

Abstract
Trichloroethene (TCE) exacerbates the development of autoimmune responses in autoimmune-prone MRL +/+ mice. Although TCE-mediated autoimmune responses are associated with an increase in serum immunoglobulins and autoantibodies, the underlying mechanism of autoimmunity is not known. To determine the progression of TCE-mediated immunotoxicity, female MRL +/+ mice were chronically exposed to TCE through the drinking water (0.5 mg/ml of TCE) for various periods of time. Serum concentrations of antinuclear antibodies increased after 36 and 48 weeks of TCE exposure. Histopathological analyses showed lymphocyte infiltration in the livers of MRL +/+ mice exposed to TCE for 36 or 48 weeks. Lymphocyte infiltration was also apparent in the pancreas, lungs, and kidneys of mice exposed to TCE for 48 weeks. Immunoglobulin deposits in kidney glomeruli were found after 48 weeks of exposure to TCE. Our results suggest that chronic exposure to TCE promotes inflammation in the liver, pancreas, lungs, and kidneys, which may lead to SLE-like disease in MRL +/+ mice.
Keywords: Trichloroethene (TCE), MRL +/+ mice, Cytokines, Immunotoxicity, Autoimmunity, Hepatitis
Introduction
Trichloroethene (trichloroethylene, TCE), a chlorinated ethene commonly used as an industrial solvent for degreasing metals, is a ubiquitous environmental contaminant (Landrigan et al.; 1987; Cohn et al., 1994). Environmental and occupational exposure to TCE has been linked to the development of autoimmune diseases, such as systemic lupus erythematosus (SLE), scleroderma, systemic sclerosis, and fasciitis (Lagakos et al., 1986; Byers et al., 1988; Kilburn and Warshaw, 1992; Waller et al., 1994; Reinl, 1957; Saihan et al., 1978; Phoon et al., 1984; Flindt-Hansen and Isager, 1987; Lockey et al., 1987; Brasington and Thorpe-Swenson, 1991; Goon et al., 2001; Brautbar, 2004).
An animal model, female autoimmune-prone MRL +/+ mice, to study the potential of TCE to induce/exacerbate autoimmunity was developed in our laboratory (Khan et al., 1995). We hypothesize that TCE reactive metabolites bind to self-proteins to form protein adducts, which then escape normal tolerance to self-proteins and act as neo-antigens, elicit autoreactive T cells resulting in B cell activation, induce autoimmune responses, and eventually lead to autoimmune diseases similar to SLE. This hypothesis is supported by our studies in MRL +/+ mice, which provided evidence that TCE induces or accelerates autoimmune responses (Khan et al., 1995), and that treatment with a reactive TCE metabolite, dichloroacetyl chloride (DCAC), increases serum levels of total IgG, DCAC-specific antibodies, and antinuclear antibodies, which is a measure of systemic autoimmunity (Khan et al., 1997; Cai et al., 2006). Protein-adducts of reactive metabolites of TCE also cause autoimmunity in MRL +/+ mice (Cai et al., 2007). However, very little is known about the effects of chronic exposure to TCE on autoimmune responses. Here, using our in vivo model, we show that TCE-accelerated autoimmunity in female MRL +/+ mice is associated with characteristics of SLE-like disease such as increased antinuclear antibodies, activated T cells, multi-organ infiltration of lymphocytes, and immunoglobulin deposits in kidney glomeruli.
Materials and methods
Animal treatment
Five-week-old female MRL +/+ mice (body weight 23 to 26g) were purchased from the Jackson Laboratory (Bar Harbor, ME) and acclimatized in the University of Texas Medical Branch’s (UTMB) humidity- and temperature-controlled animal care facility, which is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International, for one week and provided lab chow and drinking water ad libitum. All experiments were performed in accordance with the guidelines of the National Institutes of Health and were approved by the Institutional Animal Care and Use Committee of UTMB. Mice were randomly divided into groups of five, representing the control and the treatment groups. Trichloroethene (purity > 99.5%), obtained from Sigma-Aldrich (St. Louis, MO), was suspended in drinking water at a concentration of 0.5 mg/ml using the emulsifier Alkamuls EL-620 (Rhodia Chemicals, Cranbury, NJ) at 1% (w/v). This dose corresponds to the Occupational Safety and Health Administration’s permissible exposure limit of 100 ppm (~ 0.4 mg/ml) for an eight-hour exposure. The water was changed twice weekly. The average water intake was ~ 0.13 ml/g/d for controls and ~ 0.12 ml/g/d for TCE-treated mice, representing an average TCE intake of ~ 60 µg/g/d in the treatment groups. At the termination of each experiment, mice were euthanized and blood was withdrawn from the heart. Major organs (liver, pancreas, large intestine, heart, lungs, kidneys, and spleen) were removed immediately, and fixed in neutral buffered formalin. A portion of the spleen was used for the isolation of splenocytes (Cai et al., 2006). Serum was isolated from the blood and stored in small aliquots at −80°C for further analysis.
Serum aminotransferases
Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were measured using kits from Biotron Diagnostics (Hemet, CA) with slight modification of the protocol suitable for small amounts of serum (12.5 µl) (Cai et al., 2007). OD values were read using a microplate reader (Bio-Rad, Hercules, CA).
Antinuclear antibodies
Antinuclear antibodies were measured using a semi-quantitative screening kit from The Binding Site. The diluted sera (1:10) were first incubated in microwells precoated with purified antigens for dsDNA, histones, SSA/Ro (52 and 60kD), SSB/La, Sm, Sm/RNP, Scl-70, Jo-1, and Centromere B for 30 min, followed by washing and a 30-min incubation with goat anti-mouse IgG antibodies (Fc-specific, 1:2000 dilution, from Bethyl) conjugated to horseradish peroxidase. The substrate, 3,3,5,5-tetra-methyl-benzidine (TBM, from Bethyl) was then added for 15 min, and the absorbance was measured at 450 nm using a microplate reader (Bio-Rad) (Cai et al., 2006).
Serum cytokines
The T helper type I (Th1) cytokines IL-1α, IL-6, GM-CSF and TNF-α; and Th2 cytokines IL-2, IL-4, IL-5, IL-10, IL-12 (p40/p70) and IFN-γ and G-CSF and KC chemokines were measured in the serum of mice treated with TCE and age-matched controls in duplicate using multiplex immunoassay kits from Biosource (Camarillo, CA). The fluorescence was measured using a Luminex-100 luminometer (Bio-Rad) (Cai et al., 2006, 2007).
Cytokines secreted by splenocytes ex vivo
Splenocytes were isolated from control and treated mice and cultured (Cai et al., 2006). Splenocyte supernatants were harvested after 48 h in medium alone, or medium with 25 µg/ml of dichloroacetylated-albumin or unmodified mouse albumin. Dichloroacetylated albumin was synthesized as described (Cai et al., 2007). Cytokines (IL-2, IL-4, IL-5, IL-10, IL-12, IFN-γ, GM-CSF, and TNF-α) in the supernatants were measured using a multiplex assay as described (Cai et al., 2006).
Histopathology
The liver, pancreas, spleen, heart, lungs, kidneys, and the large intestine, were fixed in 10% neutral-buffered formalin, and stained with hematoxylin and eosin (H & E) for morphological evaluation. For histological scoring, H & E-stained liver, kidney, lung, and pancreatic sections were randomized, blinded, and given to a pathologist for histological assessment and scoring of the lesions. After initial review of all the specimens, a grading system for the lesions was devised based on lymphocytic infiltration. Liver: Grade 0 = no change; Grade 1 = focal periportal lymphocytic infiltration; Grade 2 = infiltration surrounding portal and central veins; Grade 3 = diffuse periportal and central vein infiltration with areas of mild hepatocellular necrosis or apoptosis. Kidney: Grade 0 = no change; Grade 1 = minimal perivasular infiltration; Grade 2 = perivascular, periglomerular, and focal interstitial infiltration; Grade 3 = diffuse perivascular, periglomerular, and interstitial infiltration. Lung: Grade 0 = no change; Grade 1 = minimal perivascular infiltration; Grade 2 = perivascular and peribronchial infiltration; Grade 3 = diffuse perivascular, peribronchiolar, and interstitial infiltration. Pancreas: Grade 0 = no change; Grade 1 = perivascular and focal periductal infiltration; Grade 2 = acinar infiltration; Grade 3 = acinar infiltration with atrophy. Anti-CD3 antibody-staining: Formalin-fixed, paraffin-embedded sections were also stained for the presence of T cells using a polyclonal rabbit anti-mouse CD3 antibody (Dako, Carpinteria, CA). Target retrieval solution (pH 9.0) (Dako) was used for heat-induced target retrieval on formalin-fixed, paraffin-embedded tissue sections mounted on glass slides prior to immunohistochemical staining procedures. Sections of the spleen were used as a positive control for T cell-staining.
Detection of proliferating cell nuclear antigen (PCNA)
Formalin-fixed, paraffin-embedded sections of the liver, lung, and kidney were stained for the presence of PCNA using a biotinylated anti-PCNA antibody (ZYMED Laboratories Inc., San Francisco, CA) according to the manufacturer’s instructions. Positive hepatocyte nuclei were counted under a microscope (Nikon, Japan) at 200x magnification. Four fields per slide were counted for each sample.
Statistical analysis
The data are presented as mean ± standard error of the mean (SEM). For determining statistical significance, the data were subjected to a two-tailed Student’s t-test and p values < 0.05 were considered statistically significant. When appropriate, analysis of variance (ANOVA) followed by Student-Newman-Keuls post-hoc test was performed.
Results
Water consumption
No statistically significant difference was observed in the consumption of water between untreated and TCE-treated MRL +/+ mice (data not shown).
Body weight gain
To determine if prolonged exposure to TCE via the drinking water affected the general health of MRL +/+ mice, body weight was recorded. After eleven weeks of TCE treatment, a statistically significant decrease in body weight gain (26% lower than age-matched, controls) was observed. For the remainder of the experimental exposure (total exposure time of 48 weeks), the average body weight gain of treated MRL +/+ mice was lower than that of control mice, but the difference did not reach statistical significance (data not shown).
Serum aminotransferases
To determine whether long-term exposure to TCE induced liver injury, we measured serum levels of aminotransferases. Serum ALT and AST enzyme activities in the TCE-exposed mice and did not differ from age-matched controls (data not shown).
Serum antinuclear antibodies
As a measure of systemic autoimmunity, we determined serum levels of antinuclear antibodies (ANA). The average levels of ANA in sera from both control and TCE-treated MRL +/+ mice were < 10 U/ml for the first 11 weeks of exposure. After 24 weeks of TCE serum ANA was elevated, but statistical significance in comparison to the age-matched control mice was not achieved. In 42- and 54-week-old mice (36 weeks and 48 weeks of exposure to TCE, respectively), antinuclear antibodies were increased in both control and TCE-treated MRL +/+ mice but was always higher on the average in TCE-treated mice (Fig. 1).
Figure 1.
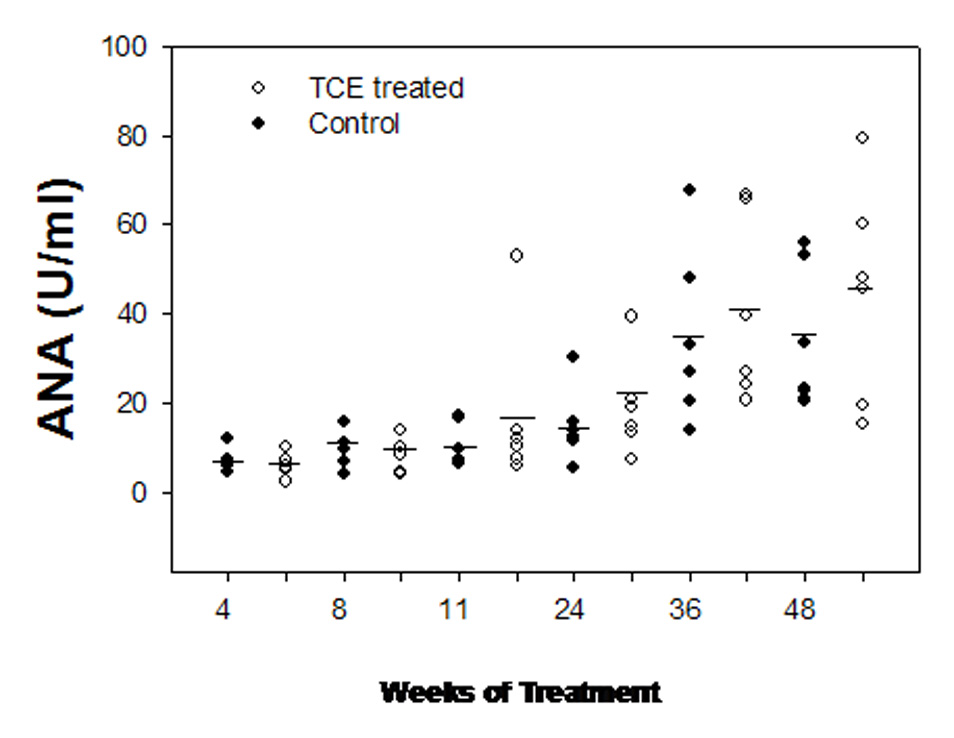
Antinuclear antibodies (ANA) in the serum of female MRL +/+ mice exposed to vehicle (filled circles) or TCE (open circles) in the drinking water. Each circle is an individual measurement and the horizontal bars indicate the means (n = 6).
Serum cytokines
To determine whether TCE exposure would lead to a general inflammatory condition, we measured serum levels of several cytokines and chemokines (see materials and methods for list of cytokines). The inflammatory cytokine IL-6 was decreased after 36 and 48 weeks of TCE exposure, and TNF-α, another inflammatory cytokine, was decreased after 48 weeks. Conversely, G-CSF was increased after 36 weeks of exposure to TCE (data not shown).
Cytokine secretion by splenocytes ex vivo
To determine whether exposure to TCE induced the differentiation of specific T cell subsets and promoted activation of T cells specific for haptenized self-proteins, isolated splenocytes from TCE-treated and age-matched control MRL +/+ mice were cultured in the absence or the presence of 25 µg/ml of dichloroacetylated-albumin or unmodified albumin. After 48 h, culture supernatants were collected and cytokine levels measured. In mice treated for 36 weeks with TCE, splenocytes from three out of five mice secreted higher levels of IFN-γ than did splenocytes from the control mice (Fig. 2). The differences did not achieve statistical significance due to the large variance within the treatment group.
Figure 2.
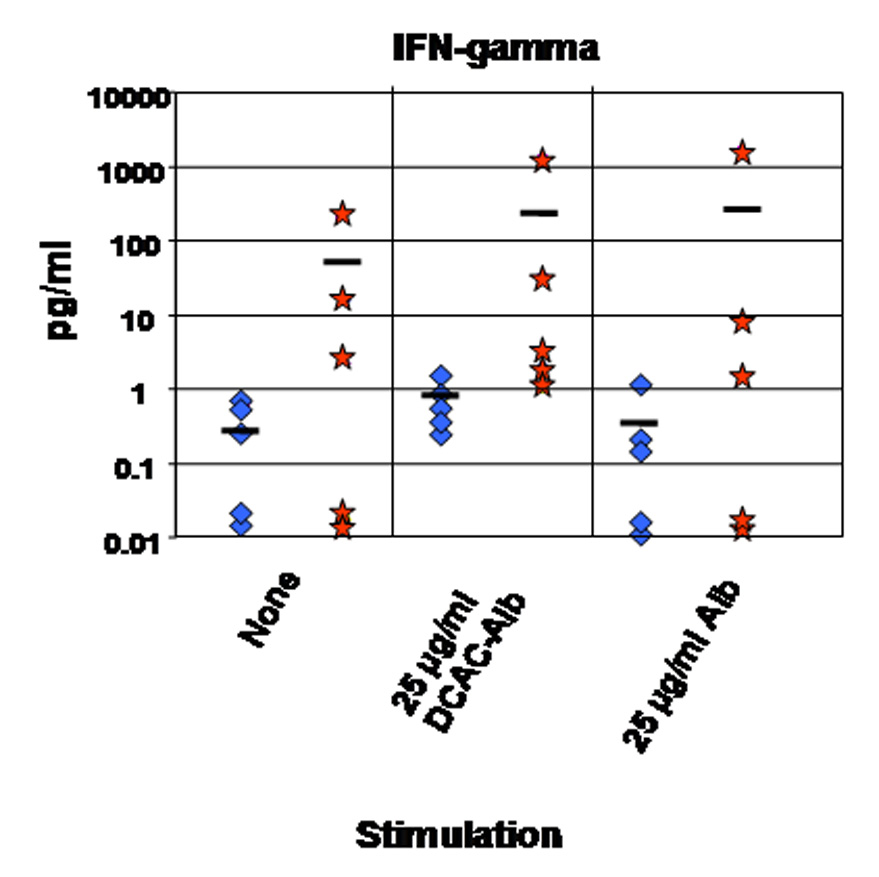
IFN-γ secretion by ex vivo cultured splenocytes from untreated, age-matched control MRL +/+ mice (diamonds) or MRL +/+ mice treated with TCE for 36 weeks (stars). IFN-γ was measured in supernatants after 48 h of culture without stimulation (none) or following stimulation with 25 µg/ml of dichloroacetylated-albumin (DCAC-Alb) or unmodified albumin (Alb). The horizontal bars indicate the means (n = 5).
Liver histopathology and hepatocyte proliferation
TCE exposure to (0.5 or 2.5 mg/ml of TCE in drinking water) in MRL +/+ mice for 32 weeks induced histopathological changes similar to autoimmune hepatitis (Griffin et al., 2000). Recently, we found that exposure to formyl-albumin (putative protein adduct resulting from TCE metabolism) causes autoimmune hepatitis in MRL +/+ mice (Cai et al., 2007). Therefore, we investigated whether long-term TCE exposure altered liver histopathology in MRL +/+ mice. We found that exposure to TCE for 36 and 48 weeks caused hepatocyte necrosis and leukocyte infiltration into liver lobules (shown for 48-week exposure in Fig. 3D). These changes were statistically significant compared to observations in control mice (Table 1). Leukocyte infiltration included CD3+ T lymphocytes and also differed significantly between TCE-treated and control mice (Fig. 3F and Table 2). After 48 weeks of continuous exposure to TCE, hepatocyte proliferation was also increased in treated MRL +/+ mice as determined by PCNA-staining. PCNA is a marker of S-phase, and thus can serve as a measure for hepatocellular proliferation (Brown et al., 2006). In treated mice, the number of PCNA-positive nuclei was significantly increased compared to age-matched control MRL +/+ mice (Fig. 3A and B, and Table 2).
Figure 3.
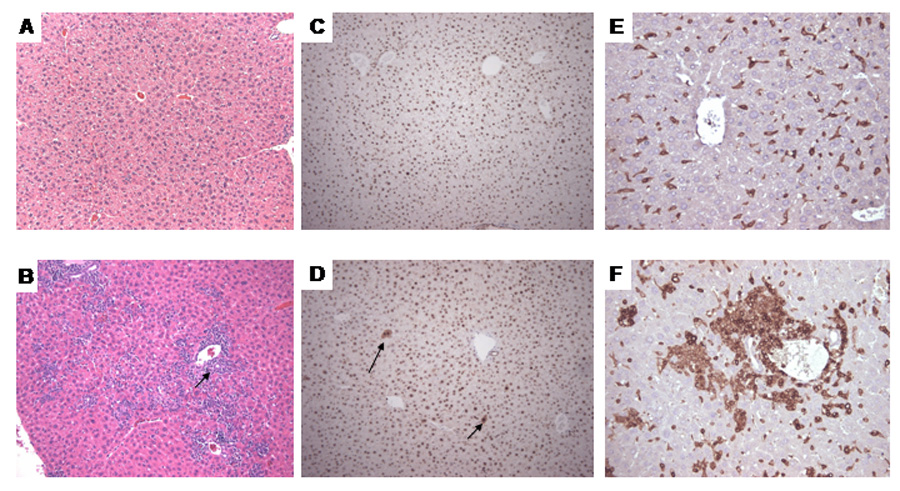
Morphological changes in the liver of female MRL +/+ mice treated with TCE for 48 weeks. A. Control, H&E stain; B. TCE-treated: the arrow indicates inflammatory infiltration of portal triads extending into lobules; also present was focal piece-meal necrosis of hepatocytes; C. Control PCNA stain; D. TCE-treated: the arrows indicate PCNA-positive hepatocellular nuclei. E. Control CD3-stain; F. TCE-treated: note the infiltrate of CD3-positive lymphocytes. (A–D, x100; E–F, x200).
Table 1.
Histopathological changes in tissues of MRL+/+ mice treated with TCE for 48 weeks.
| Control | Treated | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample | 1 | 2 | 3 | 4 | 5 | 6 | Avg±SEM | 7 | 8 | 9 | 10 | 11 | 12 | Avg±SEM | P value * |
| Tissue | |||||||||||||||
| Liver | 0 | 0 | 0 | 0 | 1 | 0 | 0.16±0.16 | 0 | 1 | 3 | 2 | 1 | 1 | 1.33±0.42 | < 0.05 |
| Kidney | 1 | 1 | 1 | 1 | 2 | 2 | 1.33±0.21 | 3 | 3 | 2 | 3 | 3 | 2 | 2.66±0.21 | < 0.05 |
| Lung | 0 | 1 | 1 | 0 | 1 | 0 | 0.5±0.22 | 1 | 1 | 2 | 3 | 3 | 1 | 1.83±0.40 | < 0.05 |
| Pancreas | 0 | 2 | 1 | 0 | 1 | 2 | 1.00±0.36 | 2 | 2 | 1 | 3 | 3 | 0 | 1.83±0.47 | n.s |
See methods for histological scoring.
Significant when treated samples were compared to age-matched controls by ANOVA.
n.s Statistically not significant as compared to controls.
Table 2.
Immunological (PCNA and CD3) staining in tissues of MRL+/+ mice treated with TCE for 48 weeks.
| Control | Treated | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CD3 | |||||||||||||||
| Sample | 1 | 2 | 3 | 4 | 5 | Avg±SEM | 7 | 8 | 9 | 10 | 11 | Avg±SEM | P value * | ||
| Tissue | |||||||||||||||
| Liver | 0 | 0 | 0 | 0 | 1 | 0.20±0.20 | 1 | 3 | 3 | 2 | 0 | 1.80±0.58 | < 0.05 | ||
| Kidney | 1 | 2 | 1 | 1 | 1 | 1.20±0.20 | 2 | 3 | 3 | 2 | 1 | 2.20±0.37 | < 0.05 | ||
| Lung | 1 | 1 | 2 | 2 | 0 | 1.20±0.37 | 3 | 2 | 2 | 3 | 1 | 2.20±0.37 | n.s | ||
| Pancreas | 0 | 1 | 0 | 0 | 0 | 0.20±0.20 | 2 | 1 | 1 | 3 | 0 | 1.40±0.50 | < 0.05 | ||
| PCNA | |||||||||||||||
| Sample | 1 | 2 | 3 | 4 | 5 | 6 | Avg±SEM | 7 | 8 | 9 | 10 | 11 | 12 | Avg±SEM | P value * |
| Tissue | |||||||||||||||
| Liver | 0 | 0 | 1 | 0 | 0 | 0 | 0.16±0.16 | 0 | 1 | 2 | 3 | 3 | 1 | 1.66±0.49 | < 0.05 |
| Kidney | 1 | 1 | 0 | 1 | 1 | 1 | 0.83±0.16 | 2 | 3 | 2 | 2 | 2 | 2 | 2.16±0.16 | < 0.05 |
| Lung | 1 | 0 | 1 | 2 | 1 | 0 | 0.83±0.30 | 2 | 2 | 1 | 2 | 3 | 2 | 2.00±0.25 | < 0.05 |
0 = minimal; 1 = mild; 2 = moderate; 3 = severe infiltration.
Significant when treated samples were compared to age-matched controls by ANOVA.
n.s: Statistically not significant as compared to controls.
Histopathology of pancreas, lungs, and kidneys
We previously reported that a six-week treatment with DCAC caused thickening of the alveolar septa and lymphocytic interstitial infiltrates in the lungs of MRL +/+ mice (Cai et al., 2006). Therefore, we investigated the toxic effects of long-term exposure to TCE on various organs and tissues (pancreas, large intestine, heart, lungs, and kidneys). In MRL +/+ mice treated with TCE for 48 weeks, necrosis and extensive infiltration of leukocytes, including CD3+ T lymphocytes, in the pancreas were noticed. However, in two out of six age-matched, untreated control mice, we found spontaneously occurring autoimmune pancreatitis, albeit to a lesser extent than in TCE-treated mice, and thus, statistical significance could not be achieved (Table 2).
Only in mice treated for 48 weeks with TCE, but not in age-matched controls, leukocytes including CD3+ T lymphocytes infiltrated the perivascular and peribronchial regions of the lungs (Fig. 4 and Table 1 and Table 2). In addition, we observed thickening of the alveolar septa in lungs of TCE-exposed mice. Exposure to TCE for 36 or 48 weeks also induced massive perivascular infiltration of leukocytes, including CD3+ T lymphocytes, in the kidneys of MRL +/+ mice (shown for 48-week exposure in Fig. 5; statistics are shown in Table 1 and Table 2). We also found intense immunoglobulin deposits in the glomeruli of MRL+/+ mice exposed to TCE for 48 weeks (Fig. 6). No histopathological changes were found in the large intestine or the heart (data not shown).
Figure 4.
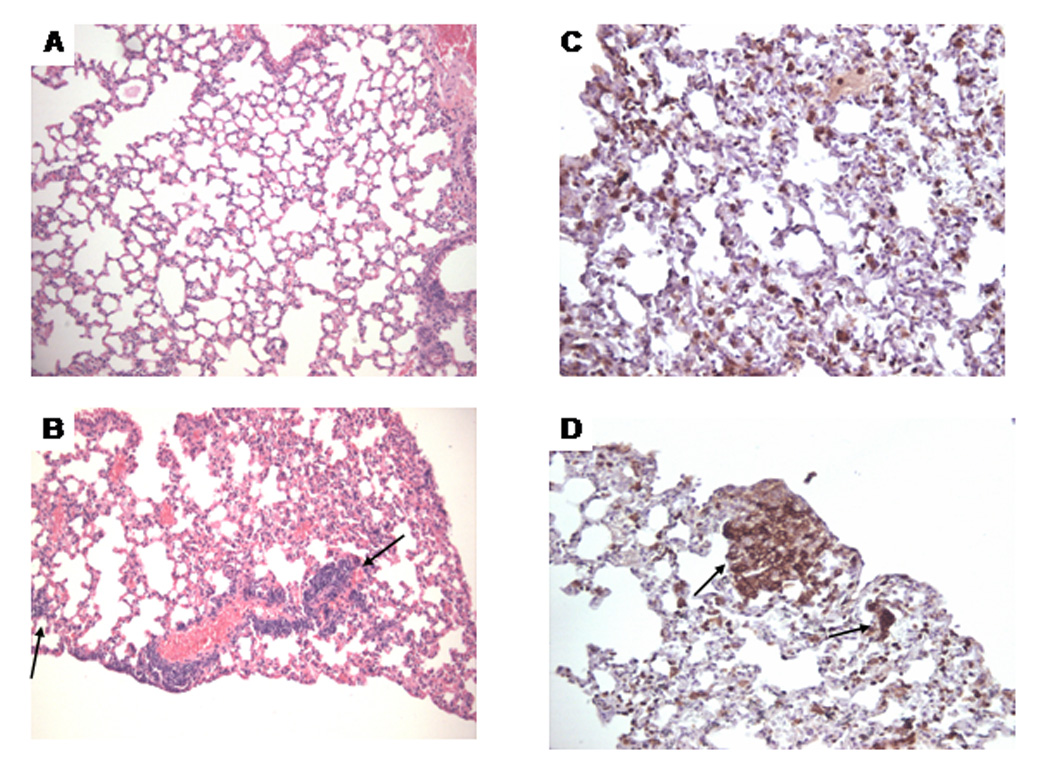
Morphological changes in the lungs of MRL +/+ mice treated with TCE for 48 weeks. A. Control, H&E stain; B. TCE-treated: the arrow indicates perivascular inflammatory infiltrates. C. Control CD3-stain; D. TCE-treated: the arrow indicates CD3-positive lymphocytes (A–B, x100; C–D, x200).
Figure 5.
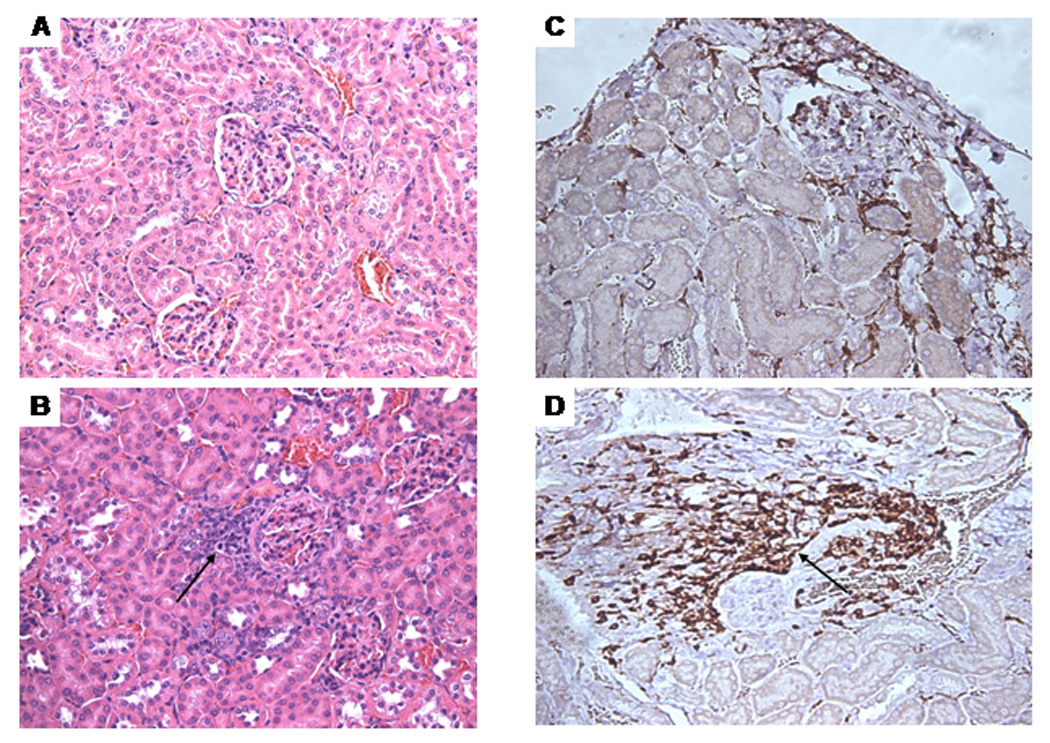
Morphological changes in the kidneys of MRL +/+ mice treated with TCE for 48 weeks. A. Control, H&E stain. B. TCE-treated: the arrow indicates perivascular inflammatory infiltrates. C. Control, CD3-stain. D. TCE-treated: the arrow indicates CD3-positive lymphocytes (A–D, x200).
Figure 6.
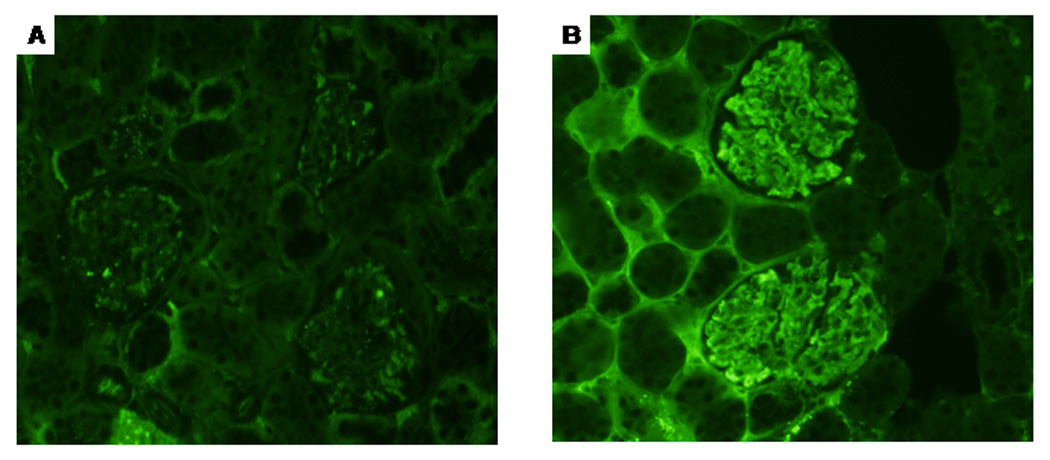
Immunoglobulin deposits in the kidney glomeruli (in the mesangial areas and the walls of adjacent capillaries) of mice treated with TCE for 48 weeks (B). In contrast, vehicle control-treated mice did not show immunoglobulin deposits in the glomeruli (A).
Discussion
In this report, we demonstrate that chronic TCE exposure may lead to SLE-like disease. The disease may be caused by reactive metabolites (intermediates) of TCE (Khan et al., 1997; Cai et al., 2006, 2007). TCE can be metabolized either oxidatively or through glutathione conjugation pathways. The glutathione-mediated metabolism is of minor significance in humans (Bloemen et al., 2001), and thus, we focused on the oxidative metabolic pathway. TCE can be oxidized to TCE-oxide, which can either rearrange to DCAC or be hydrolyzed to dichloroacetic acid. In aqueous solution, TCE-oxide forms lysine adducts with proteins (Cai and Guengerich, 1999, 2000). As small molecules, TCE or its metabolites will not be antigenic by themselves, but may haptenize to self-proteins, and thus result in structural modifications that render self-proteins antigenic, potentially leading to autoimmune responses. A trifluoro-analogue of DCAC (trifluoroacylchloride) has been shown to acylate proteins, producing antigens involved in halothane-induced hepatitis (Martin et al., 1991; Phol et al., 1991; Kenna et al., 1993; Chrsiten et al., 1994) and also, 2,2-dichlorotrifluoroethane-related liver disease (Hoet et al., 1997; White et al. 2001). Similarly, covalent binding of TCE in vivo has been observed in rodents (Bolt et al., 1977; Bolt and Filser, 1977; Stot et al., 1982; Mazzullo et al., 1992; Stevens et al., 1992; Kautiainen et al., 1997). TCE-oxide and DCAC can covalently modify proteins directly to form protein adducts (Bloemen et al., 2001; Halmes et al., 1996, 1997).
Recently, we have shown that reactive intermediates of TCE induce autoimmune responses similar to TCE (Cai et al., 2007). Here, we demonstrate chronic autoimmune effects of TCE similar to SLE-like disease. Specifically, we observed increasing antinuclear antibodies and ex vivo IFN-γ secretion by lymphocytes, multi-organ infiltration of lymphocytes, and immunocomplex deposition in kidney glomeruli, thus mimicking SLE.
Histopathological changes in liver, lung, and kidneys indicated accelerated autoimmunity in TCE-treated MRL +/+ mice as shown in Table 1. Autoimmune hepatitis, manifested by an infiltration of CD3+ lymphocytes, was observed after 36 weeks of treatment, similar to an earlier report (Griffin et al., 2000). In addition, PCNA-positive nuclei indicated hepatocellular proliferation (Brown et al., 2006). The observed autoimmune hepatitis could be a result of the formation of formyl-protein adducts, which caused autoimmune hepatitis (Cai et al., 2007). The infiltration of lymphocytes into the lungs could also be due to the formation of dichloroacetyl-protein adducts. This hypothesis is supported by Forkert et al. (2006), who showed that TCE-induced toxicity was mediated via dichloroacetylated protein adducts.
Although we observed an increase in the occurrence of autoimmune pancreatitis following 36 and 48 weeks of TCE treatment, this difference was not statistically significant because of spontaneously occurring autoimmune pancreatitis in age-matched control mice. A previous report also suggested that MRL +/+ mice spontaneously develop autoimmune pancreatitis after seven months of age (Kahno et al., 1992).
Importantly, we found perivascular infiltration of CD3+ lymphocytes in the kidneys and a strong deposition of immunoglobulins in the glomeruli in TCE-treated MRL +/+ mice, but not in untreated, age-matched controls. This finding is consistent with nephritis. Autoimmune nephritis together with multi-organ involvement and an increased level of antinuclear antibodies suggested that chronic exposure to TCE induced SLE-like disease. This observation supports the finding that SLE occurs in increased frequency in humans exposed to TCE through contaminated drinking water (Kilburn and Warshaw, 1992).
Although the data did not achieve statistical significance due to the large variance in TCE-treated samples, it is interesting that splenocytes from TCE-exposed MRL +/+ mice secreted more IFN-γ than did splenocytes from untreated, age-matched controls. Increasing secretion of IFN-γ was observed in unstimulated cultures as well as in cultures stimulated with dichloroacetylated or unmodified albumin. In particular, two of the treated mice achieved very high levels of IFN-γ, levels of a magnitude that could only be achieved in splenocytes from untreated control mice after stimulation with antibodies against CD3 and CD28 (not shown). This suggests that in some of the TCE-treated mice, T cells have the tendency to differentiate into an inflammatory phenotype, supporting the histopathological changes observed in several organs. In this context, it is important that in humans occupationally exposed to TCE, serum IFN-γ is increased (Iavicoli et al., 2005), and IFN-γ transgenic mice spontaneously develop SLE-like disease (Seery, 2000).
In conclusion, our data demonstrate that chronic exposure to TCE promoted systemic and organ-specific autoimmunity involving activation and infiltration of inflammatory lymphocytes. The present findings and earlier work from our laboratory (Khan et al., 1995, 1997; Cai et al., 2006, 2007) support the hypothesis that chronic exposure to TCE can cause autoimmune disease similar to SLE in female MRL +/+ mice.
Acknowledgments
This publication was made possible by grant ES11584 from the National Institute of Environmental Health Sciences (NIEHS), and its content are solely the responsibility of the authors and do not necessarily represent the views of the NIH or NIEHS. We gratefully acknowledge the Organic Synthesis Core at UTMB for the preparation of dichloroacetyl-albumin adducts supported by NIEHS center grant, P30ES06676.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bloemen LT, Monster AC, Kezic S, et al. Study on the cytochrome P-450- and Glutathione-dependent biotransformation of trichloroethylene in humans. Int. Arch. Occup. Environ. Health. 2001;74:102–108. doi: 10.1007/s004200000198. [DOI] [PubMed] [Google Scholar]
- Bolt HM, Buchter A, Wolowski L, Gil DL, Bolt W. Incubation of 14C-trichloroethylene vapor with rat liver microsomes. Uptake of radioactivity and covalent protein binding of metabolites. Arch. Occup. Environ. Health. 1977;39:103–111. doi: 10.1007/BF00380890. [DOI] [PubMed] [Google Scholar]
- Bolt HM, Filser JG. Irreversible binding of chlorinated ethylenes to macromolecules. Environ. Health Persp. 1977;21:107–112. doi: 10.1289/ehp.7721107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasington RD, Jr, Thorpe-Swenson AJ. Systemic sclerosis associated with cutaneous exposure to solvent: case report and review of the literature. Arthritis Rheum. 1991;34:631–363. doi: 10.1002/art.1780340516. [DOI] [PubMed] [Google Scholar]
- Brautbar N. Industrial solvents and kidney diseases. Int. Occup. Environ. Health. 2004;10:79–83. doi: 10.1179/oeh.2004.10.1.79. [DOI] [PubMed] [Google Scholar]
- Brown KE, Mathahs MM, Broadhurst KA, Weydert J. Chronic iron overload stimulates hepatocyte proliferation and cyclin D1 expression in rodent liver. Transl. Res. 2006;148:55–62. doi: 10.1016/j.trsl.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Byers VS, Levin AS, Ozonoff DM, Baldwin RW. Association between clinical symptoms and lymphocyte abnormalities in a population with chronic domestic exposure to industrial solvent contaminated domestic water supply and a high incidence of leukemia. Cancer Immunol. Immunother. 1988;27:77–81. doi: 10.1007/BF00205762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Guengerich P. Mechanism of aqueous decomposition of trichloroethylene oxide. J. Am. Chem. Soc. 1999;121:1165–11663. [Google Scholar]
- Cai H, Guengerich FP. Acylation of protein lysines by trichloroethylene oxide. Chem. Res. Toxicol. 2000;13:327–335. doi: 10.1021/tx000003p. [DOI] [PubMed] [Google Scholar]
- Cai P, Khan MF, Kaphalia BS, Ansari GAS. Immunotoxic response of oleic acid anilide and its hydrolysis products in female MRL +/+ mice. J. Immunotox. 2005;2:231–236. doi: 10.1080/15476910500362960. [DOI] [PubMed] [Google Scholar]
- Cai P, König R, Khan MF, Kaphalia BS, Ansari GAS. Immunotoxocity of dichloroacetyl chloride and dichloroacetic anhydride in MRL +/+ mice. Toxicol. Appl. Pharmacol. 2006;216:248–255. doi: 10.1016/j.taap.2006.05.010. [DOI] [PubMed] [Google Scholar]
- Cai P, König R, Khan MF, Kaphalia BS, Ansari GAS. Differential immune responses to albumin adducts of reactive intermediates of trichloroethene in MRL +/+ mice. Toxicol. Appl. Pharmacol. 2007;220:278–283. doi: 10.1016/j.taap.2007.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christen U, Quinn J, Yeaman SJ, Kenna JA, Clark JB, Gandolfi AJ, Gut J. Identification of the dihydrolipoamide acetyltransferase subunit of the human pyruvate dehydrogenase complex as an autoantigen in halothane hepatitis: Molecular mimicry of trifluoroacetyl-lysine by lipoic acid. Eur. J. Biochem. 1994;223:1035–1048. doi: 10.1111/j.1432-1033.1994.tb19082.x. [DOI] [PubMed] [Google Scholar]
- Cohn P, Klotz J, Bove F, Berkowitz M, Fagliano J. Drinking water contamination and incidence of leukemia and non-Hodgkin's lymphoma. Environ. Health Perspect. 1994;102:556–561. doi: 10.1289/ehp.94102556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flindt-Hansen H, Isager H. Scleroderma after occupational exposure to trichloroethylene and trichloroethane. Acta Derm. Venereol. 1987;67:263–264. [PubMed] [Google Scholar]
- Forkert P-G, Millen B, Lash LH, Putt DA, Ghanayem BI. Pulmonary bronchiolar cytotoxicity and formation of dichloroacetyl lysine protein adducts in mice treated with trichloroethylene. J. Pharmacol. Expt. Ther. 2006;316:520–529. doi: 10.1124/jpet.105.093062. [DOI] [PubMed] [Google Scholar]
- Goon AT, Lee L-T, Tay Y-K, et al. A case of trichloroethylene hypersensitivity syndrome. Arch. Dermatol. 2001;137:274–276. [PubMed] [Google Scholar]
- Griffin JM, Gilbert KM, Lambs LW, Pumford NP. CD4+ T-cell activation and induction of autoimmune hepatitis following trichloroethylene treatment in MRL +/+ mice. Toxicol. Sci. 2000;57:345–352. doi: 10.1093/toxsci/57.2.345. [DOI] [PubMed] [Google Scholar]
- Halmes NC, McMillan DC, Oatis JE, Jr, Pumford NR. Immunochemical detection of protein adducts in mice treated with trichloroethylene. Chem. Res. Toxicol. 1996;9:451–456. doi: 10.1021/tx950171v. [DOI] [PubMed] [Google Scholar]
- Halmes NC, Samokyszn NC, Pumford NR. Covalent binding and inhibition of cytochrome P4502E1 by trichloroethylene. Xenobiotica. 1997;27:101–110. doi: 10.1080/004982597240794. [DOI] [PubMed] [Google Scholar]
- Hoet P, Graf ML, Bourdi M, Pohl LR, Duray PH, Chen W, Peter RM, Nelson SD, Verlinden N, Lison D. Epidemic of liver disease caused by hydrochlorofluorocarbons used as ozone-sparing substitutes of chlorofloourocarbons. Lancet. 1997;350:556–559. doi: 10.1016/S0140-6736(97)03094-8. [DOI] [PubMed] [Google Scholar]
- Iavicoli I, Marinaccio A, Carelli G. Effects of occupational trichloroethylene exposure on cytokine levels in workers. J. Occup. Environ. Med. 2005;47:453–457. doi: 10.1097/01.jom.0000161728.23285.66. [DOI] [PubMed] [Google Scholar]
- Kanno H, Nose M, Itoh J, Taniguchi Y, Kyogoku M. Spontaneous development of pancreatitis in the MRL/Mp strain of mice in autoimmune mechanism. Clin. Exp. Immunol. 1992;89:68–73. doi: 10.1111/j.1365-2249.1992.tb06879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kautiainen A, Vogel JS, Turteltaub KW. Dose-dependent binding of trichloroethylene to hepatic DNA and protein at low doses in mice. Chem. Biol. Inter. 1997;106:109–121. doi: 10.1016/s0009-2797(97)00061-6. [DOI] [PubMed] [Google Scholar]
- Kenna JG, Knight LT, Van Pet FNAM. Immunity to halothane metabolite-modified proteins in halothane hepatitis. Ann. New York Acad. Sci. 1993;85:646–661. doi: 10.1111/j.1749-6632.1993.tb35930.x. [DOI] [PubMed] [Google Scholar]
- Khan MF, Kaphalia BS, Prabhakar BS, Kanz MF, Ansari GAS. Trichloroethene-induced autoimmune response in female MRL +/+ mice. Toxicol. Appl. Pharmacol. 1995;134:155–160. doi: 10.1006/taap.1995.1179. [DOI] [PubMed] [Google Scholar]
- Khan MF, Kaphalia BS, Ansari GAS. Time-dependent autoimmune response of dichloroacetyl chloride in female MRL +/+ mice. Immunopharmacol. Immunotoxicol. 1997;19:265–277. doi: 10.3109/08923979709007662. [DOI] [PubMed] [Google Scholar]
- Kilburn KH, Warshaw RH. Prevalence of symptoms of systemic lupus erythematous (SLE) and of fluorescent antinuclear antibodies associated with chronic exposure to trichloroethylene and other chemicals in well water. Environ. Res. 1992;57:1–9. doi: 10.1016/s0013-9351(05)80014-3. [DOI] [PubMed] [Google Scholar]
- Lagakos SW, Wessen BJ, Zelen M. An analysis of contaminated well water and health effects in Woburn, Massachusetts. J. Am. Stat. Assoc. 1986;81:583–596. [Google Scholar]
- Landrigan PJ, Stein GF, Kominsky JR, Ruhe RL, Watanabe AS. Common source community and industrial exposure to trichloroethylene. Arch. Environ. Health. 1987;42:327–332. doi: 10.1080/00039896.1987.9934354. [DOI] [PubMed] [Google Scholar]
- Lockey JE, Kelly CR, Cannon GW, Colby TV, Aldrich V, Livingston GK. Progressive systemic sclerosis associated with exposure to trichloroethylene. J. Occup. Med. 1987;29:493–496. [PubMed] [Google Scholar]
- Martin JL, Pumford NR, LaRossa AC, Martin BM, Gonzaga HM, Beaven MA, Pohl LR. A metabolite of halothane covalently binds to an endoplasmic reticulum protein that is highly homologous to phosphatidylinositol-specific phospholipase c-α but has no activity. Biochem. Biophys. Res. Commun. 1991;178:679–685. doi: 10.1016/0006-291x(91)90161-y. [DOI] [PubMed] [Google Scholar]
- Mazzullo M, Bartoli S, Bonora B, Colacci A, Lattanzi G, Niero A, Silingardi P, Grilli S. In vivo and in vitro interaction of trichloroethylene with macromolecules from various organs of rat and mouse. Res. Commun. Chem. Path. Pharmacol. 1992;76:192–208. [PubMed] [Google Scholar]
- Pohl LR, Thomassen D, Pumford NR, Butler LE, Satoh H, Ferrans VJ, Perrone A, Martin BM, Martin JL. Hapten carrier conjugates associated with halothane hepatitis. Adv. Exp. Biol. Med. 1991;283:111–120. doi: 10.1007/978-1-4684-5877-0_12. [DOI] [PubMed] [Google Scholar]
- Phoon WH, Chan MOY, Rajan VS, Tan KJ, Thirumoorthy T, Goh CL. Steven-Johnson syndrome associated with occupational exposure to trichloroethylene. Contact Derm. 1984;10:270–276. doi: 10.1111/j.1600-0536.1984.tb00145.x. [DOI] [PubMed] [Google Scholar]
- Reinl W. Scleroderma caused by trichloroethylene? Bull. Hygiene. 1957;32:678–679. [PubMed] [Google Scholar]
- Saihan EM, Burton JL, Heaton KW. A new syndrome with pigmentation scleroderma, gynecomastia, Raynaud’s phenomenon and peripheral neuropathy. British J. Dermatol. 1978;99:437–440. doi: 10.1111/j.1365-2133.1978.tb06184.x. [DOI] [PubMed] [Google Scholar]
- Seery JP. IFN-γ transgenic mice: clues to the pathogenesis of systemic lupus erythematosus? Arthritis Res. 2000;2:437–440. doi: 10.1186/ar124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens DK, Eyre RJ, Bull RJ. Adduction of hemoglobin and albumin in vivo by metabolites of trichloroethylene, trichloroacetate, and dichloroacetate in rats and mice. Fundam. Appl. Toxicol. 1992;19:336–342. doi: 10.1016/0272-0590(92)90171-d. [DOI] [PubMed] [Google Scholar]
- Stott WT, Quast JF, Watanabe PG. The pharmacokinetics and macromolecular interactions of trichloroethylene in mice and rats. Toxicol. Appl. Pharmacol. 1982;62:137–151. doi: 10.1016/0041-008x(82)90110-7. [DOI] [PubMed] [Google Scholar]
- Waller PA, Daniel C, Cupps T, Metcalf JS, Richard MS, Leroy EC. Fascitis (not scleroderma) following prolonged exposure to organic solvents (trichloroethylene) J. Rheumatol. 1994;21:1567–1570. [PubMed] [Google Scholar]
- White IN, Razvi N, Givvs AH, Davies AM, Manno M, Zaccaro C, DeMatteis F, Pahler A, Dekant W. Neoantigen formation and clastogenic action of HCFC-123 and perchloroethylene in human MCl-5 cells. Toxicol. Lett. 2001;124:129–138. doi: 10.1016/s0378-4274(00)00281-2. [DOI] [PubMed] [Google Scholar]