

Abstract
BACKGROUND
Opiate dependence is a result of adaptive changes in signal transduction networks in several brain regions. Noradrenergic neurons of the locus coeruleus (LC) have provided a useful model system in which to understand the molecular basis of these adaptive changes. One of most robust signaling adaptations to repeated morphine exposure in this brain region is upregulation of adenylyl cyclase (AC) activity. Earlier work revealed the selective induction of two calmodulin-dependent AC isoforms, AC1 and AC8, after chronic morphine, but their role in opiate dependence has remained unknown.
METHODS
Whole cell recordings from LC slices, behavioural paradigms for dependence, and gene array technology have been used to dissect the role of AC1 and AC8 in chronic morphine responses.
RESULTS
Both AC1 and AC8 knockout mice exhibit reduced opiate dependence based on attenuated withdrawal, however, partially distinct withdrawal symptoms were affected in the two lines. Loss of AC1 or AC8 also attenuated the electrophysiological effects of morphine on LC neurons: knockout of either cyclase attenuated the chronic morphine-induced enhancement of baseline firing rates as well as of regulation of neuronal firing by forskolin (an activator of AC’s). DNA microarray analysis revealed that both AC1 and AC8 affect gene regulation in the LC by chronic morphine and, in addition to common genes, each cyclase influences the expression of a distinct subset of genes.
CONCLUSION
Together, these findings provide fundamentally new insight into the molecular and cellular basis of opiate dependence.
Keywords: drug abuse, DNA microarray, opiate withdrawal, locus coeruleus, forskolin, knockout mice
INTRODUCTION
The locus coeruleus (LC), the major noradrenergic nucleus in brain, has served as a useful model system in which to understand the molecular and cellular mechanisms underlying the long-term effects of opiates on the brain (1). LC neurons are inhibited by acute opiate exposure, but, during a course of chronic drug administration, LC firing rates return to normal levels and increase far above normal levels upon precipitation of withdrawal (1). Several studies have shown that this withdrawal-induced activation of LC neurons contributes to opiate dependence and withdrawal (2–6), although some authors have questioned this view (7). Nevertheless, the observation that the withdrawal activation of LC neurons occurs in vitro as well as in vivo supports the involvement of factors that are intrinsic to these neurons (5, 8). Considerable evidence now supports the hypothesis that one of the intrinsic factors is upregulation of the cAMP pathway and, more specifically, induction of adenylyl cyclase (AC) (9, 10). Accordingly, LC neurons in brain slices show greater excitatory responses to forskolin (an activator of AC) after prior chronic in vivo morphine treatment (11, 12). Furthermore, drugs that inhibit the cAMP pathway, infused directly into the LC, attenuate the development and expression of opiate withdrawal, while drugs that activate the cAMP pathway worsen withdrawal and can, in drug naïve animals, even induce certain withdrawal-like behaviors (13, 14).
In earlier work, we and others demonstrated that two isoforms of AC, AC1 and AC8, are induced uniquely in the LC after chronic morphine administration, while all other isoforms of the enzyme are unaffected (11, 15, 16). AC1 and AC8 belong to a subfamily of AC’s which are activated by Ca+/calmodulin (17). In addition, several studies suggest that these isoforms are relatively insensitive to inhibition by Gαi (18, 19), which raises the possibility that their induction may help LC neurons overcome persistent morphine inhibition of AC activity and thereby contribute to the cellular forms of tolerance and dependence observed electrophysiologically.
In the present study, we characterized the behavioral, cellular, and molecular adaptations to chronic morphine in AC1 and AC8 knockout (KO) mice. Our findings implicate both AC isoforms in the long-term actions of morphine on LC neurons, with loss of each isoform causing distinct effects on opiate dependence and withdrawal, LC neuronal excitability, and changes in gene expression.
MATERIALS AND METHODS
Animals
Mice derived from heterozygous matings of AC1 or AC8 lines, N7 generation onto C57Bl/6 were used in all behavioural tests. AC1−/− mice and AC8−/− mice backcrossed 7 generations to C57Bl/6 were mated to produce individual mice heterozygous for both AC1 and AC8. These mice were then mated to generate individual mice mutant for both AC1 and AC8, as well as wildtype control animals.
For all behavioural, electrophysiological, and molecular studies, we used 2–4 month old male mice. Animals were housed in a temperature and humidity controlled environment with a 12 hr light-dark cycle and had free access to food and water. All studies were conducted according to Animal Care and Use Committees of Yale University and UT Southwestern Medical Center.
Morphine analgesia and tolerance
The hot plate test was performed on a platform heated to 52°C with a cutoff time of 40 sec (IITC Life Sciences, CA). The latency to paw lick or jump was recorded as described elsewhere (20). Also, see Suppl. Methods
Morphine withdrawal
Mice were injected i.p. with escalating morphine doses (20, 40, 60, 80, 100, 100 and 100 mg/kg) every 8 h for 2.5 days. Two hours after the last morphine injection, mice received naloxone HCl (1 mg/kg s.c., Sigma MO). Withdrawal signs (jumps, wet dog shakes, diarhhea, ptosis, tremor, weight loss) were then monitored for 25 min as described (20).
Electrophysiological recordings
Brain slices from morphine dependent animals and their controls were prepared as described previously (8, 21). Chronic morphine treatment involved the subcutaneous implantation of a sham or morphine pellet (containing 25 mg morphine base, National Institute on Drug Abuse) under light anesthesia, and mice were used 72 hr later. A 300 µm thick coronal slice containing the LC was transferred to the stage of a submerged recording chamber and secured by a fine mesh attached to a platinum wire frame and perfused with oxygenated artificial cerebrospinal fluid (ACSF). After placement of the slice, the bath temperature was raised from room temperature to 33°C. Single-unit extracellular potentials were recorded by the use patch pipettes filled with ACSF and monitored through a high-input impedance amplifier (Axoclamp 2B: Axon Instruments, Union City, CA). Detailed methods for electrophysiological recordings are provided in the Supplemental Methods File.
For statistical analysis, the firing rates of 10–14 neurons from a single LC slice were averaged and considered as a single data point. Statistical significance of differences between mutants and controls was then tested by two-tailed t-tests.
Gene expression microarrays
Microarray analysis was performed as described previously (22), using chips from Affymetrix, with few modifications. Four groups of animals, AC1 KO’s, AC8 KO’s, and each of their wildtype littermate controls, received subcutaneous sham or morphine pellets (as above) and were analyzed 72 hrs later, making a total of 6 groups (see below) for microarray comparison. To reduce variability and increase statistical power, we performed at least 3 biological replicates (triplicate Affymetrix chips) for each group (depending on the number of mice available) (23). Each biological replicate consisted of 1 µg of pooled total RNA from the LC’s of four mice
Detailed methods for gene expression arrays are provided in Supplemental Methods. To determine the optimal criteria for considering significant gene regulation, we compared the wildtype sham vs morphine dataset to our previously published microarray and real-time PCR geneset from the same brain region and same drug treatment (22). We found that the criteria that best recapitulates the previous data was to select our “significantly regulated genelist” by PLIER normalizing the data and selecting only genes with a log2 ratio of experimental/control to be >0.3 or <−0.3, and a comparison p value (using t-test) of <0.01. These criteria of generating a differentially expressed gene list using fold change primary criteria followed by a non-stringent p-value cutoff is also recommended by the largest microarray study to date, which consists of >1,300 microarrays from >50 institutions, including our own (24, 25). Our “significantly regulated genelist” and associated values for each condition were imported into Genespring (Agilent Technologies, Santa Clara CA) for data visualization and heatmap generation.
RESULTS
Behavioral Responses to Morphine in AC1 and AC8 KO mice
To determine the role of AC1 and AC8 in opiate dependence, we first monitored opiate withdrawal behavior in morphine-dependent AC1 KO mice and their respective wildtype littermate controls. Animals chronically treated with morphine received an injection of the opioid receptor antagonist, naloxone (1 mg/kg s.c.) and withdrawal behavior was monitored for 25 min. AC1 KO mice become less dependent on morphine, as several standard opiate withdrawal signs are dramatically decreased compared to wildtype controls (Fig. 1A). In particular, deletion of the AC1 gene dramatically decreases wet dog shakes, paw tremor, diarrhea, ptosis and weight loss. Jumps trended toward a decrease as well, but this did not reach statistical significance. There was no effect on general body tremor.
Figure 1. Knockout of AC1 decreases morphine dependence.

A. AC1 KO mice and wildtype littermate control mice, treated chronically with morphine, were injected with naloxone (1 mg/kg, s.c.) and opiate withdrawal signs were monitored for 25 min (n=5 per genotype). B. A different group of animals was injected with morphine once a day (20 mg/kg) and analgesic responses were monitored in a 52°C hot plate test for four consecutive days (n=8–10 per genotype). Responses are expressed as % maximal possible effect (see Materials and Methods). Data are expressed as means ± S.E.M., *p<0.05 for genotype over treatment, two-way ANOVA followed by Bonferroni test.
We next assessed the role of AC1 in morphine analgesia and analgesic tolerance by use of a 52°C hotplate assay. As shown in Fig. 1B, absence of the AC1 gene had no effect on morphine’s initial analgesic effects nor on the development of morphine tolerance after repeated drug exposures.
Behavioral responses to morphine were also evaluated in AC8 KO mice and their wildtype littermates. Interestingly, deletion of the AC8 gene caused significantly decreased jumping and general body tremor (Fig. 2A), the two signs that are not significantly affected by deletion of the AC1 gene. AC8 KO mice also exhibited a ~50% reduction in diarrhea compared to their wildtype controls, similar to the effect observed in AC1 KO mice. As with AC1 KO mice, knockout of the AC8 gene does not affect morphine analgesia or analgesic tolerance in the 52°C hot plate assay (Fig. 2B).
Figure 2. Knockout of AC8 decreases morphine dependence.

AC8 KO mice and wildtype littermate control mice were treated as described in the legend to Fig. 1. Data are expressed as means ± S.E.M., *p<0.05 for genotype over treatment, two-way ANOVA followed by Bonferroni test.
To investigate whether deletion of both AC1 and AC8 genes results in a more pronounced decrease in morphine dependence and withdrawal, we generated AC1/8 double KO mice and studied their responses to morphine. Overall, the withdrawal phenotype of the double KO’s was complex (Fig. 3A). The mice reflected features of the AC1 phenotype (dramatic reductions in paw tremor and diarrhea) as well as features of the AC8 phenotype (reduced jumps), but other signs (wet dog shakes, general body tremor, ptosis, and weight loss)—which were significantly attenuated in one or the other single KO—were no longer significantly affected in the double KO mice. These latter findings are surprising and suggest that loss of both AC isoforms may cause compensatory, developmental changes that alter their responses to morphine. The double KO’s showed no phenotype in the morphine analgesia-tolerance paradigm (Fig. 3B), as would be expected from the lack of phenotype in the two single KO lines.
Figure 3. Double knockout of AC1 and AC8 decreases morphine dependence.

AC1/8 double KO mice and wildtype littermate control mice were treated as described in the legend to Fig. 1. Data are expressed as means ± S.E.M., *p<0.05 for genotype over treatment, two-way ANOVA followed by Bonferroni test.
Electrophysiological Responses to Morphine in AC1 and AC8 KO mice
These behavioral experiments provided a framework within which we could then study the role of AC1 and AC8 in the electrophysiological responses of LC neurons to chronic morphine. Extracellular single-unit recordings from LC neurons were obtained from brain slices in six groups of sham-treated and chronic morphine-treated mice: AC1 KO, AC8 KO, AC1/8 double KO, and their respective wildtype controls. As shown in Fig. 4A, wildtype mice showed three expected properties: i) forskolin activation under sham-treated conditions, ii) elevated baseline firing rates after chronic morphine, and iii) forskolin activation to higher firing rates after chronic morphine. Each of these responses has been reported previously in rat and mouse (8, 11, 26). In contrast, in all three AC KO lines, there was a blunting of the effect of chronic morphine on baseline firing rates; thus, baseline firing rates after chronic morphine were significantly different in all three KO lines from that seen in wildtype mice, p<0.01 by one way ANOVA followed by Dunnett test. Furthermore, the ability of chronic morphine to enhance the effect of forskolin on LC firing rates was completely abolished in AC8 and AC1/8 KO’s, with a partial blunting seen in the AC1 KO (Fig. 4B–D); in all three mutant lines, the forskolin-induced firing rates after chronic morphine were significantly different from that seen in wildtype mice, p<0.01 by one way ANOVA followed by Dunnett test. These findings suggest that both AC1 and AC8 can contribute to the overall response to forskolin in these LC neurons, but that morphine upregulation of forskolin responses appears to be more dependent on AC8 than AC1. Note that, in all KO groups compared to wildtype mice, there was no reduction in baseline firing rate, indicating that baseline firing under these circumstances is not primarily dependent on AC1 or AC8.
Figure 4. Enhanced baseline and forskolin-induced firing of LC neurons after chronic morphine treatment was attenuated in AC1, AC8, and AC1/8 KO mice.
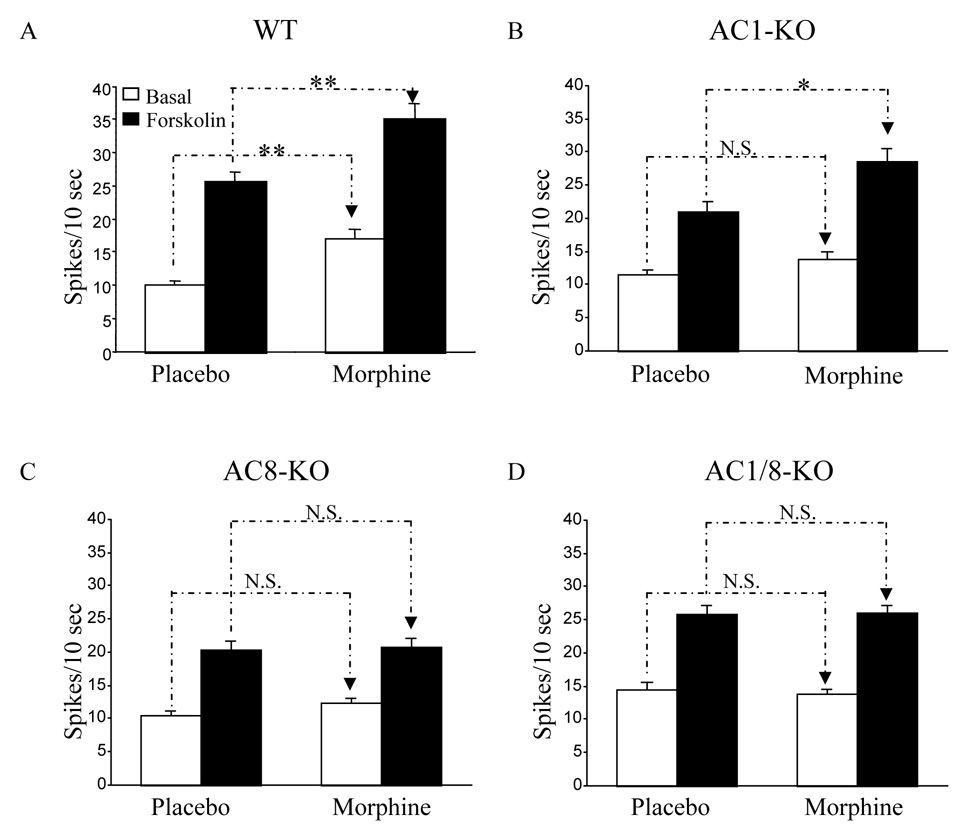
Bar graphs summarize data for LC firing rates in brain slices from placebo- and morphine-treated mice at baseline and in response to forskolin (10 µM, 10–15 min). These parameters were studied in WT mice (n=24 mice, 67 cells) (A), AC1 KO mice (n=10 mice, 34 cells) (B), AC8 KO mice (n=10 mice, 38 cells) (C), and AC1/8 double KO mice (n=11 mice, 42 cells) (D). Note that data from wildtype mice of each mutant line were combined since no differences were observed among them. As shown in the figure there is a blunting of the effect of chronic morphine on baseline firing rates in all three lines of KO and a complete block of the enhanced effect of forskolin on firing rates after chronic morphine in AC8 and AC1/8 KO’s, with a partial blunting effect in the AC 1 KO. Data shown as mean ± SEM; *p < 0.05 ; **P < 0.01 (One-way ANOVA followed by Turkey test)
Tolerance has been demonstrated toward the inhibitory effect of opiates on LC neurons when recorded ex vivo in brain slices from opiate-dependent rats (27, 28). It has been proposed that upregulation of AC activity in the LC may be one mechanism for this tolerance, since the subsequent enhanced excitability of the neurons would oppose opiate-induced inhibition (29). We examined the influence of AC1 and AC8 on the development of tolerance by recording from LC neurons of KO and wildtype. As previously shown in rats, wildtype mice exhibited a small, but significant decrease in sensitivity to the inhibitory effect of the µ opioid receptor agonist, DAMGO, after prior in vivo chronic morphine administration. This is revealed by a rightward shift of the concentration-response curve and a two-fold increase in IC50 (Fig. 5A, Table 1). In sham-treated AC1, AC8, and AC1/8 KO groups, there was a pre-existing reduction in sensitivity to DAMGO, with AC1 KO’s showing the least and AC1/8 double KO’s showing the greatest change from wildtype. In all three KO groups, there was little or no further change in sensitivity to DAMGO after chronic morphine (Fig. 5B–D, Table 1). Thus, while LC neurons from AC1, AC8, and AC1/8 KO mice are inherently less sensitive to DAMGO, the development of tolerance after chronic morphine is blunted.
Figure 5. Morphine regulation of DAMGO sensitivity in LC neurons of AC1, AC8 and AC1/8 double KO mice.

Concentration-response curves for the µ-opioid receptor agonist DAMGO were measured as percentage suppression of baseline firing rate after stepwise increases in DAMGO concentration in perfusate. Four groups each of non-dependent (placebo-treated) and dependent (morphine-treated) mice were tested: wildtype (WT) (A), AC1 KO (B), AC8 KO (C), and AC1/8 double KO’s (D). As expected from previous studies, WT mice in all three KO lines showed a rightward shift in the concentration response curve (decreased sensitivity), hence the data were collapsed into a single WT group. In contrast to WT, all three AC KO groups showed little or no rightward shift after morphine treatment, but showed decreased sensitivity to DAMGO relative to WT in the non-dependent condition (see Table for IC50 comparisons).
Table 1.
IC50 of DAMGO inhibition of LC neuronal firing in brain slices of WT, AC1, AC8, and double AC1/8 KO mice.
| Groups | Placebo | Morphine-implant | % change |
|---|---|---|---|
| Wildtype | 53 nM ± 2.2 (n=30) | 112 nM ± 9.4 (n=45) | 111% ** |
| AC1-KO | 75 nM ± 13.3 (n=10) | 86 nM ± 14.1 (n=15) | 15% |
| AC8-KO | 96 nM ± 10.8 (n=10)# | 118 nM ± 14.6 (n=14) | 22% |
| AC1/8-KO | 112 nM ± 13.5 (n=11)## | 98 nM ± 13.8 (n=15) | −14% |
Values are means ± SEM. Asterisks indicate difference at IC50 between Placebo and morphine-inplant groups
p<0.01 (t-test). Pound sings indicate difference at IC50 among wildtype and AC-KO groups (one way ANOVA followed by Dunnett)
p < 0.05
< 0.01
Morphine Regulation of Gene Expression in AC1 and AC8 KO mice
We next used DNA expression array technology to assess the role of AC1 and AC8 in changes in gene regulation induced in the LC by chronic morphine administration. This large experiment involved the analysis, at least triplicate arrays each performed on independent samples of pooled LC tissue, of AC1 KO mice, AC8 KO mice, and their respective wildtype littermate controls, under sham- and morphine-treated conditions. In both groups of wildtype mice, chronic morphine induced many of the changes in LC gene expression reported earlier in inbred C57Bl/6 mice (22). This is all the more remarkable, because our current work used Affymetrix chips that contain ~39,000 transcripts, while our earlier study used an older Affymetrix chip with ~12,000 transcripts. Examples include several enzymes in the norepinephrine biosynthetic pathway (e.g., tyrosine hydroxylase, dopamine β-hydroxylase), galanin, transthyretin, CART (cocaine- and amphetamine-regulated transcript), and FK506BP5 (FK506 binding protein 5), among many others. (See Supplementary Table S1 for complete gene lists.) The consistency of these morphine-regulated gene sets is encouraging and supports the validity of the microarray methodology and statistical analyses used.
We next compared the effects of chronic morphine in the two lines of AC KO mice. As depicted in the heatmap shown in Fig. 6A, loss of each AC isoform prevents the ability of morphine to up- or downregulate subsets of genes. The number of genes affected was roughly comparable in AC1 vs AC8 KO mice. Loss of AC1 vs AC8 disrupted morphine regulation of some of the same genes as well as some distinct genes for each enzyme. Examples of individual genes that show these various patterns of regulation are listed in Table 2 (see Supplementary Table S1 for complete gene lists). Interestingly, distinct sets of genes that are upregulated in LC by chronic morphine in wildtype mice are downregulated by morphine in LC of AC1 KO and/or AC8 KO mice. Respectively, genes downregulated by chronic morphine in wildtype LC are upregulated in AC1 and/or AC8 KO LC.
Figure 6. Morphine regulation of gene expression in LC of AC1 and AC8 KO mice.
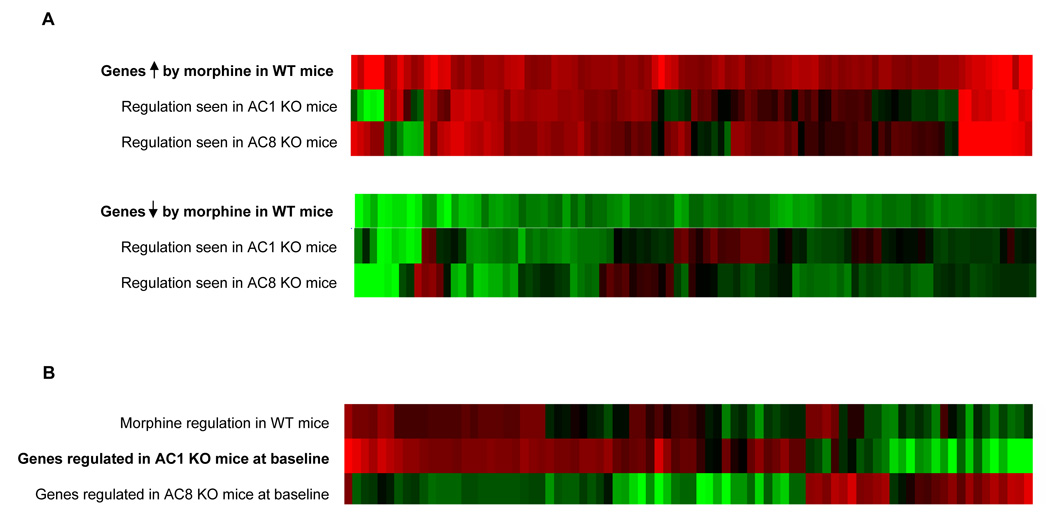
A. The upper row of the two heatmaps illustrate that genes that are significantly upregulated (red) or downregulated (green) in the LC of wildtype (WT) mice after chronic morphine administration. The two lower rows of each heatmap illustrate how those same genes were affected by chronic morphine in AC1 or AC8 KO mice. Note that while many genes are similarly regulated by morphine in WT and KO mice, roughly half of the genes normally regulated by morphine are no longer altered in each respective KO. Interestingly, certain genes are even oppositely regulated uniquely in AC1 or AC8 KO mice. B. Middle row in heatmap illustrates genes that are significantly altered in LC of sham-treated AC1 KO mice compared to WT littermate controls. The other rows illustrate how those same genes are affected by chronic morphine in WT mice (upper row) and in sham-treated AC8 KO mice compared to WT littermate controls. Note how loss of AC1 at baseline induces a regulatory pattern reminiscent of the effects of morphine in WT mice, an effect not seen in AC8 KO mice.
Table 2.
Examples of Morphine-Regulated Genes in the LC.
| No effect of AC KO | Regulation in WT LC | Effect in AC1 KO | Effect in AC8 KO |
|---|---|---|---|
| Angiotensin 2 receptor | ↑ | ↑ | ↑ |
| Calcium channel, voltage-dependent, α2, δ2 subunits | ↑ | ↑ | ↑ |
| CD24a antigen | ↑ | ↑ | ↑ |
| F-actin binding protein | ↑ | ↑ | ↑ |
| FK506BP5 | ↑ | ↑ | ↑ |
| Nicotinic α6 subunit | ↓ | ↓ | ↓ |
| Potassium inwardly-rectifying channel, subfamily J, member 15 | ↓ | ↓ | ↓ |
| Purinergic P2Y receptor, 12 | ↓ | ↓ | ↓ |
| RGS4 | ↑ | ↑ | ↑ |
| Synaptopodin | ↓ | ↓ | ↓ |
| Blockade with either AC KO | Regulation in WT LC | Effect in AC1 KO | Effect in AC8 KO |
| CART | ↑ | smaller ↑ | smaller ↑ |
| Dopamine β-hydroxylase | ↑ | ↓ | ↓ |
| Galanin | ↑ | smaller ↑ | smaller ↑ |
| Myocyte enhancer factor 2C | ↑ | no effect | ↓ |
| P21 activated kinase (PAK) | ↑ | no effect | no effect |
| Phospholipase A2 | ↑ | no effect | no effect |
| Prodynorphin | ↑ | no effect | no effect |
| Transthyretin | ↑ | ↓ | smaller ↑ |
| Tyrosine hydroxylase | ↑ | smaller ↑ | smaller ↑ |
| VMAT2 | ↑ | no effect | ↓ |
| Blockade with AC1 KO only | Regulation in WT LC | Effect in AC1 KO | Effect in AC8 KO |
| Bone morphogenetic protein 1 | ↑ | no effect | ↑ |
| Calcitonin/calcitonin-related polypeptide α | ↑ | no effect | ↑ |
| Carboxypeptidase Z | ↑ | no effect | ↑ |
| cGMP-dependent protein kinase 1 | ↓ | no effect | ↓ |
| Chemokine-like factor | ↑ | ↓ | ↑ |
| FGFR4 | ↑ | no effect | ↑ |
| Interferon inducible GTPase 1 | ↓ | no effect | ↓ |
| Blockade with AC8 KO only | Regulation in WT LC | Effect in AC1 KO | Effect in AC8 KO |
| Ankyrin 2 | ↑ | ↑ | ↓ |
| Chloride channel 1 | ↓ | ↓ | ↑ |
| Cytochrome P450, family 11, subfamily a polypeptide 1 | ↓ | ↓ | ↑ |
| Fas | ↑ | ↑ | no effect |
| GTP cyclohydrolase 1 | ↑ | ↑ | no effect |
| Protein kinase C, epsilon | ↑ | ↑ | ↓ |
| Tropomodulin 1 | ↓ | ↓ | no effect |
| Unique regulation with AC KO | Regulation in WT LC | Effect in AC1 KO | Effect in AC8 KO |
| Calcium channel, voltage-dependent, β2 subunit | no effect | ↓ | ↓ |
| C/EBP δ | no effect | ↑ | ↑ |
| Ephrin A5 | no effect | ↑ | no effect |
| G protein γ4 subunit | no effect | ↓ | ↓ |
| G protein-coupled receptor 85 | no effect | ↑ | no effect |
| Mitogen activated protein kinase kinase kinase 11 | no effect | ↑ | no effect |
| Nitric oxide synthase 1, neuronal | no effect | ↑ | ↑ |
| Nuclear factor of activated T-cells 5 | no effect | ↓ | ↓ |
| Protein phosphatase 1, catalytic subunit, γ isoform | no effect | ↑ | ↑ |
| RGS10 | no effect | ↓ | ↓ |
| VMAT1 | no effect | no effect | ↑ |
| Cyclin-dependent kinase inhibitor 1A (P21) | ↑ | larger ↑ | larger ↑ |
| Prostaglandin D2 synthase | ↑ | ↑ | larger↑ |
| Serum GC regulated kinase | ↑ | larger↑ | larger↑ |
| Tropomodulin 2 | ↑ | larger↑ | larger↑ |
Abbreviations: FK506BP5, FK506-binding protein 5; RGS, regulator of G protein signaling; CART, cocaine- and amphetamine-regulated transcript; VMAT, vesicular monoamine transporter; FGFR4, fibroblast growth factor receptor 4; C/EBPδ, CCAAT-enhancer binding protein delta.
An unexpected electrophysiological observation was the increased baseline firing rate of LC neurons from AC1 KO mice (see Fig. 4A). Since increased basal activity of LC neurons is also observed after chronic morphine treatment of wildtype mice, we used the gene array data to gain some insight into these surprising observations. As shown in the heatmap in Fig. 6B, we identified a similar pattern of adaptive changes in LC gene expression between chronic morphine-treated wildtype mice and drug-naïve AC1 KO mice. In contrast, similarities in gene expression changes were not apparent between morphine-treated wildtype mice and drug-naïve AC8 KO’s.
The gene array experiments indicated that loss of AC1 or of AC8 is not associated with compensatory changes in expression levels of other AC isoforms (data not shown). To confirm these negative findings, we performed real time PCR to assess levels of other AC family members in the LC of AC1 and AC8 KO mice. Consistent with our array data, no significant alterations in expression levels of other AC isoforms were found (data are expressed as fold change in mRNA levels over wildtype [WT] ± SEM): for AC1 KO: AC2 = 1.00 ± 0.025, AC3 = 1.10 ± 0.060, AC4 = 1.06 ± 0.080, AC5 undetectable, AC6 = 1.26 ± 0.11, AC7 = 0.986 ± 0.080, AC8 = 0.99 ± 0.070, AC9 = 1.15 ± 0090, and AC10 = 1.607 ± 0.17; for AC8 KO: AC1 = 1.18 ± 0.075, AC2 = 1.18 ± 0.060, AC3 = 1.20 ± 0.070, AC4 = 1.22 ± 0.013, AC5 undetectable, AC6 = 1.20 ± 0.032, AC7 = 1.19 ± 0.060, AC9 = 1.14 ± 0.055, and AC10 = 1.07 ± 0.18 (n=4–6)
DISCUSSION
Results of the present study contribute to our understanding of the role of AC1 and AC8 in mediating the long-term effects of morphine on the LC. Loss of either enzyme decreases the ability of forskolin to excite LC neurons under control and morphine-treated conditions and the ability of chronic morphine to increase the baseline firing rate of LC neurons. Gene expression profiling supports the importance of both AC isoforms for normal genomic responses to chronic morphine, with roughly one-half of the genes up- or downregulated by morphine in wildtype LC no longer showing significant regulation by morphine in LC from AC1 or AC8 KO mice. Interestingly, the genes influenced by AC1 or AC8 were only partially overlapping, which further reinforces the notion that the two AC isoforms, while both important, subserve distinct functions in the LC both before and after chronic morphine exposure. Consistent with these molecular and cellular observations, we also demonstrate in the present study that AC1 and AC8 KO mice exhibit attenuated morphine physical dependence and withdrawal.
Our behavioural findings are in agreement with earlier studies by Li and colleagues, who showed that AC1/8 double KO mice exhibit a milder opiate withdrawal syndrome (30). This earlier study reported that only two withdrawal signs are affected in the double KO’s. A potential concern about this study is that the authors used inbred C57Bl/6 mice as their wildtype controls. However, we, too, find a relatively small number of withdrawal behaviors affected in the double KO’s, and we used wildtype littermates which reduces concerns about the confounds of genetic background. Rather, our findings illustrate a more robust withdrawal phenotype in single AC1 KO mice, which raises the possibility that AC1/8 double KO’s may be complicated by developmental compensations, as will be discussed further below. Li et al. (30) also report that AC1/8 double KO mice are less sensitive to the analgesic actions of morphine in the tail flick test. This is in contrast to our findings in the 52°C hot plate assay. However, the tail flick test provides a measure of spinal analgesia, while the 52°C hot plate test measures supraspinal analgesia. Hence, results of these two studies would suggest no effect of AC1 or AC8 on brain mechanisms of morphine analgesia and tolerance, but important effects of the enzymes mediated at the level of the spinal cord. Indeed, AC1 and AC8 have been shown to be important for regulation of dorsal horn neurons in the spinal cord by inflammatory signals (31).
AC1 and AC8, along with AC3, belong to a subclass of AC enzymes that are activated by Ca2+/calmodulin and, as such, have been proposed to play crucial roles in integrating cAMP and Ca2+ signaling (17, 18, 32). These AC isoforms are also relatively insensitive to inhibition by Gαi, and are inhibited by Gβγ subunits (18, 19, 33, 34). All three AC isoforms are widely distributed in brain (11, 35, 36). Considerable research has been performed on AC1 and AC8 KO mice, primarily on double KO’s, which has implicated these enzymes in a wide variety of functions. In addition to their role in morphine responses described here and in spinal mechanisms mentioned above, AC1 and AC8 have been shown to be involved in hippocampal long-term potentiation and associated learning and memory, olfactory sensation, behavioral responses to stress, and allodynia (or hypersensitization) to painful stimuli (37, 38 , 39, 40). These widespread and diverse functions of AC1/8 underscore that the distinct behavioral effects reported here in AC1 vs AC8 KO mice during opiate withdrawal could well be mediated by many brain regions beyond the LC. Indeed, the lack of altered analgesic tolerance to morphine seen in AC1 and AC8 KO mice, combined with our electrophysiological findings which show blunted tolerance to the electrophysiological responses of LC neurons to DAMGO after chronic morphine treatment, suggest that other brain regions, outside the LC, are more involved in analgesic tolerance and that AC1 and AC8 are not as crucial for morphine action in these other regions.
Among the findings reported in this study are several unexpected effects observed upon deletion of AC1 and AC8, which we speculate may be due to developmental compensations. LC neurons of AC1 KO mice show higher baseline firing rates under drug-naïve conditions. This is unexpected, since it is widely known that LC neurons are activated upon stimulation of the cAMP pathway (e.g., 12, 41, 42). Even more dramatic, however, are paradoxical findings in the double KO’s. Rather than showing additive effects of the AC1 and AC8 single KO’s, AC1/8 double KO mice exhibit more severe withdrawal than the single gene KO’s . We hypothesize that loss of both AC1 and AC8 may have a dramatic impact on the formation of signal transduction complexes between opioid receptors, scaffolding proteins, and ion channels, a possibility which now warrants further study. We should also point out that the contribution of genetic background differences to the observed phenotype cannot be excluded, as it is one of the limitations of double knockout models.
The DNA microarray data indicate, that at least in the single KO’s, such changes in gene expression do not involve compensatory induction of other AC isoforms, since no such alterations in AC genes were detected. Rather, analysis of the arrays shows that a subset of genes altered upon loss of AC1 in drug-naïve animals overlap with those that are induced by chronic morphine in wildtype LC (see Fig. 6B). Further work is now needed to sift through this gene set and identify candidate genes that could contribute to the higher baseline firing exhibited by the neurons under these two experimental conditions.
Likewise, the DNA microarray data provide novel insight into the impact that loss of AC1 or AC8 has on the adaptation of LC neurons to chronic morphine. As described in Results, the most dramatic changes in gene regulation induced by chronic morphine in the LC of wildtype mice in our 2005 study (22) were also found in the present study (Table 2), which provides important validation of our new gene lists. The induction of some of these genes, e.g., FK506BP5 and RGS4 (regulator of G protein signaling 4), by morphine was unaffected by AC1 or AC8 KO, while induction of several other genes, such as galanin, tyrosine hydroxylase, dopamine β-hydroxylase, and CART (cocaine- and amphetamine-regulated transcript), was attenuated in both mutants. In contrast, morphine induction of certain genes, such as FGFR4 (fibroblast growth factor receptor 4) and protein kinase G, was blocked in one, but not the other, AC mutant. Still other genes, such as serum glucocorticoid regulated kinase, show enhanced upregulation in the absence of AC1 or AC8 compared to wildtype mice, while others (e.g., RGS10, VMAT1 [vesicular monoamine transporter 1], and ephrin A5) exhibit morphine regulation uniquely in one or both mutants, but not in wildtype mice. Several of the genes that show interesting patterns of regulation on the arrays, for example, norepinephrine synthetic enzymes, VMAT1 and 2, FK506BP5, prodynorphin, nitric oxide synthase, and RGS4, to name a few, have been related previously to LC function and opiate dependence and withdrawal (2, 21, 22, 43). Overall, our gene array findings support our hypothesis that AC1 and AC8, while both involved in mediating morphine regulation of LC neurons, play distinct roles.
While this study focused on AC1 and AC8 in the LC, these and other isoforms of AC have been implicated in the actions of chronic morphine in other regions of the central and peripheral nervous systems. AC activity has been implicated in morphine regulation of GABAergic transmission in the ventral tegmental area, a major brain reward region, although the specific enzyme isoform affected remains unknown (44). AC5, while highly enriched in striatal regions (45), does not appear to be regulated by chronic morphine (11). Nevertheless, a recent study demonstrated that loss of AC5 dramatically reduces opiate dependence, reward, and analgesic tolerance (46).
Collectively, these studies provide compelling support for the notion that upregulation of the cAMP pathway, in part through the induction of specific isoforms of AC, represents an important mechanism by which neurons adapt to chronic opiate exposure (47). The present study illustrates the cell type-specific approaches, at the behavioral, cellular, and molecular levels, which are now needed to delineate the precise role played by each AC isoform in these complex adaptations that contribute to opiate tolerance and dependence, and, ultimately, addiction.
Supplementary Material
Footnotes
Disclosure of Biomedical Financial Interests and potential conflicts of interest: Dr. Venetia Zachariou, Dr. Rongjian Liu, Mr. Quincey Laplant, Dr. Guanghua Xiao, Mr. William Renthal, Dr. Guy C. Chan, Dr. Daniel R. Storm, Dr. George Aghajanian and Dr. Eric J. Nestler report no financial Interests and potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Aghajanian GK. Tolerance of locus coeruleus neurons to morphine and suppression of withdrawal response by clonidine. Nature. 1978;276:186–188. doi: 10.1038/276186a0. [DOI] [PubMed] [Google Scholar]
- 2.Taylor JR, Elsworth JD, Garcia EJ, Grant SJ, Roth RH, Redmond DE., Jr Clonidine infusions into the locus coeruleus attenuate behavioral and neurochemical changes associated with naloxone-precipitated withdrawal. Psychopharmacology. 1988;96:121–134. doi: 10.1007/BF02431544. [DOI] [PubMed] [Google Scholar]
- 3.Koob GF, Maldonado R, Stinus L. Neural substrates of opiate withdrawal. Trends Neurosci. 1992;15:186–191. doi: 10.1016/0166-2236(92)90171-4. [DOI] [PubMed] [Google Scholar]
- 4.Maldonado R, Koob GF. Destruction of the locus coeruleus decreases physical signs of opiate withdrawal. Brain Res. 1993;605:128–138. doi: 10.1016/0006-8993(93)91364-x. [DOI] [PubMed] [Google Scholar]
- 5.Aston-Jones G, Hirata A, Akaoka H. Local opiate withdrawal in locus coeruleus in vivo. Brain Res. 1997;765:331–336. doi: 10.1016/s0006-8993(97)00682-3. [DOI] [PubMed] [Google Scholar]
- 6.Delfs JM, Zhu Y, Druhan JP, Aston-Jones G. Noradrenaline in the ventral forebrain is critical for opiate withdrawal-induced aversion. Nature. 2000;403:430–434. doi: 10.1038/35000212. [DOI] [PubMed] [Google Scholar]
- 7.Williams JT, Christie MJ, Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev. 2001;81:299–343. doi: 10.1152/physrev.2001.81.1.299. [DOI] [PubMed] [Google Scholar]
- 8.Kogan JH, Nestler EJ, Aghajanian GK. Elevated basal firing rates and enhanced responses to 8-Br-cAMP in locus coeruleus neurons in brain slices from opiate-dependent rats. Eur J Pharmacol. 1992;211:47–53. doi: 10.1016/0014-2999(92)90261-2. [DOI] [PubMed] [Google Scholar]
- 9.Duman RS, Tallman JF, Nestler EJ. Acute and chronic opiate regulation of adenylate cyclase in brain: specific effects in locus coeruleus. J Pharmacol Exp Ther. 1988;246:1033–1039. [PubMed] [Google Scholar]
- 10.Rasmussen K, Beitner-Johnson DB, Krystal JH, Aghajanian GK, Nestler EJ. Opiate withdrawal and the rat locus coeruleus: behavioral, electrophysiological, and biochemical correlates. J Neurosci. 1990;10:2308–2317. doi: 10.1523/JNEUROSCI.10-07-02308.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lane-Ladd SB, Pineda J, Boundy VA, Pfeuffer T, Krupinski J, Aghajanian GK, Nestler EJ. CREB (cAMP response element-binding protein) in the locus coeruleus: biochemical, physiological, and behavioral evidence for a role in opiate dependence. J Neurosci. 1997;17:7890–7901. doi: 10.1523/JNEUROSCI.17-20-07890.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ivanov A, Aston-Jones G. Local opiate withdrawal in locus coeruleus neurons in vitro. J Neurophysiol. 2001;85:2388–2397. doi: 10.1152/jn.2001.85.6.2388. [DOI] [PubMed] [Google Scholar]
- 13.Maldonado R, Valverde O, Garbay C, Roques BP. Protein kinases in the locus coeruleus and periaqueductal gray matter are involved in the expression of opiate withdrawal. Naunyn Schmiedebergs Arch Pharmacol. 1995;352:565–575. doi: 10.1007/BF00169392. [DOI] [PubMed] [Google Scholar]
- 14.Punch LJ, Self DW, Nestler EJ, Taylor JR. Opposite modulation of opiate withdrawal behaviors on microinfusion of a protein kinase A inhibitor versus activator into the locus coeruleus or periaqueductal gray. J Neurosci. 1997;17:8520–8527. doi: 10.1523/JNEUROSCI.17-21-08520.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsuoka I, Maldonado R, Defer N, Noel F, Hanoune J, Roques BP. Chronic morphine administration causes region-specifc increase of brain type VIII adenylyl cyclase mRNA. Eur J Pharmacol. 1994;268:215–221. doi: 10.1016/0922-4106(94)90191-0. [DOI] [PubMed] [Google Scholar]
- 16.Chao JR, Ni YG, Bolanos CA, Rahman Z, DiLeone RJ, Nestler EJ. Characterization of the mouse adenylyl cyclase type VIII gene promoter: regulation by cAMP and CREB. Eur J Neurosci. 2002;16:1284–1294. doi: 10.1046/j.1460-9568.2002.02186.x. [DOI] [PubMed] [Google Scholar]
- 17.Wang H, Storm DR. Calmodulin-regulated adenylyl cyclases: cross-talk and plasticity in the central nervous system. Mol Pharmacol. 2003;63:463–468. doi: 10.1124/mol.63.3.463. [DOI] [PubMed] [Google Scholar]
- 18.Tang WJ, Gilman AG. Type-specific regulation of adenylyl cyclase by G protein beta gamma subunits. Science. 1991;254:1500–1503. doi: 10.1126/science.1962211. [DOI] [PubMed] [Google Scholar]
- 19.Steiner D, Saya D, Schallmach E, Simonds WF, Vogel Z. Adenylyl cyclase type-VIII activity is regulated by G (betagamma) subunits. Cell Signal. 2006;18:62–68. doi: 10.1016/j.cellsig.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 20.Zachariou V, Georgescu D, Sanchez N, Rahman Z, DiLeone RJ, Berton O, Neve RL, Sim-Selley LJ, Selley DE, Gold SJ, Nestler EJ. Essential role for RGS9 in opiate action. Proc Natl Acad Sci. 2003;100:13656–13661. doi: 10.1073/pnas.2232594100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gold SJ, Han MH, Herman AE, Ni YG, Pudiak CM, Aghajanian GK, Liu RJ, Potts BW, Mumby SM, Nestler EJ. Regulation of RGS proteins by chronic morphine in rat locus coeruleus. Eur J Neurosci. 2003;17:971–980. doi: 10.1046/j.1460-9568.2003.02529.x. [DOI] [PubMed] [Google Scholar]
- 22.McClung CA, Nestler EJ, Zachariou V. Regulation of Gene Expression by Chronic Morphine and Morphine Withdrawal in the Locus Coeruleus and Ventral Tegmental Area. J Neurosci. 2005;25:6005–6015. doi: 10.1523/JNEUROSCI.0062-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peng X, Wood CL, Blalock EM, Chen KC, Landfield PW, Stromberg AJ. Statistical implications of pooling RNA samples for microarray experiments. BMC Bioinformatics. 2003;4:26. doi: 10.1186/1471-2105-4-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo L, Lobenhofer EK, Wang C, Shippy R, Harris SC, Zhang L, Mei N, Chen T, Herman D, Goodsaid FM, Hurban P, Phillips KL, Xu J, Deng X, Sun YA, Tong W, Dragan YP, Shi L. Rat toxicogenomic study reveals analytical consistency across microarray platforms. Nature Biotechnol. 2006;24:1162–1169. doi: 10.1038/nbt1238. [DOI] [PubMed] [Google Scholar]
- 25.MAQC Consortium. The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nature Biotechnol. 2006;24:1151–1161. doi: 10.1038/nbt1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Akbarian S, Bates B, Lui RJ, Skirboll SL, Pejchal T, Sun LD, Fan GP, Kucera J, Wilson MA, Kosofsky BE, Taylor JR, Bothwell M, Nestler EJ, Aghajanian GK, Jaenisch R. Neurotrophin-3 is essential for noradrenergic neuron function and opiate withdrawal. Mol Psychiatry. 2001;6:593–604. doi: 10.1038/sj.mp.4000897. [DOI] [PubMed] [Google Scholar]
- 27.Andrade R, Vandermaelen CP, Aghajanian GK. Morphine tolerance and dependence in the locus coeruleus: single cell studies in brain slices. Eur J Pharmacol. 1983;91:161–169. doi: 10.1016/0014-2999(83)90461-2. [DOI] [PubMed] [Google Scholar]
- 28.Christie MJ, Williams JT, North RA. Cellular mechanisms of opioid tolerance: studies in single brain neurons. Mol Pharmacol. 1987;32:633–638. [PubMed] [Google Scholar]
- 29.Nestler EJ, Aghajanian GK. Molecular and cellular basis of addiction. Science. 1997;278:58–63. doi: 10.1126/science.278.5335.58. [DOI] [PubMed] [Google Scholar]
- 30.Li S, Lee ML, Bruchas MR, Chan GC, Storm DR, Chavkin C. Calmodulin-Stimulated Adenylyl Cyclase Gene Deletion Affects Morphine Responses. Mol Pharmacol. 2006;70:1742–1749. doi: 10.1124/mol.106.025783. [DOI] [PubMed] [Google Scholar]
- 31.Wei F, Vadakkan KI, Toyoda H, Wu L, Zhao M, Fanny HX, Shum WF, Jia YH, Zhuo M. Calcium Calmodulin-Stimulated Adenylyl Cyclases Contribute to Activation of Extracellular Signal-Regulated Kinase in Spinal Dorsal Horn Neurons in Adult Rats and Mice. J Neurosci. 2006;26:851–861. doi: 10.1523/JNEUROSCI.3292-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xia Z, Storm DR. The role of calmodulin as a signal integrator for synaptic plasticity. Nat Rev Neurosci. 2005;6:267–276. doi: 10.1038/nrn1647. [DOI] [PubMed] [Google Scholar]
- 33.Nielsen MD, Chan GC, Poser SW, Storm DR. Differential regulation of type I and type VIII Ca2+-stimulated adenylyl cyclases by Gi coupled receptors in vivo. J. Biol. Chem. 1996;271:33308–33316. doi: 10.1074/jbc.271.52.33308. [DOI] [PubMed] [Google Scholar]
- 34.Watson EL, Jacobson KL, Singh JC, Idzerda R, Ott SM, DiJulio DH, Wong ST, Storm DR. The type 8 adenylyl cyclase is critical for Ca2+ stimulation of cAMP accumulation in mouse parotid acini. J Biol Chem. 2000;275:14691–14699. doi: 10.1074/jbc.275.19.14691. [DOI] [PubMed] [Google Scholar]
- 35.Xia Z, Choi EJ, Wang F, Blazynski C, Storm DR. Type I calmodulin sensitive adenylyl cyclase is neural specific. J Neurochem. 1993;60:305–311. doi: 10.1111/j.1471-4159.1993.tb05852.x. [DOI] [PubMed] [Google Scholar]
- 36.Cali JJ, Zwaagstra JC, Mons N, Cooper DM, Krupinski J. Type VIII adenylyl cyclase: A Ca2+/calmodulin-stimulated enzyme, expressed in discrete regions of rat brain. J Biol Chem. 1994;269:12190–12195. [PubMed] [Google Scholar]
- 37.Wu ZL, Thomas SA, Villacres EC, Xia Z, Simmons ML, Chavkin C, Palmiter RD, Storm DR. Altered behavior and long-term potentiation in type I adenylyl cyclase mutant mice. Proc Natl Acad Sci. 1995;92:220–224. doi: 10.1073/pnas.92.1.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wong ST, Athos J, Figueroa XA, Pineda VV, Schaefer ML, Chavkin CC, Muglia LJ, Storm DR. Calcium-stimulated adenylyl cyclase activity is critical for hippocampus-dependent long-term memory and late phase LTP. Neuron. 1999;23:787–798. doi: 10.1016/s0896-6273(01)80036-2. [DOI] [PubMed] [Google Scholar]
- 39.Wong ST, Trinh K, Hacker B, Chan GC, Lowe G, Gaggar A, Xia Z, Gold GH, Storm DR. Disruption of the type III adenylyl cyclase gene leads to peripheral and behavioral anosmia in transgenic mice. Neuron. 2000;27:487–497. doi: 10.1016/s0896-6273(00)00060-x. [DOI] [PubMed] [Google Scholar]
- 40.Wei F, Qiu CS, Kim SJ, Muglia L, Maas JW, Pineda VV, Xu HM, Chen ZF, Storm DR, Muglia LJ, Zhuo M. Genetic elimination of behavioral sensitization in mice lacking calmodulin-stimulated adenylyl cyclases. Neuron. 2002;36:713–726. doi: 10.1016/s0896-6273(02)01019-x. [DOI] [PubMed] [Google Scholar]
- 41.North RA, Williams JT, Surprenant A, Christie MJ. Mu and delta receptors belong to a family of receptors that are coupled to potassium channels. Proc Natl Acad Sci USA. 1987;84:5487–5491. doi: 10.1073/pnas.84.15.5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alreja M, Aghajanian GK. Pacemaker activity of locus coeruleus neurons: whole-cell recordings in brain slices show dependence on cAMP and protein kinase A. Brain Res. 1991;556:339–343. doi: 10.1016/0006-8993(91)90327-r. [DOI] [PubMed] [Google Scholar]
- 43.Pineda J, Torrecilla M, Martin-Ruiz R, Ugedo L. Attenuation of withdrawal-induced hyperactivity of locus coeruleus neurones by inhibitors of nitric oxide synthase in morphine-dependent rats. Neuropharmacology. 1998;37:759–767. doi: 10.1016/s0028-3908(98)00063-x. [DOI] [PubMed] [Google Scholar]
- 44.Bonci A, Williams JT. Increased probability of GABA release during withdrawal from morphine. J Neurosci. 1997;17:796–803. doi: 10.1523/JNEUROSCI.17-02-00796.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Glatt CE, Snyder SH. Cloning and expression of an adenylyl cyclase localized to the corpus striatum. Nature. 1993;361:536–538. doi: 10.1038/361536a0. [DOI] [PubMed] [Google Scholar]
- 46.Kim KS, Lee KW, Lee KW, Im JY, Yoo JY, Kim SW, Lee JK, Nestler EJ, Han PL. Adenylyl cyclase type 5 (AC5) is an essential mediator of morphine action. Proc Natl Acad Sci. 2006;103:3908–3913. doi: 10.1073/pnas.0508812103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nestler EJ. Historical review: Molecular and cellular mechanisms of opiate and cocaine addiction. Trends Pharmacol Sci. 2004;25:210–218. doi: 10.1016/j.tips.2004.02.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.