

Abstract
Learning and memory depend on dendritic spine actin assembly and docosahexaenoic acid (DHA), an essential n-3 (omega-3) polyunsaturated fatty acid (PFA). High DHA consumption is associated with reduced Alzheimer’s disease (AD) risk, yet mechanisms and therapeutic potential remain elusive. Here, we report that reduction of dietary n-3 PFA in an AD mouse model resulted in 80%–90% losses of the p85α subunit of phosphatidylinositol 3-kinase and the postsynaptic actin-regulating protein drebrin, as in AD brain. The loss of postsynaptic proteins was associated with increased oxidation, without concomitant neuron or pre-synaptic protein loss. N-3 PFA depletion increased caspase-cleaved actin, which was localized in dendrites ultrastructurally. Treatment of n-3 PFA-restricted mice with DHA protected against these effects and behavioral deficits and increased antiapoptotic BAD phosphorylation. Since n-3 PFAs are essential for p85-mediated CNS insulin signaling and selective protection of postsynaptic proteins, these findings have implications for neurodegenerative diseases where synaptic loss is critical, especially AD.
Introduction
While autosomal-dominant, genetically induced overproduction of amyloid β-peptide 1–42 (Aβ42) can cause Alzheimer’s disease (AD), environmental risk factors may be required for the expression of most inherited susceptibility to full-scale AD (Grant et al., 2002; Hsiao et al., 1996; Selkoe, 2001). One candidate risk factor is docosahexaenoic acid [DHA; 22:6(n-3)], an essential dietary n-3 polyunsaturated fatty acid (PFA) relevant to the maintenance of learning and memory in animal models (Hashimoto et al., 2002; Moriguchi et al., 2000; Salem et al., 2001). Epidemiological studies suggest that people who ingest higher levels of DHA are less likely to develop AD dementia (Barberger-Gateau et al., 2002; Conquer et al., 2000; Grant et al., 2002; Kyle et al., 1999; Morris et al., 2003; Tully et al., 2003). The reasons for the impact of DHA on learning and memory and the association with AD have been unclear but could result from its loss in synapses (Montine et al., 2004), which are normally enriched with DHA (Salem et al., 2001), where it is particularly central to postsynaptic signaling and neuroprotection (Bazan, 2003). Memory loss correlates better with synapse loss than with plaques or tangles, but the precise biochemical and morphological events in synaptic dysfunction in AD are not well understood. Presynaptic marker loss and dystrophic neurites are well studied in AD (Selkoe, 2002), but information about postsynaptic pathology is comparatively limited. Postsynaptic actin-rich dendritic spines are dynamically involved in synaptic plasticity, learning, and memory (Fukazawa et al., 2003; Matus, 2000; Star et al., 2002). Accumulating pathological and genetic evidence from Williams syndrome (Lim kinase 1) and seven X-linked mental retardation genes has implicated abnormalities in the regulation of dendritic spine actin as a major cause of the cognitive deficits in mental retardation (Ramakers, 2002). Further, developmentally regulated brain protein (drebrin), a dendritic spine actin-regulating protein, shows major (70%–95%) losses in Alzheimer’s disease (Harigaya et al., 1996; Hatanpaa et al., 1999; Shim and Lubec, 2002), raising the possibility that spine dysfunction also contributes to cognitive failure in AD. Dendritic proteins may be especially vulnerable because soluble toxic Aβ oligomers appear to have synaptic receptors colocalizing with PSD-95 (Gong et al., 2003), and Aβ42 accumulates in dendrites in AD patients and the human mutant amyloid precursor protein (HuAPPsw) (Tg2576) transgenic mouse AD model (Takahashi et al., 2002), where it may cause oxidative damage and caspase activation (Behl et al., 1994; Lim et al., 2001; Mattson and Duan, 1999; Pratico et al., 2001). Additional postsynaptic vulnerability to aging and oxidative stress arises from high dendritic energy consumption (Attwell and Iadecola, 2002).
Although synaptic marker loss is widely viewed as central to AD, high level expression of familial AD genes in transgenic mice (for example, APPswe in the Tg2576 model) has produced phenotypes with abundant amyloid deposition but remarkably little synaptic marker loss. While one factor limiting synaptic deficits may be the absence of human tau in the model, another may be the mouse diet, in particular, the neuroprotective role of the low n-6/n-3 essential fatty acid ratio in typical mouse chow. The purpose of the present study was to investigate the impact of dietary depletion and repletion of DHA on neuroprotective pathways, synaptic marker loss, and cognitive deficits in the Tg2576 mouse model for AD. Measurement of the impact of DHA on pre-and postsynaptic marker vulnerability revealed marked effects on dendritic structural elements and signaling pathways that could underlie behavioral deficits induced by low DHA. The results show a dramatic impact of diet on the expression of the AD-related postsynaptic marker phenotype and provide new insight into how essential fatty acid intake may modulate the expression of neurodegenerative diseases, including AD.
Results
Selective Dendritic Pathology in an Animal Model of AD
Actin is a major target of oxidative damage in AD (Aksenov et al., 2001) and is the most abundant substrate of several caspases (Yang et al., 1998). Site-specific antibodies to a caspase-cleaved fragment of actin (fractin) label puncta in the dendrites of tangle-bearing neurons, Hirano bodies, and surrounding plaques in AD (Rossiter et al., 2000; Yang et al., 1998) and in an AD mouse model (Cole et al., 1999). The schematic diagram in Figure 1G shows fractin antiserum epitope binding. To further localize fractin in AD models, we carried out an ultrastructural analysis of fractin immunostaining in aged transgenic (Tg2576) mice, which overexpress HuAPPsw and have been used as a model for memory loss and Aβ deposition in AD (Hsiao et al., 1996). Fractin immunoreactivity in 17-month-old Tg2576 mice was seen almost exclusively in dendrites, while presynaptic compartments had little or no labeling (Figures 1A–1C). Compared to young (6- to 8-month-old) Tg2576 mice (prior to amyloid accumulation), old Tg2576 mice (16- to 18-month-old) showed 48% (p < 0.001) loss of drebrin (Figure 1D). The fractin/actin ratio was increased in the cortex (+71%) of the aged Tg2576 mice compared to controls on immunoblots (Figure 1E). Since together these data indicated focal, postsynaptic caspase activation and damage to the actin cytoskeleton in Tg2576 mice, we quantified pre- and postsynaptic markers. By 22 months of age, there was a preferential loss of the actin binding protein drebrin [−62%, Tg(+) relative to Tg(−) mice] without a proportional decrease in the presynaptic marker synaptophysin (Figure 1E). Since synaptophysin loss is most profound in neurofibrillary tangle-bearing neurons (Callahan et al., 1999), the absence of tangles and persistent sprouting in Tg2576 may explain why synaptophysin deficits are not observed. There was no loss of other presynaptic markers, including synaptotagmin, synaptobrevin, and synaptosomal-associated protein-25 (SNAP-25) as a function of transgene or aging out to 22 months [mean ± SEM for SNAP-25 in the cortex: 100.0 ± 2.9 for Tg(−) and 100.3 ± 3.6 for Tg (+)]. To determine whether such asymmetric synaptic protein marker loss also occurs in AD, we compared levels of drebrin, PSD-95, and synaptophysin in the temporal cortex of AD and control brains obtained at autopsy. We found large losses of drebrin (−74%) and PSD-95 (−51%) as well as a less striking (−17%) but significant synaptophysin decrease on immunoblots (Figure 1F). These data on drebrin are consistent with reports from three different groups indicating very large decreases of drebrin that far exceed synaptophysin loss in AD (Harigaya et al., 1996; Hatanpaa et al., 1999; Shim and Lubec, 2002). Thus, while synaptophysin loss is an early event in the pathogenesis of AD (Selkoe, 2002; Terry et al., 1991), the actin regulatory protein drebrin is substantially more vulnerable in both AD and in transgenic mice modeling Aβ accumulation in AD.
Figure 1. Dendritic Caspase Activation and Selective Drebrin Loss in Tg2576 Mouse and Selective Postsynaptic Marker Loss in Alzheimer’s Disease.
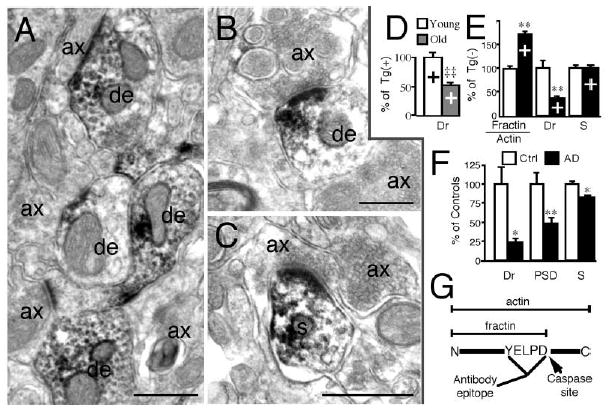
(A–C) Fractin immunoreactivity was detected in dendrites (de) and occasionally in spines (s) but not in axons (ax) in the cortex of aged Tg2576 mice. Calibration bars, 500 nm.
(D) Drebrin (Dr) in young (6–8 months) Tg(+) mice compared to old (16–18 months) Tg(+) mice in lysate from cortical membrane fractions. Values shown are percentage of young Tg(+) as the mean ± SEM. ‡‡p < 0.01 versus young Tg(+).
(E) Effect of transgene on fractin/actin ratio, drebrin (Dr), and synaptophysin (S) levels in membrane fraction of mouse cortex. Transgenic (+), n = 7–8, controls (−), n = 7–12. **p < 0.01 versus controls [Tg(−)].
(F) Drebrin (Dr), PSD-95 (PSD), and synaptophysin (S) in temporal cortex samples from AD patients (n = 10) and controls (n = 9). Values shown are percentage of control patients as the mean ± SEM. *p < 0.05, **p < 0.01 versus controls (Ctrl).
(G) Diagram illustrating the generation of fractin from cleavage of actin by caspases.
Essential Fatty Acids Protect Against Caspase Activation and Dendritic Pathology
Activation of the neuroprotective PI3-kinase/Akt pathway and elevated phospho-BAD have been invoked to explain limited synapse loss in young Tg2576 mice (Stein and Johnson, 2002). Docosahexaenoic acid [22:6(n-3)] is a n-3 PFA that comprises 15% of the fatty acids in the brain and is enriched in synapses (Breckenridge et al., 1972; Salem et al., 2001). In neuroblastoma cells, DHA directly activates the PI3-kinase/Akt pathway and prevents caspase activation (Akbar and Kim, 2002). DHA may be depleted by low dietary intake and losses from lipid peroxidation, which is elevated in AD and Tg2576 mice (Montine et al., 2004; Pratico et al., 2001). Because low DHA intake is a potential risk factor for AD (Barberger-Gateau et al., 2002; Conquer et al., 2000; Grant et al., 2002; Kyle et al., 1999; Morris et al., 2003; Tully et al., 2003) and reductions in PI3-kinase activity are observed in AD (Jolles et al., 1992; Zubenko et al., 1999), we hypothesized that DHA depletion might limit PI3-kinase and promote caspase activation in Tg2576 mice. To test this, we restricted dietary n-3 PFA at 17 months of age and found a significant decrease in the levels of DHA in the frontal cortex of aged Tg(+) mice but not in Tg(−) mice (Table 1). These observations demonstrate a transgene-dependent effect on brain DHA, consistent with increased lipid peroxidation. Levels of compensatory docosapentaenoic acid (DPA) were significantly elevated after dietary n-3 PFA restriction, confirming evidence of DHA deficiency (Table 1). Adding DHA back to the depleted diet brought cortical DHA levels even higher than in controls and decreased arachidonic acid (ARA) and DPA levels (Table 1). Western blot analysis revealed that n-3 PFA depletion induced a striking (3.2-fold) increase in the fractin/actin ratio in Tg(+) mice, which was reversed by DHA addition (Figure 2A). In contrast, the increase of this ratio in the cortex of n-3 PFA-depleted Tg(−) mice was only 2.13-fold (mean ± SEM: 213 ± 20 in low n-3 PFA group versus 100 ± 61 in controls, p < 0.05). These data demonstrate that essential fatty acid intake regulates CNS caspase cleavage of actin in vivo in aged animals–an observation of potential relevance to neurodegenerative diseases of aging.
Table 1.
Effect of Dietary Treatment on Polyunsaturated Fatty Acid Brain Content
|
Diet content |
Frontal cortex content |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mouse Diet Groups | n | DHA %w/w | n-6/n-3 ratio | Chol ppm | Total fatty acid %w/w | Tg(−) DHA(1) | Tg(−) DPA(1) | Tg(−) ARA(1) | Tg(+) DHA(1) | Tg(+) DPA(1) | Tg(+) ARA(1) |
| A) Control | 8 | 0.09 | 7:1 | 290 | 11 | 19.2±0.3 | 0.52±0.02 | 9.6±0.1 | 19.5±0.5 | 0.59±0.03 | 9.5±0.1 |
| B) Low n-3 PFA (low DHA) | 6 | <0.01 | 85:1 | 750 | 6.1 | 18.0±0.5 | 1.43±0.12†† | 9.8±0.1 | 16.4±0.8††¶ | 1.81±0.20†† | 9.9±0.2 |
| C) Low n-3 + DHA (High DHA) | 6 | 0.60 | 5:1 | 750 | 6.1 | 20.5±0.5○○ | 0.29±0.05○○ | 8.4±0.2○○ | 21.3±0.6○○ | 0.22±0.02○○ | 7.9±0.2○○ |
Control diet contained 2.45% and 0.18% of 18:2n-6 (linoleic acid) and 18:3n-3 (linolenic acid), respectively. Low n-3 PFA diet contained 4.86% and 0.06% of 18:2n-6 and 18:3n-3, respectively. Diet C is identical to diet B except for the added DHA. See Experimental Procedures for more details on the diet. ARA, arachidonic acid [20:4(n-6)]; Chol, cholesterol; DHA, docosahexaenoic acid [22:6(n-3)]; DPA, docosapentaenoic acid [22:5(n-6)]; PFA, polyunsaturated fatty acids.
Expressed as mean percentage of total fatty acid ± SEM. Total fatty acids were similar between groups, and values ± SEM were 31.7 ± 0.7, 32.6 ± 0.8, 32.8 ± 1.0, 32.0 ± 0.8, 34.0 ± 0.6, and 32.8 ± 0.7 μg/mg of tissue for Tg(−) control, Tg(−) Low n-3 PFA, Tg(−) Low n-3 PFA + DHA, Tg(+) control, Tg(+) Low n-3 PFA, and Tg(+) Low n-3 PFA + DHA, respectively.
p < 0.01 versus control diet (within transgene).
p < 0.01 versus Low n-3 PFA (within transgene).
p < 0.05 versus Tg(−) (within diet, Low n-3 PFA).
Figure 2. Western Analysis of Brains from DHA-Depleted Tg2576 Mice Show Increased Caspase-Cleaved Actin and Reduced Post-synaptic Drebrin and PSD-95, without Altering Synaptophysin.
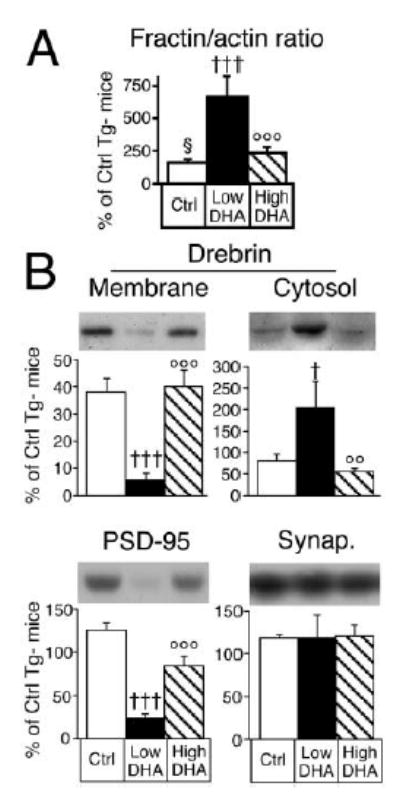
N-3 polyunsaturated fatty acid (PFA) depletion (Low DHA, n = 6) in Tg2576 mice increased the cortical fractin/actin ratio and caused a selective and massive cortical loss of drebrin and PSD-95 compared to controls (Ctrl, n = 6–7), which was prevented by the addition of docosahexaenoic acid (High DHA, n = 6). (A) The fractin/actin ratio in cortical membrane fraction. †††p < 0.001 versus Tg(+) Ctrl Diet; ○○○p < 0.001 versus Tg(+) n-3 PFA depleted (Low DHA). (B) Effect of n-3 PFA depletion (Low DHA, n = 6) and addition of DHA (High DHA, n = 6) on drebrin, PSD-95, and synaptophysin levels (Synap.) in the cortex of Tg2576 mice compared to controls (Ctrl, n = 6–7). Values shown are percentage of Tg(−) mice on control diet as the mean ± SEM. †p < 0.05, †††p < 0.001 versus Tg(+) Ctrl Diet and ○○p < 0.01, ○○○p < 0.001 versus Tg(+) Low DHA Diet.
To determine if caspase activation increased the asymmetry between pre- and postsynaptic markers, we measured the postsynaptic markers drebrin and PSD- 95 and the presynaptic markers SNAP-25 and synaptophysin. Drebrin in membrane/cytoskeletal fractions was markedly decreased by n-3 PFA depletion (−85%) in Tg(+) mice with a total transgene- and diet-dependent loss of 96% (Figure 2B). Drebrin itself did not appear to be a caspase substrate, since drebrin reduction in the membrane fraction was accompanied by a compensatory increase (+150%) in the cytosolic fraction (Figure 2B). Cytosolic release is consistent with normal drebrin retention by intact F-actin filaments (Shirao and Sekino, 2001). Depletion of n-3 PFA in Tg(+) mice also resulted in a large decrease in PSD-95 in both membrane/cytoskeletal [−77% versus Tg(−) Ctrl Diet; Figure 2B] and cytosolic fractions [−64%, mean ± SEM of cortical Tg(−) control value: 22 ± 8 in low n-3 PFA Tg(+) group versus 60 ± 4 in Tg(+) controls, p < 0.01]. PSD-95 loss can be secondary to drebrin loss because antisense suppression of drebrin expression prevents PSD-95 clustering at synapses (Takahashi et al., 2003). In contrast, the presynaptic markers SNAP-25 [mean ± SEM of cortical Tg(−) control value: 119 ± 11 in low n-3 PFA Tg(+) group versus 100 ± 4 in Tg(+) controls] and synaptophysin (Figure 2B) remained unchanged. N-3 PFA depletion and transgene expression did not cause significant neuron loss in either cortex or hippocampus, as measured by Cresyl violet (data not shown) and micrographs of NeuN immunochemistry (Figures 3A–3E) and NeuN quantification with image analysis (Figure 3F). These results show a selective synaptic phenotype in the absence of neuron loss. The synaptic protein deficits occurred in the absence of gliosis (GFAP, Westerns), increased cytokine levels (IL-1, TNF-α, ELISA), or generalized proteolysis (Coomasie blue gels). Equivalent levels of multiple presynaptic marker proteins were found across treatment groups, confirming equal protein loading. Postsynaptic marker loss was DHA dependent because DHA added back to the diet suppressed the loss of drebrin and PSD-95 (Figure 2B). Levels of drebrin and PSD-95 showed a negative correlation with the fractin/actin ratio (r2 = 0.59 and 0.64, respectively; p < 0.01), consistent with the involvement of focal postsynaptic caspase activation in selective postsynaptic marker loss.
Figure 3. No Significant Neuron Loss Was Associated with n-3 Polyunsaturated Fatty Acid Depletion in the Tg2576 Mouse.
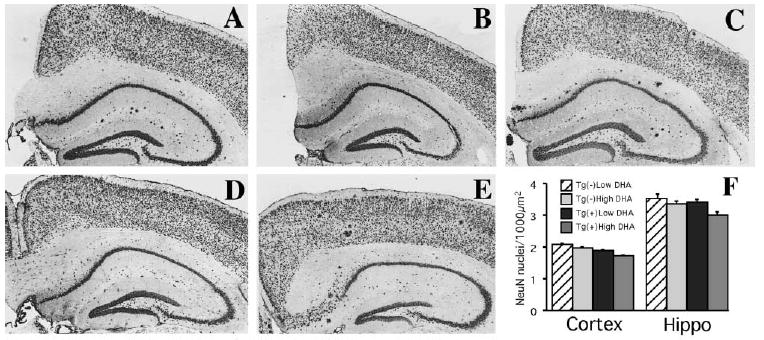
(A–E) Immunostaining with NeuN, an antibody that labels neuronal nuclei, revealed similar neuronal counts in the hippocampus or cortex between Tg2576 mice fed with low n-3 PFA diet [Tg(+) Low DHA] (A), Tg(−) fed with low n-3 PFA diet [Tg(−) Low DHA] (B), Tg(+) and Tg(−) mice fed with DHA-enriched diets (High DHA) ([C] and [D], respectively), and Tg2576 mice raised on control chow (E). Original magnification, 100×. (F) Image analysis quantification of NeuN-ir (number of neuronal nuclei per 1000 μm2) was performed on neuronal layers in cortex (entorhinal II, entorhinal III/IV, parietal II, parietal III/IV, parietal V/VI, frontal II, frontal III/IV, frontal V/VI) and hippocampus (CA1, CA2, CA3) at Bregmas −1.0 mm, 1.7mm, −2.7 mm, −3 mm (analyzed from anterior to posterior hippocampus with four consecutive sections analyzed per Bregma). There were no significant neuron density changes with APPswe transgene or with diet (n = 5 or 6 in each group). Since there were no treatment effects on neuronal nuclei densities in any layer and no interaction with regions, we show interaction bars rather than the breakdown of densities in layers and different Bregmas.
To further characterize changes in fractin and drebrin in Tg2576 mice fed with a low n-3 PFA diet, we performed simultaneous confocal microscopic analysis of fractin (Yavin et al., 2002) and drebrin (red). Figure 4 depicts the punctate plaque and neuropil fractin staining in Tg(+) mice accompanied by drebrin loss, which is prominent around plaques (Figure 4A). The KYELPD peptide antigen blocked fractin labeling in Tg(+) mice (Figure 4B). This contrasts with the absence of drebrin loss and fractin staining in Tg(−) mice fed with control (Figure 4C) or low n-3 PFA (Figure 4D) diets. Because drebrin and PSD-95 have important roles in cognitive function, the large deficits of drebrin and PSD-95 seen in AD and in Tg2576 on the low DHA diet would very likely contribute to synaptic dysfunction and cognitive decline (Hatanpaa et al., 1999; Migaud et al., 1998; Shim and Lubec, 2002; Shirao and Sekino, 2001). The findings prompted us to investigate the mechanism underlying the loss of postsynaptic markers and how it is modulated.
Figure 4. Confocal Microscopy of the Brains of Tg2576 Mice, Illustrating that Dietary DHA Depletion Increases Punctate Casapase-Cleaved Actin, which Is Associated with Reduced Drebrin.
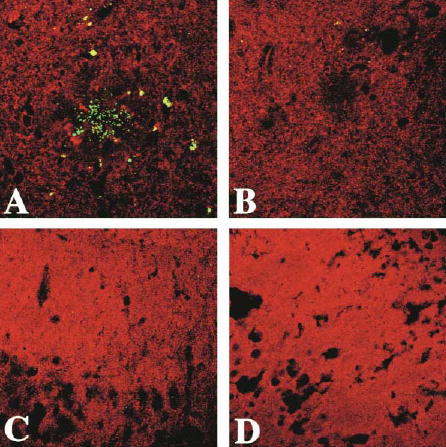
Fractin (green) and drebrin (red) double labeling in the cortex of mice showing drebrin loss and fractin labeling in the cortex of Tg2576 mice fed a low n-3 PFA diet. (A) Periplaque green punctate fractin labeling in the cortex of a Tg2576 mouse on the low n-3 polyunsaturated fatty acids diet is shown against a reduced red drebrin labeling in the vicinity of the plaque and throughout the neuropil. (B) Fractin peptide blocks fractin labeling in an adjacent section from the same Tg(+) animal. (C and D) Representative examples showing no fractin labeling and no drebrin loss in the same region of cortex in Tg(−) mice on control diet (C) or on low n-3 PFA-enriched diet (D). Magnification, 500×.
Role of Cellular Antiapoptotic Pathway and Oxidative Stress in Dendritic Pathology
A direct DHA stimulation of the PI3-kinase pathway has been observed in a neuronal cell line in vitro (Akbar and Kim, 2002), and n-3 fatty acid regulation of PI3-kinase has been reported in peripheral tissues in vivo (Taouis et al., 2002). To determine whether DHA modulates CNS PI3-kinase in vivo, we measured the levels of the p85α protein subunit of PI3-kinase protein in Tg2576 mouse cortex as well as its mRNA transcript using quantitative real-time RT-PCR (QPCR). Dietary restriction of n-3 PFA led to a very large 95% reduction of p85α in the cortex of Tg2576 mice, which was partially restored with DHA (Figure 5A). We also found a 32% decrease of p85α mRNA in DHA-depleted Tg2576 mice (Figure 5B). These data imply that essential fatty acids are powerful regulators of the CNS PI3-kinase pathway. A decrease (49%) in p85α was also observed in AD brain (Figure 5A), which may help explain the loss of PI3-kinase activity already reported in AD (Jolles et al., 1992; Zubenko et al., 1999). The increased prevalence in AD of an allelic p85α variant associated with defective insulin signaling and diabetes suggests that p85α may be in a causal pathway (Liolitsa et al., 2002). Such modulation of PI3-kinase activity can have a direct effect on cognition (see below), since electrophysiological data demonstrate that PI3-kinase is required for the induction of long-term potentiation (Opazo et al., 2003).
Figure 5. Brains of DHA-Depleted Tg2576 Mice Show Loss of p85 Expression and Phospho-BAD, Corresponding to Increased Protein Oxidation.
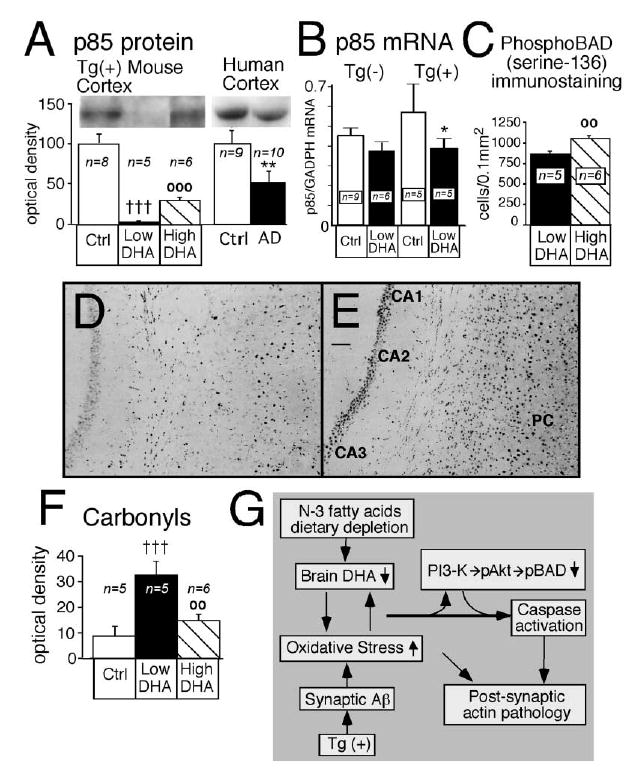
Docosahexaenoic acid (High DHA) increased p85 expression, increased phosphorylation of BAD, and reversed cortical oxidative damage induced by n-3 PFA depletion (Low DHA) in Tg2576 [Tg(+)] mice. (A) The p85α regulatory subunit of phosphatidylinositol 3 (PI3)-kinase in cortex of Tg(+) mice and in temporal cortex samples from human control and AD patients. **p < 0.01 versus human controls, †††p < 0.001 versus Tg(+) Ctrl, and ○○○p < 0.001 versus Tg(+) on n-3 PFA depletion (Low DHA). (B) Quantitative real-time RT-PCR measurements of p85α subunit mRNA in cortex of Tg(−) and Tg(+) mice fed with control diet or low DHA diet. *p < 0.05 versus Tg(+) Ctrl. (C) Image analysis quantification of phospho-BAD in cortex and hippocampus combined. ○○p < 0.01 versus Tg(+) on n-3 PFA depletion (Low DHA). (D and E) Representative examples of phospho-BAD immunostaining in hippocampus (CA1, CA2, and CA3) and parietal cortex (PC) of two mice. The two mice were depleted of N-3 (D) but the mouse in (E) received a diet that was enriched in DHA. Magnification bar, 0.3 mm. (F) The effect of DHA on cortical oxidized proteins levels. †††p < 0.001 versus Tg(+) Ctrl, and ○○p < 0.01 versus Tg(+) on n-3 PFA depletion (Low DHA). (G) Proposed mechanistic pathway for the transgene- and DHA-dependent effect on postsynaptic markers. Aβ overexpression, combined with low n-3 polyunsaturated fatty acid dietary intake, generates an autocatalytic vicious cycle in the postsynaptic dendrites leading to a further increase of oxidative stress and decrease of DHA. This could lead to decreased PI3-kinase activity, caspase activation, further oxidative damage, and consequent breakdown of dendrite spine F-actin filaments and postsynaptic damage.
To further characterize the impact of DHA on the downstream components of the PI3-kinase pathway that might regulate caspase activation, we labeled brain sections with an antibody specific for BAD phosphorylated at serine 136. BAD is a member of the Bcl-2 apoptosis-regulating protein family that is proapoptotic in the absence of serine 136 phosphorylation. Activation of the antiapoptotic PI3-kinase cascade phosphorylates BAD at serine 136 (pBAD), through Akt or PAK, promoting its binding to 14-3-3 protein and preventing subsequent caspase activation (Datta et al., 2000; Schurmann et al., 2000; Yuan and Yankner, 2000). Consistent with DHA-induced restoration of the PI3-kinase pathway, phospho-BAD immunostaining was increased in hippocampus and frontal and parietal cortex in mice fed the high DHA diet (Figures 5C and 5E) compared to the n-3 PFA-depleted group (Figures 5C and 5D). In contrast, DHA supplementation decreased total BAD [mean ± SEM of cortical Tg(−) control value: 196 ± 43 in high DHA group versus 371 ± 56 in low n-3 PFA group, p < 0.01] and left 14-3-3 protein unchanged [mean ± SEM of cortical Tg(−) control value: 88 ± 6 in high DHA group versus 82 ± 1 in low n-3 PFA group], consistent with a specific increase in phospho-BAD and reduced caspase activation. Lacking cortical homogenates prepared with phosphatase inhibitors, additional evidence of DHA regulation of PI3-K signaling through downstream phospho-Akt was sought in immunoblots of septal-striatal blocks prepared with a phosphatase inhibitor cocktail. High DHA intake increased levels of Akt phosphorylated at serine 473 measured on immunoblots by 78% compared to the n-3 PFA-depleted Tg(+) animals [mean ± SEM of cortical Tg(−) control value: 74 ± 6 in high DHA group versus 42 ± 4 in low n-3 PFA group, p < 0.05]. Collectively, these data are consistent with DHA stimulating the PI3-K pathway, leading to phosphorylation of Akt, which in turn phosphorylates BAD and limits caspase activation.
To investigate the link between oxidative stress and fatty acid modulation of caspase activation, we measured the levels of cortical oxidized proteins using Western blot analysis of dinitrophenylhydrazine (DNPH) derivatized carbonyls. We previously showed a large increase of protein carbonyls in Tg2576 mouse cortical tissue (Lim et al., 2001). In the same mouse model, n-3 PFA restriction resulted in an additional 266% increase in protein carbonyls, whereas addition of DHA was protective (Figure 5F). Because of its six double bonds, DHA is highly susceptible to lipid peroxidation (Montine et al., 2004), and increased oxidative damage due to DHA deficiency should cause greater DHA deficiency in a positive feedback loop. The major protein carbonyl in the membrane-cytoskeletal fraction comigrated with actin, and the 45 kDa carbonyl/actin ratio was 370% increased by n-3 PFA depletion [mean ± SEM of relative O.D. values: 42 ± 17 in low n-3 PFA Tg(+) group versus 9 ± 2 in Tg(+) controls, p < 0.05]. Protein carbonyl levels in cortex correlated with levels of drebrin (r2 = 0.47), PSD-95 (r2 = 0.60), and the fractin/actin ratio (r2 = 0.44). Total protein carbonyls and selective increases in oxidized β-actin have been found in vulnerable regions of AD brain (Aksenov et al., 2001), which, along with drebrin loss, could further impair actin assembly and stability (Dalle-Donne et al., 2002).
Behavioral Consequences of n-3 Depletion in Tg2576 Mice
To directly assess the role of DHA on cognition, we studied the effect of DHA supplementation on the behavior of n-3 PFA-depleted Tg2576 mice using the Morris water maze. Cohorts of Tg2576 transgene positive and negative mice received either a low n-3 fatty acid diet or a DHA-enriched diet starting at ~17 months of age. Cognitive function in mice that were capable of finding the visible platform (n = 6) was assessed in the Morris water maze at 21–22 months as described in the Experimental Procedures. This included six blocks of visible platform followed by the 12 blocks of hidden platform testing (Figure 6A) followed by a probe test without platform to assess retention of platform location. Animals from all groups were capable of learning the visible platform (Figure 6B), indicating no major sensory-motor problems. The DHA-depleted Tg(+) mice did not display increased thigmotaxis in visible platform tests (Figure 6C), arguing against a generalized increase in anxiety when challenged to swim, and there were no significant differences in swim speed (Figure 6D). However, hidden platform testing revealed profound performance deficits in Tg(+) mice on low DHA diets that were prevented by DHA supplementation (Figure 6E), with significant differences in latency during blocks 7 through 9 and 10 through 12 by 2 × 2 ANOVA (Treatment × combined block) (Figure 6F) or by repeated ANOVA (p < 0.005, data not shown). Some learning occurred as assessed by significant regressions (p < 0.005) of block on latency in all groups except Tg(+) mice on the low DHA diet, in which greater impairments were revealed by longer latencies to platform toward the end of the experiment. Although we attempted to obtain improved learning curves with more blocks, none of the groups could complete further testing because of excessive fatigue and frailty. Unlike in the visible testing paradigm, during hidden testing, deficits were associated with increased circling or thigmotaxis in Tg(+) but not Tg(−) mice placed on the low DHA diet (Figure 6G), indicating an interaction between dietary fatty acids and the HuAPPsw transgene on thigmotaxis. Retention in probe trial (percent path quadrant) revealed transgene deficits, since Tg(+) mice spent more time in the target (T) quadrant and less time in the opposite (OPP) quadrant. Despite correcting acquisition deficits, DHA did not correct the retention deficit (Figure 6H). In summary, these results from the Morris water maze trials indicate that relatively short-term DHA restriction in Tg2576 mice leads to large losses in postsynaptic proteins that correspond to a memory acquisition deficit. The lack of thigmotaxis and acquisition deficits in Tg(+) mice fed with a high DHA diet also support a beneficial effect of supplemental DHA.
Figure 6. Morris Water Maze Memory Acquisition Deficits in Tg2576 Mice Fed with a DHA-Depleted Diet.
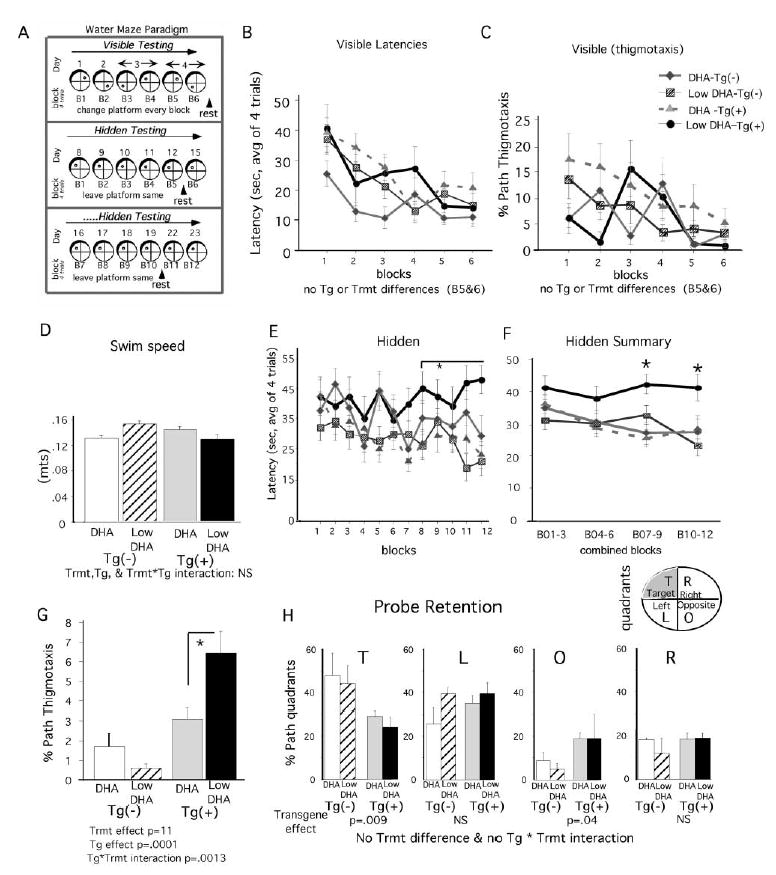
Cognitive function in Tg(+) and Tg(−) mice on low n-3 polyunsaturated fatty acid diets without DHA (Low DHA, n = 6) and with DHA (High DHA, n = 6) was assessed with the Morris water maze at 21–22 months with six blocks of visible platform followed by the 12 blocks of hidden platform testing (A). Consistent with the absence of sensorimotor deficits, there were no significant differences in the latency to find a visible platform in the latter half of visible training, although the Tg(−), DHA group performed better during the first three blocks (B). Consistent with no treatment differences in anxiety levels, there was no difference in thigmotaxis during visible training (C) nor were there differences in swim speed (D). In contrast, there were significant differences in hidden platform testing. Tg(+) mice on the low DHA diet did not appear to learn, exhibiting higher latencies from blocks 8 through 12 than the other groups (E). Learning curves of the other groups were slight due to age and fatigue, limiting further training. Nevertheless, combined block analysis (2 × 2 ANOVA: treatment × combined block) in hidden training revealed some learning and an impairment in the Tg(+) low DHA group, which was significantly different than all other groups (F). The same results were obtained by repeated ANOVA analysis p < 0.005 (data not shown). 2 × 2 ANOVA (treatment × transgene) of thigmotaxis during hidden platform testing demonstrated a significant Tg(+) effect (p = 0.0001) and a significant Tg × treatment interaction (p < 0.0013), showing that the Tg(+) mice on the low DHA diet showed excessive thigmotaxis (G). Probe retention analysis (percent path in different quadrants) confirmed a significant transgene effect (p < 0.009). Unlike the Tg(+) mice, the Tg(−) groups spent more paths in the target (T) quadrant (40%–50%) and less time in Opposite quadrant (<10%, [H]). The DHA diet failed to restore the transgene-dependent retention deficit (H).
Discussion
The search for intervention strategies for AD has been boosted by the development of transgenic mice with significant features of AD pathology (Hsiao et al., 1996; Oddo et al., 2003). One important outcome of these models is their use in identifying environmental risk factors contributing to the rate of pathogenesis and full expression of the phenotype. The present results provide evidence that the combination of genetic (mutant human APP) and environmental risk factors (dietary essential fatty acids) for AD can act synergistically to quantitatively reduce synaptic proteins, specifically dendritic scaffold proteins, that are critical for cognition as evidenced by memory deficits observed in the Morris water maze paradigm. The implication of DHA deficiency as a primary environmental risk factor in the experimental diet is supported by the correction of deleterious effects of n-3 PFA restriction with DHA supplementation. Although we cannot rule out the possibility that parallel changes in other effector pathways also contribute to synaptic protein deficits and cognitive impairment, our results argue for alterations in the PI3-kinase pathway, caspases, and oxidative stress as important mediators of DHA effects. Our data also support the idea that increased DHA intake should be considered as a potential neuroprotective strategy for AD.
Transgene- and DHA Depletion-Dependent Impairment in Behavior
With the DHA-depleting diet, the Tg(+) mice revealed poorer acquisition performance and increased thigmotaxis. This could be secondary to increased anxiety that results in thigmotaxis and impaired performance in acquisition tests. However, the same Tg(+) group on the low DHA diet did not show increased thigmotaxis in the visible platform tests, arguing against a generalized increase in anxiety induced by transgene and DHA depletion when challenged to swim and supporting a primary impairment in spatial memory. In either case, both anxiety and spatial memory deficits are relevant to early stage Alzheimer’s disease. There was a large transgene-dependent retention deficit in the probe trial that was not corrected by DHA. In the Tg2576 model, even on DHA-rich standard chow, retention deficits become severe by 20 months of age and are irreversible even with more training (Westerman et al., 2002). Acquisition deficits in Tg2576 mice are observed by 12 months of age and remain constant throughout life (Westerman et al., 2002). We show that transgene- and diet-dependent acquisition and thigmotaxis deficits, but not retention deficits, are corrected by DHA. Whether earlier intervention would have prevented the late stage retention deficits is unclear. The fact that part of the behavioral phenotype is dependent on diet is relevant to interventions in AD.
Transgene- and DHA Depletion-Dependent Insulin Resistance
Studies in animals have consistently shown that brain n-3 fatty acid content is highly dependent on dietary intake and aging (Favrelere et al., 2000; Youdim et al., 2000). Depletion of brain n-3 fatty acids up to 50%–80% is sufficient to impair neural function and is normally not observed until the second or third generation of diet-depleted rodents (Salem et al., 2001). In the present study, 3 months of safflower oil-based diet had no significant effect on brain DHA in Tg(−) mice, yet induced a small (−15%) but significant decrease of brain DHA concentration in Tg2576(+) mice, indicating transgene- dependent DHA depletion within the same generation and with short duration dietary depletion (Table 1). Dietary n-3 PFA depletion was also sufficient to induce cognitive deficits in Tg2576 mice but not in control Tg(−) mice (Figure 6). In contrast, Tg2576 mice fed with a DHA-enriched diet performed as well as the Tg(−) mice (Figure 6). This susceptibility of the mutant AD transgenics to brain n-3 PFA depletion suggests that patients bearing a genetic risk of AD may be more vulnerable to a lack of essential fatty acids. As normal aging and AD are associated with decreased brain concentrations of n-3 PFA (Favrelere et al., 2000; Guan et al., 1999; Yehuda et al., 2002; Youdim et al., 2000), older patients bearing a genetic risk for AD may be at high risk of losing the protective effect of n-3 PFA and developing dendritic and other pathology. Because deficits in DHA appear to contribute to peripheral insulin resistance, loss of p85 subunit, and adult onset diabetes (Liolitsa et al., 2002), our observations add to the growing body of data suggesting that the increased risk of AD found in diabetics reflects common causal pathways involving insulin signaling (Taubes, 2003).
DHA Loss, Not Simply Synapse Loss
Brain DHA is enriched in synapses where it is mainly found in cellular membranes esterified to phospholipids including phosphatidylserine and phosphatidylethanolamine (Breckenridge et al., 1972; Salem et al., 2001). Although the loss of DHA observed in AD brains might be interpreted as simply secondary to synaptic loss, our data would argue otherwise, since deficiency in our mice leads to compensatory increases in DPA (22:5 n-6), a well-established index of DHA deficiency in rodent brain (Salem et al., 2001). DPA was 3-fold elevated by DHA depletion and 8-fold reduced by DHA treatment (Table 1). These large compensatory changes are known to reflect the synthesis of DPA under conditions of DHA depletion and cannot be explained by synapse loss.
Because of the high double bond content, polyunsaturated fatty acids are highly vulnerable to oxidation (Montine et al., 2004). Oxidative stress, present in the brain of AD patients and in the brain of the Tg2576 mouse model, is the probable cause of the transgene-dependent loss of one of the most peroxidizable CNS lipids (DHA) and has been increasingly implicated as a significant factor in AD pathogenesis (Aksenov et al., 2001; Butterfield et al., 2002; Montine et al., 2004). This can be at least partially controlled, as the present data suggest, by increased intake of DHA and should be reduced by antioxidants from diet or supplements (Joseph et al., 2003; Lim et al., 2001). Thus, low antioxidant (Etminan et al., 2003) and low DHA (Morris et al., 2003) intake are two identified risk factors for AD that may synergize. Our data showing increased oxidative damage to proteins in DHA-depleted animals underscore this possibility. Oxidative dimerization of proteins, tau (Gamblin et al., 2000), and α-synuclein (Krishnan et al., 2003) may contribute to the chronic tau and synuclein pathology components of AD and other neurodegenerative conditions.
Selective Postsynaptic Drebrin and PSD-95 Deficits and Caspase Cleaved Actin
The absence of tangle formation and limited neuronal and presynaptic marker loss in APP transgenic mice (Irizarry et al., 1997; Suh and Checler, 2002) have allowed us to focus on the presumably more directly Aβ-dependent postsynaptic marker loss. Using image analysis of neuronal nuclei density, we confirmed the absence of neuron loss with transgene on standard chow and found no change as a function of diet. Without any evidence for neuron loss in photomicrographs or even a trend by image analysis, it is unlikely that a more stringent stereological analysis would reveal much change, since results with image analysis have been reported to closely match results with stereological methods (Everall et al., 1997). Even a 20% decrease in neurons would be far too small to explain a 70%–95% postsynaptic marker loss in the absence of any detectable presynaptic marker loss.
Consistent with this selective postsynaptic protein deficit, a selective loss of mRNA for postsynaptic proteins was recently reported in hippocampus of 18-month-old bigenic mutant PS1 × Tg2576 mice which also lack tangle formation (Dickey et al., 2003) but have a 3-fold acceleration of the amyloid cascade (Borchelt et al., 1997; Holcomb et al., 1998). We observed far more severe but still selective postsynaptic drebrin and PSD-95 loss and preservation of presynaptic markers on Westerns from Tg2576 (22-month-old) mouse cortex on the DHA-depleted diet. Preservation of presynaptic markers in aged Tg2576 mice has further been demonstrated by image analysis (King and Arendash, 2002; Suh and Checler, 2002). The selective accumulation of Aβ42 in dendritic compartments in Tg2576 and AD (Takahashi et al., 2002) and toxic Aβ oligomer binding at punctate PSD-95-positive sites (Gong et al., 2003) could drive this compartmentalized postsynaptic pathology, which was revealed at the ultrastructural level by focal caspase-cleaved actin (fractin) staining and supported by Western analysis. The concentration of actin at postsynaptic sites and the critical role of actin dynamics in synaptic plasticity and memory imply that attack by caspases and oxidative damage could have a direct impact on spine function.
Aβ Aggregates Downregulate Neuroprotective PI-3 Kinase
Increased phosphorylation of Akt and BAD through adaptive upregulation of the expression of insulin-like pathway signaling dependent on PI3-kinase has been demonstrated in Tg2576 and is proposed to limit neurodegeneration (Stein and Johnson, 2002). Like DHA depletion (Akbar and Kim, 2002), Aβ peptides can also suppress the PI3-kinase pathway and activate caspases (Takashima et al., 1996). α7 nicotinic receptors are putative Aβ42 receptors that are elevated in PS1 × 0215 Tg2576 mice (Dineley et al., 2002) and that can internalize toxic Aβ species (Nagele et al., 2002) on dendritic spines where they are associated with actin and drebrin (Shoop et al., 2000). α7 nicotinic receptors are also coupled to both p85α PI3-kinase and fyn, which is required for Aβ oligomer toxicity (Lambert et al., 1998). The PI3-kinase activity is required for α7 nicotinic neuroprotection (Kihara et al., 2001; Shaw et al., 2002). Thus, the dramatic loss of PI3-kinase p85α and reduced BAD phosphorylation in DHA-depleted Tg2576 would theoretically disable the compensatory or neuroprotective PI3-kinase mechanism preventing caspase activation, resulting in extensive postsynaptic pathology and a phenotype closer to AD. In AD, fyn kinase is preserved or increased (Shirazi and Wood, 1993) while PI3-kinase activity is markedly reduced (Jolles et al., 1992; Zubenko et al., 1999), consistent with the loss of p85α subunit, drebrin, and PSD-95 reported here.
Drebrin and PSD-95 Loss Can Cause Cognitive Deficits
Drebrin and PSD-95 are postsynaptic markers of well-known importance in synapse formation and function. They are located in dendritic spines and associated with N-methyl-d-aspartate receptors in multiprotein complexes (Husi et al., 2000). Drebrin is found in a complex with gelsolin, actin, and myosin, where it regulates actin filaments. It is critical for spine formation, and PSD-95 clustering and function are depressed by reduced drebrin expression (Shirao and Sekino, 2001; Takahashi et al., 2003). Conversely, overexpression of PSD-95 in cultured hippocampal neurons increases the number and size of dendrite spines (El-Husseini et al., 2000). Genetically induced reduction of PSD-95 also causes cognitive deficits, implying a role in synaptic plasticity, dendrite spine formation, and synaptogenesis (Migaud et al., 1998; Okabe et al., 2001). Similarly, a 40% antisense-mediated reduction in drebrin is sufficient to induce cognitive deficits (Saji et al., 2002). Consistent with these observations, cognitive deficits in Tg2576 mice bearing the familial AD gene were worsened following dietary depletion, whereas this cognitive impairment was not induced in control Tg(−) mice (Figure 6). These results support the hypothesis that the large postsynaptic deficits of drebrin and PSD-95 seen in AD brain and in Tg2576 mice on a low DHA diet may contribute to synaptic dysfunction and cognitive deficits, a finding that suggests that the importance of postsynaptic pathology in AD may have been underestimated.
Concluding Remarks
In summary, our data show that increased oxidative stress in the Tg2576 mouse model of AD is accompanied by postsynaptic caspase-mediated actin cleavage and the loss of the actin-regulating dendritic spine protein drebrin and that these processes are regulated by the levels of n-3 PFA in the brain. N-3 fatty acids, DHA in particular, mediate their protective effects at least in part by maintaining the PI3-kinase pathway (notably, p85α) to pAkt and decreasing proapoptotic BAD, consistent with in vitro data (Akbar and Kim, 2002). Because DHA is highly vulnerable to oxidation (Montine et al., 2004), the combination of low brain DHA content and APP- or Aβ-dependent lipid peroxidation may constitute a positive feedback loop leading to further synaptic DHA depletion and oxidative damage (schematic diagram, Figure 5G). This scenario generates focal caspase activation through PI3-kinase suppression, as well as actin oxidation and/or caspase cleavage of actin in the dendritic cytoskeleton. These and possibly other downstream or parallel events disrupt actin filaments, leading to the release of the actin binding protein drebrin and the impairment of normal actin dynamics in synaptic plasticity. Genetic evidence demonstrates a consistent causal link between the regulation of actin in dendritic spines and developmental cognitive deficiency (Ramakers, 2002), consistent with a significant role for the selective postsynaptic loss in the increased cognitive deficits observed here as a function of dietary DHA depletion in Tg2576 mice. The self-amplifying cycle described above could also contribute to cognitive deficits in AD, where there is decreased brain DHA (Guan et al., 1999) and PI3-kinase activity (Jolles et al., 1992; Zubenko et al., 1999), a dramatic loss of drebrin (Harigaya et al., 1996; Hatanpaa et al., 1999; Shim and Lubec, 2002) and PSD-95, along with increased oxidative stress, DHA peroxidation (Aksenov et al., 2001; Montine et al., 2004), caspase activation, and actin pathology with caspase-cleaved actin (Cole et al., 1999; Rossiter et al., 2000; Yang et al., 1998; Yuan and Yankner, 2000). Diets that are deficient in n-3 PFA, commonplace in Western societies, are a readily altered environmental risk factor for AD (Barberger-Gateau et al., 2002; Conquer et al., 2000; Grant et al., 2002; Kyle et al., 1999; Morris et al., 2003; Tully et al., 2003). Therefore, the selective vulnerability of dendritic scaffold proteins to both environmental (low DHA intake) and genetic (mutant human APP) risk factors leading to AD are likely relevant, not only to cognitive deficits in the Tg2576 mouse model, but also to AD patients.
Experimental Procedures
Material
Unless otherwise noted, reagents were obtained from Sigma (St. Louis, MO). Antibodies were purchased from the following: anti-Drebrin (clone M2F6, MBL, Nagoya, Japan), anti-PSD-95 (Upstate Biotech, Lake Placid, NY), anti-GFAP (Sigma, St. Louis, MO), anti-SNAP-25 (Sternberger Monoclonals, Lutherville, MD), anti-p85α (Stressgen, Victoria, Canada, and BD-Pharmingen, San Diego, CA), anti-neuronal nuclei (NeuN), synaptophysin (MAB368), and anti-actin (Chemicon international, Temecula, CA), anti-phosphoAkt, anti-phospho-BAD (ser136) and anti-total BAD (Cell Signaling, Beverly, MA) and anti-14-3-3 (Santa Cruz Biotechnology, Santa Cruz, CA). Affinity purified anti-fractin rabbit polyclonal antibody was developed and characterized in our laboratory (Yang et al., 1998).
Animals and Diets
Seventeen-month-old male and female Tg2576 Tg(+) and Tg(−) mice from twelve litters were randomly assigned among three treatment groups. Mice were fed for 103 ± 5 days with control diet (PMI 5015, PMI International LabDiet, St. Louis, MO), safflower oil-based diet depleted of n-3 polyunsaturated fatty acids (TD 00522, Harlan Teklad, Madison, WI), or this low DHA diet supplemented with 0.6% (w/w) DHA (Martek Bioscience, Columbia, MD) (see Table 1). The three diets were similarly supplemented in minerals and vitamins. Animals were perfused with 0.9% normal saline followed by HEPES buffer (pH 7.2) containing protease inhibitors. Brain regions were dissected from one hemisphere as previously described (Lim et al., 2001). Unless otherwise noted, biochemical measurements were performed on the residual cortex (cortex region without frontal, entorhinal, or piriform areas). A second set of similarly aged animals fed on the same diets out to 21–22 months were prepared for the behavior study.
Preparation of Tissue Samples
Tissue samples were homogenized in 10 volumes of TBS containing a cocktail of protease inhibitors [20 mg/ml each of pepstatin A, aprotinin, phosphoramidon, and leupeptin; 0.5 mM 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF); 1 mM EGTA; 5 mM fenvalerate; and 5 mM cantharidin]. Samples were sonicated briefly (2 × 10 s) and centrifuged at 100,000 × g for 20 min at 4°C to generate a TBS-soluble fraction (cytosol fraction). The TBS-insoluble pellet was sonicated in 10 volumes of lysis buffer (150 mM NaCl, 10 mM NaH2PO4, 1 mM EDTA, 1% Triton X-100, 0.5% SDS, and 0.5% deoxycholate) containing the same protease inhibitor cocktail. The resulting homogenate was centrifuged at 100,000 × g for 20 min at 4°C to produce a lysis buffer-soluble fraction (membrane fraction).
Electron Microscopy
Adult mutant mice were anesthetized with pentobarbital (60 mg/kg) and perfused with 4% para-formaldehyde (PFA) and 0.1% glutaraldehyde in PBS. After dissection, the brains were kept overnight at 4°C in 4% PFA and then kept in cold PBS. Vibratome sections (100 μm) were cryoprotected, permeabilized by freeze thawing, rinsed in PBS, and immersed for 20 min in 50 mM ammonium chloride and for 30 min in PBS with 0.1% gelatin (PBSg). Sections were incubated for 12 hr (4°C) in the anti-fractin antibody (1:100 in PBSg), and the antibody binding sites were detected using a biotinylated goat anti-rabbit antibody (1:500 in PBSg; Jackson, West Grove, PA) and avidin-biotin-HRP (Vector Laboratories. Inc., Burlingham, CA). After dehydration and osmium staining, the sections were flat embedded. Observations of ultrathin sections (pale yellow) were contrasted with uranyl acetate and lead citrate. Images were collected with a JEOL 100CXII electron microscope.
Immunoblotting
Samples (30 μg protein) were electrophoresed on 10% acrylamide gels and transferred to PVDF membranes (Immobilon, Millipore, MA) before blocking in 10% nonfat dry milk and 0.1% gelatin in PBS for 1.5 hr. Blots were immunoblotted with the appropriate primary and secondary antibody and chemiluminescence (ECL, Amersham/Pharmacia biotech, Piscataway, NJ, or Supersignal, Pierce, Rockford, IL). Band intensities were scanned and quantified with densitometric software (Molecular Analyst II). Immunoblot data were normalized to total protein load by quantification of all samples in a single assay before loading and confirmation of equal loading by image analysis of scanned Coomassie blue-stained gels after blotting.
Fatty Acid Measurement
Fatty acid analysis in frontal cortex was performed using Folch’s extraction method and gas chromatography with flame ionization detection, as previously described (Moriguchi et al., 2000). Fatty acid data in diets from Harlan were analyzed by Cornell University, Diagnostic Laboratory, Nutritional and Environmental Analytical Services (Ithaca, NY).
Protein Oxidation
Amounts of oxidized proteins containing carbonyl groups were measured in the membrane fraction of cortex samples using an Oxyblot kit (Intergen, Purchase, NY) as previously described (Lim et al., 2001).
Immunohistochemistry
Hemi-brains were fixed with 4% PFA (4°C overnight), cryoprotected with 10% and 20% sucrose-PBS, snap frozen at −70°C, and cryostat sectioned into coronal (12 μm) sections. For fractin/drebrin double labeling and NeuN labeling, slides were soaked in 75% ethanol (2 min at room temperature) and dH2O resin. Sections were processed in antigen unmasking buffer (Vector Labs, Burlingame, CA), steamed 15 min, and rinsed in 0.3%Triton X-100 in Tris-buffered saline (10 min at room temperature). Incubation with fractin (1:20) and drebrin (1:30) (or NeuN antibody [1:2000]) was performed in 0.1% Tween-20 in 3% BSA Tris-buffered saline for 1 hr at 37°C. Secondary antibodies (1:1000, 1 hr incubation at room temperature) were goat anti-rabbit-FITC and goat anti-mouse-Rhodamine (Molecular Probes, OR). For phospho-BAD, sections were exposed to three successive 3 min acetone (50%–100%–50%) washes, rinsed with 5 μg/ml Proteinase K (5 min at room temperature), 0.3% Triton X-100 (5 min at room temperature), and H2O2 in methanol (5 min at room temperature). Sections were blocked with 5% normal goat serum for 30 min at 37°C and incubated with phospho-BAD antibody (1:15) for 60 min at 37°C. After incubation with secondary antibody and ABC reagent (Vector Labs, CA), sections were developed using metal enhanced DAB kits (Pierce, Rockford, IL). Image analysis on sections (at Bregma −1.5, −2, and −3) was performed with an Optronix Engineering LX-450A CCD video system using NIH Image software. At sacrifice, there was no treatment-dependent change in brain weights, nor in Bregma-matched brain region areas, indicating the absence of major treatment-related shrinkage of brain regions. For NeuN, quantitative analysis for neuronal density was performed in all cortical layers (entorhinal II, entorhinal III/IV, parietal II, parietal III/IV, parietal V/VI, frontal II, frontal III/IV, frontal V/VI) and hippocampal pyramidal cell layers (CA1, CA2, CA3). We measured neuronal nuclei density by image analysis in these neuronal layers in four consecutive sections for each of four different Bregmas (1.0 mm, 1.7mm, −2.7 mm, and −3 mm).
Handling of Human Tissue
Postmortem tissue from temporal lobe was obtained from the USC AD Center and the UCLA AD Research Center. Nine controls were compared to ten AD patients with moderate disease. Postmortem interval, age of death, and gender were comparable between both groups. The tissue was processed as for mouse tissue except that the concentration of SDS in the lysis buffer was raised to 2% and Triton X-100 and deoxycholate were removed.
Quantitative Real-Time RT-PCR of p85α PI3-K mRNA
Total RNA was isolated from brain using the RNAqueous kit (Ambion) as per the manufacturer’s instructions and was treated with DNase. RNA (0.7 μg) was reverse transcribed using dT primers using the Retroscript kit (Ambion) and was then aliquoted. Analysis of RNA levels was performed using QPCR with the SYBR Green PCR Master Mix (Applied Biosystems) and an SDS7700 (Applied Biosystems), followed by dissociation curve analysis that verified the amplification of a single PCR species. Thermocycling parameters were standard for the SDS7700 (Applied Biosystems) instrument: 95°C for 10 min, and 40 cycles of 95°C for 15 s and 60°C for 1 min. Each sample was tested in triplicate. A standard curve was run to allow relative comparisons of sample values. The standard curve was made using cDNA (using dT primers) from brain tissue RNA reverse transcribed at two times the concentration of RNA (1.4 μg) compared to that used for reverse transcription of the sample RNA; standard curves typically had an r2 > 0.99. The primers for p85α were designed using Primer Express software (Applied Biosystems) using the mouse p85α sequence NCBI# U50413. The primer sequences were forward, ACCTGTGAACTGAGCTGCAGAA; reverse, TAGAAACGTCTGGTCATCCAACA. GAPDH mRNA levels were also measured for each cDNA sample as an internal housekeeping gene control using the primers from the TaqMan Rodent GAPDH Control Reagents (Applied Biosystems). Levels of p85α mRNA were normalized to levels of GAPDH mRNA for each sample.
Behavior
We followed Morris water maze protocols based on extensive characterization of age-dependent cognitive deficits in an APP mouse (Westerman et al., 2002). Mice were trained for six blocks (four consecutive swim trials per block, two blocks per day, one block a day in aged animals) to find a visible platform in our mouse pool (5 foot diameter swimming pool), maintained at 26°C to prevent hypothermia. There is a video camera on the ceiling, attached to a laptop with HVS image tracking capabilities, so calculation of mouse swim paths and latencies was streamlined and facilitated. Besides training mice, the visible testing phase was used to exclude mice that have visual or motor deficits (Westerman et al., 2002). If mice did not find the platform within 60 s, they were led to the platform with a fish net. If mice sank, they were picked up and prevented from drowning. After the sixth block, mice started the hidden platform trials, which consisted of 12 blocks of training. Probe trials for retention deficits (60 s swim without platform to determine percent path in target quadrant) were then performed. During swims, the staff handling the mice moved out of sight of the mice so that only distal stationary cues were present. Learning was assessed by significant regressions of block on latency, and despite weekend breaks, the number of blocks of testing was limited by excessive fatigue and their imminent mortality. At the end of each block (four consecutive swim trials), mice were towel dried and then placed back in their cages.
Statistical Analysis
Statistical comparisons of data were performed using an ANOVA followed by post hoc pairwise comparisons with Fisher’s probability of least significant difference test (PLSD). Square root transformation to establish homogeneity of variance was used for p85 data in mice. Coefficients of correlation and significance of the degree of linear relationship between parameters were determined with a simple regression model. For visible and hidden platform tests, swim latencies, paths, and swim speeds were analyzed. Regressions of latencies on blocks were calculated to determine whether learning was occurring. 2 × 2 ANOVA (Combined Group Blocks × treatment) and repeated measures ANOVA were performed to determine treatment and transgene differences in latencies. Percentage path in target and opposite quadrants in probe trial was also assessed using 2 × 2 ANOVA (transgene × treatment).
Acknowledgments
We thank the patients and families who generously donated brain tissue samples for this research via the Neuropathology and Molecular Genetics Core of the UCLA ADRC (H. Vinters, P50 AG 16570) and the USC neuropathology ADRC core (Dr. C.A. Miller [P50 AG05142]). We also thank Ping Ping Chen, Peter Kim, and Mychica Simmons for technical expertise. This work was supported by a VA Merit (G.M.C.) and NIH grants RO1 AG13741, NS43946 (G.M.C.), AG10685, and AG16793 (S.A.F.), UCLA ADRC (PO1 AG16570, G.M.C. and S.A.F.), and a Senior Research Fellowship from the Canadian Institutes of Health Research (F.C.).
References
- Akbar M, Kim HY. Protective effects of docosahexaenoic acid in staurosporine-induced apoptosis: involvement of phosphatidylinositol-3 kinase pathway. J Neurochem. 2002;82:655–665. doi: 10.1046/j.1471-4159.2002.01015.x. [DOI] [PubMed] [Google Scholar]
- Aksenov MY, Aksenova MV, Butterfield DA, Geddes JW, Markesbery WR. Protein oxidation in the brain in Alzheimer’s disease. Neuroscience. 2001;103:373–383. doi: 10.1016/s0306-4522(00)00580-7. [DOI] [PubMed] [Google Scholar]
- Attwell D, Iadecola C. The neural basis of functional brain imaging signals. Trends Neurosci. 2002;25:621–625. doi: 10.1016/s0166-2236(02)02264-6. [DOI] [PubMed] [Google Scholar]
- Barberger-Gateau P, Letenneur L, Deschamps V, Peres K, Dartigues JF, Renaud S. Fish, meat, and risk of dementia: cohort study. BMJ. 2002;325:932–933. doi: 10.1136/bmj.325.7370.932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazan NG. Synaptic lipid signaling: significance of polyunsaturated fatty acids and platelet-activating factor. J Lipid Res. 2003;44:2221–33. doi: 10.1194/jlr.R300013-JLR200. [DOI] [PubMed] [Google Scholar]
- Behl C, Davis JB, Lesley R, Schubert D. Hydrogen peroxide mediates amyloid beta protein toxicity. Cell. 1994;77:817–827. doi: 10.1016/0092-8674(94)90131-7. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–945. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- Breckenridge WC, Gombos G, Morgan IG. The lipid composition of adult rat brain synaptosomal plasma membranes. Biochim Biophys Acta. 1972;266:695–707. doi: 10.1016/0006-3002(72)90012-1. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Castegna A, Lauderback CM, Drake J. Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer’s disease brain contribute to neuronal death. Neurobiol Aging. 2002;23:655–664. doi: 10.1016/s0197-4580(01)00340-2. [DOI] [PubMed] [Google Scholar]
- Callahan LM, Vaules WA, Coleman PD. Quantitative decrease in synaptophysin message expression and increase in cathepsin D message expression in Alzheimer disease neurons containing neurofibrillary tangles. J Neuropathol Exp Neurol. 1999;58:275–287. doi: 10.1097/00005072-199903000-00007. [DOI] [PubMed] [Google Scholar]
- Cole, G.M., Yang, F., Chen, P.P., Frautschy, S., and Hsiao, K. (1999). Caspase activation in dystrophic neurites in Alzheimer’s disease and aged HuAPPsw transgenic mice. In Alzheimer’s Disease and Related Disorders, K. Iqbal, D.F. Swaab, B. Winblad, and H.M. Wisniewski, eds. (Chichester, UK: Wiley), pp. 363–369.
- Conquer JA, Tierney MC, Zecevic J, Bettger WJ, Fisher RH. Fatty acid analysis of blood plasma of patients with Alzheimer’s disease, other types of dementia, and cognitive impairment. Lipids. 2000;35:1305–1312. doi: 10.1007/s11745-000-0646-3. [DOI] [PubMed] [Google Scholar]
- Dalle-Donne I, Rossi R, Giustarini D, Gagliano N, Di Simplicio P, Colombo R, Milzani A. Methionine oxidation as a major cause of the functional impairment of oxidized actin. Free Radic Biol Med. 2002;32:927–937. doi: 10.1016/s0891-5849(02)00799-2. [DOI] [PubMed] [Google Scholar]
- Datta SR, Katsov A, Hu L, Petros A, Fesik SW, Yaffe MB, Greenberg ME. 14-3-3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Mol Cell. 2000;6:41–51. [PubMed] [Google Scholar]
- Dickey CA, Loring JF, Montgomery J, Gordon MN, Eastman PS, Morgan D. Selectively reduced expression of synaptic plasticity-related genes in amyloid precursor protein + presenilin-1 transgenic mice. J Neurosci. 2003;23:5219–5226. doi: 10.1523/JNEUROSCI.23-12-05219.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dineley KT, Xia X, Bui D, Sweatt JD, Zheng H. Accelerated plaque accumulation, associative learning deficits, and up-regulation of alpha 7 nicotinic receptor protein in transgenic mice co-expressing mutant human presenilin 1 and amyloid precursor proteins. J Biol Chem. 2002;277:22768–22780. doi: 10.1074/jbc.M200164200. [DOI] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS. PSD-95 involvement in maturation of excitatory synapses. Science. 2000;290:1364–1368. [PubMed] [Google Scholar]
- Etminan M, Gill S, Samii A. Effect of non-steroidal anti-inflammatory drugs on risk of Alzheimer’s disease: systematic review and meta-analysis of observational studies. BMJ. 2003;327:128. doi: 10.1136/bmj.327.7407.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everall IP, DeTeresa R, Terry R, Masliah E. Comparison of two quantitative methods for the evaluation of neuronal number in the frontal cortex in Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:1202–1206. doi: 10.1097/00005072-199711000-00004. [DOI] [PubMed] [Google Scholar]
- Favrelere S, Stadelmann-Ingrand S, Huguet F, De Javel D, Piriou A, Tallineau C, Durand G. Age-related changes in ethanolamine glycerophospholipid fatty acid levels in rat frontal cortex and hippocampus. Neurobiol Aging. 2000;21:653–660. doi: 10.1016/s0197-4580(00)00170-6. [DOI] [PubMed] [Google Scholar]
- Fukazawa Y, Saitoh Y, Ozawa F, Ohta Y, Mizuno K, Inokuchi K. Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron. 2003;38:447–460. doi: 10.1016/s0896-6273(03)00206-x. [DOI] [PubMed] [Google Scholar]
- Gamblin TC, King ME, Kuret J, Berry RW, Binder LI. Oxidative regulation of fatty acid-induced tau polymerization. Biochemistry. 2000;39:14203–14210. doi: 10.1021/bi001876l. [DOI] [PubMed] [Google Scholar]
- Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, Krafft GA, Klein WL. Alzheimer’s disease-affected brain: Presence of oligomeric A{beta} ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci USA. 2003;100:10417–10422. doi: 10.1073/pnas.1834302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant WB, Campbell A, Itzhaki RF, Savory J. The significance of environmental factors in the etiology of Alzheimer’s disease. J Alzheimers Dis. 2002;4:179–189. doi: 10.3233/jad-2002-4308. [DOI] [PubMed] [Google Scholar]
- Guan Z, Wang Y, Cairns NJ, Lantos PL, Dallner G, Sindelar PJ. Decrease and structural modifications of phosphatidylethanolamine plasmalogen in the brain with Alzheimer disease. J Neuropathol Exp Neurol. 1999;58:740–747. doi: 10.1097/00005072-199907000-00008. [DOI] [PubMed] [Google Scholar]
- Harigaya Y, Shoji M, Shirao T, Hirai S. Disappearance of actin-binding protein, drebrin, from hippocampal synapses in Alzheimer’s disease. J Neurosci Res. 1996;43:87–92. doi: 10.1002/jnr.490430111. [DOI] [PubMed] [Google Scholar]
- Hashimoto M, Hossain S, Shimada T, Sugioka K, Yamasaki H, Fujii Y, Ishibashi Y, Oka J, Shido O. Docosahexaenoic acid provides protection from impairment of learning ability in Alzheimer’s disease model rats. J Neurochem. 2002;81:1084–1091. doi: 10.1046/j.1471-4159.2002.00905.x. [DOI] [PubMed] [Google Scholar]
- Hatanpaa K, Isaacs KR, Shirao T, Brady DR, Rapoport SI. Loss of proteins regulating synaptic plasticity in normal aging of the human brain and in Alzheimer disease. J Neuropathol Exp Neurol. 1999;58:637–643. doi: 10.1097/00005072-199906000-00008. [DOI] [PubMed] [Google Scholar]
- Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, et al. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Husi H, Ward MA, Choudhary JS, Blackstock WP, Grant SG. Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat Neurosci. 2000;3:661–669. doi: 10.1038/76615. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT. APPSw transgenic mice develop age-related A beta deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol. 1997;56:965–973. doi: 10.1097/00005072-199709000-00002. [DOI] [PubMed] [Google Scholar]
- Jolles J, Bothmer J, Markerink M, Ravid R. Phosphatidylinositol kinase is reduced in Alzheimer’s disease. J. Neurochem. 1992;58:2326–2329. doi: 10.1111/j.1471-4159.1992.tb10981.x. [DOI] [PubMed] [Google Scholar]
- Joseph JA, Denisova NA, Arendash G, Gordon M, Diamond D, Shukitt-Hale B, Morgan D. Blueberry supplementation enhances signaling and prevents behavioral deficits in an Alzheimer disease model. Nutr Neurosci. 2003;6:153–162. doi: 10.1080/1028415031000111282. [DOI] [PubMed] [Google Scholar]
- Kihara T, Shimohama S, Sawada H, Honda K, Nakamizo T, Shibasaki H, Kume T, Akaike A. alpha 7 nicotinic receptor transduces signals to phosphatidylinositol 3-kinase to block A beta-amyloid-induced neurotoxicity. J Biol Chem. 2001;276:13541–13546. doi: 10.1074/jbc.M008035200. [DOI] [PubMed] [Google Scholar]
- King DL, Arendash GW. Maintained synaptophysin immunoreactivity in Tg2576 transgenic mice during aging: correlations with cognitive impairment. Brain Res. 2002;926:58–68. doi: 10.1016/s0006-8993(01)03294-2. [DOI] [PubMed] [Google Scholar]
- Krishnan S, Chi EY, Wood SJ, Kendrick BS, Li C, Garzon-Rodriguez W, Wypych J, Randolph TW, Narhi LO, Biere AL, et al. Oxidative dimer formation is the critical rate-limiting step for Parkinson’s disease alpha-synuclein fibrillogenesis. Biochemistry. 2003;42:829–837. doi: 10.1021/bi026528t. [DOI] [PubMed] [Google Scholar]
- Kyle DJ, Schaefer E, Patton G, Beiser A. Low serum docosahexaenoic acid is a significant risk factor for Alzheimer’s dementia. Lipids. 1999;34:S245. doi: 10.1007/BF02562306. [DOI] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim GP, Chu T, Yang F, Beech W, Frautschy SA, Cole GM. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J Neurosci. 2001;21:8370–8377. doi: 10.1523/JNEUROSCI.21-21-08370.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liolitsa D, Powell J, Lovestone S. Genetic variability in the insulin signalling pathway may contribute to the risk of late onset Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2002;73:261–266. doi: 10.1136/jnnp.73.3.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Duan W. Apoptotic biochemical cascades in synaptic compartments: roles in adaptive plasticity and neurodegenerative disorders. J Neurosci Res. 1999;58:152–166. [PubMed] [Google Scholar]
- Matus A. Actin-based plasticity in dendritic spines. Science. 2000;290:754–758. doi: 10.1126/science.290.5492.754. [DOI] [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, et al. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature. 1998;396:433–439. doi: 10.1038/24790. [DOI] [PubMed] [Google Scholar]
- Montine KS, Quinn JF, Zhang J, Fessel JP, Roberts LJ, 2nd, Morrow JD, Montine TJ. Isoprostanes and related products of lipid peroxidation in neurodegenerative diseases. Chem Phys Lipids. 2004;128:117–124. doi: 10.1016/j.chemphyslip.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Moriguchi T, Greiner RS, Salem N., Jr Behavioral deficits associated with dietary induction of decreased brain docosahexaenoic acid concentration. J Neurochem. 2000;75:2563–2573. doi: 10.1046/j.1471-4159.2000.0752563.x. [DOI] [PubMed] [Google Scholar]
- Morris MC, Evans DA, Bienias JL, Tangney CC, Bennett DA, Wilson RS, Aggarwal N, Schneider J. Consumption of fish and n-3 fatty acids and risk of incident Alzheimer disease. Arch Neurol. 2003;60:940–946. doi: 10.1001/archneur.60.7.940. [DOI] [PubMed] [Google Scholar]
- Nagele RG, D’Andrea MR, Anderson WJ, Wang HY. Intracellular accumulation of beta-amyloid(1–42) in neurons is facilitated by the alpha 7 nicotinic acetylcholine receptor in Alzheimer’s disease. Neuroscience. 2002;110:199–211. doi: 10.1016/s0306-4522(01)00460-2. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s disease with plaques and tangles. Intracellular abeta and synaptic dysfunction Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Okabe S, Miwa A, Okado H. Spine formation and correlated assembly of presynaptic and postsynaptic molecules. J Neurosci. 2001;21:6105–6114. doi: 10.1523/JNEUROSCI.21-16-06105.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opazo P, Watabe AM, Grant SG, O’Dell TJ. Phosphatidylinositol 3-kinase regulates the induction of long-term potentiation through extracellular signal-related kinase-independent mechanisms. J Neurosci. 2003;23:3679–3688. doi: 10.1523/JNEUROSCI.23-09-03679.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratico D, Uryu K, Leight S, Trojanoswki JQ, Lee VM. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J Neurosci. 2001;21:4183–4187. doi: 10.1523/JNEUROSCI.21-12-04183.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakers GJ. Rho proteins, mental retardation and the cellular basis of cognition. Trends Neurosci. 2002;25:191–199. doi: 10.1016/s0166-2236(00)02118-4. [DOI] [PubMed] [Google Scholar]
- Rossiter JP, Anderson LL, Yang F, Cole GM. Caspase-cleaved actin (fractin) immunolabelling of Hirano bodies. Neuropathol Appl Neurobiol. 2000;26:342–346. doi: 10.1046/j.1365-2990.2000.00252.x. [DOI] [PubMed] [Google Scholar]
- Saji, M., Akiba, L., Tanaka, S., Suzuki, N., Sekino, Y., and Shirao, T. (2002). Impaired spatial learning and intact adaptation in rats with antisense hippocampal knockdown of drebrin A. Paper presented at: Society for Neuroscience meeting.
- Salem N, Jr, Litman B, Kim HY, Gawrisch K. Mechanisms of action of docosahexaenoic acid in the nervous system. Lipids. 2001;36:945–959. doi: 10.1007/s11745-001-0805-6. [DOI] [PubMed] [Google Scholar]
- Schurmann A, Mooney AF, Sanders LC, Sells MA, Wang HG, Reed JC, Bokoch GM. p21-activated kinase 1 phosphorylates the death agonist bad and protects cells from apoptosis. Mol Cell Biol. 2000;20:453–461. doi: 10.1128/mcb.20.2.453-461.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Shaw S, Bencherif M, Marrero MB. Janus kinase 2, an early target of alpha 7 nicotinic acetylcholine receptor-mediated neuroprotection against Abeta-(1–42) amyloid. J Biol Chem. 2002;277:44920–44924. doi: 10.1074/jbc.M204610200. [DOI] [PubMed] [Google Scholar]
- Shim KS, Lubec G. Drebrin, a dendritic spine protein, is manifold decreased in brains of patients with Alzheimer’s disease and Down syndrome. Neurosci Lett. 2002;324:209–212. doi: 10.1016/s0304-3940(02)00210-0. [DOI] [PubMed] [Google Scholar]
- Shirao T, Sekino Y. Clustering and anchoring mechanisms of molecular constituents of postsynaptic scaffolds in dendritic spines. Neurosci Res. 2001;40:1–7. doi: 10.1016/s0168-0102(01)00209-7. [DOI] [PubMed] [Google Scholar]
- Shirazi SK, Wood JG. The protein tyrosine kinase, fyn, in Alzheimer’s disease pathology. Neuroreport. 1993;4:435–437. doi: 10.1097/00001756-199304000-00024. [DOI] [PubMed] [Google Scholar]
- Shoop RD, Yamada N, Berg DK. Cytoskeletal links of neuronal acetylcholine receptors containing alpha 7 subunits. J Neurosci. 2000;20:4021–4029. doi: 10.1523/JNEUROSCI.20-11-04021.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Star EN, Kwiatkowski DJ, Murthy VN. Rapid turnover of actin in dendritic spines and its regulation by activity. Nat Neurosci. 2002;5:239–246. doi: 10.1038/nn811. [DOI] [PubMed] [Google Scholar]
- Stein TD, Johnson JA. Lack of neurodegeneration in transgenic mice overexpressing mutant amyloid precursor protein is associated with increased levels of transthyretin and the activation of cell survival pathways. J Neurosci. 2002;22:7380–7388. doi: 10.1523/JNEUROSCI.22-17-07380.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh YH, Checler F. Amyloid precursor protein, presenilins, and alpha-synuclein: molecular pathogenesis and pharmacological applications in Alzheimer’s disease. Pharmacol Rev. 2002;54:469–525. doi: 10.1124/pr.54.3.469. [DOI] [PubMed] [Google Scholar]
- Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002;161:1869–1879. doi: 10.1016/s0002-9440(10)64463-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Sekino Y, Tanaka S, Mizui T, Kishi S, Shirao T. Drebrin-dependent actin clustering in dendritic filopodia governs synaptic targeting of postsynaptic density-95 and dendritic spine morphogenesis. J Neurosci. 2003;23:6586–6595. doi: 10.1523/JNEUROSCI.23-16-06586.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima A, Noguchi K, Michel G, Mercken M, Hoshi M, Ishiguro K, Imahori K. Exposure of rat hippocampal neurons to amyloid beta peptide (25–35) induces the inactivation of phosphatidyl inositol-3 kinase and the activation of tau protein kinase I/glycogen synthase kinase-3 beta. Neurosci Lett. 1996;203:33–36. doi: 10.1016/0304-3940(95)12257-5. [DOI] [PubMed] [Google Scholar]
- Taouis M, Dagou C, Ster C, Durand G, Pinault M, Delarue J. N-3 polyunsaturated fatty acids prevent the defect of insulin receptor signaling in muscle. Am J Physiol Endocrinol Metab. 2002;282:E664–E671. doi: 10.1152/ajpendo.00320.2001. [DOI] [PubMed] [Google Scholar]
- Taubes G. Neuroscience. Insulin insults may spur Alzheimer’s disease. Science. 2003;301:40–41. doi: 10.1126/science.301.5629.40. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Tully AM, Roche HM, Doyle R, Fallon C, Bruce I, Lawlor B, Coakley D, Gibney MJ. Low serum cholesteryl ester-docosahexaenoic acid levels in Alzheimer’s disease: a case-control study. Br J Nutr. 2003;89:483–490. doi: 10.1079/BJN2002804. [DOI] [PubMed] [Google Scholar]
- Westerman MA, Cooper-Blacketer D, Mariash A, Kotilinek L, Kawarabayashi T, Younkin LH, Carlson GA, Younkin SG, Ashe KH. The relationship between Abeta and memory in the Tg2576 mouse model of Alzheimer’s disease. J Neurosci. 2002;22:1858–1867. doi: 10.1523/JNEUROSCI.22-05-01858.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Sun X, Beech W, Teter B, Wu S, Sigel J, Vinters HV, Frautschy SA, Cole GM. Antibody to caspase-cleaved actin detects apoptosis in differentiated neuroblastoma and plaque-associated neurons and microglia in Alzheimer’s disease. Am J Pathol. 1998;152:379–389. [PMC free article] [PubMed] [Google Scholar]
- Yavin E, Brand A, Green P. Docosahexaenoic acid abundance in the brain: a biodevice to combat oxidative stress. Nutr Neurosci. 2002;5:149–157. doi: 10.1080/10284150290003159. [DOI] [PubMed] [Google Scholar]
- Yehuda S, Rabinovitz S, Carasso RL, Mostofsky DI. The role of polyunsaturated fatty acids in restoring the aging neuronal membrane. Neurobiol Aging. 2002;23:843–853. doi: 10.1016/s0197-4580(02)00074-x. [DOI] [PubMed] [Google Scholar]
- Youdim KA, Martin A, Joseph JA. Essential fatty acids and the brain: possible health implications. Int J Dev Neurosci. 2000;18:383–399. doi: 10.1016/s0736-5748(00)00013-7. [DOI] [PubMed] [Google Scholar]
- Yuan J, Yankner BA. Apoptosis in the nervous system. Nature. 2000;407:802–809. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- Zubenko GS, Stiffler JS, Hughes HB, Martinez AJ. Reductions in brain phosphatidylinositol kinase activities in Alzheimer’s disease. Biol Psychiatry. 1999;45:731–736. doi: 10.1016/s0006-3223(98)00073-0. [DOI] [PubMed] [Google Scholar]