

Abstract
Nuclease resistance and RNA affinity are key criteria in the search for optimal antisense nucleic acid modifications, but the origins of the various levels of resistance to nuclease degradation conferred by chemical modification of DNA and RNA are currently not understood. The 2′-O-aminopropyl (AP)-RNA modification displays the highest nuclease resistance among all phosphodiester-based analogues and its RNA binding affinity surpasses that of phosphorothioate DNA by 1°C per modified residue. We found that oligodeoxynucleotides containing AP-RNA residues at their 3′ ends competitively inhibit the degradation of single-stranded DNA by the Escherichia coli Klenow fragment (KF) 3′-5′ exonuclease and snake venom phosphodiesterase. To shed light on the origins of nuclease resistance brought about by the AP modification, we determined the crystal structure of an A-form DNA duplex with AP-RNA modifications at 1.6-Å resolution. In addition, the crystal structures of complexes between short DNA fragments carrying AP-RNA modifications and wild-type KF were determined at resolutions between 2.2 and 3.0 Å and compared with the structure of the complex between oligo(dT) and the D355A/E357A KF mutant. The structural models suggest that interference of the positively charged 2′-O-substituent with the metal ion binding site B of the exonuclease allows AP-RNA to effectively slow down degradation.
Keywords: antisense, cationic oligonucleotides, exonuclease inhibitor, hydration, x-ray crystallography
Second-generation oligonucleotide analogues for use as antisense (AS) therapeutics are now emerging (1, 2). Two improvements over the first-generation DNA phosphorothioates (PS-DNA) are of particular importance. First, several of the modifications confer increased affinity toward target RNA. Second and perhaps more importantly, they have increased ability to evade nuclease degradation. In the quest to achieve both goals, a large number of compounds with modifications located in the backbone, sugar, or base portion of the nucleic acid framework were introduced (3). Among the sites available for modification, the 2′ position of the deoxyribose sugar has shown particular promise (4). Attachment of electron withdrawing moieties at the 2′ carbon shifts the conformational equilibrium of the sugar toward the C3′-endo pucker, thus preorganizing the AS oligonucleotide for binding to the RNA target (5–7). The 2′-modified class of compounds also has proven to be valuable because of its enhanced nuclease resistance (4). Examples are the 2′-O-alkoxyalkyl compounds (8–11) and 2′-O-aminopropyl (AP) RNA (4, 12) (Fig. 1).
Figure 1.

Chemical structures of DNA, PS-DNA, AP-RNA, and 2′-O-butyl RNA.
Protection of AS oligodeoxynucleotides (ODNs) against 3′ exonucleases appears to be of particular concern in view of practical applications in vivo or in cell cultures (9). The nuclease resistance conferred by simple 2′-O-alkyl substituents is correlated with the length of the alkyl chain (3, 13, 14). Longer and more bulky substituents lead to improved protection and the resistance of the 2′-O-pentyl modification is similar to that of PS-DNA. However, the AP modification leads to much better protection than the 2′-O-propyl one, despite similar lengths of the substituents. ODNs containing short stretches of AP-RNA at their 3′ ends display extraordinary resistance against degradation by 3′ exonucleases (12) (Fig. 2). The unusual stability of AP-RNA may be related to the zwitterionic nature of the analogue. The amino group in the 2′-O-substituent has a pKa of 9.4 and therefore can be expected to carry a positive charge under neutral conditions (12).
Figure 2.

PAGE gel depicting the time course (in min) of SVPD-catalyzed degradation of (A) d(T)19, (B) d(T)15-2′-O-butyl-r(U)4, and (C) d(T)15-AP(U)4.
PS-DNA emerged as a suitable modification for AS therapy primarily because of its resistance to degradation by nucleases and relatively facile synthetic preparation. All AS compounds with antiviral, anticancer, and anti-inflammatory indications currently being tested in the clinic are full-length PS-ODNs (4, 15). However, the accumulated data concerning the pharmacology, pharmacokinetics, and toxicology of all first-generation AS-ODNs, including PS-DNA, suggests that these compounds have certain limitations (summarized in ref. 4). Further structural changes will be necessary to overcome these and design compounds with more optimal properties. Modifications with improved nuclease resistance may affect the dosing frequency, will reduce potential metabolite toxicity, and will be essential for future oral administration of AS oligomers.
The origins of the various degrees of nuclease resistance displayed by nucleic acid analogues currently are not understood. Here, we show that an oligo(dT) strand with AP-U modifications at its 3′ flank is a competitive inhibitor of oligo(dT) degradation by snake venom phosphodiesterase (SVPD) and Escherichia coli DNA polymerase I Klenow fragment (KF) 3′ exonuclease. Based on a high-resolution crystal structure of an A-DNA 10-mer duplex with incorporated AP-uridines, the structure of the complex between a mutated (mt) KF and oligo(dT) as well as the structures of complexes between the wild-type (wt) KF and oligonucleotides containing either one or multiple AP- ribonucleotides we have investigated how the 2′-O-aminopropyl substituent can effectively interfere with the phosphoryl transfer reaction catalyzed by the exonuclease.
Methods
Oligonucleotide Synthesis and Purification.
All oligonucleotides containing AP-ribonucleosides (Table 1) were synthesized on a Millipore Expedite nucleic acid synthesizer, by using previously described methods (12). The d(T)19 oligomer was synthesized by the standard phosphoramidite method. Oligonucleotides used for crystallization experiments were purified by ion exchange chromatography and RP-HPLC, and those used for degradation and inhibition assays were PAGE-purified.
Table 1.
Oligonucleotides used for either crystallization (c) or enzyme degradation/inhibition (d) assays
| Oligonucleotide | Purpose |
|---|---|
| d(GCGTA)-AP(U)-d(ACGC) | c |
| d(GCG)-AP(U)-d(ATACGC) | c* |
| d(GCGT)-AP(A)-d(TACGC) | c* |
| d(GCG)-AP(U)-d(A)-AP(U)-d(ACGC) | c* |
| d(TCG)-AP(AUC) | c† |
| d(TT)-AP(U)-d(T) | c† |
| d(T)19 | c†/d |
| d(T)15-AP(U)4 | d |
| d(T)15-2′-O-butyl-r(U)4 | d |
Not of sufficient quality for crystal structure determination.
† Cocrystallization with mt- or wt-KF.
Oligonucleotide Crystallization, Structure Determination, and Refinement.
Optimal crystallization conditions for the modified DNA 10-mer d(GCGTA)-AP(U)-d(ACGC) were determined with a sparse matrix crystallization screen (16). Equal volumes of the 10 mer (2 mM in water) and a buffer containing 40 mM potassium cacodylate (pH 7.0), 80 mM KCl, 12 mM spermine and 10% (vol/vol) 2-methyl-2,4-pentanediol (MPD) were mixed, and the hanging drop was equilibrated against a reservoir of 1 ml of 35% MPD. Selected crystal data are listed in Table 2. Data were collected on a shock-frozen crystal (120 K), by using in-house x-ray equipment [R-axis IIc/Rigaku (Tokyo) RU-200, Table 2]. Data processing and scaling were performed in the denzo/scalepack program suite (17). The structure was determined with the molecular replacement method, by using the program amore (18) and a standard A-form DNA duplex model. Refinement was carried out with the program x-plor (19), by using updated nucleic acid parameters (20) and setting 10% of the data aside for calculating Rfree (21). Final refinement statistics are listed in Table 2.
Table 2.
Crystal data and selected data collection and refinement parameters
| Structure | d(GCGTA)- AP(U)-d(ACGC) | mt-KF + oligo(-dT) | wt-K + d(TCG)-AP(AUC) | wt-KF + d(TT)-AP(U)-d(T) |
|---|---|---|---|---|
| Crystal data | ||||
| Space group | P212121 | P43 | P43 | P43 |
| a Å | 25.17 | 101.88 | 102.55 | 102.65 |
| b Å | 42.77 | 101.88 | 102.55 | 102.65 |
| c Å | 45.18 | 85.18 | 86.28 | 86.30 |
| Data collection | ||||
| Resolution, Å | 1.60 | 2.08 | 2.18 | 3.00 |
| Completeness, % | 94.8 | 94.6 | 98.6 | 99.9 |
| Rmerge | 0.086 | 0.050 | 0.075 | 0.078 |
| Refinement statistics | ||||
| Resolution, Å | 8-1.6 | 20-2.08 | 20-2.18 | 20-3.00 |
| Unique refls, F > 2 σ(F) | 6,234 | 44,062 | 38,672 | 16,291 |
| Rfree, 10%/R factor | 24.3/18.7 | 23.3/21.7 | 23.8/21.5 | 24.3/22.4 |
| rms bonds, Å | 0.007 | 0.008 | 0.008 | 0.009 |
| rms angles, degs. | 1.07 | 1.33 | 1.58 | 1.63 |
KF Overexpression, Purification, Crystallization, and Structural Analysis of KF-Oligonucleotide Complexes.
Cell stabs for wt-KF and D355A/E357A mt-KF were obtained from Catherine M. Joyce, Yale University, New Haven, CT. The wt and mt proteins were overexpressed and purified according to published procedures (22). Crystals for both were obtained from high gradients of sodium citrate (23), by using the sitting drop vapor diffusion method. The crystals were stabilized in 60% ammonium sulfate, buffered at pH 6, and later soaked in solutions containing zinc sulfate, magnesium sulfate (24) and the d(T)19 (mt-KF) or d(TT)-AP(U)-d(T) and d(TCG)-AP(AUC) oligomers (wt-KF). All data were collected at 120 K on the 5 ID beamline of the DuPont-Northwestern-Dow Collaborative Access Team at the Advanced Photon Source, Argonne, IL. A previously published KF structure served as the initial model for refinement of the protein portions (25). The refinements were carried out with the program cns (26). Nucleotides were built into areas of difference electron density, the final models representing those with optimal fit and R factor. Crystal data and refinement statistics for the three structures are listed in Table 2.
Nuclease Digestion.
The relative protections against degradation by SVPD (Merck) of DNA and two oligonucleotides carrying four 2′-O-modified uridines (Table 1) at the 3′ end were analyzed by using a previously described protocol (12). Aliquots of the reaction mixture were removed at the indicated times (Fig. 2), quenched by addition to an equal volume of 80% formamide gel loading buffer and heated for 2 min at 95°C before PAGE. Quantitation was performed by reading intensities on a PhosphorImager (Molecular Dynamics, Storm 860).
Inhibition Studies.
For inhibition of the KF 3′-5′ exonuclease function, 100 pmol d(T)19 was 5′ radioactively labeled, gel eluted, and taken up in 100 μl of H2O; 1-μl aliquots then were added per reaction. Inhibition was assessed in the presence of the following concentrations of the AP-RNA-modified oligodeoxynucleotide: 0, 0.015, 0.15, 1.5, and 15 μM. The 20-μl reaction contained 1.2 units of enzyme in 50 mM Tris⋅HCl (pH 8.0), 5 mM MgCl2, and 1 mM DTT. The incubation time was 7 min at 37°C. The reaction was stopped by adding an equal volume of 80% formamide and heated for 5 min at 90°C, and the mixture was analyzed by 20% denaturing PAGE. Inhibition of SVPD was performed likewise with the exception that 2 × 10−4 units enzyme were used in 50 mM Tris⋅HCl (pH 8.5), 72 mM NaCl, and 14 mM MgCl2 buffer. Quantitation was performed by reading intensities on a PhosphorImager.
Coordinates.
Final coordinates and structure factors of the AP-RNA/DNA duplex and the three complex structures were deposited in the Protein Data Bank.
Results and Discussion
Resistance of 2′-O-Butyl and 2′-O-AP-Modified Oligonucleotides to Degradation by SVPD.
Nineteen-mer oligonucleotides containing either four AP-uridines or four 2′-O-butyl-uridines at their 3′ ends (Table 1) were exposed to SVPD, and the degradation patterns were assayed with gel electrophoresis at seven time points over the course of 72 hr (Fig. 2). This assay demonstrates the superior exonuclease resistance of the zwitterionic AP-RNA modification compared with the roughly isosteric 2′-O-butyl modification and clearly shows that steric or conformational features alone cannot account for the extraordinary stability of the former modification.
Conformation of the 2′-O-AP Substituents.
The DNA decamer with sequence GCGTATACGC was used as a crystallization template for investigating the conformational properties of AP-modified residues (5). Four oligomers were synthesized, three containing single AP residues in place of either T4, A5, or T6 and a fourth one with two incorporated AP-Us (Table 1). Well-diffracting crystals were obtained only for the decamer with a single AP-U residue at position 6, and its structure was determined at a resolution of 1.6 Å (Table 2, see Fig. 8, which is published as supplementary material on the PNAS web site, www.pnas.org, for an electron density map). The decamer duplex adopts a standard A-type geometry, and the minor groove environment of the substituents therefore can be expected to be similar to that in a hybrid duplex between an RNA and a 2′-O-modified RNA. The average helical rise and twist values are 2.81 Å and 32.8°, respectively, and all sugars adopt C3′-endo pucker.
The conformations of the AP substituents of residues U6 and U16 are different (Fig. 3). In the case of the former the conformations of the torsion angles around the C-C bonds fall into the t and g ranges. The latter substituent has an extended all-trans conformation. Both protrude into the shallow groove and are relatively far removed from the phosphate groups on either the 5′ or the 3′ side (Fig. 3). Thus, the AP moiety cannot effectively shield the adjacent phosphate groups. This finding demonstrates that the protection against nuclease degradation conferred by the AP modification is not brought about by an electrostatic interaction (e.g., salt bridge formation) between the primary amino group of the AP substituent and backbone phosphates. Our findings are consistent with the results of earlier molecular dynamics simulations of an AP(UT) dinucleotide model system (12). Over the time course of the simulation, the distance between the AP nitrogen and the nonbridging phosphate oxygens varied between 3.2 and 6.5 Å. Similarly, no hydrogen bonding between the nitrogen and base carbonyl oxygens or ribose 4′-oxygens was observed.
Figure 3.

Conformation and hydration of the AP residues U16 (Left) and U6 (Right). Views of base pair step A5pU6-A15pU16 (A) along the helix axis (upper and lower base pair in cyan and yellow, respectively, and selected torsion angles in degrees), and (B) roughly perpendicular to the helix axis, into the minor groove (with hb distances in Å). Atoms are colored green, red, blue, and magenta for carbon, oxygen, nitrogen, and phosphorus, respectively. Atoms of a backbone portion from a neighboring duplex contacting the modified step in the minor groove are colored gray and red for carbon and oxygen, respectively. Hydrogen bonds are thin solid lines and water molecules are cyan spheres.
Hydration.
The amino groups of AP substituents form three hydrogen bonds each (Fig. 3). Two of them involve water molecules with both residues U6 and U16. For U6 the third hydrogen bond is formed to a 4′ ribose oxygen from a neighboring duplex. The corresponding contact in the case of residue U16 occurs to the phosphate oxygen from the same residue of the symmetry mate. As pointed out above, the distances between the amino group of the substituents and adjacent intra-strand phosphate groups are fairly large. Consequently, the coordinated water molecules cannot directly bridge the amino nitrogen to phosphate oxygens. The shortest distance between a water molecule coordinated to the amino nitrogen of the AP substituent and a phosphate oxygen is 4.36 Å (residue U6 and 3′ phosphate, see Fig. 9, which is published as supplementary material on the PNAS web site, www.pnas.org). Thus, at least two water molecules are required to link the positively charged amino group to an adjacent phosphate moiety. The presence of the positively charged moities in the minor groove improves the hydration of the modified duplex compared with a DNA duplex. This water structure may contribute to the enhanced overall thermodynamic stability of AP-RNA relative to PS-DNA and native DNA.
AP-RNA Competitively Inhibits DNA Degradation.
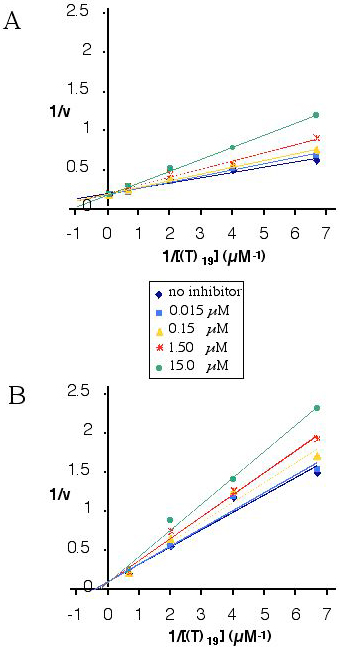
To examine whether an ODN carrying AP-modified residues at its 3′ end binds at the active site of 3′ exonucleases, the degradations by the KF 3′ exonuclease and SVPD of an oligodeoxynucleotide d(T)19 were assayed in the presence of various concentrations of d(T)15-AP(U)4. The KF exonuclease is highly specific for DNA whereas SVPD degrades both single-stranded DNA and RNA (27). Similar results were obtained for both enzymes, demonstrating competitive inhibition of DNA degradation by an ODN bearing an AP-uridine stretch at its 3′ flank (see Fig. 10, which is published as supplementary material on the PNAS web site, www.pnas.org, for 1/velocity vs. 1[d(T)19] plots). Although this result is not surprising in the case of SVPD, given its specificity for both DNA and RNA substrates, it is quite unexpected for the KF exonuclease. However, whether or not the binding modes are similar for the DNA and AP-RNA substrates requires a more detailed structural analysis.
Structural Origins of 3′-Exonuclease Inhibition.
Because AP-RNA is a competitive inhibitor of DNA degradation catalyzed by KF, we turned to this enzyme as a model system for investigating the structural origins of exonuclease resistance with a phosphodiester-based analogue. Structure, polymerase, and exonuclease functions as well as mechanistic aspects of DNA polymerase I KF were studied extensively in the past (28–30). These studies showed that for this nuclease and many others to function properly they require two bound metal ions at their active sites (31). A recently published crystal structure of a complex between wt-KF and DNA supports this conclusion and established that in the case of KF exonuclease, metal ion A is Zn2+ and metal ion B is Mg2+ (30). Mutation of amino acids known to coordinate to either one of the two metal ions reduced activity or even rendered the exonuclease inactive (e.g., D424A) (32, 33). Recent functional studies have provided further evidence that the exonuclease uses a divalent metal ion for leaving group stabilization (34) (site B, see Figs. 6 and 7). Preventing binding of this metal ion undoubtedly would lower the rate of the phosphoryl transfer reaction.
Figure 6.

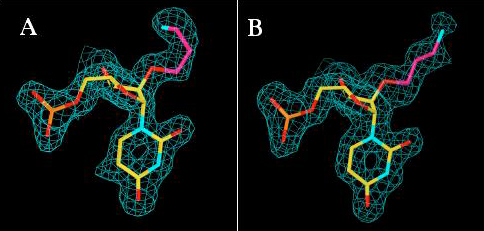
The two orientations and resulting interactions of AP(AUC)-3′ at the wt-KF exonuclease active site. (A) The positively charged nitrogen of the AP substituent of U5 is hydrogen-bonded to the carboxyl group of D424 and interferes with the binding of the B site metal ion. This ion is absent in the structure, but its normal location is indicated by a small pink sphere labeled Mg2+. (B) Alternative orientation of the modified hexamer with only two residues bound at the active site and loss of coordination between the phosphate group and the Zn2+ ion. The AP(AUC) and AP(UC) fragments are shown as stick models in yellow and atoms of phosphate groups are colored red and magenta for oxygen and phosphorus, respectively. Carbon atoms of the AP substituents of residues U5 and C6 are highlighted in green, and side-chain carbon, nitrogen and oxygen atoms of key amino acids are highlighted in cyan, blue, and red, respectively.
Figure 7.

The two orientations and resulting interactions of d(T)-AP(U)-d(T)-3′ at the wt-KF exonuclease active site. Only one of the orientations is compatible with a direct phosphate-metal ion interaction (A). However, the Zn2+ … 0 distance is longer (2.49 Å) compared with the hexamer complex (1.81 Å, Fig. 6A). See Fig. 6 for color scheme and further explanations.
The crystal structures of three different complexes between KF and oligonucleotide fragments were determined. The complex between the D355A/E357A mt-KF and oligo(dT) served as a reference and exhibits close structural similarities to the complex between wt-KF and d(GCTTAGC) at low pH (30) (Fig. 4), but lacks both metal ions at the active site. The structures of the complexes between wt-KF and d(TCG)-AP(AUC) and d(TT)-AP(U)-d(T) were refined to 2.2 and 3.0 Å, respectively (Table 2). In both cases electron density maps are consistent with the adoption of two orientations by the oligonucleotide at the exonuclease active site (Fig. 5). In the hexamer complex, the phosphate group between the two modified 3′ terminal residues for one of these orientations is coordinated to the Zn2+ ion at site A (Fig. 6A). In principle, the observed configuration is compatible with metal-ion catalyzed phosphoryl transfer. In the alternative orientation, the hexamer is shifted up and the phosphate group is relatively far removed from the Zn2+ ion (Fig. 6B). Similarly, at the active site of the other complex, the two arrangements adopted by the tetramer feature distinct distances between the terminal phosphate and Zn2+, and only one of them allows direct ion-phosphate coordination (Fig. 7).
Figure 4.

The 3′ exonuclease active site of the D355A/E357A mt-KF fragment with the bound oligo(dT). Only the three 3′ terminal residues are defined in the electron density maps. The enzyme is shown in a surface representation, the d(T)3 fragment is shown as a stick model in yellow, and atoms of phosphate groups are colored red and magenta for oxygen and phosphorus, respectively. Protein side chains involved in the binding of Zn2+ (site A) and Mg2+ (site B) are highlighted.
Figure 5.

Alternative orientations of the 5′-d(TCG)-AP(AUC)-3′ hexamer [only AP(AUC) portion visible] at the wt-KF exonuclease active site, surrounded by difference electron density before incorporation of the substrate model (2.2 Å, 1 σ level). Orientation 1 (carbon atoms in yellow) with all three modified 3′ terminal residues present at the active site. The AP substituent of residue A4 (top) was not defined in the electron density map and the substituent of residue C6 adopts two alternative conformations (bottom). The arrow highlights the position of the amino nitrogen of the AP substituent from residue U5 that is located at ca. 1.5 Å from the former position of metal ion B in the wt-KF DNA complex (ref. 30, indicated by a small gray sphere). Orientation 2 (carbon atoms in green) with only two 3′ terminal residues bound at the active site. The AP substituent of residue U5 was not defined in this arrangement. Atoms are colored red, blue, and orange for oxygen, nitrogen, and phosphorus, respectively, and the Zn2+ ion at site A is depicted as a purple sphere.
Despite similar orientations of the terminal phosphate groups at the active sites of the mt-KF and wt-KF (hexamer case) complexes (Figs. 4 and 6A), sugar and base of the AP-uridine 5 in the latter are shifted and rotated compared with the reference structure, most likely a direct consequence of the presence of the bulky 2′-O-substituents. Conversely, the 3′ terminal residue adopts very similar orientations at the active sites in the three complexes. In the complex between wt-KF and the modified hexamer, the AP-substituent of C6 is sandwiched between the side chains of residues L361 and P473, leaving the orientation of the base moiety virtually unaltered compared with the mt-KF oligo(dT) and wt-KF tetramer complexes (Figs. 4, 6A, and 7). In both complexes, a Zn2+ ion occupies the A site, but the site B metal ion is absent. In the hexamer complex the amino group of U5 forms hydrogen bonds to the carboxylate of D424 (Table 3, Fig. 6A). Similarly, in the tetramer complex the amino group of U3 forms a hydrogen bond to that side chain (Table 3, Fig. 7A). These interactions most likely prevent binding of the metal ion B (Mg2+, Figs. 6 and 7), because D424 normally is coordinated to that ion (30). In summary, the nuclease resistance of AP-RNA involves an electrostatic (Figs. 6A and 7A) and a steric (Figs. 6B and 7B) component, the former based on direct interference of a positively charged 2′-O-substituent with metal ion binding at the exonuclease active site.
Table 3.
Interactions of AP substituents at the wt-KF active site
| Donor | Acceptor | Distance, Å |
|---|---|---|
| Orientation 1 wt-KF + d(TCG)-AP(AUC) (Fig. 6A) | ||
| ND′ [AP(U5)] | OD1 [D424] | 3.1 |
| OD2 [D424] | 3.0 | |
| ND′ [AP(C6)] | OG1 [T485] | 3.0 |
| Orientation 2 (Fig. 6B): | ||
| ND′ [AP(C6)] | O3′ [AP(C6)] | 3.0 |
| O1P [AP(C6)] | 3.2 | |
| Orientation 1 wt-KF + d(TT)-AP(U)-d(T) (Fig. 7A) | ||
| ND′ [AP(U3)] | OD1 [D355] | 3.1 |
| OD2 [D424] | 2.6 | |
| Orientation 2 (Fig. 7B) | ||
| ND′ [AP(U3)] | O2 [T(2)] | 2.9 |
| OG [S658] | 3.3 | |
| O [P354] | 2.9 | |
Conclusions
Structure-based analysis of the complex formed by an oligonucleotide analogue and an exonuclease can yield novel insights into the mechanism of resistance. In the case of zwitterionic AP-RNA, the analysis shows that the modification prevents binding of a metal ion that is known to be required for the enzyme to efficiently catalyze the phosphoryl transfer reaction. A short modified stretch at the 3′ end can provide sufficient protection because only two or three residues are actually bound at the exonuclease active site (e.g., in the case of KF). The relevance of the present work on E. coli KF-oligonucleotide interactions to mammalian exonucleases is borne out by the similarities (including the specificity of metal ion cofactors) reported recently between the nucleases present in both systems (see for example ref. 35). In the case of zwitterionic nucleic acid analogues it remains to be determined how the substituent length affects the nuclease resistance [e.g., aminopentyl (36) or aminohexyl (37)]. Using structural information that takes into account both the nuclease and the nucleic acid analogue should be helpful in the design of new modifications with improved nuclease resistance.
Supplementary Material
Acknowledgments
We thank Dr. Bruce S. Ross and Mr. Giopal B. Inamati, Isis Pharmaceuticals Inc. for providing the phosphoramidites and the 2′-o-butyl-modified 19-mer, respectively, and Mr. Scott Ware for technical support. This work was supported by the National Institutes of Health (Grant R01 GM-55237 to M.E.) and the Davee Foundation. S.T.W. was a visiting graduate student and a recipient of an International Ph.D. Fellowship from the Austrian Science Foundation FWF (No. W001). The DuPont-Northwestern-Dow Collaborative Access Team is supported by the E.I. DuPont de Nemours & Co., The Dow Chemical Company, the U.S. National Science Foundation, and the State of Illinois.
Abbreviations
- AP
aminopropyl
- PS
phosphorothioate
- KF
Klenow fragment
- AS
antisense
- ODN
oligodeoxynucleotide
- SVPD
snake venom phosphodiesterase
- mt
mutated
- wt
wild type
Footnotes
Data deposition: The structural coordinates have been deposited in the Protein Data Bank, www.rcsb.org [PDB ID codes: 1d9h (10mer), 1d8y (mt KF), 1d9d (wt KF + hexamer), and 1d9f (wt KF + tetramer)].
References
- 1.De Mesmaeker A, Häner R, Martin P, Moser H E. Acc Chem Res. 1995;28:366–374. [Google Scholar]
- 2.Crooke S T. In: Handbook of Experimental Pharmacology: Antisense Research and Applications. Crooke S T, editor. Vol. 131. Heidelberg: Springer; 1998. pp. 1–50. [Google Scholar]
- 3.Freier S M, Altmann K-H. Nucleic Acids Res. 1997;25:4429–4443. doi: 10.1093/nar/25.22.4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cook P D. Annu Med Rep Chem. 1998;33:313–325. [Google Scholar]
- 5.Egli M. Angew Chem Intl Ed Engl. 1996;35:1894–1909. [Google Scholar]
- 6.Herdewijn P. Liebigs Ann. Chem. 1996. 1337–1348. [Google Scholar]
- 7.Egli M. Antisense Nucleic Acid Drug Dev. 1998;8:123–128. doi: 10.1089/oli.1.1998.8.123. [DOI] [PubMed] [Google Scholar]
- 8.Martin P. Helv Chim Acta. 1995;78:486–504. [Google Scholar]
- 9.Altmann K-H, Fabbro D, Dean N M, Geiger T, Monia B P, Müller M, Nicklin P. Biochem Soc Trans. 1996;24:630–637. doi: 10.1042/bst0240630. [DOI] [PubMed] [Google Scholar]
- 10.Tereshko V, Portmann S, Tay E C, Martin P, Natt F, Altmann K-H, Egli M. Biochemistry. 1998;37:10626–10634. doi: 10.1021/bi980392a. [DOI] [PubMed] [Google Scholar]
- 11.Teplova M, Minasov G, Tereshko V, Inamati G B, Cook P D, Manoharan M, Egli M. Nat Struct Biol. 1999;6:535–539. doi: 10.1038/9304. [DOI] [PubMed] [Google Scholar]
- 12.Griffey R H, Monia B P, Cummins L L, Freier S M, Greig M J, Guinosso C J, Lesnik E, Manalili S M, Mohan V, Owens S R, et al. J Med Chem. 1996;39:5100–5109. doi: 10.1021/jm950937o. [DOI] [PubMed] [Google Scholar]
- 13.Lesnik E A, Guinosso C J, Kawasaki A M, Sasmor H, Zounes M, Cummins L L, Ecker D J, Cook P D, Freier S M. Biochemistry. 1993;32:7832–7838. doi: 10.1021/bi00081a031. [DOI] [PubMed] [Google Scholar]
- 14.Cummins L L, Owens S R, Risen L M, Lesnik E A, Freier S M, McGee D, Guinosso C J, Cook P D. Nucleic Acids Res. 1995;23:2019–2024. doi: 10.1093/nar/23.11.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Akhtar S, Agrawal S. Trends Pharmaceut Sci. 1997;18:12–18. doi: 10.1016/s0165-6147(96)01002-4. [DOI] [PubMed] [Google Scholar]
- 16.Berger I, Kang C H, Sinha N, Wolters M, Rich A. Acta Crystallogr D. 1996;52:465–468. doi: 10.1107/s0907444995013564. [DOI] [PubMed] [Google Scholar]
- 17.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 18.Navaza J. Acta Crystrallogr A. 1994;50:157–163. [Google Scholar]
- 19.Brünger A T. x-plor: A System for X-Ray Crystallography and NMR. New Haven, CT: Yale Univ. Press; 1992. , Version 3.1. [Google Scholar]
- 20.Parkinson G, Vojtechovsky J, Clowney L, Brünger A T, Berman H M. Acta Crystallogr D. 1996;52:57–64. doi: 10.1107/S0907444995011115. [DOI] [PubMed] [Google Scholar]
- 21.Brünger A T. Nature (London) 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 22.Joyce C M, Grindley N D F. Proc Natl Acad Sci USA. 1983;80:1830–1834. doi: 10.1073/pnas.80.7.1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brick P, Ollis D, Steitz T A. J Mol Biol. 1983;166:453–456. doi: 10.1016/s0022-2836(83)80095-3. [DOI] [PubMed] [Google Scholar]
- 24.Freemont P S, Friedman J M, Beese L S, Sanderson M R, Steitz T A. Proc Natl Acad Sci USA. 1988;85:8924–8928. doi: 10.1073/pnas.85.23.8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ollis D L, Brick P, Hamlin R, Xuong N G, Steitz T A. Nature (London) 1985;313:762–766. doi: 10.1038/313762a0. [DOI] [PubMed] [Google Scholar]
- 26.Brünger A T. cns: Crystallography & NMR System. New Haven, CT: Yale Univ.; 1998. , Version 0.3. [Google Scholar]
- 27.Linn S M, Lloyd R S, Roberts R J, editors. Nucleases. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1993. [Google Scholar]
- 28.Joyce C M, Steitz T A. J Bacteriol. 1995;177:6321–6329. doi: 10.1128/jb.177.22.6321-6329.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brautigam C A, Steitz T A. Curr Opin Struct Biol. 1998;8:54–63. doi: 10.1016/s0959-440x(98)80010-9. [DOI] [PubMed] [Google Scholar]
- 30.Brautigam C A, Steitz T A. J Mol Biol. 1998;277:363–377. doi: 10.1006/jmbi.1997.1586. [DOI] [PubMed] [Google Scholar]
- 31.Steitz T A, Steitz J A. Proc Natl Acad Sci USA. 1993;90:6498–6502. doi: 10.1073/pnas.90.14.6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Derbyshire V, Freemont P S, Sanderson M R, Beese L, Friedman J M, Joyce C M, Steitz T A. Science. 1988;240:199–201. doi: 10.1126/science.2832946. [DOI] [PubMed] [Google Scholar]
- 33.Derbyshire V, Grindley N D F, Joyce C M. EMBO J. 1991;10:17–24. doi: 10.1002/j.1460-2075.1991.tb07916.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Curley J F, Joyce C M, Piccirilli J A. J Am Chem Soc. 1997;119:12691–12692. [Google Scholar]
- 35.Couture C, Chow T Y K. Nucleic Acids Res. 1992;20:4355–4361. doi: 10.1093/nar/20.16.4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Manoharan M, Guinosso C J, Cook P D. Tetrahedron Lett. 1991;32:7171–7174. [Google Scholar]
- 37.Manoharan M, Tivel K L, Anrade L K, Cook P D. Tetrahedron Lett. 1995;36:3647–3650. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}