

Abstract
Protein folding speeds are known to vary over more than eight orders of magnitude. Plaxco, Simons, and Baker (see References) first showed a correlation of folding speed with the topology of the native protein. That and subsequent studies showed, if the native structure of a protein is known, its folding speed can be predicted reasonably well through a correlation with the “localness” of the contacts in the protein. In the present work, we develop a related measure, the geometric contact number, N α, which is the number of nonlocal contacts that are well-packed, by a Voronoi criterion. We find, first, that in 80 proteins, the largest such database of proteins yet studied, N α is a consistently excellent predictor of folding speeds of both two-state fast folders and more complex multistate folders. Second, we show that folding rates can also be predicted from amino acid sequences directly, without the need to know the native topology or other structural properties.
Keywords: protein folding, geometric contact number, zippers model, folding rate
In 1998, Plaxco and colleagues made the important observation that the folding rates of two-state-folding proteins correlate with the native topologies of those proteins: Faster-folding proteins tend to have more local α-helical structure, and slower-folding proteins tend to have more nonlocal β-sheet structure. Plaxco and colleagues characterized the native topology using the average relative contact order (RCO), a measure of the relative fraction of local vs. nonlocal noncovalent contacts (Plaxco et al. 1998).
Many variations of this idea have since been studied, indicating that folding rates also correlate with the long-range order (LRO) (Gromiha and Selvaraj 2001), the effective contact order (ECO) (Dill et al. 1993; Fiebig and Dill 1993), the total contact distance (TCD) (Zhou and Zhou 2002), a chain topology parameter (CTP) (Nolting et al. 2003), and the effective length of the protein, Leff (Ivankov and Finkelstein 2004). A few of these quantities, such as the absolute contact order (ACO), have predictive power beyond two-state-folding rates (Ivankov et al. 2003); they also predict the rates of more complex multistate folders as well. Interestingly, although the protein's chain length was originally found to be poorly correlated with rates of two-state folders, chain length (sometimes with a fractional exponent) was later shown to correlate well with the folding rates in more recent studies (Thirumalai 1995; Finkelstein and Badretdinov 1997; Koga and Takada 2001; Galzitskaya et al. 2003; Shao et al. 2003; Naganathan and Munoz 2005).
However, these results were obtained with relatively small data sets and often begin from knowledge of the native structure of the protein (Plaxco et al. 1998; Gromiha and Selvaraj 2001; Zhou and Zhou 2002; Ivankov et al. 2003; Nolting et al. 2003). There have been several reports of predicting folding rates from protein sequences (Shao and Zeng 2003; Kuznetsov and Rackovsky 2004; Gromiha 2005; Punta and Rost 2005; Galzitskaya and Garbuzynskiy 2006), but these all require some level of information of protein structures, for example, knowledge of the structural class, or are based on prior predictions of the native secondary structures.
Our aim here is to develop a general method that can predict the folding rates for proteins of diverse classes based only on the amino acid sequence of the protein, without knowledge of the tertiary or secondary structures, or information of structural class, and without the aid of any other computational prediction of structural properties (e.g., secondary structures or contact order). We first use the concept of “geometric contact” (defined below) to study the correlation between native structure and folding rate (Li et al. 2003). Using a large set of proteins, including both two-state and multistate folders, we find that folding rates correlate well with the number of residues that form geometric contacts. The correlation coefficients are −0.86, −0.86, and −0.83 for two-state proteins, multistate proteins, and all proteins combined, respectively. Using a reduced alphabet of only two types of amino acids which are weighted differently, these correlation coefficients are all improved. The folding rate predicted from structure has a correlation coefficient of −0.86 with measured folding rate in leave-one-out jackknife tests. Based on estimated propensity values of different residues to form geometric contacts from a protein structural database, we further develop a simple algorithm that predicts folding rates from amino acid sequences alone, without any additional structural information. The predicted values correlate well with the experimental values, with a coefficient of −0.82. Our results suggest that both simple and complex proteins, over all the fold classes, may fold by a single mechanism in which spatial packing and zipping interactions are important determinants of the folding rate.
Materials and Methods
Model and data
Data set
A collected data set of experimentally determined folding rates for 80 proteins, of which 45 are two-state folders and 35 are multistate folders, were a generous gift from Ken Dill and Dr. Ke Fan (University of California at San Francisco). We have slightly modified this data set and have removed structures that contain large hetero groups, such as iron protoporphyrins, or irregular amino acids. We have also incorporated additional data from the literature. These proteins belong to different structural classes: 18 are all-α proteins, 32 are all-β proteins, and 30 are αβ proteins. We took the slowest rate for multistate folders, since the faster rates are due to kinetic traps; the slowest rate corresponds to the appearance of native protein and is therefore most directly comparable with the folding rate of two-state folders. The folding rates of these proteins range over more than eight orders of magnitude, from lnkf = −6.9 for ribonucleotide isomerase (1qo2) to lnkf = 12.9 for albumin-binding domain (1prb). Tables 1 and 2 give the Protein Data Bank names and experimentally measured folding-rate values for two-state and multistate proteins, respectively. Supplemental material is available at (http://gila.bioengr.uic.edu/resources/folding/Rate.html).
Table 1.
The set of 45 two-state proteins

Table 2.
The set of 35 multistate proteins

Defining geometric contacts
In most studies, pairwise contacts are typically declared if two residues are within a specific cutoff distance. Such definitions can include residue pairs that have no steric interactions (Taylor 1997; Bienkowska et al. 1999). We take the view here that a more refined definition of geometric contact may be more useful (Li et al. 2003).
We used a contact definition based on a Voronoi criterion. Voronoi diagrams have been widely used in protein structure and folding analysis (Richards 1977; Poupon 2004). Here we illustrate our contact definition using a simple two-dimensional picture of a molecule formed by a collection of disks of uniform size (Fig. 1A). In the diagram, each Voronoi cell contains one atom, and every point inside a Voronoi cell is closer to this atom than to any other atom. A Voronoi cell is defined by its boundary edges (shown as broken lines in Fig. 1A), which are perpendicular bisectors of the line segments connecting two atom centers. For each Voronoi edge, this line segment is called the corresponding Delaunay edge (Fig. 1B). In this study, residues i and j are defined to form a geometric contact if they are connected by a Delaunay edge, and the corresponding Voronoi edge intersects with the protein body. In addition, we require that contacting residue pairs must be at least four residues apart in the primary sequence, and their spatial distance is no greater than 6.5 Å. Our parameter N α, the geometric contact number, is simply the total number count of residues in a protein with such contacts. We first test N α as a predictor of folding rates against other measures. The RCO was introduced by Plaxco et al. (1998):
Figure 1.

Voronoi diagram of a simple 2D molecule. (A) The molecule is formed by disks of uniform size. The dashed lines represent the Voronoi diagram, in which each region contains one atom. (B) The Delaunay edges of the molecule.
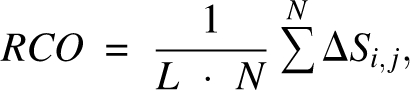 |
where N is the total number of contacts, ΔSi,j is the sequence separation between residue i and j, and L is the total number of residues. RCO measures the relative importance of local and distant contacts. The ACO was also introduced by Plaxco et al. (2000):
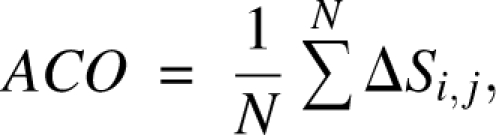 |
where ACO is the average sequence separation of contacting residues, not normalized by the chain length as RCO is. Finally, chain length (L) has also been used for correlating with folding rates (Thirumalai 1995; Finkelstein and Badretdinov 1997; Koga and Takada 2001; Galzitskaya et al. 2003; Ivankov et al. 2003; Naganathan and Munoz 2005).
Results and Discussion
The Voronoi-based geometric contact definition gives an improved correlation with protein folding rates
The results of correlating folding rates lnkf with N α and other measures of native topology are summarized in Table 3. As others have found previously (Ivankov et al. 2003), we find that the RCO correlates poorly with folding rates for this set of 80 proteins. A better measure is the ACO. Its correlation with folding rates is R = −0.83 for two-state proteins, R = −0.64 for more complex proteins, and R = −0.76 for both sets combined. Previous results suggested that the protein's chain length correlates well with the folding rate (Naganathan and Munoz 2005). We found using this enlarged data set protein chain length has a strong correlation for multistate proteins (R = −0.79), but a weaker correlation for two-state proteins (R = −0.72). Although using fractional powers of the length (e.g., L 1/2, L 2/3, or L 3/5) or the logarithm ln(L) can lead to improved correlations with multistate proteins (Naganathan and Munoz 2005), they introduce little improvement for two-state proteins (see Table 3). On the other hand, the quantity N α introduced here, correlates well in all cases (R = −0.86 for two-state proteins, R = −0.86 for multistate, and R = −0.83 for all 80 proteins). Figure 2 shows how these various measures correlate with folding rates of the combined set of proteins. These data indicate that an accurate description of geometric contacts improves the correlation of native protein structures with folding rates.
Table 3.
Correlation coefficients of structure-derived parameters with protein folding rates

Figure 2.

Relationship between different structural parameters and folding rates of two-state (open squares) and multistate (solid squares) proteins. (A) Relative contact order, RCO (R = −0.15); (B) absolute contact order, ACO (R = −0.77); (C) chain length (R = −0.72); and (D) N α (R = −0.83).
Comparing our geometry-based contact definition with distance-based definitions
We compare our measure using the geometric definition of contact with the following distance-based measure: We declare a pair of residues to be in contact if the distance between their Cα atoms is no greater than 6.5 Å. The results are shown in Table 4. The geometry-based definition gives a slightly better correlation than the distance-based definition for relative contact order and for our parameter of total contact number N α, and gives the same correlation as the distance measure when using absolute contact order. More importantly, there are 8384 and 5234 pairwise contacts by the distance-based and geometry-based measures, respectively, hence 38% of the distance-based contacts either are unnecessary or degrade the correlation.
Table 4.
Comparing distance-based and geometry-based definitions of contacts, for correlating with folding rates

N α is a better predictor of folding rate than chain length. Although chain length and N α are highly correlated (R = 0.91), we find via subset testing that N α is better than simple chain length at correlating with folding rates. We randomly selected a subset of 30 proteins from the 80 proteins, and carried out the correlation analysis on this subset. The correlation coefficients between the folding rate lnkf and the geometric contact number N α, between lnkf and the chain length L, are recorded, respectively. This is repeated seven times. As can be seen in Figure 3, the chain length L is not a consistently good predictor of protein folding rates: The correlation R is better than −0.50 only for two subsets, and the best R-value is −0.67. Depending on the class of proteins, the R-value can be as little as −0.04. In contrast, N α gives consistently good correlations: All are better than −0.58, with the best value being −0.79. These results suggest that N α is more informative than chain length for understanding protein folding mechanisms.
Figure 3.

The geometric contact number N α, is more robust than chain length L in correlating with protein folding rate. Results of a subset testing where 30 proteins are drawn from the original data set to form a subset. Correlation coefficients of folding rates with N α and with the chain length L for seven such subsets are plotted.
Different geometric contacts contribute differently to folding rates
Here, we allowed each residue type i to have a weighted contribution w i, leading to the following model for protein folding rates:
where lnkf is the folding rate of a protein, a is a constant, n α is a 20-dimensional vector recording the number counts of the 20 residue types in geometric contact, and w is the 20-dimensional weight vector whose values are to be determined. Using singular value decomposition for the data set of 80 proteins, we obtain the optimal weight vector w, and the baseline constant a, that minimize the residual error of the predicted lnkf with the experimentally determined lnkf values, by a Euclidean distance measure (Noble and Daniel 1988). The optimal weights for the 20 amino acid types are listed in Table 5. Interestingly, Val, Ile, Trp, and Tyr appear to slow down folding by the greatest extent, whereas Glu and Phe accelerate folding.
Table 5.
The weight parameters for the different residue types in determining protein folding rates

Upper bound of protein folding speed
Based on the 20 optimized weight parameters, we can estimate an upper bound for the folding speeds of the fastest proteins. In general, small proteins are fast folders: A foldable protein sequence with only 20 residues has been reported (Qiu et al. 2002). If we: (1) consider such a 20 mer, and (2) take our predicted fastest-folding residue, Glu (recognizing, however, that Glu would not lead to a stable fold), it suggests that no protein or peptide is likely to fold faster than lnkf = 10.29 + 20 × 0.451 ≈ 19.3, according to Equation 1, or roughly 4 nsec.
Folding rates and structures on a reduced alphabet of amino acids
In order to avoid overfitting, we use a reduced alphabet of amino acids containing only two types of residues, and allow these two types of residues to contribute differently to the folding rate. After exhaustive tests using different combination of residue types, we choose the following grouping of amino acids as our reduced alphabet A = (A1, A2), with A1 = (A, C, E, F, M, N, R, G, H, K, L, P, T) and A2 = (D, I, Q, S, V, W, Y). When the number counts (n1, n2) of residues with geometric contacts for these two reduced residue types are weighted differently with w1 = 0.015 and w2 = −0.324, the correlation coefficients for folding rates improves to R = −0.87, −0.87, and −0.87 for two state, multistate, and combined set, respectively.
The resulting model lnkf = 10.192 + n α · w also predicts protein folding rates well. Here n α = (n1, n2) is the vector of number counts of geometric contact, w = (w1, w2) is the vector of weights. Results from jackknife tests show that predicted and measured folding rates are strongly correlated, with a correlation coefficient of 0.86 (Fig. 5A, see below).
Figure 5.

Scatter plots of the predicted and experimentally measured values of lnkf in jackknife leave-one-out tests: (A) using weighted geometric contact number; (B) using sequence information only, and (C) using chain length.
Predicting protein folding rates from sequences
As proteins are generally tightly packed, one may assume, to first approximation, that each residue of a specific type has the same probability of a geometric contact as any other residues of the same type. With this assumption, the folding rate of a protein can be determined from knowledge of its sequence and each amino acid's general ability to form geometric contact.
The geometric contacting propensity can be estimated from known protein structures. Here, we used PDB-SELECT (2002 version), a nonredundant protein structure data set containing 1670 structures with pairwise sequence identity <25% (Boberg et al. 1992). The distribution of geometric native contacts for the 20 amino acid types and the corresponding relative values are shown in Figure 4A, and the propensity values are obtained after correction for residue composition (Fig. 4B). These propensity values collectively form the 20-dimensional contact propensity vector p. We can derive the following model for correlating protein folding rates:
Figure 4.

Propensity of residues for forming geometric contact. (A) Distribution of the number of native geometric contacts of 20 amino acids in the PDB select data set; (B) the propensity values of residues for forming geometric contact.
where n 2 is the two-dimensional vector of the simple number counts of two different simplified residue types for a protein, p is the 20-dimensional geometric contacting propensity vector, w is conceptually the 20-dimensional weight vector of different contributions of the residues, “∘” denotes component-wise vector product, and P (p ∘ w) denotes the “projection” of the 20-vector of component-wide product p ∘ w to the two-dimensional space of reduced alphabet, namely,
 |
We can denote the projection of component-wise vector product as: w s = (p ∘ w). It integrates both the propensity of a residue type to form geometric contact and its relative contribution to folding rate. The resulting model for predicting protein folding rates is:
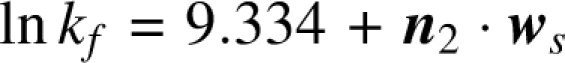 |
where lnkf is the folding rate of a protein, and n 2 is the two-dimensional vector of number count of reduced residue types in the sequence of the given protein. The optimal reduced two-alphabet and values of w s are listed in Table 6.
Table 6.
Predicting protein folding rates using reduced alphabets of amino acids

We find an excellent agreement between experimentally determined and predicted folding rates. The effectiveness of the model can be demonstrated in a jackknife test, in which the coefficients w s of the model omitting one protein were calculated and the folding rate of the omitted protein is computed. The result is shown in Figure 5B, which is significantly better (R = 0.82) than prediction results using chain length (R = 0.69, Fig. 5C). As can be seen from the large amount of scattering at the right portion of Figure 5B, chain length correlates with folding rate poorly for fast folders, as folding rates of proteins of similar length (X-axis) can differ significantly. This is a phenomenon well studied in a recent theoretical work (Kachalo et al. 2006).
The deviation of sequence weights, w s, from structural weights, w, can be thought of as an implicit correction by assuming some average structural information for specific residue types. Our results suggest that even models with two residue types can capture a significant amount of information about protein folding rates. This is reminiscent of the well-known HP model for studying protein stability and folding (Chan and Dill 1989; Ozkan et al. 2001; Kachalo et al. 2006).
Conclusions
We introduce here a quantity, N α, which is a count of the number of well-packed nonlocal contacts in the native structure of a protein, where “well packed” is defined by a Voronoi criterion. The quantity N α, is highly anti-correlated with the folding rates of 80 proteins, both two-state and multistate folders. This quantity gives a better and more consistent correlation with folding rates over this broad set of proteins than several other quantities, including the RCO, ACO, and the chain length L. For example, simple chain length does not correlate well with two-state folders. In addition, the correlation is not robust, as a different choice of protein samples results in a large variation in correlation (Fig. 3). The overall correlations using either RCO or ACO are not as good as that obtained from N α. The measure N α is not biased against shorter loops as long as their lengths are longer than a threshold of three residues, while both RCO and ACO weight more for contacts with long loops. We believe that the physical basis for this correlation is that proteins fold via a mechanism of zipping and assembly. Contacts among monomers that are more widely separated in the sequence are more difficult to form because their conformational search is more costly in chain entropy, and folding is likely to proceed through a local zipping mechanism (Dill et al. 1993; Fiebig and Dill 1993; Weikl and Dill 2003a,b; Weikl et al. 2004; Merlo et al. 2005).
The present work goes beyond predicting folding rates from known native structures or from known/predicted secondary structures (Ivankov and Finkelstein 2004; Gromiha 2005), and predicts rates, instead, just from the amino acid sequences of these proteins. Our prediction works even when protein sequences are based on alphabets of only two residue types. Although several previous studies can correlate protein folding rates with sequences well, they are based on a smaller data set, and they require additional structural knowledge of proteins in the form of general structural class (Kuznetsov and Rackovsky 2004; Gromiha 2005), or secondary structure information (Ivankov and Finkelstein 2004). We find that different amino acids have different propensities for folding speed. Proteins are most slowed down by Val, Ile, Trp, and Tyr forming geometric contacts, and most speeded up by Glu and Phe.
Acknowledgments
This work is supported by grants from the National Science Foundation (DBI-0646035) and the National Institutes of Health (GM079804-01A1 and GM081682 GM68958). We thank Dr. Ken Dill for stimulating discussions and for sharing the collected data of folding rates, and Dr. Martin Gruebele for helpful discussions.
Footnotes
Reprint requests to: Jie Liang, Department of Bioengineering, University of Illinois at Chicago, 851 South Morgan Street, Room 218, Chicago, IL 60607, USA; e-mail: jliang@uic.edu; fax: (312) 996-5921.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.034660.108.
References
- Bienkowska, J.R., Rogers, R.G., Smith, T.F. Filtered neighbors threading. Proteins. 1999;37:346–359. [PubMed] [Google Scholar]
- Boberg, J., Salakoski, T., Vihinen, M. Selection of a representative set of structures from Brookhaven Protein Data Bank. Proteins. 1992;14:265–276. doi: 10.1002/prot.340140212. [DOI] [PubMed] [Google Scholar]
- Chan, H.S., Dill, K.A. Compact polymers. Macromolecules. 1989;22:4559–4573. [Google Scholar]
- Dill, K.A., Fiebig, K.M., Chan, H.S. Cooperativity in protein folding kinetics. Proc. Natl. Acad. Sci. 1993;90:1942–1946. doi: 10.1073/pnas.90.5.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiebig, K.M., Dill, K.A. Protein core assembly processes. J. Chem. Phys. 1993;98:3475–3487. [Google Scholar]
- Finkelstein, A.V., Badretdinov, A.Y.A. Rate of protein folding near the point of thermodynamic equilibrium between the coil and the most stable chain fold. Fold. Des. 1997;2:115–121. doi: 10.1016/s1359-0278(97)00016-3. [DOI] [PubMed] [Google Scholar]
- Galzitskaya, O.V., Garbuzynskiy, S.O. Entropy capacity determines protein folding. Proteins. 2006;63:144–154. doi: 10.1002/prot.20851. [DOI] [PubMed] [Google Scholar]
- Galzitskaya, O.V., Garbuzynskiy, S.O., Ivankov, D.N., Finkelstein, A.V. Chain length is the main determinant of the folding rate for proteins with three-state folding kinetics. Proteins. 2003;51:162–166. doi: 10.1002/prot.10343. [DOI] [PubMed] [Google Scholar]
- Gromiha, M.M. A statistical model for predicting protein folding rates from amino acid sequence with structural class information. J. Chem. Inf. Model. 2005;45:494–501. doi: 10.1021/ci049757q. [DOI] [PubMed] [Google Scholar]
- Gromiha, M.M., Selvaraj, S. Comparison between long-range interactions and contact order in determining the folding rate of two-state proteins: Application of long-range order to folding rate prediction. J. Mol. Biol. 2001;310:27–32. doi: 10.1006/jmbi.2001.4775. [DOI] [PubMed] [Google Scholar]
- Ivankov, D.N., Finkelstein, A.V. Prediction of protein folding rates from the amino acid sequence-predicted secondary structure. Proc. Natl. Acad. Sci. 2004;101:8942–8944. doi: 10.1073/pnas.0402659101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivankov, D.N., Garbuzynskiy, S.O., Alm, E., Plaxco, K.W., Baker, D., Finkelstein, A.V. Contact order revisited: Influence of protein size on the folding rate. Protein Sci. 2003;12:2057–2062. doi: 10.1110/ps.0302503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kachalo, S., Lu, H.M., Liang, J. Protein folding dynamics via quantification of kinematic energy landscape. Phys. Rev. Lett. 2006;96:058106. doi: 10.1103/PhysRevLett.96.058106. [DOI] [PubMed] [Google Scholar]
- Koga, N., Takada, S. Roles of native topology and chain-length scaling in protein folding: A simulation study with a Go-like model. J. Mol. Biol. 2001;313:171–180. doi: 10.1006/jmbi.2001.5037. [DOI] [PubMed] [Google Scholar]
- Kuznetsov, I.B., Rackovsky, S. Class-specific correlations between protein folding rate, structure-derived, and sequence-derived descriptors. Proteins. 2004;54:333–341. doi: 10.1002/prot.10518. [DOI] [PubMed] [Google Scholar]
- Li, X., Hu, C., Liang, J. Simplicial edge representation of protein structures and α contact potential with confidence measure. Proteins. 2003;53:792–805. doi: 10.1002/prot.10442. [DOI] [PubMed] [Google Scholar]
- Merlo, C., Dill, K.A., Weikl, T.R. φ Values in protein folding kinetics have energetic and structural components. Proc. Natl. Acad. Sci. 2005;102:10171–10175. doi: 10.1073/pnas.0504171102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naganathan, A.N., Munoz, V. Scaling of folding times with protein size. J. Am. Chem. Soc. 2005;127:480–481. doi: 10.1021/ja044449u. [DOI] [PubMed] [Google Scholar]
- Noble, B., Daniel, J.W. Applied linear algebra. 3rd ed. Prentice-Hall; New Jersey: 1988. [Google Scholar]
- Nolting, B., Schalike, W., Hampel, P., Grundig, F., Gantert, S., Sips, N., Bandlow, W., Qi, P.X. Structural determinants of the rate of protein folding. J. Theor. Biol. 2003;223:299–307. doi: 10.1016/s0022-5193(03)00091-2. [DOI] [PubMed] [Google Scholar]
- Ozkan, S.B., Bahar, I., Dill, K.A. Transition states and the meaning of φ-values in protein folding kinetics. Nat. Struct. Biol. 2001;8:765–769. doi: 10.1038/nsb0901-765. [DOI] [PubMed] [Google Scholar]
- Plaxco, K.W., Simons, K.T., Baker, D. Contact order, transition state placement and the refolding rates of single domain proteins. J. Mol. Biol. 1998;227:985–994. doi: 10.1006/jmbi.1998.1645. [DOI] [PubMed] [Google Scholar]
- Plaxco, K.W., Simons, K.T., Ruczinski, I., Baker, D. Topology, stability, sequence, and length: Defining the determinants of two-state protein folding kinetics. Biochemistry. 2000;39:11177–11183. doi: 10.1021/bi000200n. [DOI] [PubMed] [Google Scholar]
- Poupon, A. Voronoi and Voronoi-related tessellations in studies of protein structure and interaction. Curr. Opin. Struct. Biol. 2004;14:233–241. doi: 10.1016/j.sbi.2004.03.010. [DOI] [PubMed] [Google Scholar]
- Punta, M., Rost, B. Protein folding rates estimated from contact predictions. J. Mol. Biol. 2005;348:507–512. doi: 10.1016/j.jmb.2005.02.068. [DOI] [PubMed] [Google Scholar]
- Qiu, L., Pabit, S.A., Roitberg, A.E., Hagen, S.J. Smaller and faster: The 20-residue Trp-cage protein folds in 4 micros. J. Am. Chem. Soc. 2002;124:12952–12953. doi: 10.1021/ja0279141. [DOI] [PubMed] [Google Scholar]
- Richards, F.M. Areas, volumes, packing, and protein structures. Annu. Rev. Biophys. Bioeng. 1977;6:151–176. doi: 10.1146/annurev.bb.06.060177.001055. [DOI] [PubMed] [Google Scholar]
- Shao, H., Zeng, Z.H. A sequence function reveals new features in β-protein folding. Protein Pept. Lett. 2003;10:435–439. doi: 10.2174/0929866033478690. [DOI] [PubMed] [Google Scholar]
- Shao, H., Peng, Y., Zeng, Z.H. A simple parameter relating sequences with folding rates of small α-helical proteins. Protein Pept. Lett. 2003;10:277–280. doi: 10.2174/0929866033478906. [DOI] [PubMed] [Google Scholar]
- Taylor, W.R. Multiple sequence threading: An analysis of alignment quality and stability. J. Mol. Biol. 1997;269:902–943. doi: 10.1006/jmbi.1997.1008. [DOI] [PubMed] [Google Scholar]
- Thirumalai, D. From minimal models to real proteins: Timescales for protein folding kinetics. J. Mol. Biol. 1995;5:1457–1469. [Google Scholar]
- Weikl, T.R., Dill, K.A. Folding kinetics of two-state proteins: Effect of circularization, permutation, and crosslinks. J. Mol. Biol. 2003a;332:953–963. doi: 10.1016/s0022-2836(03)00884-2. [DOI] [PubMed] [Google Scholar]
- Weikl, T.R., Dill, K.A. Folding rates and low-entropy-loss routes of two-state proteins. J. Mol. Biol. 2003b;329:585–598. doi: 10.1016/s0022-2836(03)00436-4. [DOI] [PubMed] [Google Scholar]
- Weikl, T.R., Palassini, M., Dill, K.A. Cooperativity in two-state protein folding kinetics. Protein Sci. 2004;13:822–829. doi: 10.1110/ps.03403604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, H., Zhou, Y. Folding rate prediction using total contact distance. Biophys. J. 2002;82:458–463. doi: 10.1016/S0006-3495(02)75410-6. [DOI] [PMC free article] [PubMed] [Google Scholar]