in CT processes. The CT in such antenna/emitter pairs can become very probable because the lifetimes of triplet emitters are long (tens of microseconds). In addition, quenching of the porphyrin triplet state can occur via the backward triplet—triplet ET to the antenna. To avoid both of these possibilities the antenna chromophores should be located far enough from the phosphorescent core, so that the probability of the CT and/or the triplet—triplet ET is minimal, while the efficiency of the forward antenna-to-core resonance ET is high. Moreover, if it is the CT that is responsible for the core quenching, the distance between the antenna and the core should not be regulated by means of a rigid conjugated linker, which would serve as a conducting wire, not preventing, but facilitating the CT. Instead, the supporting dendrimer should be used to regulate the distance.
As mentioned in the Introduction, the dendrimer in the construct shown in Figure 1 is not only required to protect the core from oxygen and to solubilize the molecule, but also to provide a supporting matrix, in which the “active sites” of the device (antenna and the core) are positioned at appropriate distances from one another to facilitate the energy exchange. For the purpose of distance tuning it seems feasible to embed the 2P absorbing antenna dyes in the body of an insulating dendritic matrix,56 instead of directly linking them to the phosphorescent core. Substantial literature exists on incorporation of functional motifs in the interior of dendrimers.57 By choosing which dendritic layer (generation) in the structure is functionalized with the 2P dyes, the antenna array can be positioned within the Förster distance R0, beyond the range that would permit the CT. Synthesis of such constructs will be the next challenge in the design of amplified dendritic nanosensors.
Acknowledgment
Support of the Grant Nos. NIH EB003663-01 (S.A.V.) and NIH PO1-RR001348 (R.M.H.) is gratefully acknowledged.
Footnotes
Supporting Information Available: Full experimental details, synthetic details and characterization data for the compounds described in this paper. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.a Berkowitz BA, Kowluru RA, Frank RN, Kern TS, Hohman TC, Prakash M. Invest. Ophthalmol. Visual Sci. 1999;40:2100–2105. [PubMed] [Google Scholar]; b Linsenmeier RA, Braun RD, McRipley MA, Padnick LB, Ahmed J, Hatchell DL, McLeod DS, Lutty GA. Ophthalmol. Visual Sci. 1998;39:1647–1657. [PubMed] [Google Scholar]
- 2.a Vannucci SJ, Hagberg H. J. Exp. Biol. 2004;207:3149–3154. doi: 10.1242/jeb.01064. [DOI] [PubMed] [Google Scholar]; b Brunel H, Girard N, Confort-Gouny S, Viola A, Chaumoitre K, D'Ercole C, Figarella-Branger D, Raybaud C, Cozzone P, Panuel M. J. Neuroradiology. 2004;31:123–137. doi: 10.1016/s0150-9861(04)96979-9. [DOI] [PubMed] [Google Scholar]; c Johnston MV, Nakajima W, Hagberg H. Neuroscientist. 2002;8:212–220. doi: 10.1177/1073858402008003007. [DOI] [PubMed] [Google Scholar]
- 3.a Evans SM, Koch CJ. Cancer Lett. 2003;195:1–16. doi: 10.1016/s0304-3835(03)00012-0. [DOI] [PubMed] [Google Scholar]; b Ziemer LS, Lee WMF, Vinogradov SA, Sehgal C, Wilson DF. J. Appl. Physiol. 2005;98:1503–1510. doi: 10.1152/japplphysiol.01140.2004. [DOI] [PubMed] [Google Scholar]
- 4.Rajendran JG, Krohn KA. Radiol. Clin. North Am. 2005;43:169–187. doi: 10.1016/j.rcl.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 5.Subramanian S, Matsumoto KI, Mitchell JB, Krishna MC. NMR Biomed. 2004;17:263–294. doi: 10.1002/nbm.897. [DOI] [PubMed] [Google Scholar]
- 6.a Piert M, Machulla HJ, Picchio M, Reischl G, Ziegler S, Kumar P, Wester HJ, Beck R, McEwan AJB, Wiebe LI, Schwaiger M. J. Nucl. Med. 2005;46:106–113. [PubMed] [Google Scholar]; b Apisarnthanarax S, Chao KSC. Rad. Res. 2005;163:1–25. doi: 10.1667/rr3279. [DOI] [PubMed] [Google Scholar]
- 7.a Fenton BM, Paoni SF, Lee J, Koch CJ, Lord EM. Brit. J. Cancer. 1999;79:464–471. doi: 10.1038/sj.bjc.6690072. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Liu HL, Song YL, Worden KL, Jiang X, Constantinescu A, Mason RP. Appl. Opt. 2000;39:5231–5243. doi: 10.1364/ao.39.005231. [DOI] [PubMed] [Google Scholar]
- 8.a Ballinger JR. Sem. Nucl. Med. 2001;31:321–329. doi: 10.1053/snuc.2001.26191. [DOI] [PubMed] [Google Scholar]; b Foo SS, Abbott DF, Lawrentschuk N, Scott AM. Mol. Imag. Biol. 2004;6:291–305. doi: 10.1016/j.mibio.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 9.a Vanderkooi JM, Maniara G, Green TJ, Wilson DF. J. Biol. Chem. 1987;262:5476–5483. [PubMed] [Google Scholar]; b Wilson DF, Vinogradov SA. In: Handbook of Biomedical Fluorescence. Mycek M-A, Pogue BW, editors. Marcel Dekker; New York: 2003. Ch. 17. [Google Scholar]
- 10.a Rumsey WL, Vanderkooi JM, Wilson DF. Science. 1988;241:1649–1652. doi: 10.1126/science.241.4873.1649. [DOI] [PubMed] [Google Scholar]; b Vinogradov SA, Lo L-W, Jenkins WT, Evans SM, Koch C, Wilson DF. Biophys. J. 1996;70:1609–1617. doi: 10.1016/S0006-3495(96)79764-3. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Shonat RD, Kight AC. Annal. Biomed. Eng. 2003;31:1084–1096. doi: 10.1114/1.1603256. [DOI] [PubMed] [Google Scholar]
- 11.a Rietveld IB, Kim E, Vinogradov SA. Tetrahedron. 2003;59:3821–3831. [Google Scholar]; b Finikova OS, Cheprakov AV, Beletskaya IP, Carroll PJ, Vinogradov SA. J. Org. Chem. 2004;69:522–535. doi: 10.1021/jo0350054. [DOI] [PubMed] [Google Scholar]
- 12.a Soloviev V, Wilson D, Vinogradov S. Appl. Opt. 2003;42:113–123. doi: 10.1364/ao.42.000113. [DOI] [PubMed] [Google Scholar]; b Soloviev VY, Wilson DF, Vinogradov SA. Appl. Opt. 2004;43:564–574. doi: 10.1364/ao.43.000564. [DOI] [PubMed] [Google Scholar]
- 13.Sevick-Muraca EM, Godavarty A, Houston JP, Thompson AB, Roy R. In: Handbook of Biomedical Fluorescence. Mycek M-A, Pogue BW, editors. Marcel Dekker; New York: 2003. Ch. 14. [Google Scholar]
- 14.Denk W, Strickler JH, Webb WW. Science. 1990;248:73–76. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]; b Williams RM, Zipfel WR, Webb WW. Curr. Opin. Chem. Biol. 2001;5:603–608. doi: 10.1016/s1367-5931(00)00241-6. [DOI] [PubMed] [Google Scholar]; c Stephens DJ, Allan VJ. Science. 2003;300:82–86. doi: 10.1126/science.1082160. [DOI] [PubMed] [Google Scholar]
- 15.Göppert-Mayer M. Ann. Phys. 1931;9:273. [Google Scholar]
- 16.Tirlapur UK, Konig K, Peuckert C, Krieg R, Halbhuber KJ. Exp. Cell Res. 2001;263:88–97. doi: 10.1006/excr.2000.5082. [DOI] [PubMed] [Google Scholar]
- 17.Kruk M, Karotki A, Drobizhev M, Kuzmitsky V, Gael V, Rebane A. J. Lumines. 2003;105:45–55. [Google Scholar]
- 18. GM (Göppert-Mayer) = 10–50 cm4 s photon–1molecule–1.
- 19.Mik EG, van Leeuwen TG, Raat NJ, Ince C. J. Appl. Physiol. 2004;97:1962–1969. doi: 10.1152/japplphysiol.01399.2003. [DOI] [PubMed] [Google Scholar]
- 20.a Goyan RL, Cramb DT. Photochem. Photobiol. 2000;72:821–827. doi: 10.1562/0031-8655(2000)072<0821:nitpeo>2.0.co;2. [DOI] [PubMed] [Google Scholar]; b Karotki A, Kruk M, Drobizhev M, Rebane A, Nickel E, Spangler CW. IEEE J. Select. Top. Quantum Electron. 2001;7:971–975. [Google Scholar]
- 21.a Karotki A, Drobizhev M, Kruk M, Spangler C, Nickel E, Mamardashvili N, Rebane A. J. Opt. Soc. Am. B. 2003;20:321–332. [Google Scholar]; b Drobizhev M, Karotki A, Kruk M, Mamardashvili NZ, Rebane A. Chem. Phys. Lett. 2002;361:504–512. [Google Scholar]; c Drobizhev M, Karotki A, Kruk M, Rebane A. Chem. Phys. Lett. 2002;355:175–182. [Google Scholar]
- 22.a Karotki A, Drobizhev M, Dzenis Y, Taylor PN, Anderson HL, Rebane A. Phys. Chem. Chem. Phys. 2004;6:7–10. doi: 10.1021/jp044261x. [DOI] [PubMed] [Google Scholar]; b Drobizhev M, Stepanenko Y, Dzenis Y, Karotki A, Rebane A, Taylor PN, Anderson HL. J. Am. Chem. Soc. 2004;126:15352–15353. doi: 10.1021/ja0445847. [DOI] [PubMed] [Google Scholar]
- 23. For examples, see: [Google Scholar]; a Stewart GM, Fox MA. J. Am. Chem. Soc. 1996;118:4354–4360. [Google Scholar]; b Devadoss C, Bharathi P, Moore JS. J. Am. Chem. Soc. 1996;118:9635–9644. [Google Scholar]; c Gilat SL, Adronov A, Fréchet JMJ. Angew. Chem., Int. Ed. End. 1999;38:1422–1427. doi: 10.1002/(SICI)1521-3773(19990517)38:10<1422::AID-ANIE1422>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]; d Adronov A, Gilat SL, Fréchet JMJ, Ohta K, Neuwahl FVR, Fleming GR. J. Am. Chem. Soc. 2000;122:1175–1185. [Google Scholar]; e Freeman A, Fréchet JMJ, Koene S, Thompson ML. Macromol. Symp. 2000;154:163–169. [Google Scholar]; f Kleiman VD, Melinger JS, McMorrow D. J Phys. Chem. B. 2001;105:5595–5598. [Google Scholar]; g Vögtle F, Gorka M, Vicinelli V, Ceroni P, Maestri M, Balzani V. ChemPhysChem. 2001;2:769. doi: 10.1002/1439-7641(20011217)2:12<769::AID-CPHC769>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]; h Ortiz W, Roitberg AE, Krause JL. J. Phys. Chem. B. 2004;108:8218–8225. [Google Scholar]; i Balzani V, Ceroni P, Maestri M, Vicinelli V. Curr. Opin. Chem. Biol. 2003;7:657–665. doi: 10.1016/j.cbpa.2003.10.001. [DOI] [PubMed] [Google Scholar]; j Ranasinghe MI, Wang Y, Goodson T. J. Am. Chem. Soc. 2003;125:5258–5259. doi: 10.1021/ja029242k. [DOI] [PubMed] [Google Scholar]; k Thompson AL, Gaab KM, Xu JJ, Bardeen CJ, Martinez TJ. J. Phys. Chem. A. 2004;108:671–682. [Google Scholar]; l Ortiz W, Adrian ER, Krause JL. J. Phys. Chem. 2004;108:8218–8225. [Google Scholar]; m Goodson TG. Acc. Chem. Res. 2005;38:99–107. doi: 10.1021/ar020247w. [DOI] [PubMed] [Google Scholar]; n Thomas KRJ, Thompson AL, Sivakumar AV, Bardeen CJ, Thayumanavan S. J. Am. Chem. Soc. 2005;127:373–383. doi: 10.1021/ja044778m. [DOI] [PubMed] [Google Scholar]
- 24. Here we deal only with ET in bulk phase, whereas there is a large area of research on the ET in dendritic multichromophoric systems on the single molecule level. For examples, see. [Google Scholar]; a Tinnefeld P, Hofkens J, Herten DP, Masuo S, Vosch T, Cotlet M, Habuchi S, Mullen K, De Schryver FC, Sauer M. ChemPhysChem. 2004;5:1786–1790. doi: 10.1002/cphc.200400325. [DOI] [PubMed] [Google Scholar]; b Masuo S, Vosch T, Cotlet M, Tinnefeld P, Habuchi S, Bell TDM, Oesterling I, Beljonne D, Champagne B, Mullen K, Sauer M, Hofkens J, De Schryver FC. J. Phys. Chem. B. 2004;108:16686–16696. [Google Scholar]; c Vosch T, Cotlet M, Hofkens J, Van der Biest K, Lor M, Weston K, Tinnefeld P, Sauer M, Latterini L, Mullen K, De Schryver FC. J. Phys. Chem. A. 2003;107:6920–6931. and references therein. [Google Scholar]
- 25.a Jiang DL, Aida T. J. Am. Chem. Soc. 1998;120:10895–10901. [Google Scholar]; b Kimura M, Shiba T, Muto T, Hanabusa K, Shirai H. Macromolecules. 1999;32:8237–8239. [Google Scholar]; c Harth EM, Hecht S, Helms B, Malmstrom EE, Fréchet JMJ, Hawker CJ. J. Am. Chem. Soc. 2002;124:3926–3938. doi: 10.1021/ja025536u. [DOI] [PubMed] [Google Scholar]; d Choi MS, Yamazaki T, Yamazaki I, Aida T. Angew. Chem., Int. Ed. 2004;43:150–158. doi: 10.1002/anie.200301665. [DOI] [PubMed] [Google Scholar]; e Onitsuka K, Kitajima H, Fujimoto M, Iuchi A, Takei F, Takahashi S. Chem. Commun. 2002:2576–2577. [Google Scholar]; f Hania PR, Heijs DJ, Bowden T, Pugzlys A, van Esch J, Knoester J, Duppen K. J. Phys. Chem. B. 2004;108:71–81. [Google Scholar]; g Larsen J, Andersson J, Polivka T, Sly J, Crossley MJ, Sundstrom J, Akesson E. Chem. Phys. Lett. 2005;403:205–210. [Google Scholar]
- 26.a Plevoets M, Vögtle F, De Cola L, Balzani V. New J. Chem. 1999;23:63–69. [Google Scholar]; b Zhou XL, Tyson DS, Castellano FN. Angew. Chem., Int. Ed. 2000;39:4301–4305. doi: 10.1002/1521-3773(20001201)39:23<4301::AID-ANIE4301>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]; c Tyson DS, Luman CR, Castellano FN. Inorg. Chem. 2002;41:3578–3586. doi: 10.1021/ic011298c. [DOI] [PubMed] [Google Scholar]
- 27.a Joshi MP, Swiatkiewicz J, Xu FM, Prasad PN. Optics Lett. 1998;23:1742–1744. doi: 10.1364/ol.23.001742. [DOI] [PubMed] [Google Scholar]; b Chung SJ, Lin TC, Kim KS, He GS, Swiatkiewicz J, Prasad PN, Baker GA, Bright FV. Chem. Mater. 2001;13:4071–4076. [Google Scholar]; c Chiang LY, Padmawar PA, Canteenwala T, Tan LS, He GS, Kannan R, Vaia R, Lin TC, Zheng QD, Prasad PN. Chem. Commun. 2002:1854–1855. doi: 10.1039/b202681c. [DOI] [PubMed] [Google Scholar]; d Brousmiche DW, Serin JM, Fréchet JMJ, He GS, Lin TC, Chung SJ, Prasad PN. J. Am. Chem. Soc. 2003;125:1448–1449. doi: 10.1021/ja0288688. [DOI] [PubMed] [Google Scholar]; e He GS, Lin TC, Cui YP, Prasad PN, Brousmiche DW, Serin JM, Fréchet JMJ. Optics Lett. 2003;28:768–770. doi: 10.1364/ol.28.000768. [DOI] [PubMed] [Google Scholar]; f Brousmiche DW, Serin JM, Fréchet JMJ, He GS, Lin TC, Chung SJ, Prasad PN, Kannan R, Tan LS. J. Phys. Chem. B. 2004;108:8592–8600. [Google Scholar]
- 28.Dichtel WR, Serin JM, Edder C, Fréchet JMJ, Matuszewski M, Tan LS, Ohulchanskyy TY, Prasad PN. J. Am. Chem. Soc. 2004;126:5380–5381. doi: 10.1021/ja031647x. [DOI] [PubMed] [Google Scholar]
- 29.a Adronov A, Fréchet JMJ, He GS, Kim KS, Chung SJ, Swiatkiewicz J, Prasad PN. Chem. Mater. 2000;12:2838. [Google Scholar]; b Drobizhev M, Karotki A, Rebane A, Spangler CW. Optics Lett. 2001;26:1081–1083. doi: 10.1364/ol.26.001081. [DOI] [PubMed] [Google Scholar]
- 30.a Chung SJ, Kim KS, Lin TH, He GS, Swiatkiewicz J, Prasad PN. J. Phys. Chem. B. 1999;103:10741–10745. [Google Scholar]; b Katan C, Terenziani F, Mongin O, Werts MHV, Porres L, Pons T, Mertz J, Tretiak S, Blanchard-Desce M. J. Phys. Chem. A. 2005;109:3024–3037. doi: 10.1021/jp044193e. [DOI] [PubMed] [Google Scholar]
- 31.Andrews DL, Bradshaw DS. J. Chem. Phys. 2004;121:2445–2454. doi: 10.1063/1.1769354. [DOI] [PubMed] [Google Scholar]
- 32.Turro NJ. Modern Molecular Photochemistry. University Science Books; Sausalito, CA: 1991. [Google Scholar]
- 33.Saltiel J, Atwater BW. In: Advances in Photochemistry. Volman DH, Hammond GS, Gollnick K, editors. Vol. 14. Wiley; New York: 1988. pp. 1–90. [Google Scholar]
- 34. The values of quantum yields above 5% are typically considered high for room-temperature phosphorescence and are adequate for oxygen measurements.
- 35.Rumi M, Ehrlich JE, Heikal AA, Perry JW, Barlow S, Hu ZY, McCord-Maughon D, Parker TC, Rockel H, Thayumanavan S, Marder SR, Beljonne D, Bredas JL. J. Am. Chem. Soc. 2000;122:9500–9510. [Google Scholar]
- 36.For examples of such dyes, see: refs 27d, 28, 35 and references therein.
- 37.a Xu C, Webb WW. J. Opt. Soc. Am. B. 1996;13:481–491. [Google Scholar]; b Albota MA, Xu C, Webb WW. Appl. Opt. 1998;37:7352–7356. doi: 10.1364/ao.37.007352. [DOI] [PubMed] [Google Scholar]
- 38.Hecht S, Vladimirov N, Fréchet JMJ. J. Am. Chem. Soc. 2001;123:18–25. doi: 10.1021/ja003304u. [DOI] [PubMed] [Google Scholar]
- 39.Callis JB, Gouterman M, Jones YM, Henderson BH. J. Mol. Spectrosc. 1971;39:410–420. [Google Scholar]
- 40.Eastwood D, Gouterman M. J. Mol. Spectrosc. 1970;35:359–3575. [Google Scholar]
- 41. Quadratic dependence of emission on the excitation power under 2P excitation was confirmed for all the compounds studied in this paper.
- 42.Jones G, Jackson WR, Choi C. J. Phys. Chem. 1985;89:294–300. [Google Scholar]
- 43.a Kirkwood JC, Scheurer C, Chernyak V, Mukamel S. J. Chem. Phys. 2001;114:2419–2429. [Google Scholar]; b Lupton JM, Samuel IDW, Burn PL, Mukamel S. J. Phys. Chem. B. 2002;106:7647–7653. [Google Scholar]; c Raychaudhuri S, Shapir Y, Mukamel S. J. Lumines. 2005;111:343–347. [Google Scholar]
- 44. For examples, see. [Google Scholar]; a Gentemann S, Medforth CJ, Forsyth TP, Nurco DJ, Smith KM, Fajer J, Holten D. J. Am. Chem. Soc. 1994;116:7363–7368. [Google Scholar]; b Gentemann S, Nelson NY, Jaquinod L, Nurco DJ, Leung SH, Medforth CJ, Smith KM, Fajer J, Holten D. J. Phys. Chem. B. 1997;101:1247–1254. [Google Scholar]
- 45.For example, see ref 23u and references therein.
- 46.Bergamini G, Ceroni P, Maestri M, Balzani V, Lee SK, Vögtle F. Photochem., & Photobiol. Sci. 2004;3:898–905. doi: 10.1039/b408659g. [DOI] [PubMed] [Google Scholar]
- 47.Blanchard GJ, McCarthy PK. J. Phys. Chem. 1993;97:12205–12209. [Google Scholar]
- 48. For examples, see. [Google Scholar]; a Reinhardt BA, Brott LL, Clarson SJ, Dillard AG, Bhatt JC, Kannan R, Yuan LX, He GS, Prasad PN. Chem. Mater. 1998;10:1863–1874. [Google Scholar]; b Lin TC, Chung SJ, Kim KS, Wang XP, He GS, Swiatkiewicz J, Pudavar HE, Prasad PN. Polym. Photon. Appl. II. 2003;161:157–193. [Google Scholar]; c Porres L, Mongin O, Katan C, Charlot M, Bhatthula BKC, Jouikov V, Pons T, Mertz J, Blanchard-Desce M. J. Nonlinear Opt. Phys., & Mater. 2004;13:451–460. [Google Scholar]
- 49.Rozhkov VV, Wilson DF, Vinogradov SA. Macromolecules. 2002;35:1991–1993. [Google Scholar]
- 50.a Vinogradov SA, Wilson DF. Adv. Exp. Med. Biol. 1997;428:657–662. [PubMed] [Google Scholar]; b Vinogradov SA, Lo LW, Wilson DF. Chem.- Eur. J. 1999;5:1338–1347. [Google Scholar]; c Dunphy I, Vinogradov SA, Wilson DF. Anal. Biochem. 2002;310:191–198. doi: 10.1016/s0003-2697(02)00384-6. [DOI] [PubMed] [Google Scholar]
- 51.Hecht S, Fréchet JMJ. Angew. Chem., Int. Ed. 2001;40:74–91. doi: 10.1002/1521-3773(20010105)40:1<74::aid-anie74>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 52. For examples, see. [Google Scholar]; a Jin RH, Aida T, Inoue S. J. Chem. Soc. Chem. Commun. 1993:1260–1262. [Google Scholar]; b Dandliker PJ, Diedrich F, Gross M, Knobler CB, Louati A, Sanford E. Angew. Chem., Int. Ed. Engl. 1994;33:1739–1742. [Google Scholar]; c Bhyrappa P, Young JK, Moore JS, Suslick KS. J. Am. Chem. Soc. 1996;118:5708–5711. [Google Scholar]; d Jiang DL, Aida T. Chem. Commun. 1996:1523–1760. [Google Scholar]; e Collman JP, Fu L, Zingg A, Diederich F. Chem. Commun. 1997:193–194. [Google Scholar]; f Pollak KW, Leon JW, Fréchet JMJ, Maskus M, Abruna HD. Chem. Mater. 1998;10:30–38. [Google Scholar]; g Bhyrappa P, Vaijayanthimala G, Suslick KS. J. Am. Chem. Soc. 1999;121:262–263. [Google Scholar]; h Matos MS, Hofkens J, Verheijen W, De Schryver FC, Hecht S, Pollak KW, Fréchet JMJ, Forier B, Dehaen W. Macromolecules. 2000;33:2967–2973. [Google Scholar]; i Kimura M, Shiba T, Yamazaki M, Hanabusa K, Shirai H, Kobayashi N. J. Am. Chem. Soc. 2001;123:5636–5642. doi: 10.1021/ja004312d. [DOI] [PubMed] [Google Scholar]; j Rajesh CS, Capitosti GJ, Cramer SJ, Modarelli DA. J. Phys. Chem. B. 2001;105:10175–10188. [Google Scholar]; k Van Doorslaer S, Zingg A, Schweiger A, Diederich F. ChemPhysChem. 2002;3:659–667. doi: 10.1002/1439-7641(20020816)3:8<659::AID-CPHC659>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]; l Zhang JL, Zhou HB, Huang JS, Che CM. Chem-Eur. J. 2002;8:1554–1562. doi: 10.1002/1521-3765(20020402)8:7<1554::aid-chem1554>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]; m Finikova OS, Galkin AS, Rozhkov VV, Cordero MC, Hägerhäll C, Vinogradov SA. J. Am. Chem. Soc. 2003;125:4882–4893. doi: 10.1021/ja0341687. [DOI] [PubMed] [Google Scholar]; n Zimmerman SC, Zharov I, Wendland MS, Rakow NA, Suslick KS. J. Am. Chem. Soc. 2003;125:13504–13518. doi: 10.1021/ja0357240. [DOI] [PubMed] [Google Scholar]
- 53.a Newkome GR, Lin X. Macromolecules. 1991;24:957. [Google Scholar]; b Newkome GR, Lin X, Weis C. Tetrahedron: Asymmetry. 1991;2:957–960. [Google Scholar]
- 54.Vinogradov SA. Organic Lett. 2005;7:1761–1764. doi: 10.1021/ol050341n. [DOI] [PubMed] [Google Scholar]
- 55.Cardona CM, Gawley RE. J. Org. Chem. 2002;67:1411–1413. doi: 10.1021/jo0161678. [DOI] [PubMed] [Google Scholar]
- 56.a Cameron CS, Gorman CB. Adv. Funct. Mater. 2002;12:17–20. [Google Scholar]; b Chasse TL, Yohannan JC, Kim N, Li Q, Li ZM, Gorman CB. Tetrahedron. 2003;59:3853–3861. [Google Scholar]
- 57.Hecht S. J. Polym. Sci. A. 2003;41:1047–1058. [Google Scholar]