

Summary
Microtubules are a significant therapeutic target for the treatment of cancer, where suppression of microtubule dynamicity by drugs such as paclitaxel forms the basis of clinical efficacy. Peloruside A, a macrolide isolated from New Zealand marine sponge Mycale hentscheli, is a microtubule stabilizing agent that synergizes with taxoid drugs through a unique site, and is an attractive lead compound in the development of combination therapies. We report here unique allosteric properties of microtubule stabilization via peloruside A, and present a structural model of the peloruside binding site. Using a strategy involving comparative hydrogen-deuterium exchange mass spectrometry (HDX-MS) of different microtubule stabilizing agents, we suggest that taxoid-site ligands epothilone A and docetaxel stabilize microtubules primarily through improved longitudinal interactions centered on the interdimer interface, with no observable contributions from lateral interactions between protofilaments. The mode by which peloruside A achieves microtubule stabilization also involves the interdimer interface, but includes contributions from the α/β-tubulin intradimer interface and protofilament contacts, both in the form of destabilizations. Using data-directed molecular docking simulations, we propose that peloruside A binds within a pocket on the exterior of β-tubulin at a previously unknown ligand site, rather than on α-tubulin as suggested in earlier studies.
Keywords: Microtubules, peloruside A, mass spectrometry, hydrogen-deuterium exchange, simulations
Introduction
The success of taxanes in cancer treatment demonstrates the relevance of microtubules as a therapeutic target, however numerous suboptimal pharmacological properties of these compounds have spurred the discovery and development of taxoid mimetics. While the taxoid binding site appears able to accommodate a growing variety of chemical scaffolds1, distinct and separately addressable microtubule stabilization sites may have greater potential to overcome clinical challenges such as chemoresistivity and toxicity, in part through combination therapy2 and reduced reliance on toxic drug solubilizers3; 4; 5. Peloruside A6 and laulimalide7 are two compounds that may offer the foundation for a new generation of therapeutics with these characteristics.
The macrolide peloruside A possesses an activity profile similar to the taxanes in that cells are arrested in G2/M phase and undergo apoptosis8. It appears to occupy a site distinct from the taxanes9, and synergize with other microtubule stabilizing agents at the level of tubulin assembly10; 11 and function in cell proliferation assays12. Most interestingly, peloruside A retains activity in cell lines overexpressing P-glycoprotein and in those with induced resistance to taxol and the taxoid-mimic epothilone A9. This is also true for laulimalide7; 13. The two compounds have been shown to compete in binding assays9, suggesting they bind to the same or an overlapping site.
For these ligands, the existence of a distinct binding site raises the possibility of a unique mechanism of microtubule stabilization. Typically, the α-β tubulin dimer self-assembles into a tubular arrangement of 13 head-to-tail protofilaments in vivo, although this number is dependent on many factors in vitro14; 15. A dimeric unit binds two molecules of GTP, one at a non-exchangeable site between α and β tubulin, and the other at an exchangeable site on β-tubulin. In the assembled state, the hydrolysis of GTP at the exchangeable introduces significant lattice strain, which manifests through stochastic depolymerizations16. During these events, the protofilaments peel away, most likely due to the inherent outward curvature in the dimeric unit17. The mechanism by which microtubule stabilizing agents (MSA’s) such as taxol alleviate this strain is thought to involve contributions from improved longitudinal contacts along the protofilament as well as lateral contacts between protofilaments18. Thus, a unique MSA binding site may alter the relative importance of these contacts and a comparative study of MSA’s should shed light on the mechanisms of stabilization overall.
Much of our molecular-level understanding of MSA-induced stability has derived from cryoelectron microscopy of MSA and zinc-stabilized tubulin sheets, rather than from microtubules themselves18. Unfortunately, creating stable zinc-sheets has not been successful for laulimalide-treated tubulin and presumably would also fail for peloruside19. In any case, it may be advantageous to apply a technique that can offer a differential analysis of microtubules in both MSA-free and MSA-stabilized forms, to derive a picture of the structural impact of stabilization and identify the peloruside binding site. As an alternative to electron crystallographic data, we propose to use data from hydrogen-deuterium exchange mass spectrometry (HDX-MS). In the HDX-MS method, deuteration levels are determined from enzymatically-generated peptides via LC-MS technology20. This technique been used to study protein polymers such as actin21 and most recently also microtubules22. HDX-MS is a very sensitive probe for fluctuations in hydrogen bonding networks, reflective of protein dynamics around a low-energy structure and commonly described in terms of “tightening” or “loosening”23. At a minimum, this method is useful for interrogating similarities and differences in the modes of microtubule stabilization induced by different ligands22. Further, while it is simplistic to correlate ligand-induced alterations in deuteration with a “footprint” of a binding site, it may be reasonable to expect that the footprint is at least partially represented by the ligand-altered deuteration levels. Non-covalent protein-ligand interactions are characterized by an ethalpy/entropy compensation phenomenon24, which can be expected to influence labeling at the binding site provided that the underlying secondary structure offers sufficient dynamic range in hydrogen bond fluctuations. For example, HDX-MS studies of ligand-bound PPARγ have shown that the binding pocket is represented in the full set of structural stabilizations measured by the technique25; 26. This partial correspondence has encouraged the use of HDX-MS data for scoring simulations of protein-protein interactions27, but this approach has not yet been applied to the discovery of binding sites for low molecular weight compounds.
In the present study, we discuss the mechanistic basis for microtubule stabilization imparted by epothilone A, docetaxel and peloruside A. This work demonstrates a previously unknown mechanism, which involves reduced reliance on lateral contacts between protofilaments. We then demonstrate a correlation between the HDX-MS data and known taxoid-site molecules (epothilone A and docetaxel), and on the basis of this evidence describe a coarse localization of the peloruside A binding site using HDX-MS data. We then apply a data-directed ligand docking strategy to suggest a high resolution model of the peloruside A binding site.
Results
Generation of tubulin peptide map
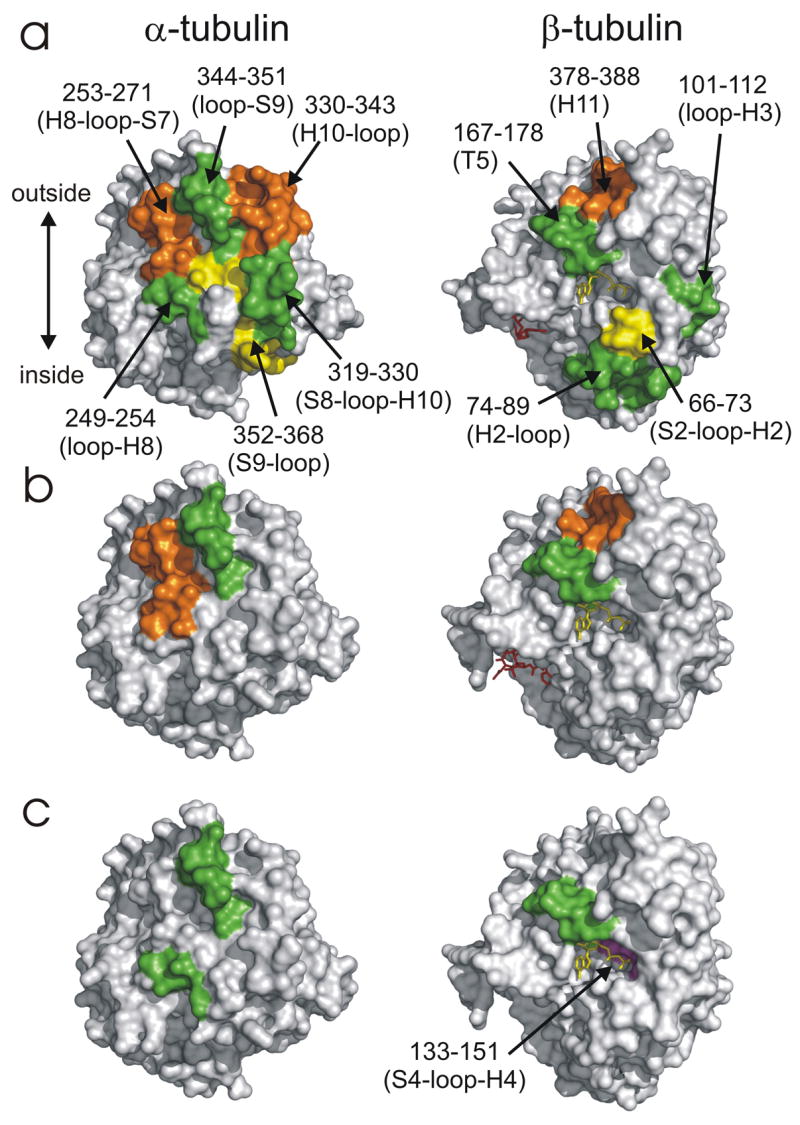
A map of bovine brain tubulin was generated from a pepsin digest, to determine the set of peptides available for monitoring deuteration levels. Bovine tubulin contains multiple isotypes28, however isotype I-C for α-tubulin and isotype II-B for β-tubulin were used to represent the map in Fig. 1. Several peptides were detected that additionally represent other isotypes. For α-tubulin, this includes two peptides for isotype αIV-A (containing the amino acids C54, S340), and four peptides for isotype αIV-A where no corresponding peptides for αI-C are detected (containing the amino acids I122, I231-S232, A34 and I384). Two other peptides represent isotype αIII (containing the amino acids G232 and V437). For β-tubulin, evidence for the presence of isotype βIII is indicated by peptides flanking the amino acids V170, S239, A275 and V351, in addition to the corresponding peptides for βII-B. A single peptide with no correspondence to βII-B contains A55, which is of unspecified bovine sequence but equivalent to porcine β-tubulin (UniProt P02554). Overall, 84 nonredundant peptides for α-tubulin were identified, leading to a sequence coverage of 88.4% and 86 nonredundant peptides for β-tubulin for a sequence coverage of 92% (inclusive of all isotypes).
1.

Peptide map displayed in red on α-tubulin, top sequence (UniProt P81948, bovine I-C chain) and in green on β-tubulin, bottom sequence (UniProt Q6B856, bovine, II-B chain). Yellow highlighted residues represent deviations from these sequences detected in the MS/MS peptide sequence data, arising from a different isotype. Red highlighted residues represent the detection of an additional isotypes in the sequence data.
One advantage of the MS-based analysis is that selective detection of isotypes is possible. As an example, isotype βII bears a serine at position 275 in the M-loop critical to taxane binding, whereas isotype βIII has an alanine in this position (Fig. 1). The MS data show equivalent, strong reductions in labeling for the peptides that span this position. Therefore in the context of the mixed isotype preparation, isotype βIII appears to bind both taxoid-site ligands as well as isotype βII. No cases of isotype-specific labeling were found in our study for the set of unique peptides found, and as with the published structural studies, the data for peptides common to all isotypes are most appropriately viewed as averaging any potential isotype-specific contributions to drug-induced stability.
GMPCPP stabilized bovine brain microtubules as a model system
It has been recognized that the isotype diversity of bovine brain tubulin preparations differ from that of humans, particularly in non-neuronal cells28. Much of the sequence difference is relegated to the C-terminii of the α and β-tubulin and not in regions critical to the taxoid binding site or to assembly in general, although the kinetics of dynamic instability are isotype-dependent29. As most of the existing experimental structural studies have been conducted on tubulin isolated from bovine brain, we chose to do the same, recognizing that bovine brain tubulin preserves 1:1 binding stochiometry (dimer:drug) irrespective of isotypes30.
To promote a comparison of the assembled state with ligand-stabilized microtubules we used the GTP analog guanosine-5′-[(a,β)-methylene]triphosphate (GMPCPP) at the exchangeable nucleotide binding site within β-tubulin. GMPCPP is a GTP analogue that promotes stability of the resulting microtubule by reducing the rate of nucleotide hydrolysis17; 31; 32. As the source of microtubule instability arises from nucleotide hydrolysis at this site, incorporating a slowly hydrolyzed form leads to a preparation that does not exhibit dynamic instability31. This is critical for differential studies, as unassembled pools of free α-β dimer will exist in GTP-loaded microtubules. Upon the addition of stabilizers, this pool will diminish in concentration, in which case the HDX-MS results will be strongly influenced by the exchange properties of the free dimer. A recent study with chicken erythrocyte tubulin has suggested that there are few labeling differences between free dimer and GTP-microtubules22. It is possible that assembly dynamics are different for these two model systems, although we note that the Xiao et al. study involved dilution of GTP-microtubules during deuteration to below the critical assembly concentration. This may offer an alternative explanation as to why few differences between free dimer and GTP-microtubules were found. In the current study, we have shown that free dimer concentrations are not detectable in either the GMPCPP-microtubule or the drug-saturated states (see below).
Equilibrium deuterium exchange of ligand free and ligand treated microtubules
To determine an appropriate deuterium labelling time for conducting the HDX-MS analysis, equilibrium deuterium in-exchange experiments33 were initiated with the addition of D2O to ligand-free microtubule preparations, and sampled over multiple time-points for HDX-MS analysis (see methods). Deuterium levels reached a plateau at 8 minutes of labelling time, representing ~20% deuterium incorporation (uncorrected for back-exchange). Experiments were then conducted at 4 minutes, where 90% of the plateau-level deuteration was retained, to ensure that rapidly exchanging regions retain sensitivity as structural probes of binding. As determination of the absolute value of deuteration is not required for comparative binding studies, all deuterium levels are uncorrected for back-exchange34.
Equilibrium deuterium in-exchange experiments were conducted for each microtubule state using the non-liganded microtubule as a control, and average deuterium incorporation was measured using software developed in-house. The data set can be found in supplementary Tables 1a,b. A subset of the exchange data (Fig. 2a,b) represents all peptides bearing a significant difference between one or more ligand-stabilized microtubule preparations and the microtubule control (a ligand-free preparation). This subset of data was used in subsequent structural representations (figures 3–6, 8). To avoid the complexity of mixed states evident in earlier studies22, all microtubule preparations were generated with GMPCPP. Ligand-free microtubules were prepared well above critical concentrations for assembly and the absence of contaminating free dimer was confirmed by monitoring peptides exhibiting large reductions in deuterium labeling upon assembly, but no further reductions upon ligand binding at the same tubulin concentration. As an example, β74–89 experiences a reduction of 788 ± 50 mmu upon assembly (i.e., dimer to GMPCPP-stabilized microtubules) and no further significant reduction upon docetaxel labeling (Fig. 2b). This indicates full assembly has occurred, as docetaxel leads to a significant decrease in the critical concentration of assembly35. Thus, ligand binding does not significantly alter the population of free vs. assembled dimer.
2.

Ligand-induced alterations in deuteration referenced against GMPCPP-stabilized microtubules for (a) α-tubulin (b) β-tubulin. Data indicate the mean +/− pooled standard deviation of three separate experiments. Peptides sequence numberings are based on UniProt entries P81948 (α-tubulin) and Q6B856 (β-tubulin) as in Fig. 1. Altered deuteration levels are expressed in millimass units (mmu).
3.
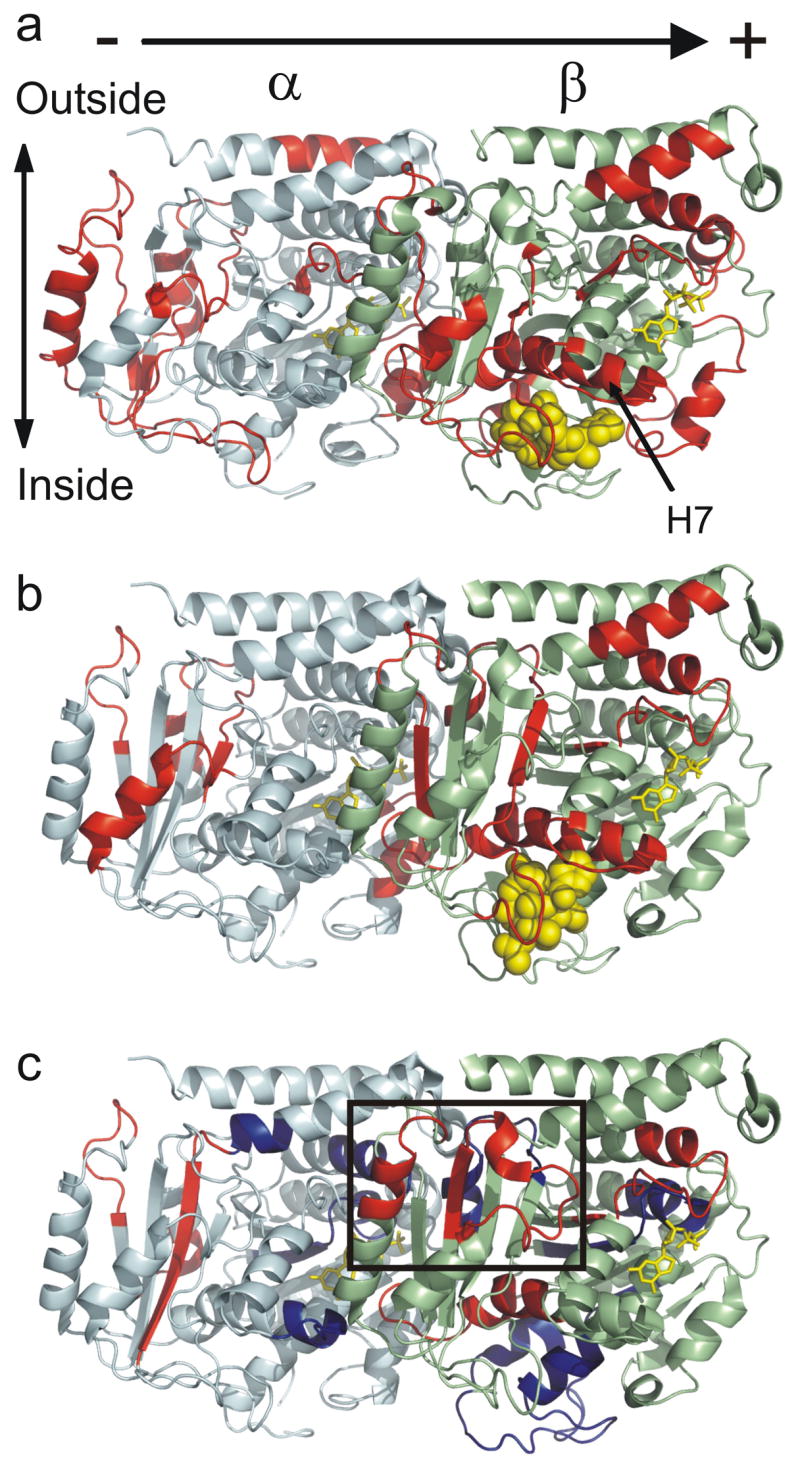
Global mapping of the ligand-induced alterations in deuteration onto α/β-tubulin oriented as in a microtubule, with polarity indicated. (a) Epothilone A induced changes modeled on PDB 1TVK, (b) docetaxel induced changes modeled on PDB 1JFF, and (c) peloruside A induced changes modeled on PDB 1JFF. Red indicates statistically significant reductions in labeling upon binding, blue indicates statistically significant increases in labeling upon binding (see methods). Tubulin monomers are labeled in pale cyan (α) and pale green (β). The exchangeable (β-tubulin) and non-exchangeable nucleotides (α-tubulin) are labelled in yellow, and ligands in yellow spheres. The rectangle highlights the proposed peloruside A binding site, which can be found at higher magnification in Fig. 8.
6.

Expanded view of the taxoid binding site, overlaid with labeling data. (a) Epothilone A and (b) taxol, in lieu of docetaxel. Individual peptides showing reduced labeling displayed in color, and corresponding secondary structure indicated (see table 1).
8.
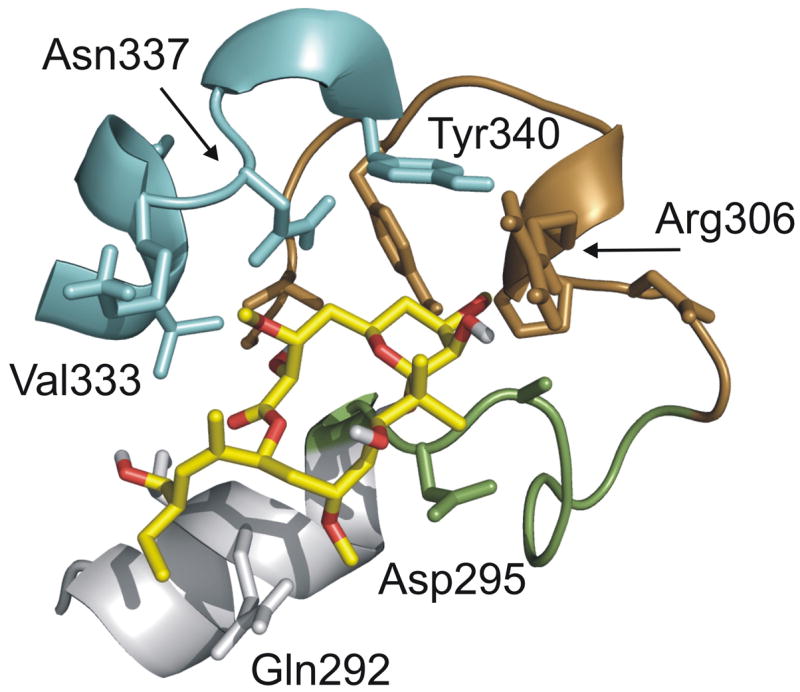
Proposed peloruside A binding site as determined by combination of labeling data and local docking simulations. Individual peptides detected by HDX-MS are indicated in color (green: β294–301, H9-H9′ loop; brown: β302–314, H9′-S8; cyan: β332–340, H10-loop). Helix H9 is in gray (β286–293). Select side-chains suggested to be significant in defining the binding pocket are labeled. Oxygens of peloruside A colored in red.
Mapping the HDX-MS data – global view
The significant differences in labeling between GMPCPP microtubules and the drug-bound forms were mapped onto representative tubulin structures to indicate regions of decreased labeling (red) or increased labeling (blue) due to ligand binding, and displayed globally in Fig. 3a–c. Graduating the degree of altered labeling via color coding was not implemented, for clarity. The taxoid-site ligands generate reductions in labeling clustering at the interfacial regions defined by the longitudinal protofilament axis of the microtubule, as well as the taxoid site itself. The effects observed for docetaxel are largely a subset of those generated by epothilone A whereas the map for peloruside A shows considerable differences. While most of the changes cluster in the same longitudinal regions, there is a significant destabilization at the intradimer interface, and at putative lateral contacts between protofilaments. Additionally, there is a mapped region of reduced labeling on the exterior of β-tubulin that is unique to peloruside A. These will be presented in greater detail.
Mapping the HDX-MS data – interdimer region
Fig. 4a–c represents the subset of exchange data in the vicinity of the interdimer interface, that is, between adjacent α-β dimers on the protofilament axis of microtubules. Secondary structure designations are made on the basis of the map in Löwe et al.18 Epothilone A extensively reduces exchange dynamics at this interface, as evidenced by six peptides on α and five peptides on β tubulin (Fig. 4a). The impact of docetaxel on this region appears less strong (Fig. 4b), however the peptides with significant labeling differences are a subset of those highlighted by epothilone A, and a number of common peptides not labeled for docetaxel generate reductions just below the chosen threshold for significance (Fig. 2). Reduced labeling at this interface is also true for peloruside A, with one exception on β-tubulin (Fig. 4c). Peptide β133–151 undergoes an increase in labeling upon peloruside binding and is immediately adjacent to the nucleotide phosphates of GMPCPP. Overall, the alterations in labeling at the interdimer interface represent the largest changes not directly associated with the taxoid site, particularly for peptides on both α and β in contact with the exchangeable nucleotide (see discussion).
4.
Opposing views of the interdimer interface, showing peptides in this region altered in deuteration level as a result of ligand binding. All other affected peptides are removed for clarity. Reductions in deuterium labeling shown in orange/green/yellow to delineate detected peptides, not degree of labeling. The increase in deuterium labeling is in magenta. The color scheme is retained for all ligands, to promote comparison between (a) epothilone A (b) docetaxel (c) peloruside A. The exchangeable nucleotide is displayed in yellow and ligands in red. Orientation is ±90° in the horizontal relative to Fig. 3.
Mapping the HDX-MS data – intradimer region
Fig. 5a,b represents an comparison view of the α-β intradimer region centered on the non-exchangeable GTP binding site. As the changes for docetaxel are again a subset of those induced by epothilone A, only the map of the latter is shown relative to peloruside A. All ligands reduce labeling for peptides β231–239 and β240–246, but the magnitude of change is significantly lower for peloruside A (Fig. 2b). These peptides represent the H7–H8 loop (β240–246) and the C-terminal end of H7 (β231–239). The latter peptide is part of the known taxoid site18. While these changes are common to all three ligands, there are notable reductions in labeling that are unique to the taxoid-site ligands. Across the intradimer interface on α-tubulin, reduced dynamics are in evidence for peptides encompassing the nonexchangeable nucleotide, specifically the sugar-binding T5 loop (α170–180). Other peptides involved in stabilization of the intradimer region are also reduced in labeling (α68–77 and β341–353). Peloruside A induces a significantly different effect, where the non-exchangeable nucleotide site undergoes an increase in labeling centered on the regions adjacent to the nucleotide phosphates and involved in stabilization of the intradimer contact18. This includes the T4 loop (α135–149), H5 (α181–189) and the H8-loop (β251–265).
5.

Expanded view at the non-exchangeable nucleotide intradimer interface for microtubules stabilized on (a) epothilone A (on 1TVK) and (b) peloruside A (on 1JFF). Ligand-induced alterations in deuterium labeling are mapped in red (reduction), blue (increase) and light blue (increase, near global significance threshold). Other aspects of the color scheme are as in Fig. 3.
Mapping the HDX-MS data for lateral contacts
Lateral interactions between protofilaments are thought to be mediated by contacts between the M-loops on α and β tubulin with the corresponding H1-S2 loops on the adjacent α and β tubulin36. Both taxoid site ligands reduce labelling at the M-loop on β-tubulin, but no changes are found for the corresponding H1-S2 loops, and no changes are observed for α-tubulin. Peloruside A induces an increase in labeling in both the H1-S2 loop and the M loop on b-tubulin; although the latter is just outside the defined significance level (see Fig 2b, peptide β266–280), it is supported by an overlapping peptide (see supplementary Table 1b). As with the taxoid site ligands, however, no changes are observed in the proposed α-α lateral contact region.
Mapping the HDX-MS data to the binding sites
For both epothilone A and docetaxel, the largest reduction in labeling in the dataset is found at the M-loop (β266–280). A second large reduction is found at the H6–H7 loop region (β213–230) with smaller changes at the core helix βH7 (β231–239) previously mentioned in the context of the intradimer interface. In addition to these changes, epothilone A induces a small reduction in labeling for loop-H9 (β281–293). Fig. 6a,b shows the labeling data associated with the known taxoid-site ligands. These peptides encompass many of the key residues involved in the binding to both epothilone A and the taxanes (see Table 1). The M-loop undergoes strong reductions in labeling and brackets critical residues in the stabilization of the oxetane ring of the taxanes18. Identifying H7 as part of the binding site is principally through β231–239, which encompasses residues for the stabilization of the 3′-phenyl ring. The N-terminal end of H7 does not show a significant reduction in labeling (peptide β236–230, data not shown), even though this peptide contains a residue critical for the stabilization of the 2-phenyl ring. Thus, although there is a peptide which bridges this region (β213–230), the associated reduction in deuteration is due to the H6-H7 loop (β213–225). Together with βH7, this loop has been shown to stabilize the 2-phenyl ring18. A further under-reporting of helical regions involved in binding may occur at H1 (β10–25). Residues that are involved in the stabilization of the 3′-phenyl ring as well as the N′-phenyl for taxol (t-butyl for docetaxel) are found in this helix. Surprisingly, the loop between βS9 and βS10 does not show significant labeling, even though close contact is made with docetaxel. This may be due in part to the lower resolution of the HDX-MS data for this region (a single 22 amino acid peptide encompasses this loop). A 2.5 times greater reduction in labeling for docetaxel over epothilone A is noted for this peptide, but the error in the measurement prevents a claim of significance. The participation of P358 in the actual ligand binding site could also reduce the ability of this region to report alteration in labeling, as prolines contain no readily exchangeable amide hydrogen. Epothilone A similarly engages residues within the M-loop and H7 helix (Table 1). The direct participation of residues in the H6-H7 loop in the binding site is less clear1, but reduced dynamics of H7 could stabilize this loop and promote interactions through space with the stabilized M-loop.
Table 1.
Correspondence between measured reductions in deuterium labelling and residues participating in the stabilization of the ligand-tubulin binding site.
| Peptic peptide# | Reduction in labeling
(docetaxel/epothilone) |
Key Residues in Ligand Binding‡ | |
|---|---|---|---|
| taxol* | epothilone | ||
| β21–31 (H1) | no/no | Val21 | - |
| Asp24 | - | ||
| β213–230 (H6–H7 loop) | yes/yes | Leu215 | - |
| Leu217 | - | ||
| β226–230 (H7) | no/no | His227 | His227 |
| Leu228 | - | ||
| β231–239 (H7) | yes/yes | Ala231 | Ala231 |
| Ser234 | - | ||
| β266–280 (S7 – M loop) | yes/yes | Phe270 | - |
| Pro272 | - | ||
| Leu273 | - | ||
| Thr274 | Thr274 | ||
| Ser275 | - | ||
| Arg276 | Arg276 | ||
| β281–293 (loop-H9) | no/yes | - | Arg282 |
| - | Gln292 | ||
| β341–363 (S9–S10) | no/no | Pro358 | - |
| Arg359 | - | ||
| Gly360 | - | ||
| Leu361 | - | ||
Clustering most of the remaining significant reductions in labeling due to peloruside A binding leads to the formation of a patch on the exterior surface of β-tubulin, comprised of peptides β294–301 (H9-H9′ loop), β302–314 (H9′-S8) and β332–340 (H10-loop). With the exception of peptide β302–314, these are large reductions in deuteration, similar to what was observed for docetaxel binding. This represents a strong candidate region for the peloruside A binding site (encircled in Fig. 3c), which was further explored through docking studies.
Data-independent and Data-driven docking of peloruside
We conducted molecular docking using a global search with the previously determined bound conformation of peloruside37 and a reconstruction of an α-β-α protofilament based on PBD entry 1TVK. Using blind docking, three sites of comparable docking energies were discovered: the α-tubulin site proposed by Jimenez-Barbero et al.37, the β-tubulin site described in this study, and a site bridging the interdimer interface. As the α-tubulin site was not represented in the HDX data, it was not considered for refined docking studies. A second partially restricted docking exercise was conducted on what is the exterior face of the MT of α-β-α protofilament segment (i.e., minus the lumenal surface). In this run, the candidate peloruside binding site on β-tubulin was identified from 5 distinct clusters consisting of 169 poses (based on a total of 500 poses), with 82 representing docking energies below −10 kCal/mol (Fig. 7a). A site within the interdimer interface was identified from 4 distinct clusters containing a total of 106 poses with 58 representing docking energies below −10 kCal/mol. As no other sites were uncovered within the regions encompassing the footprinting data, a final refined docking exercise was restricted to only these regions. For the candidate β-tubulin site, 64 of 200 poses presented with docking energies of −10 kCal/mol or better (Fig. 7b), with 40 of these poses clustering within 2Å of the orientation displayed in Fig 8. For the interdimer site, 29 of 200 poses presented with a docking energy of −9.6 kCal/mol (Fig. 7c).
7.
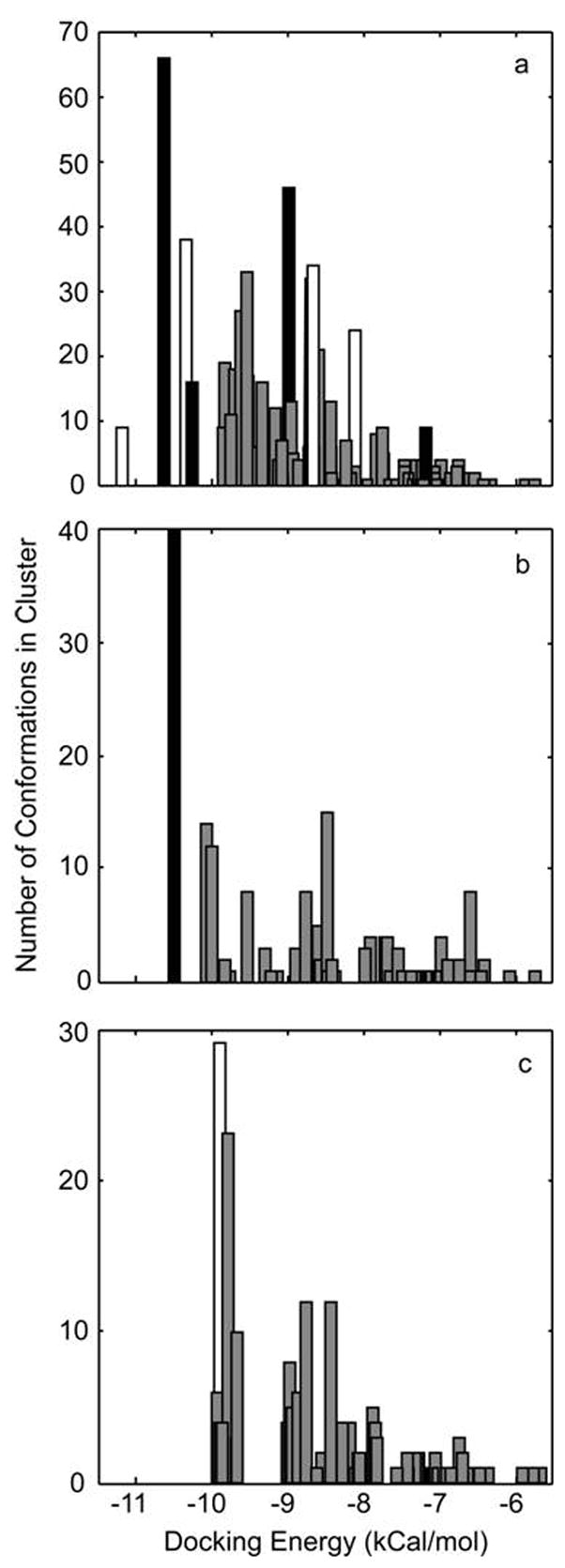
Histogram of docking energies returned from the ligand docking simulations, using the coordinates for bound peloruside and 1TVK. (a) Results from a blind docking exercise targeting the exterior surface of an α-β-α protofilament with clustering of poses at the 5Å level, where black represents poses within the region defined by the HDX data at the β-tubulin site, white the region defined by the interdimer site and gray the binding poses outside of the regions indicated by the HDX data; (b) Results from the directed docking to the general region of the exterior of b-tubulin with clustering of poses at the 2Å level, where black represents the pose displayed in Fig. 8 and gray alternative poses within or around site identified by HDX data; (c) results from the directed docking to the general region of the exterior of the α-β interface with clustering of poses at the 2Å level where white represents the best-fit to the HDX data and gray alternative poses within the interfacial region.
Molecular dynamics for binding energy estimation
Molecular Dynamics (MD) simulations were then conducted for peloruside within the β-tubulin and the interdimer sites, using the low energy docked poses as initial conformations (see methods). For the β-tubulin site, binding energies fell between −21 and −37 kcal/mol, depending on the particular binding pose or solvation model that was implemented, and suggesting a preference for association with β294–301. For the interdimer site, binding energies fell between −13 and −32 kcal/mol, also depending on the pose and solvation model used.
Discussion
MSA’s stabilize protofilaments at the interdimer interface
Stabilization of the interdimer interface appears to be a hallmark of ligand induced microtubule stabilization, and aside from changes at the actual binding site, the greatest reductions in exchange are found in this region (Fig. 4a–c). Although the extent of reduced labeling is ligand dependent, it appears to center on β167–178 for all three MSA’s investigated, which straddles the T5 loop immediately adjacent to the ribose of the exchangeable nucleotide. The taxoid-site ligands likely induce this perturbation through the action of the core helix βH738; 39. This helix comprises the bottom of the cleft defining the taxoid binding site (Fig. 6a,b), and site occupancy may reduce the dynamics of H7 to affect the adjacent T5 loop and the corresponding peptide α344–351 across the interface. This latter peptide is also stabilized by all three ligands. If interdimer stabilization is indeed a primary feature of all MSA’s, it is obvious this can be induced in different ways, as βH7 is largely unaffected by peloruside A (Fig. 3c). The most likely source of this perturbation arises from the proposed binding site for peloruside A on the exterior of β-tubulin, specifically peptides β294–301 and β302–314 (Fig. 8). These peptides comprise the H9-S8 loop that is also adjacent to the T5 loop. Thus we suggest that there are at least two routes to the stabilization of the T5 loop: through helix H7 and through the H9-S8 loop. MSA’s induce a conformational change in the T5 loop of β-tubulin and promote improved interactions across the interdimer interface, returning a stability to the interface that may be lost upon nucleotide hydrolysis. Such an effect would be consistent with our understanding of stability in the dimer itself, where the T5 loop of α-tubulin participates in extensive monomer-monomer contacts18.
It is interesting to note a subtle difference between between peloruside A and the taxoid site ligands in this region. The extensive reduction in exchange for the T5 loop seen with this peloruside A is accompanied by an increase in the exchange dynamics of the peptide adjacent to the nucleotide phosphates. This suggests that the nucleotide binding site has become strained under the action of the T5 conformational change, perhaps indicating poorer binding of the nucleotide. Whether the increased flexibility in this region is due to the partial incompatibility of the methylene bridge of GMPCPP is not known at this time.
MSA’s exert a differential effect on protofilaments at the intradimer interface
At the intradimer region, there is a significant departure between the taxoid-site ligands and peloruside A. As shown in Fig. 5, epothilone A and docetaxel reduce the exchange dynamics around the nonexchangeable GTP, but peloruside generates an increase. Both taxoid-site ligands act upon βH7 through His227 in a manner that may “lock” the H7–H8 loop at the intradimer interface, and stabilize the βT5 loop as described above. Across the intradimer interface, we observe a corresponding reduction in exchange for the αT5 loop (peptide α170–180). As in β-tubulin, this loop encompasses the nucleotide ribose, and thus it appears that βH7 communicates reduced dynamics to both interfaces along the longitudinal protofilament axis. It should be stressed that the magnitude of labeling changes at the intradimer interface are generally lower than seen at the interdimer interface, suggesting that drug binding exerts a greater influence at the latter. In general, epothilone A induces greater changes than docetaxel at both interfaces. This is interesting given the similarities in free energy of binding for these two ligands35, and that saturating levels of ligand were applied. It may be that marked differences in binding energy between these ligands and the various tubulin isotypes is occurring leading to unequal binding site occupancy. However, the magnitude of this difference would have to be great given that saturating ligand levels are used and thus this argument does not seem reasonable. The greater stabilization induced by epothilone A appears to correlate with enthalpy of binding35. Further studies are underway to explore the relationships between HD exchange properties and MSA thermodynamic properties.
In the case of peloruside A, the C-terminal end of H7 (β231–239) and the H7–H8 loop (β240–246) experience reduced labeling in common with epothilone A and docetaxel, but to a lesser extent (Fig. 2b). This degree of stabilization is insufficient to induce the associated stabilization of the αT5 loop seen with the taxoid-site ligands. In the absence of such stabilization, the intradimer interface becomes more labeled around the nonexchangeable nucleotide phosphates at loops T4 and T5, and likely also the base (Fig. 5b; peptide α219–227 at loop-H7 shows an increase just outside of the stated significance threshold). Whether this is directly through the action of an adjacent peloruside binding site or indirectly through a stabilized interdimer interface is unclear, however it suggests a significant departure in stability mechanisms, discussed below.
Peloruside preferentially destabilizes lateral protofilament contacts
A second region of significant destabilization accompanies peloruside A binding. An increase in labeling is found at the N-terminal H1-S2 loop (β21–42, β45–65) at the proposed lateral contact between protofilaments18. A similar effect on the corresponding M-loop of the adjacent β-tubulin might be expected, and indeed a change of similar magnitude is in evidence (β266–280) although the difference is also just outside of our defined significance level. However, peloruside binding returns the deuteration of this peptide to a level equivalent to the free dimer (data not shown) and thus is likely significant. This observation can be explained as a reduced interaction between protofilaments, and strongly suggests peloruside A reduces dependency on lateral contacts for microtubule stabilization, at least between β-tubulin monomers. Interestingly, neither taxoid-site ligand affects the exchange properties of the proposed lateral contacts, suggesting that stability may be an exclusively longitudinal phenomenon under the action of these ligands17. This is in contrast to the findings of Xiao et al.22, who demonstrate that taxol binding stabilizes the lateral interaction at α-tubulin but not at β-tubulin. As taxol defines a microtubule with 12 protofilaments as opposed to 13 for docetaxel40, this difference may reflect an alternative lattice configuration but it could also represent an artefact arising from the presence of free dimer.
Proposed models for ligand-induced stabilization
On the basis of these findings, we suggest that an improved interdimer interaction is the primary mode of microtubule stabilization, and that at least two distinct conformational responses to interdimer stabilization accompany MSA binding. In one, all longitudinal contacts are stabilized with no obvious improvement in lateral contacts. This is exemplified by the taxoid-site ligands used in this study. In the other, for peloruside A, the assembled dimer appears to adopt a configuration with greater flexibility at the intradimer interface and reduced interactions between protofilaments at β-tubulin. This may be driven by an alternative organization at the interdimer interface, or possibly the absense of a stabilized βH7. Both conformational responses clearly have the potential to counteract the natural curvature of the protofilaments that is the primary source of lattice strain in the assembled form41. It is interesting to speculate on the nature of the structural perturbation that may accompany the altered labeling described for these ligands. The taxoid site compounds could favor a simple “tightening” along the protofilament axis, whereas peloruside A may be described by a “plate tectonic” model. In the latter, a discontinuity in the protofilament at the intradimer interface could expose regions of this interface to improved solvent exchange while still promoting strengthened interactions at the interdimer interface. Such an effect rationalizes the increased labeling between β-tubulin across protofilaments, particularly if β-tubulin moves in the lumenal direction. This model might be expected to induce a compression in α-tubulin; we note a large reduction in labeling at α373–383 for the internal beta sheet S10 (Figs. 2a, 3c), which may be evidence of this.
Labeling data encompass critical features of the taxoid site
To precisely identify the peloruside A binding site, it is useful to first consider the congruence between the exchange data and the known taxoid site for epothilone A and docetaxel. There is general agreement between the HDX data and the known binding sites for these ligands (see Table 1 and Fig. 6a,b). Indeed, the sensitivity of the HDX data to site stabilization can be seen when comparing the results for epothilone A with docetaxel in the vicinity of the M-loop. Based on the epothilone A-tubulin structure 1TVK, Q292 of helix H9 participates in the stabilization of the M-loop through hydrogen bonding42, and a segment spanning this residue and the C-terminal region of the M-loop undergoes a reduction in labeling. Q292 does not participate in the docetaxel-induced stabilization, and no HDX labeling changes are observed (Fig. 6b). Although not all residues critical to ligand binding are revealed in the data, this may be expected because binding will not distort hydrogen bonding in a universal manner. For example the N-terminal end of βH7 is “silent” most probably because internal hydrogen bonding is little influenced by ligand binding, Overall, it is clear that such data may be used to identify binding sites for other microtubule stabilizing agents.
Data-directed ligand docking suggests a novel peloruside A binding site
A strong candidate binding site for peloruside A is the region of reduced labeling on the exterior of β-tubulin (Fig. 3c), as mapped by peptides β294–301 (H9-H9′ loop), β302–314 (H9′-S8) and β332–340 (H10-loop). To test this location as the peloruside binding site and generate a high resolution ligand binding model, we implemented both blind and data-directed docking simulations. Blind binding site predictions using tools such as AutoDock have been implemented on several systems43; 44, including tubulin38. However, application of a blind docking exercise using the known conformation of bound peloruside was insufficient to identify the binding site with high confidence (Fig. 7a). The precision of these simulations increase when transitioning from a blind to a data-led dock. In these experiments, docking simulations were focused on grids fully encompassing labeling data for the proposed binding site, as well as the exterior surface flanking the interdimer interface. The interdimer region was added since upon assembly of an α-β-α protofilament model, a surface map of the labeling data suggested the possibility of a contiguous region bridging this interface (not shown). These directed docking experiments allowed extensive sampling of local regions of the microtubule surface and improved discrimination between poses. The simulations (Fig. 7b) support peloruside A binding at the β-site as localized by the labeling data and provide a high resolution binding model of peloruside within the site (Fig. 8). Subsequent MD simulations returned reasonable binding energies for the orientations clustered within 2Å of the binding model shown in Fig. 8. All poses at this resolution reveal the macrolactone within the pocket defined by D295, A296, P305, R306, N337 and Y340, oriented such that the modified pyran group is deeply embedded. These amino acid residues are contained in peptides detected in the labeling experiment, indicating a good correlation. In this model, peloruside A appears to interact with Q291 on a helical segment (H9) leading into the pocket, where no overlap with labeling data is seen. This is likely due to the relative insensitivity of the helix as a reporter of binding, much as βH7 was for the taxoid-site ligands (Fig. 6).
While the region spanning the interdimer interface cannot be ruled out as a secondary binding site (Fig. 7c), it is less likely for four reasons. First, the labeling reductions observed in this region are also seen in both epothilone A and docetaxel stabilized microtubules, suggesting that this region represents improved dimer-dimer interactions (i.e., the T5 loop reductions discussed above). Second, in the case of peloruside, the labeling data offer a poor overlap with the ligand docking results particularly as the largest reduction in deuterium labeling is fully within the interdimer interface (data not shown). Also, peptide α330–343 is within the site identified by localized docking, however it does not does not show a decrease in labeling upon peloruside binding, even though it is a sensitive reporter of deuteration change (Fig. 2a). Third, MD simulations demonstrate higher binding energies than those generated at the primary site and finally, the related compound laulimalide has been demonstrated to possess 1:1 binding stoichiometry with tubulin thus occupation of a second site is unlikely.
The binding site described in this work is at odds with recently published studies proposing a site centered at the M-loop of α-tubulin (α272–286)37; 45 in addition to partial occupation of the taxoid site45. Both studies utilized blind-docking procedures in AutoDock similar to the current study, and refined their simulations around the α-tubulin site discovered in this fashion. A similar candidate site was found in the current study during blind docking, but as there was no significant reduction in labeling in this region, it was not considered in further simulations (see Fig. 9 for a comparison of proposed binding sites). Several of the residues proposed by Jimenez-Barbero et al. that would be critical to stabilizing the peloruside A binding site on α-tubulin are located in regions that would be expected to show significant reductions in labeling upon binding, particularly the αH1-loop, the αS9–S10 loop, the αH7–H8 loop and the αS8-loop. Reduced labeling is seen for peptide α352–368 (Fig. 2a) corresponding to the αS9–S10 loop, but the reduction is not significant. Further, this peptide is only strongly affected by epothilone A; given that it also spans the interdimer interface, we interpret the reduction as arising from stabilization in this region. It remains possible that a translocation in this loop to expose the binding site (leading to an increase in labeling) is counterbalanced by the binding event itself (leading to a decrease in labeling). This is in fact suggested by Pineda et al.45, where the movement of this loop was required for lowest energy binding. However, it is difficult to see how binding at the α-tubulin site could rationalize the large reduction in labeling seen in the proposed β-tubulin site. Resolution of the conflicting models awaits further study.
9.
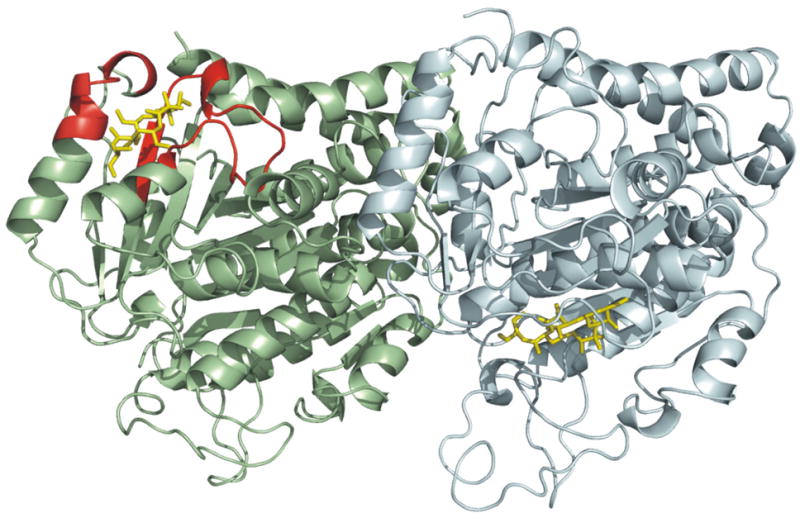
Comparison between the data-directed docked pose of peloruside A (this study) and that proposed by Jimenez-Barbero et al.37 This rendering depicts the interdimer interface between β-tubulin (pale green) from one dimer and α-tubulin (pale cyan) from an adjacent dimer. HDX data aligning with the docked pose of current study is displayed in red, and peloruside A in yellow.
In conclusion, it has been shown that stabilizing agents can affect overall stability of the microtubule through a mechanism primarily involving improved longitudinal contacts across the interdimer interface, and that improved lateral protofilament contacts may not be a requirement. Peloruside A ligation presents a significant mechanistic departure from the taxoid-site ligands, whereby a relaxation of the intradimer interface and β–β interaction across the lateral interface accompanies binding. Data-directed docking strongly suggests the identification of a novel stabilizer site on β-tubulin independent of the taxoid site, and a model is proposed that can be tested in mutational and lead optimization studies.
Materials and Methods
MT preparation and labeling
Purified bovine brain tubulin (Cytoskeleton Inc., cat. no. TL238-A) was reconstituted in nucleotide-free buffer (20 mM KCl, 10 mM K-PIPES, pH 6.9) to 20 mg/ml and incubated at 37°C for 30 min to initiate polymerization and hydrolysis of GTP present in storage buffer. The resulting microtubules were pelleted and washed with a small amount of assembly buffer (1 mM GMPCPP, 100 mM KCl, 10 mM K-PIPES, 1 mM MgCl2, pH 6.9), then depolymerized on ice to a concentration of >60 μM. Prior to conducting HDX-MS experiments, an aliquot of this cold solution was incubated at 37°C for 30 min to induce polymerization. The solution was brought to room temperature and labeled by addition of an equal volume of D2O (50% labeling, to minimize the influence on D2O assembly and dynamics seen at higher levels46). These exchange conditions were preserved for 4 minutes prior to quenching. Deuterium labeling experiments were conducted at several timepoints, and it was determined that dimer labeling was essentially complete after 4 minutes (data not shown). This time-period was used for all subsequent comparisons of assembled and drug-stabilized MT states. To prepare drug-saturated microtubules, the procedure was repeated with the addition of docetaxel (200 μM), peloruside (125 μM) or epothilone A (125 μM) to the assembly buffer. Docetaxel and epothilone A were purchased from Sigma-Aldrich and peloruside was synthesized as described elsewhere 47.
The termination of labeling and the initiation of pepsin digestion was achieved by adding the labeled sample to a chilled slurry of immobilized pepsin (Applied Biosystems Inc.) in 0.1 M glycine-HCl (pH 2.3), and digested for 2.5 minutes on ice. Digestion was terminated by centrifugation of the immobilized pepsin and an aliquot of the supernatant (containing ~30 pmol of digest) was injected into the LC/MS system for analysis. All analyses were performed in triplicate.
HDX LC/MS system
The LC-MS system consisted of an injection valve, a column loading pump, a prototype splitless low flow gradient pump (Upchurch Scientific Inc.) and a QStar Pulsar i mass spectrometer (AB/Sciex Inc.) fitted with a turboionspray source. Chilled digest was injected onto a 150 μm ID × 65 mm C18 column prepared in-house. The valve, column and fluid lines were housed in a chilled container (~0 °C) to minimize the back-exchange of deuterium label during analysis. A rapid gradient separation was performed, and the total time for analysis (from digestion to analysis) was 16 minutes. The mass spectrometer was operated in positive polarity and TOF-MS mode (m/z range from 300 – 1200).
Peptide identification
Non-labeled tubulin was digested with pepsin as described and analyzed by LC-MS/MS at room temperature. MS/MS spectra were obtained via recursive information-dependent acquisitions and manual product ion acquisitions to maximize the number of peptides detected. Spectra were searched against a custom database assembled to capture the sequence diversity present in the mixture of bovine brain tubulin, using MASCOT. To create the database, porcine sequences for α- and β-tubulin (P02550 and P02554), were searched against the TIGR cattle EST database (v. 11.0) to generate a number of contigs. The same procedure was applied to the bovine Ensembl database (v.37). A total of 31 contigs were compiled, to which was added the porcine sequences and recent bovine entries to the SwissProt database. Applying this workflow, 84 nonredundant peptides for α-tubulin (88.4% sequence coverage) and 86 nonredundant peptides for β-tubulin (92% sequence coverage) were identified. Sequencing results were manually verified.
Data analysis and presentation
Average deuterium incorporation for all verified peptide sequences was determined using software developed by our group. Standard deviations were determined from triplicate analyses of ligand-saturated and ligand-free microtubules, on a per-peptide basis. A labelling difference between states for a given peptide is reported as significant if it passed two criteria: first, a two-tailed t-test (P<0.02) using pooled standard deviations from the two triplicate analyses (pooling was deemed acceptable on the basis of per-peptide F-tests); second, a visual inspection of the isotopic distributions to guard against spectral overlap in conjunction with manual inspection of tandem MS data for peptide purity. Levels of ΔD were color coded per peptide on PDB entries 1JFF and 1TVK. Tubulin structures were rendered in all figures using Pymol (http://pymol.sourceforge.net).
Molecular Modeling - Docking
Ligand docked was conducted with Autodock 3 (http://autodock.scripps.edu/). To perform the automated ligand docking search, it was essential to obtain an accurate representation of the bound peloruside molecule as Autodock does not search torsions within ring structures. Following the construction of an initial peloruside model using the Prodrg server48, dihedral angles similar those reported for the B solvated confomer37 were assigned using the Leap module of AMBER849. The system was then optimized with Gaussian03, at the Hartree Fock level with a 6–31G(d,p) basis set, producing our final macrolide ring conformation. Following QM optimization, the electrostatic potential fit charges were obtained using the Merz-Singh-Kollman method within Gaussian03. These charges were then entered into the Autodock (version 3) implementation of the AMBER forcefield and the molecule imported into Autodock50. The αand β chains of the 1TVK PDB file, without bound nucleotides or epothilone, was used to construct an α-β-α protofilament to act as the receptor for subsequent blind docking runs. The tubulin complex was then imported into Autodock 3 and default Kollman’s united-atoms partial charges and solvent parameters were added.
In the blind docking exercise, the entire exterior surface of the α-β-α protofilament (i.e., inclusive of the interdimer interface) was interrogated using a grid box with a coarse spacing of 0.825Å and dimensions of 126×62×72 points. Using the Lamarckian genetic algorithm, we performed 500 trials of 1 × 107 energy evaluations for each trial and docked poses were clustered using RMSD tolerances of 5Å. In the refined docking study, we focused the search on the general area of the exterior of β-tubulin centered on the site indicated by the HDX data, and the interdimer region. A default grid spacing of 0.325Å and box dimensions of 96×86×96 points were used in both cases. We then performed 200 trials of 1 × 107 energy evaluations each. Docked poses were clustered using RMSD tolerances of 2Å.
Molecular Modeling – Molecular Dynamics
After identification of the preferred binding site, peloruside was imported into AMBER8 and parameters were assigned using Antechamber and charges obtained as above. Parameters for the tubulin dimer were taken from the AMBER99 forcefield. A short Steepest Decents/Conjugate gradient minimization was performed, producing our minimized input structures for molecular dynamics (MD) simulations. MD simulations were performed both in vacuo and using the generalized Born implicit solvation model. In both cases, the system was restrained and thermalized from 0K to 300K over approximately 100 picoseconds. Unrestrained MD was then performed for an additional nanosecond, after which binding energies were evaluated over the last three-quarters of each trajectory. Using MM-PBSA the binding energy was evaluated using vacuum electrostatics, and solvation was approximated using generalized Born model.
Supplementary Material
Acknowledgments
We acknowledge Tyler Luchko for assisting in the creation of the alpha-beta-alpha protofilament from periodic images from the crystal structure of epothilone-bound tubulin. This work was supported by the Alberta Cancer Board, the Alberta Heritage Foundation for Medical Research, the Canadian Institutes of Health Research and intramural funds from the National Institute of Child Health and Human Development, NIH, USA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nettles JH, Li HL, Cornett B, Krahn JM, Snyder JP, Downing KH. The binding mode of epothilone A on alpha,beta-tubulin by electron crystallography. Science. 2004;305:866–869. doi: 10.1126/science.1099190. [DOI] [PubMed] [Google Scholar]
- 2.Bergstralh DT, Ting JP. Microtubule stabilizing agents: their molecular signaling consequences and the potential for enhancement by drug combination. Cancer Treat Rev. 2006;32:166–79. doi: 10.1016/j.ctrv.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 3.Chao TC, Chu Z, Tseng LM, Chiou TJ, Hsieh RK, Wang WS, Yen CC, Yang MH, Hsiao LT, Liu JH, Chen PM. Paclitaxel in a novel formulation containing less Cremophor EL as first-line therapy for advanced breast cancer: a phase II trial. Invest New Drugs. 2005;23:171–7. doi: 10.1007/s10637-005-5863-8. [DOI] [PubMed] [Google Scholar]
- 4.Weiss RB, Donehower RC, Wiernik PH, Ohnuma T, Gralla RJ, Trump DL, Baker JR, Jr, Van Echo DA, Von Hoff DD, Leyland-Jones B. Hypersensitivity reactions from taxol. J Clin Oncol. 1990;8:1263–8. doi: 10.1200/JCO.1990.8.7.1263. [DOI] [PubMed] [Google Scholar]
- 5.Gelderblom H, Verweij J, Nooter K, Sparreboom A. Cremophor EL: the drawbacks and advantages of vehicle selection for drug formulation. Eur J Cancer. 2001;37:1590–8. doi: 10.1016/s0959-8049(01)00171-x. [DOI] [PubMed] [Google Scholar]
- 6.West LM, Northcote PT, Battershill CN. Peloruside A: a potent cytotoxic macrolide isolated from the new zealand marine sponge Mycale sp. J Org Chem. 2000;65:445–9. doi: 10.1021/jo991296y. [DOI] [PubMed] [Google Scholar]
- 7.Mooberry SL, Tien G, Hernandez AH, Plubrukarn A, Davidson BS. Laulimalide and isolaulimalide, new paclitaxel-like microtubule-stabilizing agents. Cancer Res. 1999;59:653–60. [PubMed] [Google Scholar]
- 8.Hood KA, West LM, Rouwe B, Northcote PT, Berridge MV, Wakefield SJ, Miller JH. Peloruside A, a novel antimitotic agent with paclitaxel-like microtubule- stabilizing activity. Cancer Res. 2002;62:3356–60. [PubMed] [Google Scholar]
- 9.Gaitanos TN, Buey RM, Diaz JF, Northcote PT, Teesdale-Spittle P, Andreu JM, Miller JH. Peloruside A does not bind to the taxoid site on beta-tubulin and retains its activity in multidrug-resistant cell lines. Cancer Res. 2004;64:5063–7. doi: 10.1158/0008-5472.CAN-04-0771. [DOI] [PubMed] [Google Scholar]
- 10.Hamel E, Day BW, Miller JH, Jung MK, Northcote PT, Ghosh AK, Curran DP, Cushman M, Nicolaou KC, Paterson I, Sorensen EJ. Synergistic effects of peloruside A and laulimalide with taxoid site drugs, but not with each other, on tubulin assembly. Mol Pharmacol. 2006;70:1555–64. doi: 10.1124/mol.106.027847. [DOI] [PubMed] [Google Scholar]
- 11.Gapud EJ, Bai R, Ghosh AK, Hamel E. Laulimalide and paclitaxel: a comparison of their effects on tubulin assembly and their synergistic action when present simultaneously. Mol Pharmacol. 2004;66:113–21. doi: 10.1124/mol.66.1.113. [DOI] [PubMed] [Google Scholar]
- 12.Wilmes A, Bargh K, Kelly C, Northcote PT, Miller JH. Peloruside A synergizes with other microtubule stabilizing agents in cultured cancer cell lines. Mol Pharm. 2007;4:269–80. doi: 10.1021/mp060101p. [DOI] [PubMed] [Google Scholar]
- 13.Pryor DE, O’Brate A, Bilcer G, Diaz JF, Wang Y, Wang Y, Kabaki M, Jung MK, Andreu JM, Ghosh AK, Giannakakou P, Hamel E. The microtubule stabilizing agent laulimalide does not bind in the taxoid site, kills cells resistant to paclitaxel and epothilones, and may not require its epoxide moiety for activity. Biochemistry. 2002;41:9109–15. doi: 10.1021/bi020211b. [DOI] [PubMed] [Google Scholar]
- 14.Diaz JF, Valpuesta JM, Chacon P, Diakun G, Andreu JM. Changes in microtubule protofilament number induced by Taxol binding to an easily accessible site. Internal microtubule dynamics. J Biol Chem. 1998;273:33803–10. doi: 10.1074/jbc.273.50.33803. [DOI] [PubMed] [Google Scholar]
- 15.Wade RH, Chretien D, Job D. Characterization of microtubule protofilament numbers. How does the surface lattice accommodate? J Mol Biol. 1990;212:775–86. doi: 10.1016/0022-2836(90)90236-F. [DOI] [PubMed] [Google Scholar]
- 16.Desai A, Mitchison TJ. Microtubule polymerization dynamics. Annu Rev Cell Dev Biol. 1997;13:83–117. doi: 10.1146/annurev.cellbio.13.1.83. [DOI] [PubMed] [Google Scholar]
- 17.Wang HW, Nogales E. Nucleotide-dependent bending flexibility of tubulin regulates microtubule assembly. Nature. 2005;435:911–5. doi: 10.1038/nature03606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lowe J, Li H, Downing KH, Nogales E. Refined structure of alpha beta-tubulin at 3.5 A resolution. J Mol Biol. 2001;313:1045–57. doi: 10.1006/jmbi.2001.5077. [DOI] [PubMed] [Google Scholar]
- 19.Thepchatri P, Cicero DO, Monteagudo E, Ghosh AK, Cornett B, Weeks ER, Snyder JP. Conformations of laulimalide in DMSO-d6. J Am Chem Soc. 2005;127:12838–46. doi: 10.1021/ja042890e. [DOI] [PubMed] [Google Scholar]
- 20.Maier CS, Deinzer ML. Protein conformations, interactions, and H/D exchange. Methods Enzymol. 2005;402:312–60. doi: 10.1016/S0076-6879(05)02010-0. [DOI] [PubMed] [Google Scholar]
- 21.Chik JK, Schriemer DC. Hydrogen/deuterium exchange mass spectrometry of actin in various biochemical contexts. J Mol Biol. 2003;334:373–85. doi: 10.1016/j.jmb.2003.09.044. [DOI] [PubMed] [Google Scholar]
- 22.Xiao H, Verdier-Pinard P, Fernandez-Fuentes N, Burd B, Angeletti R, Fiser A, Horwitz SB, Orr GA. Insights into the mechanism of microtubule stabilization by Taxol. Proc Natl Acad Sci U S A. 2006;103:10166–73. doi: 10.1073/pnas.0603704103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maity H, Lim WK, Rumbley JN, Englander SW. Protein hydrogen exchange mechanism: Local fluctuations. Protein Science. 2003;12:153–160. doi: 10.1110/ps.0225803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Williams DH, Stephens E, O’Brien DP, Zhou M. Understanding noncovalent interactions: Ligand binding energy and catalytic efficiency from ligand-induced reductions in motion within receptors and enzymes. Angewandte Chemie-International Edition. 2004;43:6596–6616. doi: 10.1002/anie.200300644. [DOI] [PubMed] [Google Scholar]
- 25.Chalmers MJ, Busby SA, Pascal BD, He YJ, Hendrickson CL, Marshall AG, Griffin PR. Probing protein ligand interactions by automated hydrogen/deuterium exchange mass spectrometry. Analytical Chemistry. 2006;78:1005–1014. doi: 10.1021/ac051294f. [DOI] [PubMed] [Google Scholar]
- 26.Hamuro Y, Coales SJ, Morrow JA, Molnar KS, Tuske SJ, Southern MR, Griffin PR. Hydrogen/deuterium-exchange (H/D-Ex) of PPAR gamma LBD in the presence of various modulators. Protein Science. 2006;15:1883–1892. doi: 10.1110/ps.062103006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anand GS, Law D, Mandell JG, Snead AN, Tsigelny I, Taylor SS, Ten Eyck LF, Komives EA. Identification of the protein kinase A regulatory RIalpha-catalytic subunit interface by amide H/2H exchange and protein docking. Proc Natl Acad Sci U S A. 2003;100:13264–9. doi: 10.1073/pnas.2232255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Banerjee A, Roach MC, Trcka P, Luduena RF. Preparation of a monoclonal antibody specific for the class IV isotype of beta-tubulin. Purification and assembly of alpha beta II, alpha beta III, and alpha beta IV tubulin dimers from bovine brain. J Biol Chem. 1992;267:5625–30. [PubMed] [Google Scholar]
- 29.Derry WB, Wilson L, Khan IA, Luduena RF, Jordan MA. Taxol differentially modulates the dynamics of microtubules assembled from unfractionated and purified beta-tubulin isotypes. Biochemistry. 1997;36:3554–62. doi: 10.1021/bi962724m. [DOI] [PubMed] [Google Scholar]
- 30.Diaz JF, Andreu JM. Assembly of purified GDP-tubulin into microtubules induced by taxol and taxotere: reversibility, ligand stoichiometry, and competition. Biochemistry. 1993;32:2747–55. doi: 10.1021/bi00062a003. [DOI] [PubMed] [Google Scholar]
- 31.Hyman AA, Salser S, Drechsel DN, Unwin N, Mitchison TJ. Role of GTP hydrolysis in microtubule dynamics: information from a slowly hydrolyzable analogue, GMPCPP. Mol Biol Cell. 1992;3:1155–67. doi: 10.1091/mbc.3.10.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang HW, Long S, Finley KR, Nogales E. Assembly of GMPCPP-bound tubulin into helical ribbons and tubes and effect of colchicine. Cell Cycle. 2005;4:1157–60. doi: 10.4161/cc.4.9.2042. [DOI] [PubMed] [Google Scholar]
- 33.Krishna MM, Hoang L, Lin Y, Englander SW. Hydrogen exchange methods to study protein folding. Methods. 2004;34:51–64. doi: 10.1016/j.ymeth.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 34.Ehring H. Hydrogen exchange electrospray ionization mass spectrometry studies of structural features of proteins and protein/protein interactions. Analytical Biochemistry. 1999;267:252–259. doi: 10.1006/abio.1998.3000. [DOI] [PubMed] [Google Scholar]
- 35.Buey RM, Barasoain I, Jackson E, Meyer A, Giannakakou P, Paterson I, Mooberry S, Andreu JM, Diaz JF. Microtubule interactions with chemically diverse stabilizing agents: thermodynamics of binding to the paclitaxel site predicts cytotoxicity. Chem Biol. 2005;12:1269–79. doi: 10.1016/j.chembiol.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 36.Nogales E, Whittaker M, Milligan RA, Downing KH. High-resolution model of the microtubule. Cell. 1999;96:79–88. doi: 10.1016/s0092-8674(00)80961-7. [DOI] [PubMed] [Google Scholar]
- 37.Jimenez-Barbero J, Canales A, Northcote PT, Buey RM, Andreu JM, Diaz JF. NMR determination of the bioactive conformation of peloruside A bound to microtubules. J Am Chem Soc. 2006;128:8757–65. doi: 10.1021/ja0580237. [DOI] [PubMed] [Google Scholar]
- 38.Magnani M, Ortuso F, Soro S, Alcaro S, Tramontano A, Botta M. The betaI/betaIII-tubulin isoforms and their complexes with antimitotic agents. Docking and molecular dynamics studies. Febs J. 2006;273:3301–10. doi: 10.1111/j.1742-4658.2006.05340.x. [DOI] [PubMed] [Google Scholar]
- 39.Reese M, Sanchez-Pedregal VM, Kubicek K, Meiler J, Blommers MJ, Griesinger C, Carlomagno T. Structural Basis of the Activity of the Microtubule-Stabilizing Agent Epothilone A Studied by NMR Spectroscopy in Solution. Angew Chem Int Ed Engl. 2007;46:1864–1868. doi: 10.1002/anie.200604505. [DOI] [PubMed] [Google Scholar]
- 40.Correia JJ, Lobert S. Physiochemical aspects of tubulin-interacting antimitotic drugs. Curr Pharm Des. 2001;7:1213–28. doi: 10.2174/1381612013397438. [DOI] [PubMed] [Google Scholar]
- 41.Nogales E, Wang HW. Structural mechanisms underlying nucleotide-dependent self-assembly of tubulin and its relatives. Curr Opin Struct Biol. 2006;16:221–9. doi: 10.1016/j.sbi.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 42.He LF, Yang CPH, Horwitz SB. Mutations in beta-tubulin map to domains involved in regulation of microtubule stability in epothilone-resistant cell lines. Molecular Cancer Therapeutics. 2001;1:3–10. [PubMed] [Google Scholar]
- 43.Laederach A, Dowd MK, Coutinho PM, Reilly PJ. Automated docking of maltose, 2-deoxymaltose, and maltotetraose into the soybean beta-amylase active site. Proteins-Structure Function and Genetics. 1999;37:166–175. doi: 10.1002/(sici)1097-0134(19991101)37:2<166::aid-prot3>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 44.Hetenyi C, van der Spoel D. Blind docking of drug-sized compounds to proteins with up to a thousand residues. FEBS Lett. 2006;580:1447–50. doi: 10.1016/j.febslet.2006.01.074. [DOI] [PubMed] [Google Scholar]
- 45.Pineda O, Farras J, Maccari L, Manetti F, Botta M, Vilarrasa J. Computational comparison of microtubule-stabilising agents laulimalide and peloruside with taxol and colchicine. Bioorg Med Chem Lett. 2004;14:4825–9. doi: 10.1016/j.bmcl.2004.07.053. [DOI] [PubMed] [Google Scholar]
- 46.Panda D, Chakrabarti G, Hudson J, Pigg K, Miller HP, Wilson L, Himes RH. Suppression of microtubule dynamic instability and treadmilling by deuterium oxide. Biochemistry. 2000;39:5075–81. doi: 10.1021/bi992217f. [DOI] [PubMed] [Google Scholar]
- 47.Jin M, Taylor RE. Total synthesis of (+)-peloruside A. Org Lett. 2005;7:1303–5. doi: 10.1021/ol050070g. [DOI] [PubMed] [Google Scholar]
- 48.Schuttelkopf AW, van Aalten DMF. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallographica Section D-Biological Crystallography. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 49.Case DA, Cheatham TE, 3rd, Darden T, Gohlke H, Luo R, Merz KM, Jr, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–88. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. Journal of Computational Chemistry. 1998;19:1639–1662. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.