

Abstract
Tyrosyl-tRNA synthetase (TyrRS) is able to catalyze the transfer of both l- and d-tyrosine to the 3′ end of tRNATyr. Activation of either stereoisomer by ATP results in formation of an enzyme-bound tyrosyl-adenylate intermediate and is accompanied by a blue shift in the intrinsic fluorescence of the protein. Single turnover kinetics for the aminoacylation of tRNATyr by d-tyrosine were monitored using stopped-flow fluorescence spectroscopy. Bacillus stearothermophilus tyrosyl-tRNA synthetase binds d-tyrosine with an 8.5-fold lower affinity than that of l-tyrosine (K D-Tyrd = 102 μm) and exhibits a 3-fold decrease in the forward rate constant for the activation reaction (k D-Tyr3 = 13 s–1). Furthermore, as is the case for l-tyrosine, tyrosyl-tRNA synthetase exhibits “half-of-the-sites” reactivity with respect to the binding and activation of d-tyrosine. Surprisingly, pyrophosphate binds to the TyrRS·d-Tyr-AMP intermediate with a 14-fold higher affinity than it binds to the TyrRS·l-Tyr-AMP intermediate (K PPid = 0.043 for TyrRS·d-Tyr-AMP·PPi). tRNATyr binds with a slightly (2.3-fold) lower affinity to the TyrRS·d-Tyr-AMP intermediate than it does to the TyrRS·l-Tyr-AMP intermediate. The observation that the K Tyrd and k3 values are similar for l- and d-tyrosine suggests that their side chains bind to tyrosyl-tRNA synthetase in similar orientations and that at least one of the carboxylate oxygen atoms in d-tyrosine is properly positioned for attack on the α-phosphate of ATP.
Tyrosyl-tRNA synthetase (TyrRS)2 catalyzes the transfer of tyrosine to the 3′ end of tRNATyr in a two-step reaction (Fig. 1). In the first step, tyrosine is activated by ATP, forming the enzyme-bound tyrosyl-adenylate intermediate. In the second step, the tyrosyl moiety is transferred to the 3′ end of tRNATyr. The observations that the two steps of the reaction can be run independently of each other, and that formation of the tyrosyl-adenylate intermediate is accompanied by a change in the intrinsic fluorescence of the enzyme, make it possible to use stopped-flow fluorescence to monitor single turnover kinetics for each step in the reaction (2, 3).
FIGURE 1.

Reaction diagram for the aminoacylation of tRNATyr by tyrosine. The activation of tyrosine by ATP and subsequent transfer of the tyrosyl moiety from the enzyme-bound tyrosyl-adenylate intermediate to the 3′ end of tRNATyr are shown. Dissociation and rate constants are shown above or below the step with which they are associated. Noncovalent interactions are indicated by “·” and covalent interactions are indicated by “-”.
Tyrosyl-tRNA synthetase is composed of two identical 47-kDa subunits, each of which consists of a Rossmann fold domain containing the active site, a helical anticodon binding domain, and a carboxyl-terminal domain that binds the variable loop in tRNATyr. Tyrosyl-tRNA synthetase exhibits an extreme form of negative cooperativity with respect to tyrosine binding, known as “half-of-the-sites” reactivity, in which the unliganded subunit is completely inactivated. This behavior has been rationalized by the observation that, in solution, tyrosyl-tRNA synthetase binds only one molecule of tRNATyr and therefore has no need for two functional active sites. Discrimination between l-tyrosine and other amino acids is achieved solely on the basis of binding affinity (i.e. there is no editing domain in tyrosyl-tRNA synthetase). Surprisingly, Calendar and Berg (4) observed that tyrosyl-tRNA synthetase is able to aminoacylate tRNA with either the l-or d-stereoisomer of tyrosine, although activation is more efficient for l-tyrosine than it is for d-tyrosine. Hydrolysis of d-Tyr-tRNATyr is catalyzed by d-tyrosyl-tRNA deacylase in vivo, as tyrosyl-tRNA synthetase does not have an editing mechanism to prevent formation of d-Tyr-tRNATyr (5–7).
Recognition of tRNATyr differs between bacteria and eukaryotes, with the bacterial and eukaryotic (or archaeal) tyrosyl-tRNA synthetases being unable to efficiently aminoacylate each others' tRNATyr substrates (8, 9). This property has been exploited to introduce unnatural amino acids into proteins in both bacterial and eukaryotic systems. For example, Schultz and co-workers (10) have modified the tyrosyl-tRNA synthetase:tRNATyr pair from Methanococcus jannaschii so that it is completely nonorthologous to that of Escherichia coli. By replacing the anticodon in tRNATyr with one that is complementary to a stop codon, they have been able to introduce unnatural amino acids at specific positions in recombinant proteins expressed from E. coli (10). The observation that tyrosyl-tRNA synthetase catalyzes the aminoacylation of tRNATyr by d-tyrosine raises the possibility that tyrosyl-tRNA synthetase variants designed to incorporate unnatural l-amino acids into proteins can be adapted to selectively incorporate the d-analogs of the unnatural amino acids. As a first step toward this goal, we have characterized the binding, activation, and transfer of d-tyrosine to tRNATyr by Bacillus stearothermophilus tyrosyl-tRNA synthetase using pre-steady-state kinetics.
EXPERIMENTAL PROCEDURES
Materials—Reagents were purchased from the following sources: d-[14C]tyrosine (American Radiolabeled Chemicals Inc.); l-[14C]tyrosine (Moravek Biochemicals); β-mercaptoethanol, l-amino acid oxidase, and inorganic pyrophosphatase (Sigma); nitrocellulose filters (Schleicher & Schuell); Source 15Q-Sepharose anion exchange resin and NAP-25 columns (GE Healthcare); and DispoEquilibrium biodialyzer (The Nest Group, Inc.). All other reagents were purchased from Fisher. Grafit version 5.0.6 (Erithacus Software Ltd.) and Kaleidograph version 3.6 (Synergy Software) were used to fit the kinetic data.
Purification of Recombinant Tyrosyl-tRNA Synthetase—Purification of the wild-type tyrosyl-tRNA synthetase was performed as described previously (1, 11–16). Briefly, the purification consists of the following: 1) expression of tyrosyl-tRNA synthetase in E. coli Tg2 cells (17); 2) lysis of the E. coli cells and incubation of the extract at 56 °C for 40 min, followed by centrifugation to remove contaminating E. coli proteins; 3) dialysis of the remaining supernatant against three changes of 20 mm Tris buffer, pH 7.78, containing 1 mm EDTA, 5 mm β-mercaptoethanol, and 0.1 mm pyrophosphate to remove any tyrosyl-adenylate bound to the tyrosyl-tRNA synthetase, followed by dialysis against 20 mm BisTris, pH 6.0, 1 mm EDTA, 5 mm β-mercaptoethanol; and 4) high pressure liquid chromatography purification of the B. stearothermophilus tyrosyl-tRNA synthetase variants on a Source 15Q-Sepharose anion exchange column using a gradient from 20 mm BisTris, pH 6.0, to 20 mm BisTris, pH 6.0, 1 m NaCl. A peak eluting at 180 mm NaCl was collected and dialyzed overnight against 20 mm Tris, pH 7.78, 1 mm EDTA, 5 mm β-mercaptoethanol. This protein was then repurified on a Source 15Q-Sepharose column using a gradient from 20 mm Tris, pH 7.78, to 20 mm Tris, pH 7.78, 1 m NaCl. A peak eluting at 220 mm NaCl was collected and dialyzed overnight against 20 mm Tris, pH 7.78, 1 mm EDTA, 5 mm β-mercaptoethanol, and 10% glycerol (v/v). Typical yields were 20–30 mg/liter. Purified protein was stored at –70 °C. A single band corresponding to the B. stearothermophilus tyrosyl-tRNA synthetase was observed on SDS-PAGE. The concentration of the tyrosyl-tRNA synthetase was determined using a filter-based activesite titration assay, in which the incorporation of [14C]tyrosine into the enzyme-bound tyrosyl-adenylate intermediate is monitored (18). Comparison of the tyrosyl-tRNA synthetase concentration determined by active site titration with that determined by A280 (19) indicated that >95% of the purified protein was active tyrosyl-tRNA synthetase.
l-Amino Acid Oxidase Treatment of d-Tyrosine—Treatment of d-tyrosine with l-amino acid oxidase was performed as described by Calender and Berg (4). Briefly, d-tyrosine (2.0 mm) was treated with l-amino acid oxidase (0.1 unit/ml) in 144 mm Tris buffer, pH 7.78, at 37 °C. The reaction was terminated by boiling for 2 min.
Purification of tRNATyr—In vitro transcription of tRNATyr was performed using the procedure described by Xin et al. (20). In vitro transcribed tRNATyr was purified by a modification of the procedure described by Uter et al. (21). The in vitro reaction was loaded onto a 5-ml DE52 (Whatman) column, eluted with elution buffer (100 mm HEPES-KOH, pH 7.5, 12 mm MgCl2, 600 mm NaCl). Fractions containing tRNATyr were pooled and desalted on a NAP-25 column. Fractions from the NAP-25 column that contained tRNATyr were pooled and precipitated by adding 2 volumes of 100% ethanol and incubating at –20 °C overnight. After centrifugation, the tRNA pellet was dried and resuspended in 100 μl of 10 mm MgCl2. Annealing of tRNATyr was achieved by incubation at 80 °C for 10 min, followed by slow cooling overnight. A nitrocellulose filter assay, in which the incorporation of [14C]tyrosine into the Tyr-tRNATyr product is monitored, was used to determine the concentration of tRNATyr (2).
Steady-state Fluorescence Spectra—Steady-state fluorescence emission measurements were performed at 25 °C using a TimeMaster fluorescence spectrometer (Photon Technology International). The intrinsic fluorescence of the B. stearothermophilus tyrosyl-tRNA synthetase was measured in the absence and presence of substrates (λex = 295 nm, λem = 300–400 nm) in 144 mm Tris, pH 7.78, 10 mm β-mercaptoethanol, 10 mm MgCl2, and 1 unit/ml inorganic pyrophosphatase (Buffer A). Specifically, aliquots of either MgATP or d-or l-tyrosine were added to the B. stearothermophilus enzyme (0.5 μm) and 1 unit/ml inorganic pyrophosphatase in either Buffer A alone, Buffer A + 200 μml-tyrosine, Buffer A + 500 μmd-tyrosine, or Buffer A + 10 mm MgATP. After allowing the reaction to equilibrate for 2 min at 25 °C, the intrinsic fluorescence of the enzyme was determined by exciting the protein at 295 nm, and the relative intensities of the fluorescence emission spectra were determined by integrating the area under the emission curve from 320 to 400 nm.
Fluorescence spectra for samples containing MgATP were corrected to eliminate inner filter effects. This was done by multiplying the spectra by a scalar determined from Equation 1,
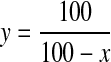 |
(Eq. 1) |
where y is the correction factor, and x is the percent decrease in the total fluorescence that is observed on the addition of an equivalent amount of MgATP to the unliganded enzyme.
Equilibrium Binding Studies—Equilibrium dialysis was performed using a modification of the method previously described by Fersht (22). Briefly, one chamber of each equilibrium dialysis cell contained 40 μm tyrosyl-tRNA synthetase and 1 unit/ml inorganic pyrophosphatase in buffer composed of 144 mm Tris, pH 7.78, 10 mm β-mercaptoethanol, and 10 mm MgCl2 (chamber A). The other chamber (chamber B) of each equilibrium dialysis cell contained concentrations of d-[14C]tyrosine ranging from 40 to 1300 μm in the same buffer. A dialysis membrane with a molecular mass cutoff of 10,000 daltons separated the chambers. After overnight dialysis at 4 °C, the amount of d-[14C]tyrosine present in each chamber was determined by removing 40-μl aliquots, adding each aliquot to 5 ml of Cytoscint scintillation mixture, and counting in a Beckman LS 6500 scintillation counter. The concentration of tyrosine in each chamber was calculated from the specific activity of the stock d-[14C]tyrosine. The concentrations of enzyme-bound and free tyrosine were calculated by subtracting the tyrosine concentration in chamber B ([Tyr]free) from that in chamber A ([Tyr]bound + [Tyr]free). The data were then fit to Equations 2 and 3 (23–25),
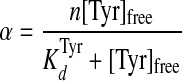 |
(Eq. 2) |
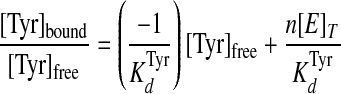 |
(Eq. 3) |
where α = [Tyr]bound/[E]T; K Tyrd is the dissociation constant for tyrosine; n is the total number of binding sites, and [E]T is the total enzyme concentration.
Kinetic Procedures—All kinetic analyses were performed in 144 mm Tris buffer, pH 7.78, 10 mm β-mercaptoethanol, and 10 mm MgCl2 (Buffer B) at 25 °C unless otherwise indicated. ATP was added as the Mg2+ salt to maintain the free concentration of Mg2+ at 10 mm.
Tyrosine Activation—Formation of the enzyme-bound tyrosyl-adenylate complex is accompanied by a decrease in the intrinsic fluorescence of tyrosyl-tRNA synthetase (3). This allows the kinetics of the tyrosine activation reaction to be monitored using stopped-flow fluorescence methods (2, 3). An Applied Photophysics SX-18.MV stopped-flow spectrophotometer was used to monitor the decrease in the intrinsic fluorescence of B. stearothermophilus tyrosyl-tRNA synthetase on formation of the TyrRS·Tyr-AMP intermediate (λex = 295 nm, λem > 320 nm). The rate constant (k3; where k3 is the forward rate constant for the activation of tyrosine) and equilibrium constant for the dissociation of tyrosine (K′ Tyrd) from the TyrRS·Tyr·ATP complex were calculated from the variation of kobs with respect to tyrosine concentration in the presence of 10 mm ATP. Under these conditions ∼70% of the enzyme has ATP bound to it. The equilibrium constants for the dissociation of tyrosine and ATP from the TyrRS·Tyr and TyrRS·ATP complexes were calculated in the same manner as K′ Tyrd, except that ATP and tyrosine concentrations are kept at 0.5 mm and 10 μm, respectively. Under these conditions, >90% of the enzyme was present as the unliganded enzyme. For determination of K Tyrd and K′ Tyrd, the concentration of tyrosine was varied from 10 to 1200 μm. For determination of K ATPd (where K ATPd is the equilibrium constant for the dissociation of ATP from the TyrRS·ATP complex), the concentration of ATP was varied from 0.5 to 50 mm.
In general, the experimental setup for determining the rate and equilibrium constants is as follows: syringe 1 contains 0.3–0.5 μm tyrosyl-tRNA synthetase, 1 unit/ml inorganic pyrophosphatase, and the substrate that is not being varied in Buffer B. Syringe 2 contains 1 unit/ml inorganic pyrophosphatase and the substrate whose dissociation constant is being determined in Buffer B. After mixing equal volumes from each syringe, the decrease in the intrinsic fluorescence of the protein was monitored. The addition of inorganic pyrophosphatase prevents the reverse reaction from occurring once the TyrRS·Tyr-AMP complex has formed.
Pyrophosphorolysis and Pyrophosphate Release—The kinetics for pyrophosphorolysis of the ATP moiety were determined by monitoring the reverse reaction for tyrosine activation. The conversion of TyrRS·Tyr-AMP + pyrophosphate to TyrRS + Tyr + ATP is accompanied by an increase in the intrinsic fluorescence of tyrosyl-tRNA synthetase (3). This allows stopped-flow fluorescence methods to be used to monitor the reverse rate constant (k–3) and the equilibrium constant for the dissociation of pyrophosphate from the TyrRS·Tyr-AMP·PPi complex (K PPid). The TyrRS·Tyr-AMP intermediate was prepared by incubating tyrosyl-tRNA synthetase with saturating concentrations of MgATP and tyrosine and 1 unit/ml inorganic pyrophosphatase in Buffer B for 30 min at 25 °C. The TyrRS·Tyr-AMP complex was separated from free tyrosine and MgATP by gel filtration on a NAP-25 column (26). The experimental setup for monitoring the reverse reaction is similar to that described above for the activation of tyrosine, except that syringe 1 contains the TyrRS·Tyr-AMP complex (0.3 μm) in Buffer B and syringe 2 contains 0.1–0.8 mm disodium pyrophosphate.
Pre-steady-state Kinetic Measurement of tRNATyr Aminoacylation—Formation of the TyrRS·[Tyr-tRNATyr·AMP]‡ complex is accompanied by an increase in the intrinsic fluorescence of the protein (2). An Applied Photophysics model SX 18. MV stopped-flow spectrophotometer was used to monitor changes in the intrinsic fluorescence of the TyrRS·Tyr-AMP intermediate on the addition of tRNATyr as described by Xin et al. (20). Briefly, the TyrRS·Tyr-AMP intermediate is mixed with various concentrations of in vitro transcribed tRNATyr in the stopped-flow spectrophotometer and the change in the intrinsic fluorescence of the protein is monitored over time using an excitation wavelength of 295 nm and an emission filter with cutoff above 320 nm.
Analysis of Kinetic Data—All kinetic data were fit to a single exponential floating end point equation using the Applied Photophysics stopped-flow software package to determine the observed rate constants (kobs). The Kaleidagraph software was used to plot kobs versus the substrate concentrations and to fit these plots to the following hyperbolic function shown in Equation 4,
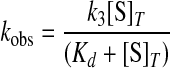 |
(Eq. 4) |
where k3 is the forward rate constant for the formation of tyrosyl-adenylate; [S]T is the total substrate concentration, and Kd is the dissociation constant for the substrate of interest (27). Goodness of fit was determined from the Eadie-Hofstee transformation of Equation 4 to Equation 5,
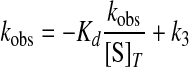 |
(Eq. 5) |
where kobs, k3, [S]T, and Kd are as described above.
The forward rate constant for the transfer of the tyrosyl moiety to tRNATyr (k4) and the equilibrium constant for the dissociation of tRNATyr from the TyrRS·Tyr-AMP·tRNATyr complex (K tRNAd) are calculated using equations that are analogous to Equations 4 and 5.
Calculation of Standard Free Energies of Binding—For the activation and transfer of d-tyrosine to tRNATyr, the relative standard free energies for each state along the reaction pathway were calculated from the rate and dissociation constants using Equations 6, 7, 8, 9, 10, 11, 12, assuming standard states of 1 m for ATP, tyrosine, pyrophosphate, and tRNATyr,
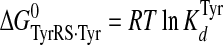 |
(Eq. 6) |
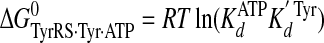 |
(Eq. 7) |
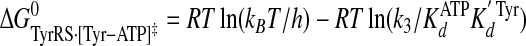 |
(Eq. 8) |
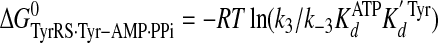 |
(Eq. 9) |
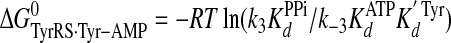 |
(Eq. 10) |
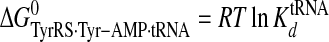 |
(Eq. 11) |
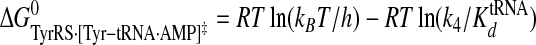 |
(Eq. 12) |
where ΔG0 is the standard Gibbs free energy change; R is the gas constant; T is the absolute temperature; kB is the Boltzmann constant; h is Planck's constant; “·” and “-” represent noncovalent and covalent bonds, respectively, and ‡ denotes the transition state complex. These equations are a modification of those previously used to calculate the standard free energies for the activation of l-tyrosine, with the K ATPdK′ Tyrd term replacing the K TyrdK′ ATPd term used by Fersht (28) (where K′ ATPd is dissociation of ATP from the TyrRS·Tyr·ATP complex). Because the binding of tyrosine and ATP is a random order process (29), the K′ ATPd K Tyrd and K′ Tyrd K ATPd terms are equivalent, and substituting one for the other will not affect the standard free energy value for each state. Standard free energies for each complex are calculated relative to the standard free energy of either the unliganded enzyme (Equations 6, 7, 8, 9, 10) or the TyrRS·Tyr-AMP intermediate (Equations 11 and 12). The Gibbs activation energy for the formation of tyrosyl-adenylate was calculated by taking the difference in free energies between the transition state (Equation 8) and the TyrRS·Tyr·ATP complex immediately preceding the transition state (Equation 7) which gives Equation 13,
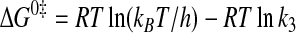 |
(Eq. 13) |
where ΔG0‡ is the activation energy; R is the gas constant; T is the absolute temperature; kB is the Boltzmann constant; h is Planck's constant; and k3 is the forward rate constant for the activation of tyrosine (28).
Ligand Docking—The tyrosyl- and tryptophanyl-tRNA synthetase coordinates used in ligand docking were taken from the 4TS1 and 1MB2 PDB files (30, 31). Only the coordinates for chain A were used in ligand docking. Similarly, the coordinates for l-Tyr and l-Trp were extracted from the 4TS1 and 1MB2 PDB files, respectively. The coordinates for d-Tyr were generated as follows: 1) the l-Tyr coordinates were reflected through the z axis to generate d-Tyr; 2) the four possible rotamers of d-Tyr were generated using Coot (32, 33); 3) LSQKAB (in the CCP4 software suite) was used to superpose the Cβ and side chain atoms for each of the d-Tyr rotomers onto the original l-Tyr coordinates, and the rotomer that superposed best was selected by visual inspection (32, 34). The coordinates for d-Trp were generated from the l-Trp coordinates in an analogous manner.
Ligand docking was performed using Autodock 4.0 (35). Protein coordinates were fixed during docking. For docking of tyrosine and tryptophan, the ligand was flexible and allowed to move on a grid centered on the l-Tyr and l-Trp coordinates from the 4TS1 and 1MB2 PDB files, respectively. An initial population of 300 starting structures was used for energy optimization, and 100 docking runs were performed to find the optimal ligand conformation. A maximum of 2.5 × 106 energy evaluations was used. Default settings were used for all other parameters, including the grid spacing, which was set to 0.375 Å. Grid searching was performed using a Lamarckian genetic algorithm. Molprobity was used to analyze atomic clashes between the docked ligand and protein (36).
RESULTS
Formation of the Enzyme-bound d-Tyrosyl-Adenylate Complex Is Accompanied by a Blue Shift in the Intrinsic Fluorescence of the Enzyme—In B. stearothermophilus tyrosyl-tRNA synthetase, formation of the enzyme-bound l-tyrosyl-adenylate complex results in an 8 nm shift in the intrinsic fluorescence emission of the enzyme (3). To determine whether a similar fluorescence change also occurs in the enzyme on formation of the TyrRS·Tyr-AMP complex with d-tyrosine, the fluorescence emission spectra of the enzyme in the presence of d-tyrosine, MgATP, and d-tyrosine + MgATP were monitored.
In the absence of substrates, B. stearothermophilus exhibits a relative fluorescence emission maximum at 349 (±3) nm when excited at 295 nm (Fig. 2, solid line in panels A–D). The addition of 500 μmd-tyrosine to the enzyme causes the relative fluorescence emission to be blue-shifted by 8 (±2) nm, resulting in an 8% decrease in the fluorescence emission of the enzyme above 320 nm (Fig. 2, panel C). As is the case for l-tyrosine, inner filter effects due to the presence of d-tyrosine are negligible. The addition of 10 mm MgATP to the enzyme results in a 14% decrease in the total fluorescence emission of the enzyme (Fig. 2, panels B and D). The decreased fluorescence of tyrosyl-tRNA synthetase on addition of MgATP exhibits a linear dependence with respect to the concentration of MgATP, indicating that the fluorescence decrease is because of inner filter effects (data not shown). With the exception of the TyrRS·ATP spectra (Fig. 1, panels B and D), all of the steady-state fluorescence emission spectra in which MgATP is present are corrected for this inner filter effect. The addition of 500 μm d-tyrosine and 10 mm MgATP together produces a 2(±1) nm enhancement of the blue shift observed in the presence of tyrosine alone. Integrating the areas under the curves indicates that there is an additional 8% (after correcting for the inner filter effect) decrease in the relative fluorescence of the enzyme above 320 nm. These changes in the emission spectrum of B. stearothermophilus tyrosyl-tRNA synthetase upon formation of the TyrRS·d-Tyr and TyrRS·d-Tyr-AMP complexes are similar to the intrinsic fluorescence changes observed for the formation of the TyrRS·l-Tyr and TyrRS·l-Tyr-AMP complexes, suggesting that the conformation of the enzyme-ligand complex is similar in both cases.
FIGURE 2.

Steady-state fluorescence emission spectra for B. stearothermophilus tyrosyl-tRNA synthetase in the presence of d-or l-tyrosine. The fluorescence emission spectra for B. stearothermophilus tyrosyl-tRNA synthetase in the absence and presence of d-tyrosine, l-tyrosine, MgATP, and disodium pyrophosphate are shown (λex = 295 nm, λem = 300–400 nm). The emission spectra of the enzyme (0.5 μm) in the presence of 200 μml-tyrosine and 10 mm MgATP (panels A and B), and 500 μmd-tyrosine and 10 mm MgATP (panels C and D), respectively, are shown. Panels A and C, tyrosine is added first followed by MgATP, and in panels B and D the respective orders of addition are reversed. With the exception of the TyrRS·ATP spectra (panels B and D), all of the emission spectra in which MgATP is present are corrected for the inner filter effects of MgATP by multiplying the spectra by a factor of 1.19. Panels E and F show changes in the relative fluorescence intensity (λex = 295 nm, λem = 300–400 nm) for the TyrRS·l-Tyr-AMP and TyrRS·d-Tyr-AMP complexes in the presence of 0.8 mm disodium pyrophosphate. The concentrations of TyrRS·l-Tyr-AMP and TyrRS·d-Tyr-AMP were 1 μm for these experiments. To ensure that formation of the enzyme-bound tyrosyl-adenylate complex is stoichiometric, all of the steady-state emission spectra were determined in the presence of inorganic pyrophosphatase.
If the blue-shift in the fluorescence emission spectrum of tyrosyl-tRNA synthetase is because of formation of the TyrRS·Tyr-AMP complex, then the addition of pyrophosphate to this complex should produce a corresponding red-shift in the fluorescence emission spectrum. In the absence of pyrophosphate, the purified B. stearothermophilus TyrRS·d-Tyr-AMP complex exhibits a fluorescence emission maximum at 341 (±2) nm. The addition of 0.8 mm disodium pyrophosphate to this TyrRS·d-Tyr-AMP complex produces a 7nm red-shift in the fluorescence emission spectrum (Fig. 2, panel F). Similar changes in fluorescence were observed with the B. stearothermophilus TyrRS·l-Tyr-AMP intermediate (Fig. 2, panel E).
Recognition of d-Tyrosine by Tyrosyl-tRNA Synthetase—The observation that formation of the TyrRS·Tyr-AMP intermediate produces changes in the fluorescence emission spectrum for both l- and d-tyrosine suggests that stopped-flow fluorescence spectroscopy can be used to monitor the activation of d-tyrosine by tyrosyl-tRNA synthetase. To verify that this is the case, the change in the intrinsic fluorescence of tyrosyl-tRNA synthetase with respect to time was monitored using stopped-flow methods. When MgATP is mixed with enzyme that has been preincubated with d-tyrosine, a rapid single exponential decrease in the intrinsic fluorescence of tyrosyl-tRNA synthetase is observed (Fig. 3, panel A). A similar change in the intrinsic fluorescence of tyrosyl-tRNA synthetase is observed when d-tyrosine is mixed with enzyme that had been preincubated with MgATP (data not shown). For the reverse reaction, a rapid single exponential increase in relative fluorescence is observed when disodium pyrophosphate is mixed with the purified TyrRS·d-Tyr-AMP complex (Fig. 3, panel B). This observed increase in fluorescence mirrors the decrease observed when free enzyme was mixed with d-tyrosine and MgATP and corresponds to the conversion of TyrRS·d-Tyr-AMP + pyrophosphate to TyrRS + d-Tyr + ATP. A single exponential increase in the intrinsic fluorescence of tyrosyl-tRNA synthetase is also observed when tRNATyr is added to the TyrRS·d-Tyr-AMP intermediate (data not shown). This increase in intrinsic fluorescence is identical to that observed for the aminoacylation of tRNATyr by the TyrRS·l-Tyr-AMP intermediate (2).
FIGURE 3.

Stopped-flow fluorescence emission spectra for the formation and pyrophosphorylation of the TyrRS·Tyr-AMP complex. Panel A shows the reaction trace for the formation of the TyrRS·d-Tyr-AMP complex, as determined by monitoring the decrease in the fluorescence emission above 320 nm. Tyrosyl-tRNA synthetase (0.5 μm) was preincubated in the presence of d-tyrosine and subsequently mixed with MgATP. Panel B shows the conversion of 0.25 μm TyrRS·d-Tyr-AMP + pyrophosphate to TyrRS + d-Tyr + ATP, determined by monitoring the increase in the fluorescence emission above 320 nm. Data acquisition for both curves was split, with 500 data points measured during the initial 20% of each reaction trace, and 500 data points measured during the remainder of the reaction trace.
Activation of d-Tyrosine by Tyrosyl-tRNA Synthetase—The equilibrium constants for the dissociation of d-tyrosine from the TyrRS·Tyr and the TyrRS·Tyr·ATP complexes (K D-Tyrd and K′ D-Tyrd, respectively) were determined by measuring the observed rate constant for the formation of TyrRS·d-Tyr-AMP as a function of the concentration of d-tyrosine. For determination of K D-Tyrd (Fig. 4, panel A), the concentration of ATP was 0.5 mm (∼1/10 K ATPd), whereas for determination of K′ D-Tyrd (Fig. 4, panel B), the concentration was 10 mm (∼3 K ATPd). Comparison of the dissociation and rate constants for d- and l-tyrosine indicates that the enzyme has an 8.5-fold lower affinity and 3-fold lower forward rate constant when d-tyrosine is the substrate of the reaction than when l-tyrosine is the substrate (Table 1). Pretreatment of the d-tyrosine stock solution with l-amino acid oxidase does not alter the kinetics, indicating that the observed results are not due to l-tyrosine contamination (data not shown).
FIGURE 4.

Dissociation of tyrosine from the TyrRS·Tyr and TyrRS·Tyr·ATP complexes. The equilibrium constants for the dissociation of tyrosine from the TyrRS·Tyr (K Tyrd) and TyrRS·Tyr·ATP (K′ Tyrd) complexes were determined by fitting plots of kobs versus the concentration of d-tyrosine to Equation 4. For determination of K Tyrd, the ATP concentration was 0.5 mm, whereas for K′ Tyrd, it was 10 mm (K ATPd = 3.5 mm). Panels A and B show typical plots for kobs versus d-tyrosine concentration at 0.5 mm and 10 mm MgATP, respectively. The inset shows the data fit to the Eadie-Hofstee transformation of Equation 4.
TABLE 1.
Rate and dissociation constants for the activation of l- and d-tyrosine
Experimental errors are indicated in parentheses.
| l-Tyrosinea | d-Tyrosine | |
|---|---|---|
| KdTyr (μm) | 12 | 102 (±4) |
| K′dTyr (μm) | NDb | 288 (±8) |
| KdATP (mm) | 3.5 | 4.9 (±0.4) |
| k3 (s-1) | 38 | 13 (±1) |
| KdPPi (mm) | 0.61 | 0.043 (±0.007) |
| k-3 (s-1) | 16.6 | 4.6 (±0.6) |
| k-3/KdPPi (s-1m-1) | 27,200 | 110,000 |
| KdtRNA (μm) | 0.39 | 0.91 (±0.02) |
| k4 (s-1) | 31 | 21.6 (±0.2) |
| k4/KdtRNA (s-1m-1) | 7.9 × 107 | 2.37 (±0.02) × 107 |
Equilibrium dialysis was used to determine whether the enzyme displays half-of-the-sites reactivity with respect to d-tyrosine binding. The equilibrium dialysis data were fit to Equation 2 to calculate both the dissociation constant for d-tyrosine (K D-Tyrd) and the number of d-tyrosine binding sites per tyrosyl-tRNA synthetase dimer (Fig. 5). Analysis of the equilibrium dialysis data indicates that the enzyme binds 1.2 (±0.1) molecules of d-tyrosine per enzyme dimer. This is consistent with the observation that comparing the enzyme concentration determined by A280 measurements with that determined by active site titration also shows formation of a single tyrosyl-adenylate per tyrosyl-tRNA synthetase dimer (data not shown). The K D-Tyrd value obtained from equilibrium dialysis (58 ± 4 μm) is approximately half the K D-Tyrd determined from the stopped-flow fluorescence experiment. To determine whether the difference in K D-Tyrd values obtained from these two methods is because of the difference in the temperatures at which the equilibrium dialysis (6–8 °C) and stopped-flow fluorescence (25 °C) experiments were done, stopped-flow fluorescence experiments were performed at 4 °C. The K D-Tyrd value obtained from these experiments is 34 (±3) μm, consistent with the hypothesis that the lower K D-Tyrd obtained from equilibrium dialysis is because of the temperature at which it was determined.
FIGURE 5.

Analysis of d-tyrosine binding by equilibrium dialysis. The binding of d-[14C]tyrosine to tyrosyl-tRNA synthetase during a typical equilibrium dialysis experiment is shown. Data are fit to the Langmuir isotherm (Equation 3) (24). The inset shows the data fit to the Scatchard equation (Equation 2) (23).
The equilibrium constant for the dissociation of ATP from the TyrRS·ATP complex (K ATPd) was determined by measuring the observed rate constant for the formation of TyrRS·d-Tyr-AMP as a function of the ATP concentration. In these experiments, the concentration of d-tyrosine was 10 μm (∼1/10 K D-Tyrd). Under these conditions, the enzyme displayed typical hyperbolic kinetics with a K ATPd of 3.8 (±0.4) mm (Fig. 6). In contrast, measuring the ATP dependence of the observed rate constant for formation of the TyrRS·d-Tyr-AMP complex at saturating concentrations of d-tyrosine gives sigmoidal kinetics (see accompanying paper (45)).
FIGURE 6.

Analysis of MgATP binding to unliganded tyrosyl-tRNA synthetase. A typical plot for the dependence of kobs with respect to the concentration of MgATP in the presence of 10 μmd-tyrosine is shown. At this concentration of d-tyrosine, less than 10% of the enzyme contains d-tyrosine bound to the active site. The dissociation constant determined under these conditions corresponds to the dissociation of ATP from the TyrRS·ATP intermediate (i.e. K ATPd) (26).
Pyrophosphorolysis and Pyrophosphate Release—The kinetics for cleavage of the scissile bond between the α- and β-phosphates of ATP, and the subsequent release of pyrophosphate, were determined by monitoring the conversion of TyrRS·Tyr-AMP + pyrophosphate to TyrRS + Tyr + ATP (i.e. the reverse of the tyrosine activation reaction). In these experiments, the equilibrium strongly favors formation of the free enzyme, as the total concentrations of d-tyrosine and MgATP released from the TyrRS·d-Tyr-AMP complex are well below their dissociation constants. As discussed previously, the addition of pyrophosphate to the purified TyrRS·Tyr-AMP complex results in a time-dependent increase in the intrinsic fluorescence of the enzyme that can be fit to a single exponential equation (Fig. 4). Surprisingly, pyrophosphate was found to bind to the TyrRS·d-Tyr-AMP complex with a 14-fold higher affinity than it binds to the TyrRS·l-Tyr-AMP complex (Fig. 7 and Table 1). The rate constant for the conversion of TyrRS·Tyr-AMP + pyrophosphate to TyrRS + Tyr + ATP (k–3) is 3.6-fold lower for the TyrRS·d-Tyr-AMP intermediate than it is for TyrRS·l-Tyr-AMP.
FIGURE 7.

Analysis of pyrophosphorolysis and pyrophosphate dissociation. A typical plot for the conversion of TyrRS·Tyr-AMP + pyrophosphate to TyrRS + Tyr + ATP is shown. The dissociation constant determined from this assay corresponds to the dissociation of pyrophosphate from the TyrRS·Tyr-AMP·PPi complex (K PPid), and the rate constant (k–3) corresponds to the rate constant for formation of the bond between the α- and β-phosphates of ATP.
Aminoacylation of tRNATyr by d-Tyrosine—Both the forward rate constant for the transfer of the tyrosyl moiety to tRNATyr and the equilibrium constant for the dissociation of tRNATyr from the TyrRS·Tyr-AMP·tRNATyr complex (K tRNAd) were determined by measuring the observed rate constant for the transfer of the tyrosyl moiety from the TyrRS·Tyr-AMP·tRNATyr complex to tRNATyr as a function of the tRNATyr concentration (Fig. 8). As shown in Table 1, the forward rate constant for the transfer of the d-tyrosyl moiety to tRNATyr (k4) is not significantly different from the forward rate constant for the transfer of the l-tyrosyl moiety to tRNATyr. In contrast, the binding of tRNATyr to the TyrRS·d-Tyr-AMP intermediate is 2.3-fold weaker than the binding of tRNATyr to the TyrRS·l-Tyr-AMP intermediate (Fig. 8 and Table 1).
FIGURE 8.

Analysis of the tRNATyr aminoacylation reaction. A typical hyperbolic plot for the transfer of d-tyrosine from the TyrRS·Tyr-AMP intermediate to in vitro transcribed B. stearothermophilus tRNATyr substrate is shown. The forward rate constants (k4) and the tRNATyr dissociation (K tRNAd) constants were determined from a plot of initial rate versus tRNATyr concentration.
Analysis of the Free Energy Profile for the Activation of d-Tyrosine—The Gibbs standard free energy values (ΔG0) for each bound state in the reaction pathway were calculated relative to the free energy of the unliganded enzyme (Fig. 9). Values shown for l-tyrosine are taken from previously published data and were calculated using K′ ATPd and K L-Tyrd values (26). It is not possible to calculate the stability of the TyrRS·d-Tyr·ATP complex from the values for K D-Tyrd and K′ ATPd because formation of the TyrRS·d-Tyr-AMP complex displays sigmoidal dependence with respect to the concentration of ATP at saturating tyrosine concentrations (accompanying paper, Ref. 45). It is possible, however, to determine the stability of this complex using the lower pathway for TyrRS·d-Tyr-AMP formation shown in Fig. 1. In this case, K ATPd and K′ D-Tyrd are used to calculate ΔG0E·Tyr·ATP. Each of the steps up through formation of the TyrRS·[Tyr-AMP]‡ complex is destabilized when d-tyrosine is substituted for the l-stereoisomer (ΔΔG0TyrRS·Tyr = 5.3 kJ/mol, ΔΔG0TyrRS·Tyr·ATP = 8.7 kJ/mol, and ΔΔG0 TyrRS·[Tyr-ATP]‡ = 11.3 kJ/mol). This effect is offset, however, by an increase in the affinity of the enzyme for pyrophosphate (K PPid) and a decreased reverse rate constant (k–3) when d-tyrosine is present. The net result is that the stability of the TyrRS·Tyr-AMP intermediate is nearly identical for l- and d-tyrosine (ΔΔG0TyrRS·Tyr-AMP =–2.2 kJ/mol).
FIGURE 9.

Standard free energy diagram for the aminoacylation of tRNATyr by l- and d-tyrosine for B. stearothermophilus tyrosyl-tRNA synthetase. Solid and dashed lines indicate the standard free energy changes during the course of the reaction for the activation of d-tyrosine and l-tyrosine, respectively, and their subsequent transfer to tRNATyr. Standard free energy values for the enzyme with l-tyrosine are taken from Refs. 2 and 26. For the activation of l-tyrosine, K TyrdK′ ATPd was used in place of the K ATPdK′ Tyrd term in Equations 7, 8, 9, 10.
Modeling of d-Tyrosine Binding—To gain further insight into the physical basis of d-tyrosine activation, Autodock 4.0 was used to dock d-tyrosine to the B. stearothermophilus tyrosyl-tRNA synthetase. As shown in Fig. 10, panel A, d-tyrosine binds in a manner similar to that of l-tyrosine. In particular, the side chain and amino groups of l- and d-tyrosine are located in similar positions. In addition, the carboxylate oxygens of d-tyrosine are located in close proximity to those of l-tyrosine, suggesting that they are in an appropriate position for attack on the α-phosphate of ATP. Analysis of all atom contacts by Molprobity confirmed that there were no steric clashes between tyrosyl-tRNA synthetase and d-tyrosine.
FIGURE 10.

Modeling the TyrRS·d-Tyr and TrpRS·d-Trp complexes. Panel A, docking of l- and d-tyrosine to B. stearothermophilus tyrosyl-tRNA synthetase is shown. l- and d-tyrosine are shown as stick models with oxygen atoms in red, nitrogen atoms in blue, and carbon atoms in green (l-tyrosine) and cyan (d-tyrosine). B. stearothermophilus tyrosyl-tRNA synthetase is shown as a schematic representation (magenta). Docking was done using Autodock 4.0 as described in the text. Panel B, docking of l- and d-tryptophan to B. stearothermophilus tryptophanyl-tRNA synthetase is shown. l- and d-tryptophan are shown as stick models with oxygen atoms in red, nitrogen atoms in blue, and carbon atoms in green (l-tryptophan) and cyan (d-tryptophan). B. stearothermophilus tryptophanyl-tRNA synthetase is shown as a schematic representation with the Gln-147 side chain shown as a stick model (magenta). Modeling was done by superimposing the d-tryptophan coordinates onto those of l-tryptophan and selecting the rotomer that most closely resembled the conformation adopted by d-tryptophan in the TrpRS·d-Trp complex as described in the text. The molecular graphics for this figure were generated using PyMol (44).
The dissociation constant calculated by Autodock 4.0 for the d-Tyr·TyrRS complex is 139 μm. Although this value is very close to that observed experimentally (Table 1), it should be noted that docking of l-tyrosine to B. stearothermophilus tyrosyl-tRNA synthetase resulted a similar value (K L-Tyrd = 133 μm). As a result, it is not clear from the docking results why there is an 8-fold difference in the binding of the two stereoisomers.
Because tryptophanyl-tRNA synthetase is a structural homolog of tyrosyl-tRNA synthetase (37), we investigated whether modeling the binding of d-tryptophan to B. stearothermophilus tryptophanyl-tRNA synthetase would reveal significant steric clashes between the ligand and enzyme. In contrast to the tyrosine docking simulations, docking of l-tryptophan to tryptophanyl-tRNA synthetase failed to produce a structure that superimposed well on the original coordinates. For this reason, modeling of d-tryptophan binding was done by superimposing the coordinates for d-tryptophan onto those of l-tryptophan from the TrpRS·l-Trp structure (PDB code 1mb2) and then selecting the rotomer that most closely resembled the conformation of d-tyrosine bound to tyrosyl-tRNA synthetase (Fig. 10, panel B). Molprobity analysis of all atom clashes for the resulting complex indicates that only the carboxyl oxygen of d-tryptophan and the ε-amine of Gln-147 in tryptophanyl-tRNA synthetase display significant steric overlap (0.519 Å). Soutourina et al. (38) have observed that E. coli tryptophanyl-tRNA synthetase is able to catalyze the formation of d-Trp-tRNATrp in vitro, suggesting that the above overlap is not sufficient to prevent d-tryptophan from binding to tryptophanyl-tRNA synthetase.
DISCUSSION
Aminoacyl-tRNA synthetases are highly specific enzymes, with misacylation of tRNA occurring less than 1 in 104–105 turnovers of the enzyme (39). Although there is some selection by EF-Tu to prevent the use of misacylated tRNA during protein synthesis (40), the accuracy of the translation process is primarily dependent on the ability of the aminoacyl-tRNA synthetases to recognize and aminoacylate their cognate tRNAs with the correct amino acid. For this reason, it was particularly surprising when Calendar and Berg (4) demonstrated that the E. coli and Bacillus subtilis tyrosyl-tRNA synthetases can aminoacylate tRNATyr with the d-stereoisomer of tyrosine. In a subsequent paper, these authors (5) identified and partially purified the E. coli d-Tyr-tRNA deacylase, which hydrolyzes the aminoacyl bond in d-Tyr-tRNATyr but not in l-Tyr-tRNATyr. Furthermore, Calendar and Berg (5) found d-tyrosyl-tRNA deacylase activity in E. coli, yeast, rabbit reticulocyte, and rat liver extracts, suggesting that it is widespread in nature. Blanquet and co-workers (6) have confirmed this finding by searching genome sequences for homologs of the E. coli and Saccharomyces cerevisiae d-tyrosyl-tRNA deacylases. d-Tyr-tRNATyr deacylase is not specific for d-Tyr-tRNATyr, but it will also catalyze the hydrolysis of other d-aminoacyl-tRNAs, including d-Trp-tRNATrp and d-Asp-tRNAAsp in E. coli and d-Leu-tRNALeu in S. cerevisiae.
In this paper, the mechanism by which B. stearothermophilus tyrosyl-tRNA synthetase recognizes d-tyrosine has been investigated using single turnover kinetics. In contrast to steady-state kinetics, single turnover kinetics allows one to determine the rate and dissociation constants for each intermediate step in the reaction. For catalysis of the tyrosine activation reaction by B. stearothermophilus tyrosyl-tRNA synthetase, replacing l-tyrosine by the d-stereoisomer increases the dissociation constant for tyrosine (K D-Tyrd) by 8.5-fold and decreases the forward rate constant (k3) by 3-fold (Table 1). This corresponds to a 24-fold decrease in the specificity constant (k3/K D-Tyrd). The observation that there is only an 8.5-fold increase in the K D-Tyrd values when l-tyrosine is replaced by the d-stereoisomer suggests that the d-tyrosine side chain binds to the enzyme in a manner that is similar to that observed for l-tyrosine. In addition, the observation that there is only a 3-fold decrease in k3 suggests that at least one of the carboxylate oxygen atoms is in the correct position for nucleophilic attack on the α-phosphate of ATP. The results of the docking studies are consistent with these hypotheses.
Surprisingly, pyrophosphate binds to tyrosyl-tRNA synthetase with a 14-fold higher affinity when d-tyrosine is activated than it does when l-tyrosine is activated (Table 1). This is particularly intriguing, as stabilization of the transition state for the l-tyrosine activation reaction is primarily due to interactions between the pyrophosphate moiety of ATP and the enzyme (1, 11–14). Specifically, seven residues in tyrosyl-tRNA synthetase have been shown to interact with pyrophosphate in the TyrRS·l-Tyr-AMP·PPi intermediate complex: Thr-40, His-45, Lys-82, Arg-86, Lys-230, Lys-233, and Thr-234 (1, 13). Four of these residues (His-45, Lys-230, Lys-233, and Thr-234) are in the HIGH and KMSKS signature sequences and are highly conserved among the class I aminoacyl-tRNA synthetases. The observation that pyrophosphate is bound more tightly in the TyrRS·d-Tyr-AMP·PPi complex when d-tyrosine is present than it is in the l-tyrosine complex suggests that it is oriented in a manner that increases the interaction between pyrophosphate and one or more of the residues discussed above. This suggests that the decreased stability of the transition state when d-tyrosine is present may be due to the altered interaction between tyrosyl-tRNA synthetase and the pyrophosphate moiety of ATP.
Although it is encouraging that the docking results predict a binding affinity for d-tyrosine similar to the experimentally observed value (K theord = 139 μm versus K exptd = 102 μm), this must be tempered by the observation that a similar value is predicted for the dissociation constant of l-tyrosine (K theord = 133 μm versus K exptd = 12 μm). Recently published molecular dynamics simulations predict that the free energy difference between the binding of l- and d-tyrosine to E. coli tyrosyl-tRNA synthetase is 13 (±8) kJ/mol (41). Both our results and those of Thompson et al. (41) are close to the experimentally determined free energy difference for the binding of l- and d-tyrosine (ΔΔG0 = 5.0 kJ/mol). Despite the success of the docking studies, however, the precise mechanism by which tyrosyl-tRNA synthetase discriminates between l- and d-tyrosine remains to be elucidated.
Several lines of evidence suggest that the binding of d-tyrosine has effects on the active site that extend beyond the tyrosine binding pocket. First, in contrast to the free enzyme, the TyrRS·d-Tyr complex exhibits sigmoidal kinetics with respect to ATP binding (see accompanying paper (45)). Second, pyrophosphate binds 14-fold more tightly when d-tyrosine is present (Table 1). Third, there is a 2.3-fold decrease in the binding affinity of tyrosyl-tRNA synthetase for tRNATyr when d-Tyr-AMP is bound (Table 1). These observations suggest that the tyrosine binding pocket is intimately connected to the ATP and tRNATyr binding pockets with apparently subtle changes to tyrosine binding affecting distal parts of the active site.
It is intriguing that Calendar and Berg (4) found that replacing l-tyrosine with its d-stereoisomer has a significantly larger effect on the value of Km Tyr for the E. coli enzyme than it does for B. subtilis tyrosyl-tRNA synthetase (Km D-Tyr /KKm L-Tyr = 23 for E. coli tyrosyl-tRNA synthetase versus 3 for B. subtilis tyrosyl-tRNA synthetase (4)). In contrast, replacing l-tyrosine with d-tyrosine results in a 10-fold decrease in the Vmax values for B. subtilis tyrosyl-tRNA synthetase, but only a 5-fold decrease in Vmax values for the E. coli enzyme (4). The effect that replacing l-tyrosine with d-tyrosine has on the K Tyrd and k3 values in B. stearothermophilus tyrosyl-tRNA synthetase is consistent with the above results obtained by Calendar and Berg (4) using steady-state kinetic methods. The observation that the stereoselectivity for tyrosine differs between the E. coli, B. subtilis, and B. stearothermophilus tyrosyl-tRNA synthetases suggests that tyrosyl-tRNA synthetase has the potential to be more stereoselective. This raises the question as to why tyrosyl-tRNA synthetases have not evolved the level of stereoselectivity that is observed in other aminoacyl-tRNA synthetases (39, 42, 43). One possibility is that the levels of d-tyrosine are sufficiently low that there is little selective pressure to discriminate between the d- and l-stereoisomers. This is unlikely, however, as the widespread distribution of d-Tyr-tRNA deacylase in nature suggests that misacylation of tRNATyr by d-tyrosine is a significant problem in organisms that are not auxotrophic for tyrosine (6). An alternative explanation is that increasing the stereospecificity of tyrosyl-tRNA synthetase may come at the expense of its catalytic activity. In this scenario, the selective advantage of increasing the catalytic activity of tyrosyl-tRNA synthetase outweighs the energetic costs associated with the editing of d-Tyr-tRNATyr in trans by d-Tyr-tRNA deacylase. It remains to be determined whether mutations that alter the stereoselectivity of tyrosyl-tRNA synthetase also affect the forward rate constant for the activation of tyrosine.
Acknowledgments
We thank Jason Manning, Chuka Ifeanyi, and Tara Andrews for technical assistance in protein purification.
This work was supported, in whole or in part, by National Institutes of Health Grant GM68070 from NIGMS. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: TyrRS, tyrosyl-tRNA synthetase; PPi, pyrophosphate; BisTris, 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol; PDB, Protein Data Bank.
References
- 1.Fersht, A. R., Knill-Jones, J. W., Bedouelle, H., and Winter, G. (1988) Biochemistry 27 1581–1587 [DOI] [PubMed] [Google Scholar]
- 2.Avis, J. M., Day, A. G., Garcia, G. A., and Fersht, A. R. (1993) Biochemistry 32 5312–5320 [DOI] [PubMed] [Google Scholar]
- 3.Fersht, A. R., Mulvey, R. S., and Koch, G. L. (1975) Biochemistry 14 13–18 [DOI] [PubMed] [Google Scholar]
- 4.Calendar, R., and Berg, P. (1966) Biochemistry 5 1690–1695 [DOI] [PubMed] [Google Scholar]
- 5.Calendar, R., and Berg, P. (1967) J. Mol. Biol. 26 39–54 [DOI] [PubMed] [Google Scholar]
- 6.Soutourina, J., Blanquet, S., and Plateau, P. (2000) J. Biol. Chem. 275 11626–11630 [DOI] [PubMed] [Google Scholar]
- 7.Soutourina, O., Soutourina, J., Blanquet, S., and Plateau, P. (2004) J. Biol. Chem. 279 42560–42565 [DOI] [PubMed] [Google Scholar]
- 8.Kleeman, T. A., Wei, D., Simpson, K. L., and First, E. A. (1997) J. Biol. Chem. 272 14420–14425 [DOI] [PubMed] [Google Scholar]
- 9.Wakasugi, K., Quinn, C. L., Tao, N., and Schimmel, P. (1998) EMBO J. 17 297–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang, L., Brock, A., Herberich, B., and Schultz, P. G. (2001) Science 292 498–500 [DOI] [PubMed] [Google Scholar]
- 11.First, E. A., and Fersht, A. R. (1993) Biochemistry 32 13658–13663 [DOI] [PubMed] [Google Scholar]
- 12.First, E. A., and Fersht, A. R. (1993) Biochemistry 32 13651–13657 [DOI] [PubMed] [Google Scholar]
- 13.First, E. A., and Fersht, A. R. (1993) Biochemistry 32 13644–13650 [DOI] [PubMed] [Google Scholar]
- 14.First, E. A., and Fersht, A. R. (1995) Biochemistry 34 5030–5043 [DOI] [PubMed] [Google Scholar]
- 15.Jones, D. H., McMillan, A. J., Fersht, A. R., and Winter, G. (1985) Biochemistry 24 5852–5857 [DOI] [PubMed] [Google Scholar]
- 16.Fersht, A. R., Wilkinson, A. J., Carter, P., and Winter, G. (1985) Biochemistry 24 5858–5861 [DOI] [PubMed] [Google Scholar]
- 17.Gibson, T. (1984) Studies on the Epstein-Barr Virus Genome. Ph.D. thesis, University of Cambridge, Cambridge, UK
- 18.Wilkinson, A. J., Fersht, A. R., Blow, D. M., and Winter, G. (1983) Biochemistry 22 3581–3586 [DOI] [PubMed] [Google Scholar]
- 19.Appel, R. D., Bairoch, A., and Hochstrasser, D. F. (1994) Trends Biochem. Sci. 19 258–260 [DOI] [PubMed] [Google Scholar]
- 20.Xin, Y., Li, W., and First, E. A. (2000) Biochemistry 39 340–347 [DOI] [PubMed] [Google Scholar]
- 21.Uter, N. T., Gruic-Sovulj, I., and Perona, J. J. (2005) J. Biol. Chem. 280 23966–23977 [DOI] [PubMed] [Google Scholar]
- 22.Fersht, A. R. (1975) Biochemistry 14 5–12 [DOI] [PubMed] [Google Scholar]
- 23.Scatchard, G. (1948) Ann. N. Y. Acad. Sci. 51 660–672 [Google Scholar]
- 24.Langmuir, I. (1916) J. Am. Chem. Soc. 38 2221–2295 [Google Scholar]
- 25.Clarke, A. R. (1996) in Enzymology Labfax (Engel, P., ed) 1st Ed., pp. 201–205, Academic Press, New York
- 26.Wells, T. N., and Fersht, A. R. (1986) Biochemistry 25 1881–1886 [DOI] [PubMed] [Google Scholar]
- 27.Ho, C. K., and Fersht, A. R. (1986) Biochemistry 25 1891–1897 [DOI] [PubMed] [Google Scholar]
- 28.Fersht, A. R., Leatherbarrow, R. J., and Wells, T. N. (1987) Biochemistry 26 6030–6038 [DOI] [PubMed] [Google Scholar]
- 29.Santi, D. V., and Pena, V. A. (1971) FEBS Lett. 13 157–160 [DOI] [PubMed] [Google Scholar]
- 30.Brick, P., and Blow, D. M. (1987) J. Mol. Biol. 194 287–297 [DOI] [PubMed] [Google Scholar]
- 31.Retailleau, P., Huang, X., Yin, Y., Hu, M., Weinreb, V., Vachette, P., Vonrhein, C., Bricogne, G., Roversi, P., Ilyin, V., and Carter, C. W., Jr. (2003) J. Mol. Biol. 325 39–63 [DOI] [PubMed] [Google Scholar]
- 32.Collaborative Computational Project, No 4. (1994) Acta Crystallogr. 50 760–763 [Google Scholar]
- 33.Emsley, P., and Cowtan, K. (2004) Acta Crystallogr. 60 2126–2132 [DOI] [PubMed] [Google Scholar]
- 34.Kabsch, W. (1976) Acta Crystallogr. Sect. A 32 922–923 [Google Scholar]
- 35.Morris, G. M., Goodsell, D. S., Halliday, R. S., Huey, R., Hart, W. E., Belew, R. K., and Olson, A. J. (1998) J. Comput. Chem. 19 1639–1662 [Google Scholar]
- 36.Davis, I. W., Leaver-Fay, A., Chen, V. B., Block, J. N., Kapral, G. J., Wang, X., Murray, L. W., Arendall, W. B., III, Snoeyink, J., Richardson, J. S., and Richardson, D. C. (2007) Nucleic Acids Res. 35 W375–W383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Doublie, S., Bricogne, G., Gilmore, C., and Carter, C. W., Jr. (1995) Structure (Lond.) 3 17–31 [DOI] [PubMed] [Google Scholar]
- 38.Soutourina, J., Plateau, P., and Blanquet, S. (2000) J. Biol. Chem. 275 32535–32542 [DOI] [PubMed] [Google Scholar]
- 39.Jakubowski, H., and Goldman, E. (1992) Microbiol. Rev. 56 412–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Asahara, H., and Uhlenbeck, O. C. (2005) Biochemistry 44 11254–11261 [DOI] [PubMed] [Google Scholar]
- 41.Thompson, D., Lazennec, C., Plateau, P., and Simonson, T. (2007) J. Biol. Chem. 282 30856–30868 [DOI] [PubMed] [Google Scholar]
- 42.Lin, S. X., Baltzinger, M., and Remy, P. (1983) Biochemistry 22 681–689 [DOI] [PubMed] [Google Scholar]
- 43.Tsui, W. C., and Fersht, A. R. (1981) Nucleic Acids Res. 9 4627–4637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Delano, W. L. (2002) The PyMol Molecular Graphics System, Delano Scientific, Palo Alto, CA
- 45.Sheoran, A., and First, E. A. (2008) J. Biol. Chem. 283 12971–12980 [DOI] [PMC free article] [PubMed] [Google Scholar]