

Abstract
We have compared Ca-dependent exocytosis in excised giant membrane patches and in whole-cell patch clamp with emphasis on the rat secretory cell line, RBL. Stable patches of 2–4 pF are easily excised from RBL cells after partially disrupting actin cytoskeleton with latrunculin A. Membrane fusion is triggered by switching the patch to a cytoplasmic solution containing 100–200 μM free Ca. Capacitance and amperometric recording show that large secretory granules (SGs) containing serotonin are mostly lost from patches. Small vesicles that are retained (non-SGs) do not release serotonin or other substances detected by amperometry, although their fusion is reduced by tetanus toxin light chain. Non-SG fusion is unaffected by N-ethylmaleimide, phosphatidylinositol-4,5-bis-phosphate (PI(4,5)P2) ligands, such as neomycin, a PI-transfer protein that can remove PI from membranes, the PI(3)-kinase inhibitor LY294002 and PI(4,5)P2, PI(3)P, and PI(4)P antibodies. In patch recordings, but not whole-cell recordings, fusion can be strongly reduced by ATP removal and by the nonspecific PI-kinase inhibitors wortmannin and adenosine. In whole-cell recording, non-SG fusion is strongly reduced by osmotically induced cell swelling, and subsequent recovery after shrinkage is then inhibited by wortmannin. Thus, membrane stretch that occurs during patch formation may be a major cause of differences between excised patch and whole-cell fusion responses. Regarding Ca sensors for non-SG fusion, fusion remains robust in synaptotagmin (Syt) VII−/− mouse embryonic fibroblasts (MEFs), as well as in PLCδ1, PLC δ1/δ4, and PLCγ1−/− MEFs. Thus, Syt VII and several PLCs are not required. Furthermore, the Ca dependence of non-SG fusion reflects a lower Ca affinity (KD ∼71 μM) than expected for these C2 domain–containing proteins. In summary, we find that non-SG membrane fusion behaves and is regulated substantially differently from SG fusion, and we have identified an ATP-dependent process that restores non-SG fusion capability after it is perturbed by membrane stretch or cell dilation.
INTRODUCTION
Mast cells of hematopoietic origin play a central role in inflammatory responses by releasing numerous substances that modulate immune responses (Metcalfe et al., 1997). Fusion of large secretory granules (SGs) to the plasma membrane underlies degranulation of mast cells, and much experimental effort has focused on the regulation of this exocytotic process (Burgoyne and Morgan, 2003; Sagi-Eisenberg, 2007). In addition to exocytosis of SGs, another type of fusion that does not result in clear stepwise changes of membrane capacitance (non-SG) was reported in rat peritoneal mast cells twenty years ago (Almers and Neher, 1987). While the step size of non-SG events is not readily resolved by capacitance recording, the increase of cell capacitance mediated by such fusion events can be of very large magnitude (Almers and Neher, 1987). The sources of membrane involved in non-SG fusion as well as the underlying fusion mechanisms remain rather enigmatic. Data from chromaffin cells indicate that non-SG fusion requires relatively high Ca (∼100 μM) and is ATP dependent, but neurotoxin insensitive. Non-SGs in chromaffin cells are not likely to represent acetylcholine-containing synaptic-like microvesicles (Xu et al., 1998), and it is striking that non-SG fusion can also be massive in CHO and 3T3 cells (Coorssen et al., 1996), as well as BHK and HEK293 cells (Yaradanakul et al., 2008), and as described in some detail in this article, in RBL, MEF, and INS-1 cells. The prevalence of large-scale Ca-activated non-SG fusion processes in both secretory and nonsecretory cell lines suggests that the non-SG fusion may be important for cell survival. Since the non-SG pool can exceed 50% of the total surface membrane area and the requirements for cytoplasmic Ca are rather high (Yaradanakul et al., 2008), it seems reasonable that this membrane pool is involved in wound repair of the plasma membrane.
Present understanding of membrane fusion relies strongly on studies of transmitter/hormone release from neurons (Sakaba et al., 2005; Bronk et al., 2007) and endocrine cells (Burgoyne and Morgan, 2003), and the homotypic fusion of yeast vacuoles (Ostrowicz et al., 2008). In these cases, the SNARE (soluble N-ethylmaleimide–sensitive factor attachment protein receptor) proteins are clearly implicated to initiate fusion, and in general are thought to do so by associating and perturbing the two membranes involved. As introduced in an accompanying article (Yaradanakul et al., 2008), phosphoinositides and their derivatives are presently thought to importantly modify SNARE-dependent fusion processes (De Matteis and Godi, 2004). Evidence from PC12 cells suggested that formation of phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) microdomains at syntaxin clusters can activate the exocytotic sites (Aoyagi et al., 2005). PI(4,5)P2 appears to be required for “priming” of vesicles in pancreatic β cells (Olsen et al., 2003) and the yeast vacuole fusion process (Mayer et al., 2000), and it regulates the releasable vesicle pool size in chromaffin cells (Milosevic et al., 2005). In dense core vesicle fusion, PI(4,5)P2 acts via the calcium-dependent activator protein for secretion (CAPS) to regulate vesicle recruitment and increase the initial rate of fusion (Loyet et al., 1998; Grishanin et al., 2004; Jockusch et al., 2007; Speidel et al., 2008). Furthermore, PI(4,5)P2 is suggested in a liposome–liposome fusion system to promote fusion directly via its interaction with the neuronal Ca sensor, synaptotagmin (Syt) I (Bai et al., 2004). Finally, PLCs, which cleave PI(4,5)P2 to produce diacylglycerol (DAG) and inositol triphosphate (IP3), are implicated to regulate some vesicle fusion processes (Fukami et al., 2001; Jun et al., 2004), and, in general, the conversion of large phospholipid head groups to smaller ones is expected to favor fusion of phospholipid vesicles. Other phosphoinositides, such as phosphatidylinositol 3,4,5-triphosphate (PIP3) and phosphatidylinositol 3-phosphate (PI(3)P) also play important roles in membrane trafficking, including events leading up to fusion (Lindmo and Stenmark, 2006). For example, inhibition of a class IA PI(3) kinase (PI(3)K), which produces PIP3, reduces receptor-mediated degranulation in mast cells (Ali et al., 2004). PI(3)K-C2α, which produces mainly PI(3)P, is also required for the ATP-dependent priming of SGs in neurosecretory cells (Meunier et al., 2005).
From the various methods used to monitor membrane fusion, membrane capacitance measurements give the highest signal and temporal resolution, and the development of improved excised patch models to analyze and manipulate fusion would have important experimental advantages of high resolution recording with free access to the cytoplasmic membrane side. Efforts to date have used chromaffin cells (Dernick et al., 2003) and the insulin-secreting INS-1 cells (MacDonald et al., 2005). In the present study, we describe the use of giant excised membrane patches (pipette diameter ∼10–15 μm, patch size ∼2–4 pF) to attempt to preserve more fusion-capable vesicles in a configuration that allows free access to the cytoplasmic side. In short, we have found that partial disruption of the actin cytoskeleton facilitates the formation of stable patches from some cell types and preserves more fusion-capable vesicles in patches from all cell types tested. Nevertheless, our experience is that SGs are easily lost from excised patches, presumably because they are not docked in a stable fashion at the plasma membrane in RBL cells. The non-SG fusion is much more robust, and we describe here several fundamental characteristics of this type of fusion, including its dependence on ATP and Ca. The results define clear differences between non-SG and SG fusion processes and provide new insights into the physical basis of non-SG fusion.
MATERIALS AND METHODS
Cell Culture
Adherent RBL-2H3 cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Mediatech) supplemented with 15% FBS. Cells were plated on uncoated Petri dishes 1–2 d before experiments and collected by treating them with 0.25% trypsin/EDTA solution. Serotonin and 5-hydroxytryptophan (0.2 mM each) were also added to cells for amperometric recording 1 d before the experiments to increase the formation of SGs (Williams et al., 1999; Mahmoud and Fewtrell, 2001). After treatment with trypsin, the cells were resuspended in the culture medium and left in the CO2 incubator for 30 min. Cells were then treated with latrunculin A (50–100 ng/ml) at 37°C for 5 min before experiments, as this treatment clearly facilitated the formation of giant excised patches with fusion-competent vesicles. Mouse embryonic fibroblasts (MEFs) were cultured in DMEM supplemented with 10% FBS and penicillin-streptomycin. They were plated on cell culture dish 1–2 d before experiment and collected as stated above. The Syt VII–deficient MEF cell line was provided by T. Südhof (University of Texas Southwestern [UTSW] Medical Center at Dallas), the PLCδ1- and PLCδ1/δ4-deficient MEF cell lines were provided by K. Fukami (Tokyo University, Tokyo, Japan), and the PLCγ1-deficient cell line was provided by G. Carpenter (Vanderbilt University, Nashville, TN)
Solutions
Unless otherwise stated, the patches were excised into a solution containing (in mM) 140 NaCl, 1 MgCl2, 0.3 EGTA, 20 HEPES, pH 7.3. ATP and GTP were added at final concentration of 2.4 and 0.3, respectively. Membrane fusion was triggered by the same solution with 0.5 CaCl2 (i.e., 0.2 mM free Ca), usually without ATP and GTP. For whole-cell recording with RBL cells, both the cytoplasmic and extracellular solutions contained (in mM) 40 NaCl, 90 N-methyl-d-glucamine (NMG), 1 MgCl2, 0.01 EGTA, 10 HEPES, pH 7.3 adjusted with MES. Relatively large-diameter pipette tips (4–6 μm i.d.) were employed in whole-cell recording to allow fast exchange of the cytoplasm via pipette perfusion. Using this low conductance solution, the cell time constants (30–60 μs) were large enough to use square wave perturbation for capacitance measurements, as described subsequently. A solution with 0.2 mM free Ca, highly buffered with nitrilotriacetic acid, was infused into the cell to induce membrane fusion. The complete composition was (in mM) 15 NaCl, 90 NMG, 3 MgCl2, 5 CaCl2, 10 nitrilotriacetic acid, 10 HEPES, pH 7.3 (adjusted with MES). All free Ca values given in this article were calculated with WEBMAXC (http://www.stanford.edu/∼cpatton/maxc.html) (Patton et al., 2004). Other solutions employed were FVPP solution (Huang et al., 1998), a phosphatase inhibitor cocktail (110 NaCl, 5 NaF, 0.1 Na3VO4, 2 EDTA, 10 Na4P2O7, 20 HEPES), EDTA buffer solution (140 NaCl, 2 EDTA, 20 HEPES), and protein dialysis solution (140 NaCl, 0.3 EGTA, 0.3 ZnSO4, 20 HEPES, 2 β-mercaptoethanol). For whole-cell experiments presented in Fig. 7, the standard solutions described previously (Yarandanakul et al., 2008) were employed. For Fig. 8, results in panel A employed the standard solution with 70 mM LiOH substituted for NMG. The cytoplasmic solution was modified by addition of 200 mM sucrose and dilution by 30% to generate the hyper- and hypoosmotic solutions, respectively. In Fig. 8 B, the standard solutions were employed with NMG (aspartate) reduced by 80 mM to generate the hypoosmotic extracellular solution. The control solution was generated by adding 160 mM sucrose to this solution. In Fig. 8 C, the solution given above for RBL cells was employed. Hyperosmotic cytoplasmic solution was generated by addition of 200 mM sucrose, and hypoosmotic extracellular solution was generated by deletion of 80 mM NMG.
Figure 7.

Non-SG fusion in whole-cell recording is wortmannin/adenosine insensitive. (A) Typical whole-cell capacitance records and composite statistics for RBL cells during cytoplasmic infusion of 200 μM Ca with control cytoplasmic solution (top) and with 5 μM wortmannin and 0.5 mM adenosine (bottom). Differences are not significant. (B) Exchange current densities (top) and capacitance responses (bottom) for BHK cells in which membrane fusion was activated by outward Na/Ca exchange current with control cytoplasmic solution (left bar graph), with 0.5 mM adenosine (middle bar graph), and with 5 μM wortmannin and 0.5 mM adenosine (right bar graph) are shown.
Figure 8.

Non-SG fusion in whole-cell recordings is strongly inhibited by cell swelling. (A and B) Bar graphs give exchange current densities (top) and capacitance responses (bottom) for two batches of BHK cells in which membrane fusion was activated by outward Na/Ca exchange current. (A) Effects of cytoplasmic osmolarity on fusion responses. The left bar graphs give results for isoosmotic solution, the middle bars for cytoplasmic solution with 200 mM sucrose, and the right bars for cytoplasmic solution diluted 30% with distilled water. (B) Effects of extracellular osmolarity on fusion responses. From left to right the bar graphs are with (1) standard extracellular solution for 2 min after cell opening, (2) standard extracellular solution with the NMG-aspartate concentration reduced by 80 mM for 2 min after cell opening, (3) standard solution reapplied for 2 min after applying hypoosmotic solution for 2 min, and (4), as in (3) with 5 μM wortmannin in all cytoplasmic solutions. (C) Capacitance responses of RBL cells for pipette perfusion of cytoplasmic solution with 200 μM free Ca. The left bar graph indicates the response magnitude for control cells, the middle graph for cells swollen with hyperosmotic cytoplasmic solution (200 mM sucrose) for 2 min, and the right bar for cells swollen with extracellular solution in which the NMG concentration was reduced by 80 mM.
Recording Software
Capacitance measurement software, Capmeter 6, was developed in MATLAB using its data acquisition toolbox (R2006b; The MathWorks). Three major on-line functions were developed: (1) a software lock-in amplifier, (2) routines for continuous cell parameter determination via square wave voltage perturbation, and (3) data smoothening and deglitching routines. To synchronize the timing of analogue output and input, a 1-mV trigger signal was added to the sine or square wave, usually at 100 Hz. For the lock-in amplifier function, the phase-sensitive detector of the program multiplied the current with either an in-phase or an orthogonal reference signal. The direct-current (DC) component of the product was extracted by averaging to cancel the non-DC noise. The double of the DC component was assigned to X or Y where the in-phase or the orthogonal reference signal was employed, respectively. The optimal phase angle, θ, was determined by small changes of the optimally adjusted capacitance compensation of the patch clamp as follows:
 |
(1) |
where θ0 (and X0, Y0) and θ (and X, Y) represent values before and after changing capacitance compensation, respectively.
For square wave perturbation (time-domain method; see Fig. 1), continuous square pulses were applied, and current transients were recorded and analyzed on-line using Capmeter 6. The average current (i.e., the DC component) was first calculated and subtracted from the total current. The resulting trace was then divided into two parts and fitted separately to exponential functions. For curve fitting, the steady-state current (I∞ = b in Fig. 1 A) was determined as the asymptote of current from the averages of three consecutive data sections of equal length (A, B, and C; Fig. 1 B dashed sections):
 |
(2) |
Figure 1.

Method to determine whole-cell capacitance via square wave perturbation. For whole-cell recording from cells with time constants >50 μs, square pulses were employed at 0.5 kHz with an amplitude of 20 mV. Model current is shown in A. Peak current, a, and the projected steady-state current, b, were determined as described in Materials and methods. (B) Half of the current from a typical recording (dots) with the fitted exponential function used to determine cell parameters is shown. The asymptote of current was determined using the averages of three equally spaced data sections (dashed sections) according to Eq. 2, given in Materials and methods. The asymptote was subtracted and the data range from the peak to a point located at ∼3τ, estimated as peak current times e−3, was used to determine the exponential constants via linear regression of the log of the decaying current transient (solid line).
This equation is the solution for the asymptote, b, of the three simultaneous functions, A = b + Y*e−t/τ, B = b + Y*e−(t+x)/τ, and C = b + Y*e−(t+2x)/τ. The steady-state current was then subtracted from the trace, and the data range from the peak current to a point located at ∼3τ (estimated as peak current times e−3; Fig. 1 B, solid section) was used for fitting via linear regression to determine the slope and intercept at zero time, namely −1/τ and ln(a − b), respectively.
Our routine to calculate cell capacitance (Cm), membrane resistance (Rm), and access resistance (Ra) can be derived as follows. Membrane voltage (V(t)) approaches a steady state (Vss),
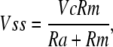 |
(3) |
as a function of the command voltage, Vc (peak-to-peak step = 2Vc), with a time constant, τ, of Cm/(1/Ra + 1/Rm). For square wave perturbation, membrane voltage during the pulse is given by the exponential function,
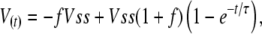 |
(4) |
where f is the fraction of Vss across the membrane at the end of the voltage step of duration, Δ. Solving for f with t = Δ,
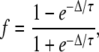 |
(5) |
and membrane voltage at the beginning of the voltage step is
 |
(6) |
From the steady-state current,
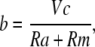 |
(7) |
and the peak current,
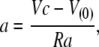 |
(8) |
the solutions for Rm, Ra, and Cm are
 |
(9) |
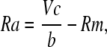 |
(10) |
 |
(11) |
Our algorithm was verified by using it to retrieve cell parameters from model cell simulations using the MATLAB component, Simulink, as well as our own routines. In the absence of noise and a filter function, the algorithm retrieved simulated cell parameters with errors of ∼1 ppm. With cell parameters that would be considered experimentally unacceptable (e.g., 200 pF, a Ra of 20 MΩ, a Rm of 50 MΩ, and voltage oscillation at 200 Hz), the algorithm still retrieved the parameters with an accuracy of 99.9%.
Signals were usually acquired at 100 kHz, and digital filtering was performed by averaging signals in an adjustable time window. Data were usually digitized at 100 Hz and a running mean/median filter was applied to the digitized data when data smoothening/deglitching was desired. Program Capmeter 1 was used with the hardware lock-in amplifier, serving as a plain data recorder with digital filtering and data smoothening/deglitching functions. The programs are available for download at http://capmeter.googlepages.com.
Patch Clamp and Data Acquisition
We used National Instruments board PCI-6052E to generate the command potential and collect signals, and we used an Axopatch-1D (Molecular Devices) for patch clamp. Electrode tips were dipped in molten hard dental wax (Kerr Corporation) before cutting and polishing to reduce stray capacitance. For excised patches, electrodes with ∼15 μm inner diameters were employed. The giant patch was excised by essentially aspirating the cell into a second pipette with a sharp, unpolished edge (Hilgemann and Lu, 1998). The patches were positioned in front of a temperature controlled (∼30°C) solution outlet immediately after excision. Membrane fusion was triggered by moving the patch to a solution outlet containing 0.2 mM free Ca. Capacitance and conductance were measured using the Lindau-Neher method (Lindau and Neher, 1988). Sine waves generated by Capmeter 6 with 20 mV peak-to-peak amplitude at 2 kHz were applied to the cell. The current output from the patch clamp was low-pass filtered at 10 kHz. When sine wave perturbation was employed, the optimal phase angle was determined as described above. When patch amperometry was employed, a hardware lock-in amplifier (SR830; Stanford Research Systems) was employed, as it allowed a higher signal-to-noise ratio at oscillation frequencies >3 kHz. Sine waves with Vrms of 20 mV at 10 kHz were usually employed. The signals were recorded by Capmeter 1.
For whole-cell recording, with ∼5 μm inner diameter pipette tips, membrane fusion was initiated via perfusion of Ca-containing (nitrilotriacetic acid-bufferd) solution through a quartz capillary with a 40 μm outlet, manipulated within the patch pipette to a distance of 50∼100 μm from the cell opening (Hilgemann and Lu, 1998). Square wave 20 mV (peak-to-peak) perturbation at 0.5 kHz was employed in all experiments presented in this article for whole-cell capacitance recording, with cell parameters determined by Capmeter 6 as described above.
Patch Amperometry
The setup was connected according to Dernick et al. (2005) with some modifications. In brief, two Axopatch-1D amplifiers were used. One of the headstages was connected to the bath for capacitance recording, the other one was connected to the carbon electrode for recording the amperometric current, and the patch pipette was the ground. The carbon electrodes were made from 7-μm carbon fibers (C005711; Goodfellow Corporation) and quartz capillaries (Polymicro Technologies). Flowable silicone windshield/glass sealer (Permatex) was used to insulate the carbon fiber, and the tip was cut to expose the carbon surface before installing (Fig. 2 A). The carbon electrode was installed through the infusion line of the pipette holder and connected to the amplifier using 3 M KCl and Ag/AgCl wire. The electrode was moved as close as possible to the patch membrane and a holding potential of 0.7 V was applied. The amperometric signals were low-pass filtered at 5 kHz by the amplifier and digitally filtered again by averaging signals acquired in a time period of 1 ms using Capmeter 1 and then digitized at 500 Hz.
Figure 2.

Amperometric and capacitance measurement in RBL cells. (A) Schematic illustration of the intra-patch pipette carbon electrode. Carbon electrodes were prepared to allow facile insertion and manipulation in the patch pipette holder employed. (B) In the cell-attached configuration, 2 μM of calcium ionophore, A23187, triggers profuse exocytosis. Two distinct vesicle pools are observed. One is the secretory granule (SG) pool with large-amplitude capacitance steps and amperometric spikes upon stimulation. The other pool (at the end of the trace) contains vesicles of much smaller size that do not release serotonin (non-SG pool). (C) Amperometric recording of SG fusion in an ∼20 μm (diameter) excised patch. (D) Expansion of C. In most cases, SGs are lost from membrane patches during the excision procedure. Occasionally, when SGs are preserved, fusion gives rise to capacitance steps of tens of fF, indicating that the diameter of the granules is close to micrometer range. A typical fusion event with fusion pore dilation is marked between two broken lines. A gradual increase of capacitance, transient increase of conductance, and the amperometric foot-signal is observed. Here and in all subsequent figures, numbers given between axis ticks indicate the tick interval.
Recombinant Proteins and Antibodies
The wild-type and mutant (E234Q) GST-tetanus toxin light chain fusion constructs were provided by T. Südhof. Recombinant proteins were purified from bacteria, BL21, according to the manufacture's protocol (GE Healthcare). Proteins were eluted from the beads using reduced glutathione and then dialyzed against buffer containing ZnSO4 (toxin final concentration, 200 nM). Anti-PI(4,5)P2 antibody (1:50) was provided by K. Fukami. Anti-PI(3)P (1:100) and anti-PI(4)P (1:100) antibodies were purchased from Echelon Biosciences. The PI-transfer protein (140 μg/ml) was generously provided by V.A. Bankaitis (The University of North Carolina, Chapel Hill, NC).
Data Analysis
Except for experiments done with MEFs, all experiments were performed in a one control vs. one test result pattern. For excised patch records, the capacitance traces were well described by a monoexponential function with a small linear drift component:
 |
(12) |
where a represents the theoretical maximal amplitude, k is the rate constant used in statistical analysis, and c represents the slow component. For patches with a < 0, the amplitude of zero was assigned. To calculate the ratio of active patches, we used a threshold of 50 fF to define active and inactive (a < 50 fF) patches. We mention that we were not able to determine the capacitance of excised patches routinely, so that normalization of results to patch size is impossible. Also, we mention that methodologically induced capacitance changes during solution changes are in the range of a few tens of femtofarad (see Fig. 3 A). Thus, we typically calculated a ratio of active patches for a group of experiments, as well as the average capacitance changes, and rate constants were collected and compared from the patches that met the “active” criterion.
Figure 3.

Non-SG fusion is SNARE dependent. (A) Two typical recordings from excised patches are shown. In the left panel, the amplitude is smaller than 50 fF, which is close to the artifact caused by moving the patch. Therefore, this patch is designated “inactive.” In the right panel, robust non-SG fusion (>50 fF) is observed in an “active” patch. (B) The same trace is fitted to a mono-exponential function (rate constant, k) with a linear “creep” component (c). (C) Incubation of patches with 200 nM tetanus toxin light chain (TeTx) for 2 min blocks fusion while the mutant toxin (E234Q) has no effect, indicating that non-SG fusion is SNARE dependent.
For whole-cell experiments, phase-sensitive detection was also used off-line to improve the signal-to-noise performance of the exponential fitting routine with square wave perturbation, as follows. The phase angle was determined at which C, the absolute capacitance determined by the exponential analysis, and Y had the highest cross-correlation. Off-line phase angle adjustment was done using equations,
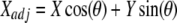 |
(13) |
 |
(14) |
The whole-cell capacitance traces were fitted with a delayed monoexponential function:
 |
(15) |
where a and b represent the vesicle pool size and the initial cell capacitance, respectively. The constants k1 and n reproduce reasonably the observed delays, and the constant k2 is the rate constant used for statistical comparison. The Ca-activated conductance increase was used as a reference for determining the t0 point.
For all bar graphs in which the amplitude and ratio of active patches are presented, numbers in the bars give the total number of patches; for other panels, numbers represent the numbers of valid data after removing outliers with Grubbs' test. Statistical significance was determined by Student's t test. All error bars in figures represent SEM.
Online Supplemental Material
The programs used to determine and monitor cell electrical parameters are included as online supplemental material (available at http://www.jgp.org/cgi/content/full/jgp.200709950/DC1) together with instructions to run the programs. Zip files are included for two programs employed in this article, Capmeter1 and Cameter6. These files can be downloaded independently for use with Matlab and National Instruments acquisition equipment.
RESULTS
Distinct Vesicle Populations in RBL Cells
To study serotonin secretion in RBL cells, carbon electrodes were prepared as described in Materials and methods and mounted in a quartz tube with a Ag/AgCl electrode that could be inserted into the patch pipette (Fig. 2 A) to detect the released serotonin. In the cell-attached configuration, application of 2 μM calcium ionophore, A23187, induced massive membrane fusion that was detected as an increase of membrane capacitance (Fig. 2 B). Two types of vesicle populations were observed; one is the secretory granule (SG) pool accompanied with big capacitance steps and amperometric spikes; the other one observed here at the end of the trace contains vesicles of much smaller size that are evidently not filled with serotonin (non-SG pool). The capacitance steps of SG fusion were in the range of tens of fF, indicating that the diameters of the SGs were in the submicro- to micrometer range, consistent with values reported by most (Spudich and Braunstein, 1995) but not all investigators (Smith et al., 2003).
Empirically, we found that treating the cells with latrunculin A facilitated both the formation of giant excised patches and the preservation of non-SGs on the patches. Nevertheless, our success rate to preserve SGs in the RBL giant patches was prohibitive for routine studies, perhaps because the SGs are not predocked at the membrane in RBL cells (Smith et al., 2003) in a stable fashion and/or because the docking is disrupted by membrane suction. We also attempted to develop the INS-1 cell line and bovine chromaffin cells for excised giant patch studies. Our experiences with the INS-1 cells were similar to those reported for RBL cells. Batch-to-batch variability was even greater, and we were not able to identify a reliable line. Bovine chromaffin cells readily allowed seal formation with large-diameter pipettes, but excised giant patches were not stable with significant solution flow, thereby greatly limiting their use. In short, the giant patch approaches did not facilitate, in our hands, excised patch analysis of SG fusion processes. Occasionally, high resolution recordings were indeed obtained with clear capacitance steps and amperometric spikes as shown in Fig. 2 (C and D) for RBL patches. The fact that only non-SGs were preserved on the great majority of excised patches implies that the non-SGs are in close vicinity to the plasma membrane and might be associated with the membrane physically.
Non-SG fusion is SNARE Dependent but NEM Insensitive in Excised Patches
The physical characteristics of non-SGs are not well established, and we therefore used the giant patch approach to analyze this process in some detail, in particular to manipulate the cytoplasmic membrane side. Capacitance traces were fitted as described in Materials and methods (Y in Fig. 3 B), defining patches with capacitance increases smaller than 50 fF as inactive (see Fig. 3 A, left), and patches with more robust non-SG fusion as active patches (see Fig. 3 A, right). Notably, the exocytotic response often appeared to be followed by an endocytotic response, even in the presence of Ca, when ATP was present on the cytoplasmic side (see Fig. 3 A)
To test whether the non-SG fusion is SNARE dependent, we treated the patches with tetanus toxin (TeTx) light chain at a concentration of 200 nM for 2 min. As shown in Fig. 3 C, both the amplitude and the ratio of active RBL patches were decreased significantly by treatment with the wild-type toxin, compared with the inactive mutant form, indicating that the fusion is indeed SNARE dependent. Notably, however, the treatment of patches with 1 mM N-ethylmaleimide (NEM) did not block non-SG fusion (Table I), implying that SNARE cycling, which is blocked by NEM (Xu et al., 1999), is not required for non-SG fusion in excised patches. As mentioned in the Introduction, it is reported that non-SG fusion in bovine chromaffin cells is toxin insensitive (Xu et al., 1998). A simple explanation for the discrepancy to our data is that RBL and chromaffin cells use different sets of SNAREs for non-SG fusion, and the SNAREs accounting for non-SG fusion in chromaffin cells are toxin resistant. Another possibility is that the SNAREs of non-SGs in chromaffin cells are complexed, such that neurotoxins cannot cleave them (Chen et al., 2001). As described later, in whole-cell recordings with RBL cells, the amplitudes of non-SG fusion usually exceed 50% of the basal cell capacitance (Figs. 6 and 7). Non-SG fusion in chromaffin whole-cell recordings is usually substantially smaller in absolute and relative terms (Xu et al., 1998), indicating that its non-SG pool is much smaller and possibly already primed and therefore toxin resistant.
TABLE I.
Effects of Various Reagents on Non-SG Fusion in Excised Patches
| Paired control/testing condition | Amplitude (fF) mean ± SEM (n) |
Ratio of active patchesa mean ± SEM (n) |
Rate constant(1/s)ab mean±SEM (n) |
|---|---|---|---|
| TeTx E234Q | 209.2 ± 60 (9) | 0.89 ± 0.11 (9) | 0.3 ± 0.04 (7) |
| TeTx WT | 10.9 ± 8.4 (10)d | 0.1 ± 0.1 (10)e | 0.36 (1) |
| ATP/GTP | 160.8 ± 42.2 (18) | 0.67 ± 0.11 (18) | 1.52 ± 0.27 (12) |
| NEM/ATP/GTP | 138.5 ± 43.7 (18) | 0.5 ± 0.12 (18) | 1.01 ± 0.21 (9) |
| ATP | 119.8 ± 31.8 (12) | 0.58 ± 0.15 (12) | 0.8 ± 0.11 (7) |
| AMP-PNP | 35.3 ± 9.4 (13)c | 0.15 ± 0.1 (13)c | 1.04 ± 0.27 (3) |
| Mg2+ buffer | 53.3 ± 19.9 (10) | 0.3 ± 0.15 (10) | 1.12 ± 2e-3 (2) |
| EDTA | 168.6 ± 70.7 (10) | 0.8 ± 0.13 (10)c | 1.5 ± 0.34 (8) |
| EDTA 1min | 221.3 ± 106.2 (5) | 0.8 ± 0.2 (5) | 0.72 ± 0.11 (4) |
| neomycin/EDTA | 102.4 ± 38.5 (5) | 0.8 ± 0.2 (5) | 0.47 ± 0.1 (3) |
| FVPP 1min | 267.3 ± 61 (8) | 1 (8) | 0.75 ± 0.29 (8) |
| PIP2Ab/FVPP | 118.3 ± 25.6 (7) | 0.86 ± 0.14 (7) | 1.12 ± 0.3 (6) |
| FVPP 2min | 78.2 ± 39 (6) | 0.5 ± 0.22 (6) | 0.32 ± 0.09 (2) |
| PIP2Ab/FVPP | 128.9 ± 40.4 (6) | 0.67 ± 0.21 (6) | 0.68 ± 0.17 (4) |
| Mg2+ buffer | 93.1 ± 44.2 (13) | 0.38 ± 0.14 (13) | 1.08 ± 0.35 (5) |
| neomycin | 71 ± 22.9 (14) | 0.29 ± 0.13 (14) | 0.62 ± 0.21 (5) |
| Mg2+ buffer | 103.1 ± 47.3 (14) | 0.43 ± 0.14 (14) | 0.92 ± 0.19 (6) |
| PIP2Ab | 92.6 ± 46.8 (14) | 0.43 ± 0.14 (14) | 1.17 ± 0.13 (5) |
| ATP | 76.6 ± 16.9 (14) | 0.64 ± 0.13 (14) | 1.06 ± 0.23 (9) |
| staurosporine/ATP | 50.3 ± 13.8 (15) | 0.47 ± 0.13 (15) | 1.25 ± 0.34 (7) |
| ATP | 62.2 ± 13.5 (20) | 0.6 ± 0.11 (20) | 0.83 ± 0.17 (11) |
| wort/adeno/ATP | 6.9 ± 3.1 (21)e | 0.05 ± 0.05 (21)e | 0.88 (1) |
| ATP | 47.3 ± 26.4 (8) | 0.38 ± 0.18 (8) | 1.04 ± 0.32 (3) |
| LY294002/ATP | 159.1 ± 70.9 (7) | 0.57 ± 0.2 (7) | 1.49 ± 0.19 (4) |
| ATP | 104.9 ± 75.1 (4) | 0.5 ± 0.29 (4) | 1.87 ± 0.51 (2) |
| PI-TP/ATP | 202 ± 50.4 (4) | 1 (4) | 1.28 ± 0.46 (4) |
TeTx, tetanus toxin light chain, 200 nM; NEM, N-ethylmaleimide, 1 mM; AMP-PNP, 2 mM; neomycin, 500 μM; PIP2Ab, 1:50; staurosporine, 200 nM; wort, wortmannin, 4 μM; adeno, adenosine, 0.5 mM; LY294002, 100 μM; PI-TP, PI-transfer protein, 140 μg/ml.
Counts patches with amplitude ≥50 fF.
Outliers are removed using Grubbs' test.
P<0.05.
P<0.01.
P<0.001.
Figure 6.

Non-SG fusion is not blocked by antibodies against PI(3)P and PI(4)P. In whole-cell recording, antibodies (1:100 dilution) were added to the cytoplasmic (pipette) solution, which was dialyzed into cells for 5 min before Ca2+ was infused to trigger fusion. (A) A typical record from an RBL cell. The Ca2+-activated conductance rise was used to define the t0 point. (B) The rise of capacitance was well described by the delayed monoexponential function given in the figure. The variable k2 is the rate constant used for statistical analysis. The number of the data points was reduced to highlight the fitted curve. Antibodies against PI(3)P (C), PI(4)P (D), and both (E) fail to block non-SG fusion.
ATP Hydrolyzing Processes Support Non-SG Fusion
It is well documented that ATP is required by the NSF (NEM-sensitive factor) to disassemble the cis-SNARE complex in the SNARE cycle (Whiteheart et al., 1994; Jahn et al., 2003), and that the addition of Vam7p (a soluble SNARE) bypasses the requirement of ATP in the yeast vacuole fusion system (Thorngren et al., 2004). Since non-SG fusion in excised patches is NEM insensitive (Table I), one might expect that the non-SG fusion would be ATP independent. In whole-cell recording, replacement of ATP with a nonhydrolyzable analogue was found to significantly reduce non-SG fusion (Yarandanakul et al., 2008), and we describe here that ATP-hydrolyzing processes clearly support non-SG fusion in excised patches. In some cases, the absence of ATP caused complete failure of fusion, and the ability to fuse was restored when ATP and GTP were added back to the solution (see Fig. 4 A). That ATP is the critical factor for restoration of fusion was verified in several experiments in which ATP was applied without GTP (Fig. 3 A, Fig. 4 B, and unpublished data). Further experiments supported the notion that ATP hydrolysis is essential to maintain fusion because the nonhydrolyzable ATP analogue, AMP-PNP (2 mM), did not substitute for ATP (Fig. 4 B).
Figure 4.

ATP hydrolysis is required for supporting non-SG fusion. (A) Without ATP, Ca2+ application fails to trigger exocytosis in this patch, but exocytosis was restored by placing the patch in ATP/GTP-containing solution for an additional minute. (B) AMP-PNP cannot preserve fusion, indicating that the hydrolysis of ATP is required to support non-SG fusion.
Since the decrease of fusion after removal of ATP, and with substitution by AMP-PNP, takes much longer than expected for washout of ATP from the patch, ATP-hydrolyzing reactions clearly are not part of the final fusion process. The ATP mechanism(s) that supports fusion in the longer term could involve the phosphorylation of target proteins/lipids and/or the use of ATP in other types of energy-dependent reactions, as in the case of NSF. In favor of the former idea, we could preserve the fusion capability in the absence of ATP by adding EDTA to the solution (see Table I). Since magnesium is the only divalent ion in our recording solution, we hypothesized that ATP is used to phosphorylate one or more targets, and that activity of the counteracting phosphatases would be magnesium dependent.
ATP-dependent Generation of PI(4,5)P2 Is Not Critical
As mentioned earlier, PI(4,5)P2 has been implicated in multiple aspects of fusion processes, and we therefore tested if the role of ATP is to maintain PI(4,5)P2 in the excised patches and, as well, if the cleavage of PI(4,5)P2 is required for triggering membrane fusion. Application of a very high concentration of neomycin (500 μM) to bind PI(4,5)P2 (Eberhard et al., 1990), and probably other anionic phospholipids, in a magnesium-free solution did not block non-SG fusion. Furthermore, application of anti-PI(4,5)P2 antibodies in FVPP solution did not block fusion (Table I), although the concentrations of antibody employed potently blocked PI(4,5)P2-senstive currents in excised patches (e.g., outward Na/Ca exchange current, unpublished data). These results suggest that neither the synthesis nor the hydrolysis of PI(4,5)P2 can be a requirement for non-SG fusion. As shown in Table I, no protecting effect of these agents was observed when they were applied in a magnesium-containing solution. Clearly, magnesium-dependent hydrolysis of PI(4,5)P2 cannot be the mechanism of rundown of the fusion process.
Non-SG Fusion Is Probably Phosphoinositide Independent
Since the ATP mechanism does not appear to involve PI(4,5)P2, we used other inhibitors to probe potential phosphorylation targets. As shown in Table I, treating the patches with staurosporine, a broad-spectrum protein kinase inhibitor, was not able to block fusion. Interestingly, treating the patches with high concentrations of wortmannin (4 μM) and adenosine (0.5 mM), which inhibit multiple classes of PI(3)Ks and PI(4)Ks (Balla et al., 2008), significantly decreased non-SG fusion (Fig. 5), implying that PI(3)P and/or PI(4)P might be responsible for the ATP dependency. Therefore, we tested for roles of individual lipids by applying specific antibodies in whole-cell recording experiments before triggering fusion via Ca infusion. Impressively, neither anti-PI(3)P nor anti-PI(4)P antibody was able to block or slow down non-SG fusion, and a combination of both antibodies was also ineffective (Fig. 6). It seems unlikely that PIP3 is involved because another PI(3)K inhibitor, LY294002, also did not block fusion in excised patches (Table I). In fact, pretreatment of the cells with a submicromolar concentration of wortmannin, which blocks PIP3 producing PI(3)K, and treatment with genistein, an inhibitor of tyrosine kinases that are typically activated in this pathway (Galetic et al., 1999), failed to block non-SG fusion in whole-cell recording (unpublished data).
Figure 5.

Non-SG fusion in excised patches is wortmannin/adenosine sensitive. Incubation of patches with the PI-kinase inhibitors wortmannin (wort; 4 μM) and adenosine (0.5 mM) significantly decreases the average fusion magnitudes and the ratio of active patches.
We also used a PI transfer protein (Mousley et al., 2007) to remove PI from excised patches to test if there is any role of PIs at all in non-SG fusion. As shown in Table I, fusion was not inhibited by a concentration that we found to inhibit the ATP-dependent stimulation of Na/Ca exchange current in excised patches, a process determined to reflect phosphorylation of PI (Nasuhoglu et al., 2002). All of these negative results support the conclusion that non-SG fusion is PI independent. The actual targets and the specificity of wortmannin and adenosine in this study must therefore be questioned, and as described next there is an important difference between excised patches and whole-cell responses in this regard.
Wortmannin/Adenosine Insensitivity of Non-SG Fusion in Whole-Cell Recording
The pronounced inhibition of non-SG fusion in excised patches by the wortmannin/adenosine combination was unexpected, as we had tested these agents in whole-cell recording of Ca-induced fusion in BHK fibroblasts and found no effect. Therefore, we reexamined this issue in whole-cell recordings from both RBL cells and from BHK cells, as shown in Fig. 7. Individual records from RBL cells are shown in Fig. 7 A with the composite statistics for the dataset. Pipette perfusion of solution containing 200 μM free Ca, as in Fig. 6, was initiated 2 min after opening cells. In control cells, the average increase of cell capacitance was 119 ± 12%, and it occurred with a time constant of 26 ± 3.1 s (n = 5). In cells that were perfused with 5 μM wortmannin and 0.5 mM adenosine for 2 min, the average increase was 114 ± 5.2%, and the time constant was 30 ± 3.1 s (n = 6). Fig. 7 B shows the equivalent results for whole-cell BHK recording using NCX1 to initiate membrane fusion. In these experiments, we tested adenosine (0.5 mM) alone, as well as the same adenosine/wortmannin combination used in RBL cells. After 2 min cytoplasmic infusion of the respective solutions, the peak exchange currents were modestly reduced (∼20%), at just the level of significance, but the percent increment of cell capacitance upon activating exchange currents (∼65% in these experiments) was unchanged by either treatment.
Non-SG Fusion in Whole-Cell Recording Is Blocked by Cell Swelling
The lack of a significant effect of these treatments in whole-cell recordings suggested that a mechanism becomes important to maintain fusion capability in the excised patch, which is not critical under the usual conditions of whole-cell recording. We reasoned that mechanical forces, exerted on the membrane during seal formation and patch excision, might disrupt the fusion machinery, and ATP-hydrolyzing processes would then become essential to restore the fusion capability. In other words, membrane stretch and/or distention (i.e., flattening of invaginations) might be an important factor, and accordingly we tested whether cell swelling might mimic effects of seal formation and excision. Fig. 8 describes two sets of results, one from BHK cells and one from RBL cells, both demonstrating strong inhibition of non-SG fusion by cell swelling. We note that different BHK batches were employed in the two datasets in A and B, and that, as often was the case, the average capacitance responses were substantially different in different batches.
In Fig. 8 A, cell swelling was induced in BHK cells by using a cytoplasmic solution with addition of 200 mM sucrose, and shrinkage was induced by using a cytoplasmic solution diluted by 30%. Exchange currents were activated 2 min after opening cells and cell shape changes had clearly occurred. As shown in the top bar graphs, the peak exchange currents were not significantly affected. Cell swelling was associated with a 76% decrease of the fusion response (P < 0.01), whereas cell shrinkage with hypoosmotic cytoplasmic solution was without effect.
Next, we tested whether cell swelling by hypoosmotic extracellular solution also reduces membrane fusion. As shown in Fig. 8 B, placement of cells in extracellular solution with NMG reduced by 80 mM (hypoosmotic outside) for 2 min before activating exchange current caused an 85% decrease of membrane fusion. To examine whether the effect of swelling is reversible, we placed cells for 2 min in hypoosmotic solution and then moved them back into isoosmotic solution for 2 min before activating exchange currents. During the protocol, swelling and shrinkage of cells was clearly visible. As shown for post-swelling, the fusion response was partially restored. In a fourth experimental group, we tested whether the restoration of fusion might be blocked by wortmannin. Using the same protocol to allow restoration of fusion, inclusion of 5 μM wortmannin in the pipette solution significantly decreased the recovery of fusion responses after swelling (P < 0.05). From three observations, we did not find the adenosine/wortmannin combination to be more effective (unpublished data). Overall, these whole-cell results support the notion that membrane perturbation and/or stretch strongly inhibits non-SG fusion, and that biochemical processes are required to restore fusion capability.
Fig. 8 C presents the equivalent experiments for RBL cells with membrane fusion induced by pipette perfusion of cytoplasmic solution with 200 μM free Ca, as in Figs. 6 and 7. Results for hyperosmotic cytoplasmic solution (with 200 mM added sucrose) and for hypoosmotic extracellular solution (with NMG reduced by 80 mM) are very similar to results with BHK cells. Membrane fusion responses are reduced by 74 and 75%, respectively. Thus, the high sensitivity of the non-SG fusion process to inhibition by cell swelling is verified across two cell lines and with different protocols to induce membrane fusion.
Synaptotagmin VII and PLCs Are Not Required for Non-SG Fusion
The non-SG pool in RBL cells can almost double the total surface membrane area (Fig. 7 A and Fig. 8 C), and it seems likely that this pool will become involved in the wound repair of the plasma membrane. Lysosomes have been suggested to be the major vesicles that undergo Ca-dependent exocytosis in nonsecretory cells (Jaiswal et al., 2002) and in wound repair (Chakrabarti et al., 2003). Furthermore, it is suggested that the Ca sensor in lysosomal fusion is the synaptotagmin VII (Syt VII) (Chakrabarti et al., 2003). To test if Syt VII is important for non-SG fusion, we examined membrane fusion in mouse embryonic fibroblasts (MEFs) because, like RBL cells, they also have robust non-SG fusion, and knockout lines are available. As shown in Fig. 9 B, non-SG fusion was still robust in syt7 knockout MEFs (six observations). Unlike results with RBL cells, it was notable that non-SG fusion in MEFs often involved fusion of large vesicles (Fig. 9, arrowheads). Whether such large vesicles can be lysosomes is unclear, but it is evident that both types of fusion are still robust when syt7 is ablated. We conclude that Syt VII cannot be an important Ca sensor for non-SG fusion.
Figure 9.

Synaptotagmin VII is not required for non-SG fusion. Whole-cell recordings from wild type (wt; A) and syt7 knockout (B) mouse embryonic fibroblasts (MEFs). Fusion remains robust in syt7 knockout MEFs. Unlike results with RBL cells, non-SG fusion in MEFs often involves fusion of large vesicles (arrowheads).
Relevant to the potential importance of phosphoinosidites, phospholipases, and alternative possible Ca sensors, we tested whether this type of membrane fusion in MEF cells was affected by deletion of three PLCs. No evident differences were found for membrane fusion episodes recorded using MEFs and excised patches from MEFs with deletion of PLCγ1, PLCδ1, and both PLCδ1 and PLCδ4 versus a control MEF cell line (n > 3 for all observations, unpublished data).
Ca Dependence of Non-SG Fusion in Excised RBL Patches
To further characterize the Ca-sensing machinery of non-SG fusion, we triggered fusion with different concentrations of free Ca in excised patches from RBL cells. The concentration–response data for free Ca versus rate of fusion shows a Hill coefficient of ∼2, an apparent KD of ∼71 μM, and a maximal rate constant of ∼2.1 s−1 (Fig. 10). We point out that the two data points at low Ca concentrations were weighted to force the fit through these points. Without weighting, these points were not well described by the fit, and the Hill coefficient was ∼3. We stress, however, that Hill coefficients determined for non-SG fusion in BHK cells were also in the range of 2 (Yaradanakul et al., 2008). It is reported that the Hill coefficient and [Ca]1/2 of Syt VII is ∼3.6 and ∼0.9 μM, respectively, in a liposome binding assay (Wang et al., 2005). Therefore, these results further support a conclusion that Syt VII is not likely to be the sensor for non-SG fusion.
Figure 10.

Ca dependence of non-SG fusion in excised patches from RBL cells. The concentration–response data are best described by a Hill equation with a slope coefficient of 2.0, a Km of 71 μM, and a maximal rate constant of 2.1 s−1.
DISCUSSION
In this article, we have described some efforts over several years to maintain and to monitor Ca-dependent exocytosis in giant excised patches. While nonsecretory vesicle fusion could be routinely monitored, in our hands neurotransmitter release was seldom maintained in the excised patches. Even in the case of non-SG fusion, we found it necessary to pretreat cells with agents that disrupt cytoskeleton to maintain fusion. That membrane stretch and distention may readily disrupt fusion has been verified in whole-cell recording with both BHK and RBL cells. We discuss first the methodological and experimental problems encountered and then the datasets on non-SG fusion.
Membrane Fusion in Excised Giant Membrane Patches
In principle, the giant patch methods should facilitate multiple types of fusion studies. Excised patches allow free access to the cytoplasmic side for a wide range of possible manipulations by exogenous factors, including proteins. Nevertheless, after several years we have not been able to establish conditions that allow routine recordings of neurotransmitter release in excised patches, including efforts with several cell types. For example, bovine chromaffin cells readily allowed seal formation with large-diameter pipettes, but excised patches are not stable with significant solution flow, thereby greatly limiting their use. Overall, our experience is that the ability to induce neurotransmitter release is very easily lost or destroyed during excision procedures, and we did not overcome this limitation for routine work. Given the strong inhibition of fusion by cell swelling documented in Fig. 8, we strongly suspect that mechanical factors are of most importance, but it also remains possible that important soluble factors are lost from the patches.
Nonsecretory Fusion in Excised Patches
In this article, we have described that non-SG membrane fusion is both robust and massive in several cell lines when studied by whole-cell voltage clamp, while responses in excised patches showed a large degree of variability. Specifically, we found that the ratio of active patches varied substantially from batch to batch of cells, as well as with the length of time after isolation. Sometimes, no fusion at all was observed in excised patches from an entire batch of cells, although whole-cell responses were robust and highly reliable in the same cell batch. Also, individual cell batches were encountered in which fusion did not run down in the absence of ATP and EDTA (unpublished data), while the loss of fusion over time in ATP-free solution was highly reliable in other batches. For these reasons, all experiments were done in a one control vs. one test result pattern, as pointed out in Materials and methods, and results can only be compared with data collected at the same time using the same batch of cells.
Our overall interpretation is that in the routine whole-cell configuration nearly all available vesicles eventually fuse to the plasma membrane, as there are no inactive cells, and the percentage increase of cell area so impressively large in relation to the number of docked vesicles observed (Yaradanakul et al., 2008). Since the fusion process is strongly inactivated by cell swelling, it seems likely that membrane stretch, or flattening of invaginations, disrupts the fusion machinery with restoration requiring ATP-dependent processes of an unknown nature. Remodeling of cytoskeleton is an attractive but unproved possibility. Our results on rundown in patches (Fig. 4 and Table I) are consistent with ATP being used to phosphorylate one or more targets that maintain the fusion capability, whereby dephosphorylation evidently occurs by a magnesium-dependent phosphatase. In this regard, it is known that the enzymatic activities of tyrosine phosphatases vary with culture conditions and ages (Pallen and Tong, 1991; Cool and Blum, 1993). It would not be surprising if activities of the kinases, as well as other phosphatases, also vary among batches of cells and the length of time after isolation. Unless AMP-PNP is added to the pipette solution, the ATP dependency of non-SG fusion in whole-cell recordings is usually not significant (Yaradanakul et al., 2008). Thus, the excised patch model naturally produces many more sources of variability that do not exist in the intact cells. This is at once an advantage for identifying important partial mechanisms of fusion, and its regulation, and a disadvantage because the reproducibility of experiments is significantly decreased.
ATP Sensitivity of Non-SG Fusion
Results from this article further support our conclusion that neither PI(4,5)P2 nor its metabolism significantly modulate non-SG fusion, when the trigger Ca concentration is high (Yaradanakul et al., 2008). Depletion of PI(4,5)P2 at the plasmalemma does not block non-SG fusion in M1 receptor-expressing BHK cells (Yaradanakul et al., 2008), and we have shown here that multiple PI(4,5)P2 ligands, namely neomycin and PI(4,5)P2 antibodies, do not affect Ca-induced fusion in the excised patches from RBL cells. While the antibodies employed might not have sufficient affinity to block very high-affinity functions of phosphoinositides in fusion, we expect that the high concentration of neomycin employed (500 μM) would bind nonselectively all phosphoinositides and other anion phospholipids, and thereby inhibit their functional role. Only one positive result concerning phosphoinositides was obtained, namely that non-SG fusion was blocked by the combined application of wortmannin and adenosine. However, both reagents can have nonspecific effects. At high concentrations, wortmannin inhibits mitogen-activated protein kinase (MAPK) (Ferby et al., 1996) and myosin light chain kinases (MLCK) (Nakanishi et al., 1992). Since non-SG fusion is not blocked by staurosporine (Table I), although staurosporine blocks the activities of MLCK, MAPK might be an interesting target to be tested in future work.
In conclusion, it does not seem surprising that non-SG membrane fusion, which is probably used for cell wound repair, is regulated in a substantially different fashion from the release of neurotransmitters, and the ATP dependence of non-SG fusion likely represents a unique regulatory mechanism that comes into play after mechanical perturbation of the fusion system.
Wound Repair and Non-SG Fusion
While we infer that the membrane fusion process examined in this study is related to the membrane wound response, the membrane compartment(s) involved in non-SG fusion are still enigmatic. In addition to lysosomes, a novel organelle, named the enlargosome, is proposed to mediate membrane repair (Borgonovo et al., 2002). Fusion of the enlargosome in rat PC12 cells is TeTx insensitive, which is different from non-SG fusion in RBL cells as described here. However, it has also been reported that the wound repair machinery is TeTx sensitive in other model systems (Togo et al., 1999), and as mentioned earlier, non-SG fusion is toxin insensitive in bovine chromaffin cells (Xu et al., 1998). One evidence supporting the notion of wound repair by non-SG fusion comes from the low Ca sensitivity of the Ca sensor, which is relevant to the ongoing debate about the role of Syt VII in membrane repair (McNeil and Kirchhausen, 2005). Syt VII is reported to be a high affinity Ca sensor in SGs of PC12 cells (Wang et al., 2005). The low Ca sensitivity of non-SG fusion could in principle ensure that these vesicles are not affected by normal Ca signaling in the cell, and that they would fuse with the plasmalemma only when bulk Ca influx comes from the wounded sites.
In addition to establishing a new approach to study non-SG fusion with giant excised patches, we have developed computer software that can be useful to other groups to implement both time-domain and frequency-domain methods (Lindau and Neher, 1988). In our experience, the time-domain method is especially useful when clamp time constants are relatively long (e.g., hundreds of microseconds in cardiac myocytes). With these approaches, we have delineated several new characteristics of non-SG fusion. First, while this type of fusion is SNARE dependent, it is not NEM sensitive in excised patches. Second, while this type of fusion is ATP dependent, it is not phosphoinositide sensitive. Thus, the ATP sensitivity comes about by a novel mechanism that might prove to be relevant to other types of membrane fusion. Third, the non-SG vesicles are presumably predocked at the plasmalemma to remain attached to excised patches, and in this regard more detailed ultrastructural analysis will be of paramount importance for further progress. Fourth, the Ca dependence of this mechanism is unlikely to represent the function of the putative Ca sensor, Syt VII. And finally, the non-SG fusion mechanism is strongly inhibited by cell swelling and, presumably, membrane stretch. Together, these results have established multiple, potentially novel directions for future studies of non-SG fusion, which is likely to be an important partial reaction of the ubiquitous membrane wound repair response.
Acknowledgments
We thank Vincenzo Lariccia (UTSW) for assistance and discussions, Marc Llaguno (UTSW) and Alp Yarandanakul (UTSW) for advice and criticism, Thomas Südhof, Kiyoko Fukami, and Graham Carpenter for generously providing MEF cell lines, Chengchen Shen (UTSW) and Mei-Jung Lin (UTSW) for assistance, and Vladislav Markin (UTSW) for discussions of mathematical methods.
This work was supported by HL0679420 and HL051323 to D.W. Hilgemann.
Lawrence G. Palmer served as editor.
Abbreviations used in this paper: CAPS, calcium-dependent activator protein for secretion; DAG, diacylglycerol; DC, direct-current; IP3, inositol triphosphate; MEF, mouse embryonic fibroblast; NEM, N-ethylmaleimide; NMG, N-methyl-d-glucamine; NSF, NEM-sensitive factor; PI(3)K, PI(3) kinase; PI(3)P, phosphatidylinositol 3-phosphate; PI(4,5)P2, phosphatidylinositol-4,5-bis-phosphate; PIP3, phosphatidylinositol 3,4,5-triphosphate; SG, secretory granule; Syt, synaptotagmin; TeTx, tetanus toxin.
References
- Ali, K., A. Bilancio, M. Thomas, W. Pearce, A.M. Gilfillan, C. Tkaczyk, N. Kuehn, A. Gray, J. Giddings, E. Peskett, et al. 2004. Essential role for the p110δ phosphoinositide 3-kinase in the allergic response. Nature. 431:1007–1011. [DOI] [PubMed] [Google Scholar]
- Almers, W., and E. Neher. 1987. Gradual and stepwise changes in the membrane capacitance of rat peritoneal mast cells. J. Physiol. 386:205–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyagi, K., T. Sugaya, M. Umeda, S. Yamamoto, S. Terakawa, and M. Takahashi. 2005. The activation of exocytotic sites by the formation of phosphatidylinositol 4,5-bisphosphate microdomains at syntaxin clusters. J. Biol. Chem. 280:17346–17352. [DOI] [PubMed] [Google Scholar]
- Bai, J., W.C. Tucker, and E.R. Chapman. 2004. PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nat. Struct. Mol. Biol. 11:36–44. [DOI] [PubMed] [Google Scholar]
- Balla, A., Y.J. Kim, P. Varnai, Z. Szentpetery, Z. Knight, K.M. Shokat, and T. Balla. 2008. Maintenance of hormone-sensitive phosphoinositide pools in the plasma membrane requires phosphatidylinositol 4-kinase IIIα. Mol. Biol. Cell. 19:711–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgonovo, B., E. Cocucci, G. Racchetti, P. Podini, A. Bachi, and J. Meldolesi. 2002. Regulated exocytosis: a novel, widely expressed system. Nat. Cell Biol. 4:955–962. [DOI] [PubMed] [Google Scholar]
- Bronk, P., F. Deák, M.C. Wilson, X. Liu, T.C. Südhof, and E.T. Kavalali. 2007. Differential effects of SNAP-25 deletion on Ca2+-dependent and Ca2+-independent neurotransmission. J. Neurophysiol. 98:794–806. [DOI] [PubMed] [Google Scholar]
- Burgoyne, R.D., and A. Morgan. 2003. Secretory granule exocytosis. Physiol. Rev. 83:581–632. [DOI] [PubMed] [Google Scholar]
- Chakrabarti, S., K.S. Kobayashi, R.A. Flavell, C.B. Marks, K. Miyake, D.R. Liston, K.T. Fowler, F.S. Gorelick, and N.W. Andrews. 2003. Impaired membrane resealing and autoimmune myositis in synaptotagmin VII-deficient mice. J. Cell Biol. 162:543–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y.A., S.J. Scales, and R.H. Scheller. 2001. Sequential SNARE assembly underlies priming and triggering of exocytosis. Neuron. 30:161–170. [DOI] [PubMed] [Google Scholar]
- Cool, D.E., and J.J. Blum. 1993. Protein tyrosine phosphatase activity in Leishmania donovani. Mol. Cell. Biochem. 127:143–149. [DOI] [PubMed] [Google Scholar]
- Coorssen, J.R., H. Schmitt, and W. Almers. 1996. Ca2+ triggers massive exocytosis in Chinese hamster ovary cells. EMBO J. 15:3787–3791. [PMC free article] [PubMed] [Google Scholar]
- De Matteis, M.A., and A. Godi. 2004. PI-loting membrane traffic. Nat. Cell Biol. 6:487–492. [DOI] [PubMed] [Google Scholar]
- Dernick, G., G. Alvarez de Toledo, and M. Lindau. 2003. Exocytosis of single chromaffin granules in cell-free inside-out membrane patches. Nat. Cell Biol. 5:358–362. [DOI] [PubMed] [Google Scholar]
- Dernick, G., L.W. Gong, L. Tabares, G. Alvarez de Toledo, and M. Lindau. 2005. Patch amperometry: high-resolution measurements of single-vesicle fusion and release. Nat. Methods. 2:699–708. [DOI] [PubMed] [Google Scholar]
- Eberhard, D.A., C.L. Cooper, M.G. Low, and R.W. Holz. 1990. Evidence that the inositol phospholipids are necessary for exocytosis. Biochem. J. 268:15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferby, I.M., I. Waga, M. Hoshino, K. Kume, and T. Shimizu. 1996. Wortmannin inhibits mitogen-activated protein kinase activation by platelet-activating factor through a mechanism independent of p85/p110-type phosphatidylinositol 3-kinase. J. Biol. Chem. 271:11684–11688. [DOI] [PubMed] [Google Scholar]
- Fukami, K., K. Nakao, T. Inoue, Y. Kataoka, M. Kurokawa, R.A. Fissore, K. Nakamura, M. Katsuki, K. Mikoshiba, N. Yoshida, and T. Takenawa. 2001. Requirement of phospholipase Cδ4 for the zona pellucida-induced acrosome reaction. Science. 292:920–923. [DOI] [PubMed] [Google Scholar]
- Galetic, I., M. Andjelkovic, R. Meier, D. Brodbeck, J. Park, and B.A. Hemmings. 1999. Mechanism of protein kinase B activation by insulin/insulin-like growth factor-1 revealed by specific inhibitors of phosphoinositide 3-kinase–significance for diabetes and cancer. Pharmacol. Ther. 82:409–425. [DOI] [PubMed] [Google Scholar]
- Grishanin, R.N., J.A. Kowalchyk, V.A. Klenchin, K. Ann, C.A. Earles, E.R. Chapman, R.R. Gerona, and T.F. Martin. 2004. CAPS acts at a prefusion step in dense-core vesicle exocytosis as a PIP2 binding protein. Neuron. 43:551–562. [DOI] [PubMed] [Google Scholar]
- Hilgemann, D.W., and C.C. Lu. 1998. Giant membrane patches: improvements and applications. Methods Enzymol. 293:267–280. [DOI] [PubMed] [Google Scholar]
- Huang, C.L., S. Feng, and D.W. Hilgemann. 1998. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gβγ. Nature. 391:803–806. [DOI] [PubMed] [Google Scholar]
- Jahn, R., T. Lang, and T.C. Südhof. 2003. Membrane fusion. Cell. 112:519–533. [DOI] [PubMed] [Google Scholar]
- Jaiswal, J.K., N.W. Andrews, and S.M. Simon. 2002. Membrane proximal lysosomes are the major vesicles responsible for calcium-dependent exocytosis in nonsecretory cells. J. Cell Biol. 159:625–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jockusch, W.J., D. Speidel, A. Sigler, J.B. Sorensen, F. Varoqueaux, J.S. Rhee, and N. Brose. 2007. CAPS-1 and CAPS-2 are essential synaptic vesicle priming proteins. Cell. 131:796–808. [DOI] [PubMed] [Google Scholar]
- Jun, Y., R.A. Fratti, and W. Wickner. 2004. Diacylglycerol and its formation by phospholipase C regulate Rab- and SNARE-dependent yeast vacuole fusion. J. Biol. Chem. 279:53186–53195. [DOI] [PubMed] [Google Scholar]
- Lindau, M., and E. Neher. 1988. Patch-clamp techniques for time-resolved capacitance measurements in single cells. Pflugers Arch. 411:137–146. [DOI] [PubMed] [Google Scholar]
- Lindmo, K., and H. Stenmark. 2006. Regulation of membrane traffic by phosphoinositide 3-kinases. J. Cell Sci. 119:605–614. [DOI] [PubMed] [Google Scholar]
- Loyet, K.M., J.A. Kowalchyk, A. Chaudhary, J. Chen, G.D. Prestwich, and T.F. Martin. 1998. Specific binding of phosphatidylinositol 4,5-bisphosphate to calcium-dependent activator protein for secretion (CAPS), a potential phosphoinositide effector protein for regulated exocytosis. J. Biol. Chem. 273:8337–8343. [DOI] [PubMed] [Google Scholar]
- MacDonald, P.E., S. Obermüller, J. Vikman, J. Galvanovskis, P. Rorsman, and L. Eliasson. 2005. Regulated exocytosis and kiss-and-run of synaptic-like microvesicles in INS-1 and primary rat β-cells. Diabetes. 54:736–743. [DOI] [PubMed] [Google Scholar]
- Mahmoud, S.F., and C. Fewtrell. 2001. Microdomains of high calcium are not required for exocytosis in RBL-2H3 mucosal mast cells. J. Cell Biol. 153:339–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer, A., D. Scheglmann, S. Dove, A. Glatz, W. Wickner, and A. Haas. 2000. Phosphatidylinositol 4,5-bisphosphate regulates two steps of homotypic vacuole fusion. Mol. Biol. Cell. 11:807–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeil, P.L., and T. Kirchhausen. 2005. An emergency response team for membrane repair. Nat. Rev. Mol. Cell Biol. 6:499–505. [DOI] [PubMed] [Google Scholar]
- Metcalfe, D.D., D. Baram, and Y.A. Mekori. 1997. Mast cells. Physiol. Rev. 77:1033–1079. [DOI] [PubMed] [Google Scholar]
- Meunier, F.A., S.L. Osborne, G.R. Hammond, F.T. Cooke, P.J. Parker, J. Domin, and G. Schiavo. 2005. Phosphatidylinositol 3-kinase C2α is essential for ATP-dependent priming of neurosecretory granule exocytosis. Mol. Biol. Cell. 16:4841–4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milosevic, I., J.B. Sørensen, T. Lang, M. Krauss, G. Nagy, V. Haucke, R. Jahn, and E. Neher. 2005. Plasmalemmal phosphatidylinositol-4,5-bisphosphate level regulates the releasable vesicle pool size in chromaffin cells. J. Neurosci. 25:2557–2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousley, C.J., K.R. Tyeryar, P. Vincent-Pope, and V.A. Bankaitis. 2007. The Sec14-superfamily and the regulatory interface between phospholipid metabolism and membrane trafficking. Biochim. Biophys. Acta. 1771:727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi, S., S. Kakita, I. Takahashi, K. Kawahara, E. Tsukuda, T. Sano, K. Yamada, M. Yoshida, H. Kase, Y. Matsuda, et al. 1992. Wortmannin, a microbial product inhibitor of myosin light chain kinase. J. Biol. Chem. 267:2157–2163. [PubMed] [Google Scholar]
- Nasuhoglu, C., S. Feng, Y. Mao, I. Shammat, M. Yamamato, S. Earnest, M. Lemmon, and D.W. Hilgemann. 2002. Modulation of cardiac PIP2 by cardioactive hormones and other physiologically relevant interventions. Am. J. Physiol. Cell Physiol. 283:C223–C234. [DOI] [PubMed] [Google Scholar]
- Olsen, H.L., M. Hoy, W. Zhang, A.M. Bertorello, K. Bokvist, K. Capito, A.M. Efanov, B. Meister, P. Thams, S.N. Yang, et al. 2003. Phosphatidylinositol 4-kinase serves as a metabolic sensor and regulates priming of secretory granules in pancreatic β cells. Proc. Natl. Acad. Sci. USA. 100:5187–5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowicz, C.W., C.T. Meiringer, and C. Ungermann. 2008. Yeast vacuole fusion: a model system for eukaryotic endomembrane dynamics. Autophagy. 4:5–19. [DOI] [PubMed] [Google Scholar]
- Pallen, C.J., and P.H. Tong. 1991. Elevation of membrane tyrosine phosphatase activity in density-dependent growth-arrested fibroblasts. Proc. Natl. Acad. Sci. USA. 88:6996–7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton, C., S. Thompson, and D. Epel. 2004. Some precautions in using chelators to buffer metals in biological solutions. Cell Calcium. 35:427–431. [DOI] [PubMed] [Google Scholar]
- Sagi-Eisenberg, R. 2007. The mast cell: where endocytosis and regulated exocytosis meet. Immunol. Rev. 217:292–303. [DOI] [PubMed] [Google Scholar]
- Sakaba, T., A. Stein, R. Jahn, and E. Neher. 2005. Distinct kinetic changes in neurotransmitter release after SNARE protein cleavage. Science. 309:491–494. [DOI] [PubMed] [Google Scholar]
- Smith, A.J., J.R. Pfeiffer, J. Zhang, A.M. Martinez, G.M. Griffiths, and B.S. Wilson. 2003. Microtubule-dependent transport of secretory vesicles in RBL-2H3 cells. Traffic. 4:302–312. [DOI] [PubMed] [Google Scholar]
- Speidel, D., A. Salehi, S. Obermueller, I. Lundquist, N. Brose, E. Renstrom, and P. Rorsman. 2008. CAPS1 and CAPS2 regulate stability and recruitment of insulin granules in mouse pancreatic beta cells. Cell Metab. 7:57–67. [DOI] [PubMed] [Google Scholar]
- Spudich, A., and D. Braunstein. 1995. Large secretory structures at the cell surface imaged with scanning force microscopy. Proc. Natl. Acad. Sci. USA. 92:6976–6980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorngren, N., K.M. Collins, R.A. Fratti, W. Wickner, and A.J. Merz. 2004. A soluble SNARE drives rapid docking, bypassing ATP and Sec17/18p for vacuole fusion. EMBO J. 23:2765–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togo, T., J.M. Alderton, G.Q. Bi, and R.A. Steinhardt. 1999. The mechanism of facilitated cell membrane resealing. J. Cell Sci. 112:719–731. [DOI] [PubMed] [Google Scholar]
- Wang, P., M.C. Chicka, A. Bhalla, D.A. Richards, and E.R. Chapman. 2005. Synaptotagmin VII is targeted to secretory organelles in PC12 cells, where it functions as a high-affinity calcium sensor. Mol. Cell. Biol. 25:8693–8702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteheart, S.W., K. Rossnagel, S.A. Buhrow, M. Brunner, R. Jaenicke, and J.E. Rothman. 1994. N-ethylmaleimide-sensitive fusion protein: a trimeric ATPase whose hydrolysis of ATP is required for membrane fusion. J. Cell Biol. 126:945–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, R.M., J.B. Shear, W.R. Zipfel, S. Maiti, and W.W. Webb. 1999. Mucosal mast cell secretion processes imaged using three-photon microscopy of 5-hydroxytryptamine autofluorescence. Biophys. J. 76:1835–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, T., T. Binz, H. Niemann, and E. Neher. 1998. Multiple kinetic components of exocytosis distinguished by neurotoxin sensitivity. Nat. Neurosci. 1:192–200. [DOI] [PubMed] [Google Scholar]
- Xu, T., U. Ashery, R.D. Burgoyne, and E. Neher. 1999. Early requirement for α-SNAP and NSF in the secretory cascade in chromaffin cells. EMBO J. 18:3293–3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaradanakul, A., T.M. Wang, V. Lariccia, M.J. Lin, C. Shen, X. Liu, and D.W. Hilgemann. 2008. Massive Ca-induced membrane fusion and phospholipid changes triggered by reverse Na/Ca exchange in BHK fibroblasts. J. Gen. Physiol. 132:29–50. [DOI] [PMC free article] [PubMed] [Google Scholar]