

Abstract
Activation of proMMP-2 and cell surface collagenolysis are important activities of membrane-type 1 matrix metalloproteinase (MT1-MMP) to promote cell migration in tissue, and these activities are regulated by homodimerization of MT1-MMP on the cell surface. In this study, we have identified the transmembrane domain as a second dimer interface of MT1-MMP in addition to the previously identified hemopexin domain. Our analyses indicate that these two modes of dimerization have different roles; transmembrane-dependent dimerization is critical for proMMP-2 activation, whereas hemopexin-dependent dimerization is important for degradation of collagen on the cell surface. Our finding provides new insight into the potential molecular arrangement of MT1-MMP contributing to its function on the cell surface.
Membrane-type 1 matrix metalloproteinase (MT1-MMP)2 is a type I transmembrane proteinase that promotes cell migration in tissue (1). MT1-MMP is implicated in many physiological and pathological conditions including wound healing (2), bone development (3, 4), lung development (5, 6), angiogenesis (3, 7, 8), cancer invasion (9) and growth (10), rheumatoid arthritis (11), and atherosclerosis (12–14). MT1-MMP promotes cellular invasion by degrading barrier extracellular matrix components including collagens I, II, III, fibronectin, laminins, vitronectin, and aggrecan (15–17); by activating other MMPs, namely proMMP-2 (9) and proMMP-13 (18); by shedding cell adhesion molecules such as CD44 (19) and syndecan 1 (20); and by activating extracellular signal-regulated kinase (ERK) through as yet undefined mechanisms (21, 22).
Having such diverse functions, MT1-MMP is regulated by different mechanisms including gene expression, activation of the zymogen (23, 24), inhibition by endogenous inhibitors, including tissue inhibitor of metalloproteinases (TIMPs) (25), RECK (26), and Testicans (27, 28), localization to the leading edge of migrating cells, including lamellipodia (29–31) and invadopodia (32), autolytic degradation and processing (33–35), endocytosis through clathrin- and caveolae-dependent mechanisms (36–38), palmitoylation at its cytoplasmic domain (39), recycling (40), and lysosomal degradation (41). Such regulation is thought to be essential to coordinate MT1-MMP activity with cellular events, enabling it to promote cell invasiveness (42).
ProMMP-2 activation is one of the MT1-MMP functions thought to be important in cancer invasion (9, 43) and growth (44), where its significance lays particularly on basement membrane degradation as MT1-MMP itself cannot degrade collagen IV, a major component of the matrix but activated MMP-2 does. In this activation process, MT1-MMP forms a complex with its endogenous inhibitor, TIMP-2 (45–47). TIMP-2 binds to the catalytic site of MT1-MMP through its inhibitory site in the N-terminal domain, leaving the exposed C-terminal domain of TIMP-2 to interact with the hemopexin (Hpx) domain of proMMP-2 (45–47). Thus the MT1-MMP-TIMP-2 complex acts as a receptor for proMMP-2. To activate proMMP-2 in this complex, a second MT1-MMP, which is free of TIMP-2, needs to be positioned in close proximity to the trimolecular complex. This is achieved by the formation of an MT1-MMP homodimer complex (31, 48).
Another important biological activity of MT1-MMP is collagen degradation
(15,
49). Among MMP family members,
at least six enzymes can degrade fibrillar type I collagen, namely MMP-1,
MMP-2, MMP-8, MMP-13, MT1-MMP
(50), and MT2-MMP
(51). MT1-MMP and MT2-MMP are
membrane-bound collagen-degrading enzymes, but MT2-MMP is the weakest of the
collagenolytic MMPs, exhibiting
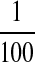 of the activity of
MT1-MMP (51). Thus MT1-MMP is
likely to be the major pericellular collagenase. MT1-MMP-null mice exhibit
phenotypes thought to be due to a lack of cellular collagenolytic activity
(3,
4). We have recently shown that
MT1-MMP dimer formation is essential for collagen degradation on the cell
surface (52), suggesting that
dimerization of MT1-MMP may be an important regulatory mechanism to activate
MT1-MMP on the cell surface for both collagenolysis and proMMP-2 activation
(52).
of the activity of
MT1-MMP (51). Thus MT1-MMP is
likely to be the major pericellular collagenase. MT1-MMP-null mice exhibit
phenotypes thought to be due to a lack of cellular collagenolytic activity
(3,
4). We have recently shown that
MT1-MMP dimer formation is essential for collagen degradation on the cell
surface (52), suggesting that
dimerization of MT1-MMP may be an important regulatory mechanism to activate
MT1-MMP on the cell surface for both collagenolysis and proMMP-2 activation
(52).
We have previously reported that dimerization of MT1-MMP is driven by homodimeric complex formation of the Hpx domains and that this interaction is crucial for proMMP-2 activation (31) and collagen degradation (52) on the cell surface. It was also reported that the Hpx and cytoplasmic domains can drive dimerization for proMMP-2 activation (48). On the other hand, it was recently reported that an Hpx domain-deleted MT1-MMP mutant retains the ability to activate proMMP-2 (53), suggesting that Hpx domain-dependent dimer formation may not play a role in this process. In this report, we have reevaluated the role of the Hpx, linker-2, transmembrane (TM), and cytoplasmic domains in proMMP-2 activation and the collagenolytic activity of MT1-MMP and found that MT1-MMP has two modes of dimer formation: Hpx domain- and TM domain-dependent dimerization. For proMMP-2 activation, TM-dependent dimerization is essential, whereas Hpx-dependent dimerization is essential for collagenolytic activity. Inhibition of Hpx domain-dependent dimerization by co-expressing the Hpx domain resulted in inhibition of TM-dependent dimer formation, proMMP-2 activation, and collagenolytic activity. Our finding reveals an additional molecular arrangement contributing to MT1-MMP function on the cell surface.
MATERALS AND METHODS
Cell Culture and Transfection—COS7 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (BioWhittaker) supplemented with 10% fetal bovine serum and penicillin/streptomycin (BioWhittaker). TIMP-2–/– fibroblasts were a kind gift from Dr. Paul Soloway at Cornell University (Ithaca, NY) and Dr. Gillian Murphy at University of Cambridge (Cambridge, UK) and were cultured in DMEM with 10% fetal bovine serum with antibiotics. For transfection, cells were cultured in 6- or 12-well plates and were transfected with expression plasmids using FuGENE 6™ (Roche Applied Science, Basel, Switzerland) for COS7 cells and Lipofectamine 2000 (Invitrogen, Paisley, UK) for TIMP-2-null fibroblasts according to the manufacturer's instructions.
Antibodies—Mouse anti-FLAG M1 and M2 monoclonal antibodies and alkaline phosphatase-conjugated goat anti-(mouse IgG) antibodies were purchased from Sigma-Aldrich (Dorset, UK). Mouse monoclonal anti-phospho-tyrosine antibody (PY-20) was purchased from ICN Biochemicals, and Alexa Fluor 488-conjugated and Alexa Fluor 568 conjugated goat anti-(mouse IgG) and anti-(rabbit IgG) antibodies were from Molecular Probes (Cambridge, UK).
Construction of MT1-MMP Mutants—FLAG (DYKDDDDK)-tagged MT1-MMP (MT1F) was constructed as described previously (54) and subcloned into pSG5 (Stratagene). A FLAG tag was inserted at the end of the propeptide (between Arg111 and Tyr112), and properly activated enzyme will have the FLAG tag at its N terminus and thus can be recognized by the anti-FLAG M1 antibody (54). MT1F-ΔCat is a FLAG-tagged mutant MT1-MMP in which the region of Tyr112 to Pro312 was deleted. MT1F-ΔHpx is a FLAG-tagged mutant MT1-MMP in which the region of Cys319–Cys508 was deleted. MT1F-ΔHpxΔL2 is an Hpx domain and a linker-2 (L2) region deletion mutant of MT1-MMP where Phe336–Gly535 was deleted. MT1F-ΔCatΔTM is a FLAG-tagged mutant MT1-MMP in which the regions of Tyr112 to Gly288 and also Ala536 to Val582 were deleted. MT1F-ΔHpxΔL2A to MT1F-ΔHpxΔL2J are MT1F-ΔHpx derivatives in which the L2 region was further deleted as follows. In MT1F-ΔHpxΔL2A, Pro509–Asp515 was further deleted; in MT1F-ΔHpxΔL2B, Pro509–Glu523 was deleted; in MT1F-ΔHpxΔL2C, Pro509–Val529 was deleted; in MT1F-ΔHpxΔL2D, Val524–Ile527 was deleted; in MT1F-ΔHpxΔL2E, Glu516–Glu523 and Asp530–Glu532 were deleted; in MT1F-ΔHpxΔL2F, Pro509–Glu532 was deleted; in MT1F-ΔHpxΔL2G, Asp530–Glu532 was deleted; in MT1F-ΔHpxΔL2H, Val524–Glu532 was deleted; in MT1F-ΔHpxΔL2I, Glu516–Glu532 was deleted; and in MT1F-ΔHpxΔL2J, Glu516–Val529 was deleted. MT1F-ΔCP and MT1F-ΔHpxΔCP are cytoplasmic domain (Arg563–Val582) deletion mutants. MT1F/NGFRTM and MT1F-ΔHpx/NGFRTM are chimera mutants in which corresponding regions of Ala536–Val582 in MT1-MMPs are replaced with Val412–Lys441 of nerve growth factor receptor (NGFR). MT1F/NGFRL2TMCP, MT1FΔCat/NGFRL2TMCP, and MT1FΔHpx/NGFRL2TMCP are chimera mutants of MT1F and its mutants in which corresponding regions of Glu516–Val582 in MT1-MMPs were replaced with Glu384–Gly790 of NGFR. MT1F/NGFRCP, MT1FΔCat/NGFRCP, and MT1FΔHpx/NGFRCP are chimera mutants of MT1F and its mutants in which corresponding regions of Arg563–Val582 in MT1-MMPs were replaced with Asn434–Gly790 of NGFR. These mutants were generated by the PCR extension method (55). All the PCR-generated fragments were confirmed by DNA sequencing and subcloned into the pSG5 vector.
Western Blotting and Zymography—Western blotting was carried out as described previously (56). Total cell lysates were prepared by the addition of 1× SDS-PAGE loading buffer containing 2-mercaptethanol to cells in the culture plate and subsequent boiling for 20 min. Zymography was carried out as described previously (54).
Surface Biotinylation and Subsequent Immuno-precipitation—Surface biotinylation and subsequent immuno-precipitation were carried our as described previously (31). Briefly, transfected COS7 cells were washed three times with chilled PBS containing 1 mm MgCl2 and 0.1 mm CaCl2. Cells were then incubated with sulfo-NHS-biotin (Pierce) in same buffer (2 mg/ml) at 4 °C for 30 min. The reaction was terminated by further incubating the cells with 25 mm lysine in PBS. The cells were lysed in the buffer of 1% Nonidet P-40, 0.1% SDS, 1% deoxycolic acid, 50 mm Tris-HCL (pH 7.5), 150 mm NaCl, 0.02% NaN3, and the biotinylated proteins were precipitated with streptavidin-agarose beads (Sigma). The samples were analyzed by Western blotting using anti-FLAG M2 antibody.
Indirect Immuno-fluorescence Staining—To localize cell surface MT1-F and its mutants, transfected COS7 cells cultured on 4-well glass slide chambers (Nalge Nunc International) coated with F-gelatin were fixed with 3% paraformaldehyde in PBS. After blocking with 5% goat serum and 3% bovine serum albumin in Tris-buffered saline for 1 h at room temperature, cells were incubated with an anti-FLAG M1 antibody (5 μg/ml) at room temperature for 2 h without permeabilizing cells. 1 mm CaCl2 was included throughout the procedure of washing and incubation for the staining with the anti-FLAG M1 antibody. Alexa Fluor 488-conjugated goat anti-mouse IgG was used to visualize the antigen signal. Note that anti-FLAG M1 antibody can only recognize FLAG tag at the N terminus of molecule (54); thus only active forms of the enzyme can be stained with this procedure. The signals were analyzed by CCD camera-equipped microscope (Nikon TE-2000) with ×10 objective lens.
In Situ Gelatin Degradation Assay—4-well chamber slides (Nunc) were coated with Alexa Fluor 488-conjugated gelatin (F-gelatin) prepared with an Alexa Fluor 488 labeling kit (Molecular Probes) as described previously (31). Transfected COS7 cells were cultured in the chamber slides for 16 h. Cells were then fixed with 3% paraformaldehyde in PBS and analyzed using CCD camera-equipped microscope (Nikon TE-2000) with either ×10 (see Fig. 1) or ×20 objective lens (the rest of the figures). The degraded area was visualized as a dark, non-fluorescent zone.
FIGURE 1.

Hpx domain is dispensable for proMMP-2 activation but important for collagenolytic activity. A, schematic representation of mutant constructs used in the experiments. S, signal peptide; Pro, propeptide; FLAG, FLAG tag (DYKDDDDK); Cat, catalytic domain; L1, linker 1 (hinge); HPX, hemopexin domain; L2, linker 2; TM, transmembrane domain; Zn, catalytic zinc ion. B, COS7 cells were transfected with empty vector (Mock), MT1F, MT1F-ΔHpx, and MT1F-ΔHpxΔL2 as indicated. Cells were then incubated with purified proMMP-2 in serum-free culture medium for 18 h. ProMMP-2 activation in the media was analyzed by zymography (upper panel), and cell lysates were analyzed for expression of the proteins by Western blotting using anti-FLAG M2 antibody (lower panel). The arrows indicate MT1-MMP mutants expressed. P, proMMP-2; A, active MMP-2. C, transfected cells were subjected to surface biotinylation as described under “Materials and Methods” and analyzed by Western blotting using anti-FLAG M2 antibody. The upper panel is biotinylated samples, and the bottom panel is whole cell lysates. D, in situ gelatin degradation assay was carried out as described under “Materials and Methods.” Transfected COS7 cells were seeded on Alexa Fluor 488-labeled gelatin-coated 4-well chamber slides and cultured for 18 h. The cell surface-localized active form of FLAG-tagged MT1-MMP mutants was visualized by staining with anti-FLAG M1 antibody in the presence of 1 mm CaCl2 without permeabilization. Green channel (F-gelatin) and red channel (FLAG M1) fluorescences were captured in each field using ×10 objective lens. Merged images are also shown in the bottom. The bar indicates 200 μm. E, in situ solid-phase collagen degradation assays were carried out as described under “Materials and Methods.” The bar indicates 100 μm.
In Situ Collagen Degradation Assay—The experiments were done as described previously (52). 6-well culture plates were coated with a thin layer of chilled neutralized PureCol™ collagen (Inamed Biomaterials, Fremont, CA) at 2.7 mg/ml in 1× RPMI medium (typically 100 μl/well) and incubated for 60 min at 37 °C for fibril formation, and COS7 cells (4 × 105/well) were then seeded on the film. 18 h later, cells were transfected with the expression plasmids in the growth medium (10% fetal bovine serum/DMEM) using FuGENE 6™ according to the manufacturer's instructions. The following day, culture medium was changed to serum-free DMEM, and cells were cultured for a further 3 days at 37 °C. The remaining collagen film was exposed by removing cells using repeated treatment with PBS containing 0.5 mg/ml trypsin and 1 mm EDTA. The collagen film was then fixed with 3% paraformaldehyde for 20 min at room temperature. Collagen was visualized by staining with Coomassie Brilliant Blue R250, and the images were captured by CCD camera-equipped microscope (Nikon TE-2000) with ×20 objective lens. Degraded areas were visualized as a white, unstained, non-collagen-containing zone. In this assay, stained collagen was trypsin-resistant, suggesting that it was intact fibrillar collagen.
RESULTS
Hpx Domain Is Dispensable for proMMP-2 Activation by MT1-MMP—We have previously reported that MT1-MMP forms a homodimer through Hpx domain, and this interaction facilitates proMMP-2 activation on the cell surface (31). In contrast, Wang et al. (53) have reported that the Hpx domain is not required for proMMP-2 activation by showing that Hpx domain-deleted mutant activates proMMP-2. To address this discrepancy, we re-evaluated the roles of the Hpx domain by analyzing the following deletion mutants. MT1-ΔHpx is the mutant lacking Hpx domain only (Cys319–Cys508), and MT1-ΔHpxΔL2 further lacks the linker-2 region (L2 region, Pro509– Gly535) linking Asn317 and Ala536 (Fig. 1A). COS7 cells were transfected with expression plasmids for these mutants and tested for proMMP-2 activation ability. In our previous study, we used MT1F-ΔHpxΔL2 to examine the role of the Hpx domain in proMMP-2 activation (31), and here we confirm that MT1F-ΔHpxΔL2 is unable to activate proMMP-2 as shown in Fig. 1B. However, MT1F-ΔHpx activates proMMP-2 as efficiently as full-length MT1F, supporting the results of Wang et al. (53). To test whether these enzymes are expressed on the cell surface, the transfected cells were subjected to surface biotinylation experiments. As shown in Fig. 1C, all of the MT1-MMPs were biotinylated in a similar manner, suggesting that they are all expressed on the cell surface. To further confirm this result, these cells were cultured on a fluorescent-labeled gelatin (F-gelatin) film and stained with anti-FLAG M1 antibody without permeabilizing the cells. As in all enzymes, the FLAG tag is inserted immediately downstream of the 108RRKR111 sequence, where proprotein convertases recognize and process (23, 57), only correctly processed enzyme can be recognized by the M1 antibody (54). In Fig. 1D, the wide view of a representative area of FLAG M1 staining/F-gelatin film degradation is shown. Anti-FLAG M1 antibody staining indicates that all the enzymes were expressed as correctly processed active forms on the cell surface and that the absence of the L2 region has no effect on this. Although active MT1F-ΔHpxΔL2 was present on the cell surface, it was unable to degrade F-gelatin film, whereas both MT1F and MT1F-ΔHpx did degrade F-gelatin effectively as evident by the numerous dark non-fluorescent patches. However, deletion of the Hpx domain did abrogate collagenolytic activity on the cell surface as reported previously (Fig. 1E) (53). These results suggest that the Hpx domain is not required for proMMP-2 activation, and the L2 region may play an important role in proMMP-2 activation and F-gelatin degradation, whereas the Hpx domain is critical for collagenolysis.
L2 Domain Is Not Responsible for proMMP-2 Activation—Based on the results above, we postulated that the L2 region may play an important role in proMMP-2 activation by MT1F-ΔHpx. We therefore made a series of deletions or mutations in the L2 region of MT1F-ΔHpx to test this possibility (Fig. 2A). According to its sequence, L2 can be divided into five parts: 509–514, which is a non-polar region, 515–523, which is a Glurich acidic region, 524–529, which is a hydrophobic region, 530–532, which is a short acidic region, and 533–535, which is a flexible region consisting of three glycines. Therefore, deletions or mutations were made to modify these different regions. As shown in Fig. 2B, among these mutants, MT1F-ΔHpxΔL2B and MT1F-ΔHpxΔL2E showed inefficient proMMP-2 activation, whereas all other variants were as good as MT1F-ΔHpx. MT1F-ΔHpxΔL2B and MT1F-ΔHpxΔL2E are expressed on the cell surface as active forms as they degraded F-gelatin, although the level of degradation seems to be lower. The level of F-gelatin degradation may not completely reflect the amount of cell surface enzyme as cells expressing MT1F-ΔHpxΔL2A, which activates proMMP-2 efficiently, also showed weaker F-gelatin degradation. The common feature of MT1F-ΔHpxΔL2B and MT1F-ΔHpxΔL2E is a lack of Glu516–Glu523, which is an acidic region immediately upstream of the hydrophobic region. However, this is not a region responsible for proMMP-2 activation as MT1F-ΔHpxΔL2I and MT1F-ΔHpxΔL2J, which lack this region, activate proMMP-2. Furthermore, MT1F-ΔHpxΔL2F, which lacks most of the L2 except the three glycines, activates proMMP-2 very well. Only further deletion of these glycines (MT1F-ΔHpxΔL2) made the enzyme inactive for proMMP-2 activation (Fig. 1). This suggests that L2 may not play a direct role in proMMP-2 activation but may be important for providing flexibility and/or correct arrangement to the ecto-domains and thereby orientation of the catalytic domain for proMMP-2 activation to take place.
FIGURE 2.

Effect of L2 region mutations on proMMP-2 activation. A, schematic representation of mutant MT1-MMPs used in the experiments. Different deletions were made in the L2 region of MT1F-ΔHpx. S, signal peptide; Pro, propeptide; FLAG, FLAG tag (DYKDDDDK); Cat, catalytic domain; L1, linker 1 (hinge); HPX, hemopexin domain; L2, linker 2; TM, TM domain; CP, cytoplasmic domain; Zn, catalytic zinc ion. B, ProMMP-2 activation ability of these mutant MT1-MMPs were analyzed as in Fig. 1B. ProMMP-2 activation in the media was analyzed by zymography (upper panel), and cell lysates were analyzed for expression of the proteins by Western blotting using anti-FLAG M2 antibody (lower panel). The arrows in the Western blot (lower panel) indicate MT1-MMP mutants expressed. P, proMMP-2; A, active MMP-2. Mock, cells transfected with empty vector. C, in situ gelatin degradation assay was carried out as described under “Materials and Methods.”
TM Domain Plays an Important Role in proMMP-2 Activation—We next examined the role of the cytoplasmic (CP) and TM domains in proMMP-2 activation. To test this, we deleted CP domain and exchanged the TM region with that of NGFR. As NGFR dimerization requires binding to the ligand in its ecto-domain, we did not expect the TM of NGFR to dimerize efficiently by itself. Indeed, our previous results support this notion (31). These mutations were incorporated into both MT1F and MT1F-ΔHpx as shown in Fig. 3A. The CP domain deletion did not affect the ability to activate proMMP-2 on the cell surface as reported previously (Fig. 3B) (37). This suggests that the CP domain is dispensable for proMMP-2 activation. On the other hand, replacing the TM domain with that of NGFR caused significant reduction in proMMP-2 activation by both full-length mutant (MT1F/NGFRTM) and the Hpx-deleted mutant (MT1F-ΔHpx/NGFRTM) (Fig. 3B). The reduction in proMMP-2 activation was not due to a reduction in cell surface localization as indicated by surface biotinylation study (Fig. 3C). Also, it is not due to a lack of activation as F-gelatin degradation was similar for all mutants (Fig. 3D).
FIGURE 3.

TM domain plays important role in proMMP-2 activation. A, schematic representation of mutant MT1-MMPs used. TM-NGFR, nerve growth factor receptor derived transmembrane domain. B, ProMMP-2 activation ability of these mutant MT1-MMPs were analyzed as in Fig. 1B. ProMMP-2 activation in the media was analyzed by zymography (upper panel), and cell lysates were analyzed for expression of the proteins by Western blotting using anti-FLAG M2 antibody (lower panel). The arrows in the Western blot (lower panel) indicate MT1-MMP mutants expressed. P, proMMP-2; A, active MMP-2. C, transfected cells were subjected to surface biotinylation as described under “Materials and Methods” and analyzed by Western blotting using anti-FLAG M2 antibody. The upper panel is biotinylated samples, and the bottom panel is whole cell lysates. The arrows in Western blot indicate MT1-MMP mutants expressed. D, in situ gelatin degradation assay was carried out as described under “Materials and Methods.”
The TM Domain Acts as Dimer Interface—One of the possible roles of TM domain in proMMP-2 activation could be that it acts as a dimer interface. To address this question, we utilized chimera mutants of MT1F and NGFR. We have previously shown that such chimera mutants can be used to test the ability of the MT1-MMP to form a dimer by monitoring tyrosine phosphorylation at their CP domain (31, 52). We created two sets of chimera mutants. The first group of the chimeras has L2, TM, and CP regions derived from NGFR with other ecto-domains from MT1-MMP, and in the second group of chimeras, the portion derived from MT1-MMP extended up to the TM domain, with only the CP domain derived from NGFR (Fig. 4A). With these two sets of chimeras, one can compare the contribution of the TM domain of MT1-MMP in its dimer formation. As shown in Fig. 4B, MT1F/NGFRL2TMCP and MT1FΔCat/ NGFRL2TMCP showed strong phospho-tyrosine signals (lanes 2 and 3), whereas MT1FΔHpx/NGFRL2TMCP showed a minimal signal (lane 4), indicating the importance of Hpx domain for dimerization among these ecto-domains. On the other hand, all the second set of chimeras including MT1F/NGFRCP and MT1FΔCat/NGFRCP, MT1FΔHpx/NGFRCP and MT1FΔHpxΔL2F/NGFRCP showed strong phospho-tyrosine signals regardless of whether or not the Hpx domain or L2 region is present (lanes 5–8). These data strongly indicate that the TM domain can induce dimerization. Furthermore, the Hpx and TM domains can dimerize independently, i.e. neither TM-dependent dimerization nor Hpx-dependent dimerization is a prerequisite for dimerization of Hpx domain or TM-dependent dimer, respectively. When the proMMP-2 activation ability of these constructs was compared, it is clear that the catalytic domain of MT1-MMP is absolutely essential (see lanes 3 and 6) and also that the presence of MT1-MMP-derived TM domain greatly increases proMMP-2 activation (compare lanes 4, 7, and 8), supporting earlier results.
FIGURE 4.

Hpx and TM domains drive dimerization of MT1-MMP. A, schematic representation of mutant MT1-MMPs used in the experiments. NGFR-L2TMCP, NGFR-derived L2, TM, and CP domains; S, signal peptide; Pro, propeptide; FLAG, FLAG tag (DYKDDDDK); Cat, catalytic domain; L1, linker 1 (hinge); HPX, hemopexin domain; L2, linker 2; Zn, catalytic zinc ion; TK, tyrosine kinase domain. B, COS7 cells were transfected with expression plasmids for mutant constructs as indicated. Cells were lysed and subjected to Western blotting analyses using anti-phospho-tyrosine antibody (PY, upper panel) and anti-FLAG M2 antibody (FLAG, lower panel). Zymo, zymography; Mock, cells transfected with empty vector; P, proMMP-2; A, active MMP-2.
Role of Hpx-dependent Dimerization in Cell Surface Collagenolytic Activity—We have recently reported that dimerization of MT1-MMP is essential for cell surface collagenolytic activity (52). Inhibition of dimerization by co-expression of membrane-bound or soluble Hpx domain inhibited collagen degradation by MT1-MMP-expressing cells (52). Therefore, we examined which mode of dimerization plays a role in cell surface collagenolytic activity. For this purpose, we tested the collagen-degrading ability of the mutants that cannot activate proMMP-2 efficiently due to the absence of the MT1-MMP TM domain, namely MT1F/NGFRTM (Fig. 5A). As shown in Fig. 5B, expression of MT1F in COS7 cells caused degradation of a collagen film, and the activity was significantly inhibited upon co-expression of a membrane-anchored or soluble Hpx domains, MT1F-ΔCatL1 or MT1F-ΔCatL1ΔTM, respectively. Deletion of the CP domain, exchanging the TM domain with the one derived from NGFR, did not affect collagenolytic activity, and all of these were inhibited by co-expression with MT1F-ΔCatL1 or MT1F-ΔCatL1ΔTM, like MT1F. These data suggest that TM domain-dependent dimer formation is not essential for cell surface collagenolytic activity, and Hpx domain-dependent dimerization is sufficient to support the activity.
FIGURE 5.

TM domain is not critical for collagenolytic activity on the cell surface. A, schematic representation of mutant MT1-MMPs used in the experiments. S, signal peptide; Pro, propeptide; FLAG, FLAG tag (DYKDDDDK); Cat, catalytic domain; L1, linker 1 (hinge); HPX, hemopexin domain; L2, linker 2; TM, transmembrane domain; Zn, catalytic zinc ion; TM-NGFR, NGFR-derived TM domain; GPI-MT4, MT4-MMP-derived sequence that contains GPI-anchoring signal peptide (Glu524–Leu603). B, in situ solid-phase collagen degradation assay were carried out as described under “Materials and Methods.” The bar indicates 100 μm. Mock, cells transfected with empty vector.
Inhibition of Hpx-dependent Dimerization Inhibits TM-dependent Dimer Formation—We have previously shown that co-expression of membrane-anchored Hpx domain (MT1F-ΔCatL1) with MT1-MMP inhibits proMMP-2 activation (31). MT1F-ΔCatL1 contains two dimer interfaces, the Hpx and the TM domains. We thus next asked whether the inhibition of Hpx domain-dependent dimerization is sufficient to cause inhibition of proMMP-2 activation. As shown in Fig. 6A, co-expression of membrane-anchored Hpx domain, MT1F-ΔCatL1, or soluble Hpx (MT1F-ΔCatL1ΔTM) with MT1F inhibits proMMP-2 activation in a dose-dependent manner, although MT1F-ΔCatL1ΔTM had weaker activity. Since soluble Hpx inhibits Hpx-dependent dimerization, we postulated that inhibition of Hpx-dependent dimerization by soluble Hpx also inhibits TM-dependent dimerization. To test this, soluble Hpx domain (MT1F-ΔCatL1ΔTM) was co-expressed with MT1F/NGFRCP, which contains two dimer interfaces, the Hpx and TM domains. As shown in Fig. 6B, co-expression of either MT1F-ΔCatL1 or MT1F-ΔCatL1ΔTM with MT1F/NGFRCP significantly decreased phospho-tyrosine signal in a similar manner. These data suggest that disruption of Hpx domain-dependent dimer formation also disrupts TM domain-dependent dimerization.
FIGURE 6.

Inhibition of Hpx-dimer by soluble Hpx abrogates TM dimer formation. A, COS7 cells were transfected with the expression plasmids of MT1F with or without either MT1F-ΔCatL1 or MT1F-ΔCatL1ΔTM as indicated and proMMP-2 activation was analyzed as in Fig. 1B. FLAG, anti-FLAG M2 antibody; P, proMMP-2; A, active MMP-2. B, COS7 cells were transfected with expression plasmids for MT1F/NGFRCP with or without MT1F-ΔCatL1 or MT1F-ΔCatL1ΔTM as indicated. Cells were lysed and subjected to Western blotting analyses using anti-phospho-tyrosine antibody (PY, upper panel) and anti-FLAG M2 antibody (lower panel). The relative intensity of the bands detected by PY-20 was analyzed with NIH Image, normalized by the intensity of FLAG bands.
TM Dimer Arranges proMMP-2 Orientation—Activation of proMMP-2 by MT1-MMP involves TIMP-2 bridging MT1-MMP and proMMP-2. To address whether deletion of the Hpx domain affect TIMP-2 requirement for the activation, TIMP-2–/– fibroblasts were transfected with the expression plasmids for MT1F, MT1F-ΔHpx, and MT1F-ΔHpxΔL2F in the presence or absence of TIMP-2 (3 nm). As shown in Fig. 7A, the MT1-MMPs expressed in TIMP-2–/– cells were not able to activate proMMP-2, whereas they activated proMMP-2 with the addition of TIMP-2 in the medium. To test the ability of proMMP-2 to bind cell surface, COS7 cells were transfected with the same plasmids and reacted with proMMP-2 since COS7 cells express TIMP-2 endogenously (data not shown). As shown in Fig. 7B, cells expressing these MT1-MMPs bound MMP-2. Major MMP-2 species found in the cell fractions are the active form in all transfected cells. These results suggest that activation of proMMP-2 by Hpx-deleted mutants occurs with the same mechanism as wild-type MT1-MMP, and TM dimer is sufficient to arrange proMMP-2 orientation for the activation.
FIGURE 7.

Involvement of TIMP-2 in ptoMMP-2 activation. A, TIMP-2-null fibroblasts were transfected with MT1-F, MT1F-ΔHpx, and MT1F-ΔHpxΔL2F as indicated. These cells were then reacted to proMMP-2 (0.5 μg/ml) in the culture medium for 24 h in the presence or absence of exogenous TIMP-2 (3 nm). ProMMP-2 activation was analyzed by zymography (upper panel), and cell lysates were analyzed for expression of the proteins by Western blotting using anti-FLAG M2 antibody (lower panel). * indicates nonspecific band as they are present throughout the samples including mock-transfected cells. The arrows in the Western blot (lower panel) indicate MT1-MMP mutants expressed. P, proMMP-2; A, active MMP-2. B, two sets of COS7 cells were transfected with MT1-F, MT1F-ΔHpx, and MT1F-ΔHpxΔL2F as indicated. One set of cells were reacted with proMMP-2 (0.5 μg/ml) in the culture medium for 24 h. Culture media were analyzed by zymography for MMP-2 activation (top panel, Sup) and cell lysates by Western blotting using anti-FLAG M2 antibody (lower panel) for the expression of the proteins. Another set of transfected cells was reacted with proMMP-2 (2 μg/ml) at room temperature for 1 h. Cells were washed with PBS three times, and cell lysates were analyzed for proMMP-2 binding by zymography (middle panel, Cell). The arrows in the Western blot (lower panel) indicate MT1-MMP mutants expressed. Sup, supernatant.
DISCUSSION
In this report, we have investigated modes of MT1-MMP dimerization and found that two domains of the enzyme can dimerize. A summary of the findings is depicted in Fig. 8. In the full-length enzyme, both the Hpx and the TM domains can form homodimer interfaces, and the enzyme shows both proMMP-2 activation and collagen-degrading activities on the cell surface. In the Hpx domain-deleted mutant, only a TM domain-dependent dimer can form. The mutant retains proMMP-2 activation ability but has lost collagen-degrading ability. The loss of collagen-degrading activity of this mutant is not only due to a loss of the Hpx domain as a dimerization domain but also to a loss of intrinsic collagenolytic activity due to lack of the Hpx domain (15, 53). The mutants lacking the MT1-MMP-derived TM domain, namely MT1F/NGFRTM, still retain the ability to form an Hpx domain-dependent dimer. This mutant does not activate proMMP-2 efficiently but retains collagenolytic activity. Formation of proMMP-2-TIMP-2-MT1-MMP complex was not affected by the lack of TM domain; thus TM dimer is likely to play an essential role to arrange proMMP-2 orientation for the activation. Taken together, TM-dependent dimerization is critical for proMMP-2 activation, but not for collagen degradation, and Hpx-dependent dimerization is critical for collagen degradation, but not essential for proMMP-2 activation.
FIGURE 8.

Schematic representation of a model of MT1-MMP dimerization on the cell surface.
The TM and Hpx domains appear to be able to dimerize independently. However, abrogation of Hpx-dependent dimer formation by co-expressing soluble Hpx domain also disrupts TM-dependent dimer formation, thereby inhibiting proMMP-2 activation. Although formation of a dimer by the TM domain does not rely on Hpx-dependent dimerization, it may be possible that Hpx domain-dependent dimer may influence TM-dependent dimerization. Previously, we have shown that expression of the constitutively active form of Rac1 small GTPase (Rac1CA) enhances Hpx domain-dependent dimerization using the MT1F/NGFRL2TMCP construct, which contains only the Hpx domain as a dimerizing domain (31). This suggests that Hpx-dependent dimer can be regulated by Rac1. Rac1CA also stimulated proMMP-2 activation by full-length MT1-MMP (31), suggesting that enhancement of Hpxdependent dimerization has resulted in enhanced TM-dependent dimerization as TM dimerization dictates proMMP-2 activation. Although each mode of dimerization can occur independently and plays a distinct roles, in full-length wild-type MT1-MMP, the dimerizations through these domains are likely to occur at the same time. It is possible that regulation of Hpx-dependent dimerization is one of the mechanisms to control TM dimerization of MT1-MMP, which in turn regulates proMMP-2 activation on the cell surface.
The L2 region does not seem to play a critical role in enzyme dimerization to form the correct complex configuration for proMMP-2 activation since co-expression of soluble Hpx domain construct containing intact L2 region (MT1F-ΔCatL1ΔTM) does not inhibit proMMP-2 activation by MT1F-ΔHpx with intact L2 region (data not shown). Also, the MT1F-ΔHpxΔL2F mutant, which lacks the majority of the L2 region, leaving only three glycines, can activate proMMP-2 as well as MT1F-ΔHpx. On the other hand, complete deletion of the L2 region makes the enzyme inactive for proMMP-2 activation (MT1ΔHpxΔL2). Interestingly, MT1F-ΔHpxΔL2 was also inactive for F-gelatin film degradation, although the enzyme is expressed on the cell surface as a correctly processed active form. Since MT1F-ΔHpxΔL2F degraded F-gelatin efficiently, flexibility given by at least three glycine residues seems important for both proMMP-2 activation and F-gelatin film degradation. It is possible that the inflexible nature of MT1F-ΔHpxΔL2 did not allow correct positioning of the catalytic domain for proMMP-2 activation or for interaction with other molecules that may also be essential to localize the enzyme to F-gelatin attachment site of the cells. Systematic deletions of L2 region revealed that the enzyme becomes ineffective for proMMP-2 activation when acidic sequence (Glu516–Glu523) immediately upstream of hydrophobic sequence (Val-524–Val-529) is deleted (MT1F-ΔHpxΔL2B). Since further deletion of this hydrophobic region (MT1F-ΔHpxΔL2C) regains proMMP-2 activation activity, it may suggest that the hydrophobic sequence negatively affects proMMP-2 activation, but this was counteracted by the presence of the acidic sequence immediately upstream. This notion is also supported by comparison of MT1F-ΔHpxΔL2E and MT1F-ΔHpxΔL2I where the presence of the hydrophobic region correlates with inefficient proMMP-2 activation (Fig. 2). Taken together, although L2 region does not positively support MT1-MMP activity, it may be important to provide flexibility to the Hpx and the catalytic domains to determine their arrangement on the cell surface.
Arrangement of wild-type MT1-MMP ecto-domains may be dictated by the presence of two dimeric interactions within the molecule. Without TM-dependent dimerization, the enzyme still forms an Hpx domain-dependent dimer, but in this form, it cannot activate proMMP-2. This suggests that the molecular arrangement of the ecto-domains with and without TM dimer are different, and Hpx dimer is not enough to allow the domains to be arranged correctly for proMMP-2 activation. It is probable that correct orientation of two catalytic domains of MT1-MMP dimer is essential for proMMP-2 activation to take place. The report by Wu et al. (58) suggests this possibility where they found that O-glycosylation at hinge (L1) region is essential for proMMP-2 activation. Glycosylation at L1 region might be important to arrange the catalytic domains in the correct orientation (1). These ideas together with our present results suggest that linker-1 and -2 may form a defined structure and provide flexibilities that determine correct arrangement of the ecto-domains.
Functionalities of biologically active proteins are often regulated by interaction with other proteins or by clustering, and this feature is found in many membrane proteins including integrins, cadherins, and growth factor receptors. MT1-MMP, as a type I transmembrane protein, is another example of a membrane protein that forms hetero- and homocomplexes to exhibit its biological activities. We have identified the TM domain as a dimer interface of MT1-MMP. This was unexpected, but such TM domain-dependent dimerization has been found in other transmembrane molecules such as E-cadherin (59) and tyrosine kinase receptors including ErbB receptors (60), discoidin domain receptors (61), and erythropoietin receptor (62). For E-cadherin, mutations that reduce TM dimerization drastically reduced cell-cell adhesion activity, suggesting that the TM-dependent dimerization is essential for biological function (59). In the case of the ErbB receptor, TM-dependent dimerization keeps the receptor in an inactive conformation, and mutation to prevent dimer formation resulted in spontaneous activation of the ErbB receptor (60). In the case of discoidin domain receptors, mutations that inhibit TM dimer formation reduced activation of the receptors (61). Thus TM-dependent dimerization has great impact on the functions of these membrane proteins.
Dimerization of MT1-MMP is essential for proMMP-2 activation (31, 48) and for collagen degradation (52). However, formation of such complexes is only required when MT1-MMP is expressed as a membrane-anchored form because proMMP-2 can be activated in a test tube by recombinant catalytic domain of MT1-MMP in the absence of TIMP-2 and dimerizing domains (25). Membrane anchoring may create spatial restrictions that make the enzyme inefficient without complex formation. Complex formation thus adds a level of spatial regulation to control MT1-MMP activity, creating a local proteolytic environment that can be coordinated with other cellular events. Understanding such mechanisms is not only important biologically but also for potential therapy development for MT1-MMP-related diseases including cancer since inhibition of MT1-MMP dimerization may provide a novel way to control MT1-MMP activity in a specific manner. Thus elucidating these interaction sites may lead us to design an allosteric inhibitor of MT1-MMP with much greater specificity.
Acknowledgments
We thank Yohei Otake for useful discussion and Rob Visse and Linda Troeberg for critical reading of the manuscript. We also thank Paul Soloway and Gill Murphy for a kind gift of TIMP-2-null fibroblasts.
This work was supported, in whole or in part, by National Institutes of Health Grant AR40994. This work was also supported by Cancer Research UK Project Grant C1507/A5541 and the Wellcome Trust equipment grant, and the Arthritis Research Campaign core grant to the Kennedy Institute of Rheumatology. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: MMP, matrix metalloproteinase; MT1-MMP, membrane-type 1 matrix metalloproteinase; Hpx domain, hemopexin domain; TM domain, transmembrane domain; CP domain, cytoplasmic domain; NGFR, nerve growth factor receptor; TIMP, tissue inhibitor of metalloproteinases; DMEM, Dulbecco's modified Eagle's medium; PBS, phosphate-buffered saline; PY, phospho-tyrosine.
References
- 1.Itoh, Y., and Seiki, M. (2004) Trends Biochem. Sci. 29 285–289 [DOI] [PubMed] [Google Scholar]
- 2.Okada, A., Tomasetto, C., Lutz, Y., Bellocq, J. P., Rio, M. C., and Basset, P. (1997) J. Cell Biol. 137 67–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou, Z., Apte, S. S., Soininen, R., Cao, R., Baaklini, G. Y., Rauser, R. W., Wang, J., Cao, Y., and Tryggvason, K. (2000) Proc. Natl. Acad. Sci. U. S. A. 97 4052–4057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holmbeck, K., Bianco, P., Caterina, J., Yamada, S., Kromer, M., Kuznetsov, S. A., Mankani, M., Robey, P. G., Poole, A. R., Pidoux, I., Ward, J. M., and Birkedal-Hansen, H. (1999) Cell 99 81–92 [DOI] [PubMed] [Google Scholar]
- 5.Atkinson, J. J., Holmbeck, K., Yamada, S., Birkedal-Hansen, H., Parks, W. C., and Senior, R. M. (2005) Dev. Dyn. 232 1079–1090 [DOI] [PubMed] [Google Scholar]
- 6.Oblander, S. A., Zhou, Z., Galvez, B. G., Starcher, B., Shannon, J. M., Durbeej, M., Arroyo, A. G., Tryggvason, K., and Apte, S. S. (2005) Dev. Biol. 277 255–269 [DOI] [PubMed] [Google Scholar]
- 7.Hiraoka, N., Allen, E., Apel, I. J., Gyetko, M. R., and Weiss, S. J. (1998) Cell 95 365–377 [DOI] [PubMed] [Google Scholar]
- 8.Chun, T. H., Sabeh, F., Ota, I., Murphy, H., McDonagh, K. T., Holmbeck, K., Birkedal-Hansen, H., Allen, E. D., and Weiss, S. J. (2004) J. Cell Biol. 167 757–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sato, H., Takino, T., Okada, Y., Cao, J., Shinagawa, A., Yamamoto, E., and Seiki, M. (1994) Nature 370 61–65 [DOI] [PubMed] [Google Scholar]
- 10.Hotary, K. B., Allen, E. D., Brooks, P. C., Datta, N. S., Long, M. W., and Weiss, S. J. (2003) Cell 114 33–45 [DOI] [PubMed] [Google Scholar]
- 11.Honda, S., Migita, K., Hirai, Y., Origuchi, T., Yamasaki, S., Kamachi, M., Shibatomi, K., Fukuda, T., Kita, M., Hida, A., Ida, H., Aoyagi, T., Kawakami, A., Kawabe, Y., Oizumi, K., and Eguchi, K. (2001) Clin. Exp. Immunol. 126 131–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rajavashisth, T. B., Xu, X. P., Jovinge, S., Meisel, S., Xu, X. O., Chai, N. N., Fishbein, M. C., Kaul, S., Cercek, B., Sharifi, B., and Shah, P. K. (1999) Circulation 99 3103–3109 [DOI] [PubMed] [Google Scholar]
- 13.Lehti, K., Allen, E., Birkedal-Hansen, H., Holmbeck, K., Miyake, Y., Chun, T. H., and Weiss, S. J. (2005) Genes Dev. 19 979–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Filippov, S., Koenig, G. C., Chun, T. H., Hotary, K. B., Ota, I., Bugge, T. H., Roberts, J. D., Fay, W. P., Birkedal-Hansen, H., Holmbeck, K., Sabeh, F., Allen, E. D., and Weiss, S. J. (2005) J. Exp. Med. 202 663–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohuchi, E., Imai, K., Fujii, Y., Sato, H., Seiki, M., and Okada, Y. (1997) J. Biol. Chem. 272 2446–2451 [DOI] [PubMed] [Google Scholar]
- 16.Fosang, A. J., Last, K., Fujii, Y., Seiki, M., and Okada, Y. (1998) FEBS Lett. 430 186–190 [DOI] [PubMed] [Google Scholar]
- 17.Koshikawa, N., Giannelli, G., Cirulli, V., Miyazaki, K., and Quaranta, V. (2000) J. Cell Biol. 148 615–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knäuper, V., Will, H., López-Otín, C., Smith, B., Atkinson, S. J., Stanton, H., Hembry, R. M., and Murphy, G. (1996) J. Biol. Chem. 271 17124–17131 [DOI] [PubMed] [Google Scholar]
- 19.Kajita, M., Itoh, Y., Chiba, T., Mori, H., Okada, A., Kinoh, H., and Seiki, M. (2001) J. Cell Biol. 153 893–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Endo, K., Takino, T., Miyamori, H., Kinsen, H., Yoshizaki, T., Furukawa, M., and Sato, H. (2003) J. Biol. Chem. 278 40764–40770 [DOI] [PubMed] [Google Scholar]
- 21.Takino, T., Miyamori, H., Watanabe, Y., Yoshioka, K., Seiki, M., and Sato, H. (2004) Cancer Res. 64 1044–1049 [DOI] [PubMed] [Google Scholar]
- 22.Gingras, D., Bousquet-Gagnon, N., Langlois, S., Lachambre, M. P., Annabi, B., and Beliveau, R. (2001) FEBS Lett. 507 231–236 [DOI] [PubMed] [Google Scholar]
- 23.Yana, I., and Weiss, S. J. (2000) Mol. Biol. Cell 11 2387–2401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sato, H., Takino, T., Kinoshita, T., Imai, K., Okada, Y., Stetler, S. W., and Seiki, M. (1996) FEBS Lett. 385 238–240 [DOI] [PubMed] [Google Scholar]
- 25.Will, H., Atkinson, S. J., Butler, G. S., Smith, B., and Murphy, G. (1996) J. Biol. Chem. 271 17119–17123 [DOI] [PubMed] [Google Scholar]
- 26.Oh, J., Takahashi, R., Kondo, S., Mizoguchi, A., Adachi, E., Sasahara, R. M., Nishimura, S., Imamura, Y., Kitayama, H., Alexander, D. B., Ide, C., Horan, T. P., Arakawa, T., Yoshida, H., Nishikawa, S., Itoh, Y., Seiki, M., Itohara, S., Takahashi, C., and Noda, M. (2001) Cell 107 789–800 [DOI] [PubMed] [Google Scholar]
- 27.Nakada, M., Yamada, A., Takino, T., Miyamori, H., Takahashi, T., Yamashita, J., and Sato, H. (2001) Cancer Res. 61 8896–8902 [PubMed] [Google Scholar]
- 28.Nakada, M., Miyamori, H., Yamashita, J., and Sato, H. (2003) Cancer Res. 63 3364–3369 [PubMed] [Google Scholar]
- 29.Mori, H., Tomari, T., Koshikawa, N., Kajita, M., Itoh, Y., Sato, H., Tojo, H., Yana, I., and Seiki, M. (2002) EMBO J. 21 3949–3959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sato, T., del Carmen Ovejero, M., Hou, P., Heegaard, A. M., Kumegawa, M., Foged, N. T., and Delaisse, J. M. (1997) J. Cell Sci. 110 589–596 [DOI] [PubMed] [Google Scholar]
- 31.Itoh, Y., Takamura, A., Ito, N., Maru, Y., Sato, H., Suenaga, N., Aoki, T., and Seiki, M. (2001) EMBO J. 20 4782–4793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakahara, H., Howard, L., Thompson, E. W., Sato, H., Seiki, M., Yeh, Y., and Chen, W. T. (1997) Proc. Natl. Acad. Sci. U. S. A. 94 7959–7964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stanton, H., Gavrilovic, J., Atkinson, S. J., d'Ortho, M. P., Yamada, K. M., Zardi, L., and Murphy, G. (1998) J. Cell Sci. 111 2789–2798 [DOI] [PubMed] [Google Scholar]
- 34.Toth, M., Hernandez-Barrantes, S., Osenkowski, P., Bernardo, M. M., Gervasi, D. C., Shimura, Y., Meroueh, O., Kotra, L. P., Galvez, B. G., Arroyo, A. G., Mobashery, S., and Fridman, R. (2002) J. Biol. Chem. 277 26340–26350 [DOI] [PubMed] [Google Scholar]
- 35.Lehti, K., Lohi, J., Valtanen, H., and Keski-Oja, J. (1998) Biochem. J. 334 345–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang, A., Lehti, K., Wang, X., Weiss, S. J., Keski-Oja, J., and Pei, D. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 13693–13698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uekita, T., Itoh, Y., Yana, I., Ohno, H., and Seiki, M. (2001) J. Cell Biol. 155 1345–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Remacle, A., Murphy, G., and Roghi, C. (2003) J. Cell Sci. 116 3905–3916 [DOI] [PubMed] [Google Scholar]
- 39.Anilkumar, N., Uekita, T., Couchman, J. R., Nagase, H., Seiki, M., and Itoh, Y. (2005) FASEB J. 19 1326–1328 [DOI] [PubMed] [Google Scholar]
- 40.Wang, X., Ma, D., Keski-Oja, J., and Pei, D. (2004) J. Biol. Chem. 279 9331–9336 [DOI] [PubMed] [Google Scholar]
- 41.Takino, T., Miyamori, H., Kawaguchi, N., Uekita, T., Seiki, M., and Sato, H. (2003) Biochem. Biophys. Res. Commun. 304 160–166 [DOI] [PubMed] [Google Scholar]
- 42.Itoh, Y., and Seiki, M. (2006) J. Cell. Physiol. 206 1–8 [DOI] [PubMed] [Google Scholar]
- 43.Stetler-Stevenson, W. G., Aznavoorian, S., and Liotta, L. A. (1993) Annu. Rev. Cell Biol. 9 541–573 [DOI] [PubMed] [Google Scholar]
- 44.Taniwaki, K., Fukamachi, H., Komori, K., Ohtake, Y., Nonaka, T., Sakamoto, T., Shiomi, T., Okada, Y., Itoh, T., Itohara, S., Seiki, M., and Yana, I. (2007) Cancer Res. 67 4311–4319 [DOI] [PubMed] [Google Scholar]
- 45.Strongin, A. Y., Collier, I., Bannikov, G., Marmer, B. L., Grant, G. A., and Goldberg, G. I. (1995) J. Biol. Chem. 270 5331–5338 [DOI] [PubMed] [Google Scholar]
- 46.Butler, G. S., Butler, M. J., Atkinson, S. J., Will, H., Tamura, T., van, W. S., Crabbe, T., Clements, J., d'Ortho, M. P., and Murphy, G. (1998) J. Biol. Chem. 273 871–880 [DOI] [PubMed] [Google Scholar]
- 47.Kinoshita, T., Sato, H., Okada, A., Ohuchi, E., Imai, K., Okada, Y., and Seiki, M. (1998) J. Biol. Chem. 273 16098–16103 [DOI] [PubMed] [Google Scholar]
- 48.Lehti, K., Lohi, J., Juntunen, M. M., Pei, D., and Keski-Oja, J. (2002) J. Biol. Chem. 277 8440–8448 [DOI] [PubMed] [Google Scholar]
- 49.Holmbeck, K., Bianco, P., Yamada, S., and Birkedal-Hansen, H. (2004) J. Cell. Physiol. 200 11–19 [DOI] [PubMed] [Google Scholar]
- 50.Visse, R., and Nagase, H. (2003) Circ. Res. 92 827–839 [DOI] [PubMed] [Google Scholar]
- 51.Morrison, C. J., and Overall, C. M. (2006) J. Biol. Chem. 281 26528–26539 [DOI] [PubMed] [Google Scholar]
- 52.Itoh, Y., Ito, N., Nagase, H., Evans, R. D., Bird, S. A., and Seiki, M. (2006) Mol. Biol. Cell 17 5390–5399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang, P., Nie, J., and Pei, D. (2004) J. Biol. Chem. 279 51148–51155 [DOI] [PubMed] [Google Scholar]
- 54.Itoh, Y., Kajita, M., Kinoh, H., Mori, H., Okada, A., and Seiki, M. (1999) J. Biol. Chem. 274 34260–34266 [DOI] [PubMed] [Google Scholar]
- 55.Ho, S. N., Hunt, H. D., Horton, R. M., Pullen, J. K., and Pease, L. R. (1989) Gene (Amst.) 77 51–59 [DOI] [PubMed] [Google Scholar]
- 56.Itoh, Y., Ito, A., Iwata, K., Tanzawa, K., Mori, Y., and Nagase, H. (1998) J. Biol. Chem. 273 24360–24367 [DOI] [PubMed] [Google Scholar]
- 57.Sato, H., Kinoshita, T., Takino, T., Nakayama, K., and Seiki, M. (1996) FEBS Lett. 393 101–104 [DOI] [PubMed] [Google Scholar]
- 58.Wu, Y. I., Munshi, H. G., Sen, R., Snipas, S. J., Salvesen, G. S., Fridman, R., and Stack, M. S. (2004) J. Biol. Chem. 279 8278–8289 [DOI] [PubMed] [Google Scholar]
- 59.Huber, O., Kemler, R., and Langosch, D. (1999) J. Cell Sci. 112 4415–4423 [DOI] [PubMed] [Google Scholar]
- 60.Mendrola, J. M., Berger, M. B., King, M. C., and Lemmon, M. A. (2002) J. Biol. Chem. 277 4704–4712 [DOI] [PubMed] [Google Scholar]
- 61.Noordeen, N. A., Carafoli, F., Hohenester, E., Horton, M. A., and Leitinger, B. (2006) J. Biol. Chem. 281 22744–22751 [DOI] [PubMed] [Google Scholar]
- 62.Kubatzky, K. F., Ruan, W., Gurezka, R., Cohen, J., Ketteler, R., Watowich, S. S., Neumann, D., Langosch, D., and Klingmuller, U. (2001) Curr. Biol. 11 110–115 [DOI] [PubMed] [Google Scholar]