

Abstract
High-copy plasmids are useful for producing large quantities of plasmid DNA, but are generally inadequate for tightly regulating gene expression. Attempts to suppress expression of genes on high-copy plasmids often results in residual or “leaky” production of protein. For stringent regulation of gene expression, it is often necessary to excise the gene of interest and subclone it into a low-copy plasmid. Here, we report a dual plasmid technique that enables tight regulation of gene expression driven by the lac promoter in a high-copy vector. A series of plasmids with varying copies of the lacIq gene have been constructed to permit titration of the LacI protein. When a high-copy plasmid is transformed along with the appropriate lacIq-containing plasmid, tight gene regulation is achieved, thus eliminating the need to subclone genes into low-copy plasmids. In addition, we show that this dual plasmid technique enables high-copy gene expression of a protein lethal to Escherichia coli, the ccdB protein. In principle, this technique can be applied to any high-copy plasmid containing the popular pUC replication of origin and provides an easier means of obtaining rigid control over gene expression.
INTRODUCTORY STATEMENT
To facilitate the manipulation of DNA, cloned genes are routinely propagated in high-copy number plasmids. Cloned genes are most commonly propagated in the gamma proteobacterium, Escherichia coli, due to its ability to grow rapidly and densely on numerous inexpensive substrates, its well-characterized genetics and cellular properties, and the availability of increasingly numerous cloning vectors and mutant strains [1, 2]. However, when recombinant proteins are required, genes are typically subcloned and expressed from medium- or low-copy plasmids to avoid residual gene expression or “leakiness” that occurs in high-copy plasmids. Low-copy plasmids provide a lower gene dosage, which minimizes background levels and maintains tight regulation of expression. Typically, a plasmid that carries a gene of interest often requires a trans-acting regulatory protein that is expressed from the genome, an episome, or a second plasmid in order to suppress its expression [3, 4]. It is especially difficult to avoid basal gene expression when using the popular lac repressor system, because four repressor molecules (forming a tetramer) are needed to bind to a single operator sequence in each cell in order to suppress gene expression.
Here, we describe a new, flexible strategy for controlled regulation of gene expression from a high-copy plasmid in E. coli. To test this strategy, we chose a high-copy plasmid that is commonly used to clone and sequence PCR fragments, pCR4-TOPO-Blunt (Invitrogen), which contains a pUC origin of replication that allows copy numbers of greater than 400 per cell [5]. This vector is popular because it exerts negative selection on vectors that do not contain an insert in its cloning site. The cloning site lies 5’ proximal to the cytotoxic ccdB gene, which is driven by the lac promoter. In the absence of an insert, the ccdB protein is expressed, poisons DNA gyrase complexes [6, 7], and kills the cell. In the presence of an insert, ccdB expression is disrupted (unless there is an open reading frame that continues expression of ccdB), permitting the cell to survive. If a gene is ligated into the cloning site, such that it is translated in the proper reading frame, the lac promoter may be utilized for the expression of that gene. This type of gene expression is constitutive, since there are insufficient repressor molecules available to suppress gene expression from a high-copy number plasmid. We have engineered a series of compatible plasmids that permit titration of the LacI repressor protein in E. coli, thereby allowing the investigator to choose a level of regulation that optimizes inducible expression from the lac promoter. This technique could be applied to expression from any high-copy plasmid with a compatible origin of replication.
MATERIALS AND METHODS
EGFP-containing Plasmids to Monitor Gene Expression
The Enhanced Green Fluorescent Protein (EGFP) gene was used as a visual indicator to monitor expression. The EGFP gene was amplified from the vector pEGFP-1 (Clontech) by PCR, and cloned via topoisomerase I into pCR4-TOPO-Blunt (Invitrogen) to create the plasmid pCR4-TOPO-Blunt-EGFP, using the manufacturer’s directions. Similarly, the amplified EGFP gene was subcloned into pET30 (Novagen) to create the plasmid pET30-EGFP.
Construction of a Series of Compatible Plasmids Containing Different Copies of the Lac Repressor Gene
We selected a plasmid containing the RSF origin of replication for co-expression of the lac repressor, because the RSF replication of origin is compatible with plasmids that contain the pUC replication of origin [8]. A series of plasmids containing the RSF replication of origin were constructed as illustrated in Fig. 1. The original plasmid, pRSFDuet-1, contains a kanamycin cassette, but this is incompatible with pCR4-TOPO-Blunt, which also contains a kanamycin resistance gene. We therefore replaced the kanamycin cassette with a chloramphenicol resistance gene from pACYCDuet-1 (Novagen), as shown in Figure 1. The resulting vector contains a single lacI gene and two operator sequences (R1).
Figure 1. Construction of a series of plasmids containing the RSF replication of origin and varying number of lacI or lacIq genes.
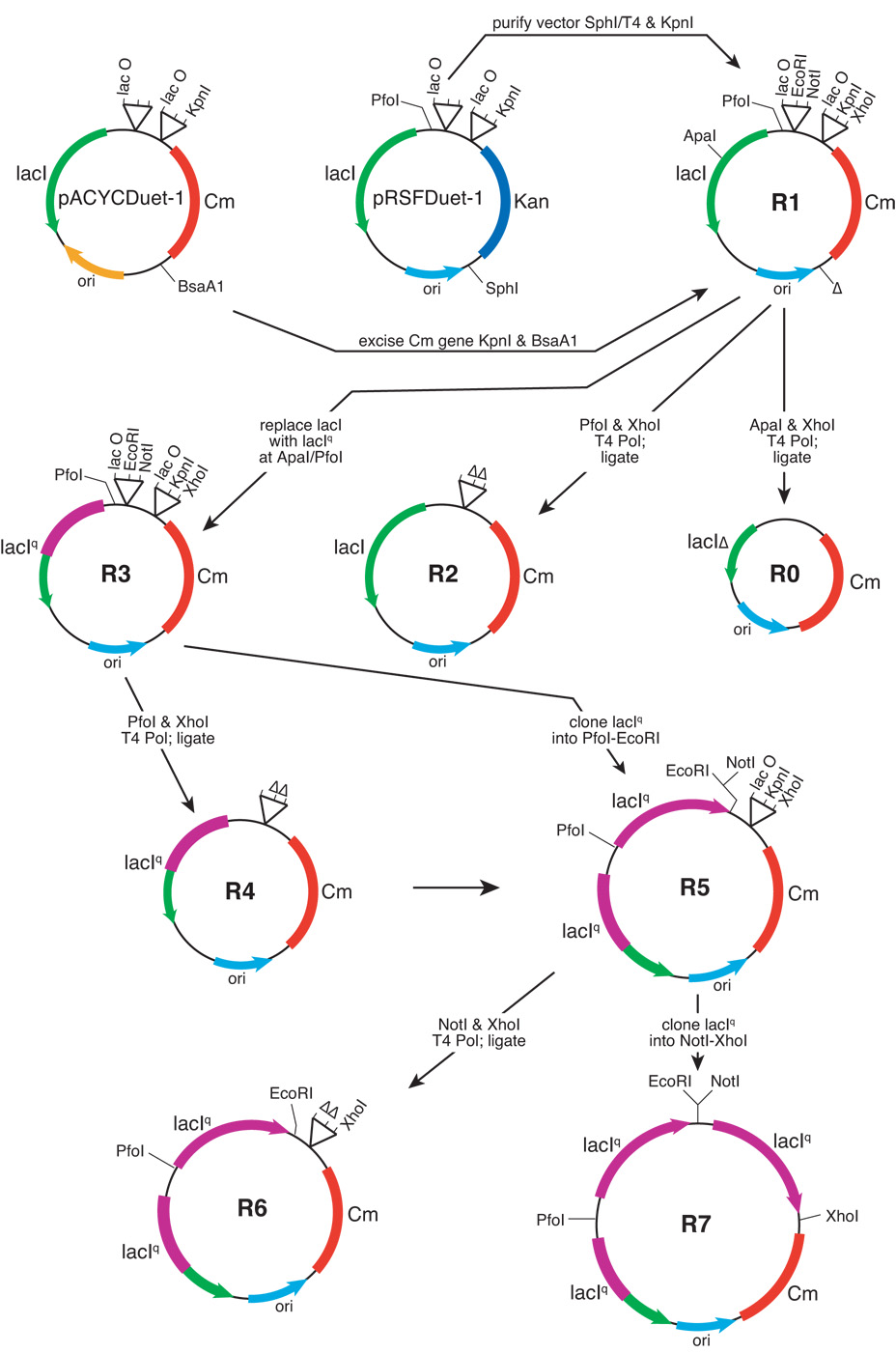
The parental vector for this series is R1, which was constructed by replacing the kanamycin cassette (Kan) of pRSFDuet-1 (excised with SphI and KpnI) with the chloramphenical cassette (Cm) of pACYCDuet-1 (excised with KpnI and BsaA1). The SphI site was made compatible with the blunt-ended BsaA1 site by treatment with T4 DNA polymerase. The replication of origin (ori), location of lac operators (lacO), deleted sequences (Δ), and various restriction enzyme sites, are schematically shown.
R0, which contains a truncated lacI gene and has no lac operator sequences, was created by digesting R1 with ApaI and XhoI, and recircularizing the vector.
R2 possesses no lac operator sequences, which were deleted from R1 by PfoI-XhoI digestion and recircularization.
In R3, the lacI gene is replaced with the lacIq gene. A portion of the lacIq gene (from pGex6P-1) was amplified using gene-specific primers, digested with ApaI and PfoI, and inserted into the ApaI-PfoI region of R1, thereby converting the lacI gene into lacIq.
R4 was constructed from R3, and is similar except that the two lac operators were deleted by PfoI-XhoI digestion and recircularization.
In R5, a second lacIq gene was inserted into the first cloning site of R3 (using PfoI and EcoRI, which deletes one of the two lac operators).
R6 was constructed from R5, and is similar except that the lac operators were deleted by NotI-XhoI digestion and recircularization.
In R7, a third lacIq gene was inserted into the second cloning site of R5 (using NotI and XhoI, which deletes the remaining lac operator).
The vector R1 was engineered to (a) delete the lac repressor gene and both operators (R0); (b) remove both operator sequences (R2); or (c) mutate lacI to lacIq (R3). The lacIq mutation results in an increase in the production of lac repressor by 10 to 20 fold [9]. Since lac operators existing on the same plasmid may titrate out the lac repressor, plasmids were constructed that eliminated the operators (R2, R4, and R5). By exploiting the multiple cloning sites of pRSFDuet-1, two and three copies of the lacIq gene were inserted into the vector (R6 and R7 respectively). R0 serves as a negative control since it lacks a functional lacI gene and contains no operators.
Monitoring Gene Expression In E. coli
Expression in the vector pCR4-TOPO-Blunt was carried out in TOP10 cells and expression in the vector pET30 was carried out in BL21 cells. Gene expression assays were conducted by growing bacteria in a rotating incubator at 37°C. An overnight culture was initiated by inoculating bacterial media (either 2 mL of CircleGrow or Luria broth) with a single colony. The media was supplemented with 20 µg/ml kanamycin, but if the colony was co-transformed with a second chloramphenicol-resistant plasmid, the media contained an additional 17 µg/ml chloramphenicol. To initiate the test induction, 200 µL of an overnight culture was inoculated into 2 mL fresh bacterial media. An aliquot (1 mL) was taken after 3–6 hours of growth in the absence of Isopropyl-beta-D-1-thiogalactopyranoside (IPTG) to obtain an uninduced sample. IPTG was then added to a final concentration of 0.5 mM after 3 hours of growth, and the cultures were incubated for a further 3 hours before being harvested for western blot analyses.
Samples (1 mL) were centrifuged at 13,000 rpm for 1 minute, bacterial media removed, and the E. coli pellet lysed in 100 µL BugBuster supplemented with 0.1 unit/µL beconase (Novagen) for 5 minutes at room temperature. An equal volume of 2× SDS sample buffer was added, and the proteins were boiled for 2 minutes. 10–20 µL of protein were resolved by SDS-PAGE and immunoblotted using procedures described by the manufacturer (BioRad).
Flag-ccdB Plasmids
To create a Flag tag for detection of the ccdB protein on western blots, the Flag oligonucleotides GACTACAAGGATGACGATGACAAG and its reverse complement were ligated into pCR4-TOPO-Blunt. Ligated products were co-transformed with the plasmid R7 (above) into TOP10 cells, and single colonies were picked for analyses. Plasmids containing in-frame cloned oligonucleotides in the correct orientation, which express the Flag epitope, were confirmed by sequencing.
Western Blot Analyses
Western blots were developed using a rabbit polyclonal antibody directed against GFP (Santa Cruz Biotechnology), or a mouse monoclonal antibody directed against LacI (Upstate) or the FLAG epitope (Scientific Imaging Systems). Immunoreactivities were revealed using the appropriate Alexa Fluor conjugated secondary antibody (Molecular Probes), followed by scanning in an Odyssey Infrared Imaging System (Licor Biosciences) or Typhoon 9410 Variable Mode Imager (Amersham Biosciences).
RESULTS
Titration of LacI using a Series of Plasmids
We took advantage of a compatible plasmid, pRSFDuet-1 (Novagen), that contains a different origin of replication, and can therefore co-exist in the same cell as pCR4-TOPO-Blunt. Plasmids were engineered to contain a different antibiotic gene (Chloramphenicol) for independent selection, and varying copies of the lacI or lacIq repressor gene as well as lac operator sequences (Fig. 1 and Table 1). The plasmids, termed R0 to R7 to reflect increasing doses of lacI repression, were used to titrate the suppression of gene expression driven by the lac promoter in pCR4-TOPO-Blunt. To monitor gene expression, we used EGFP as our test gene since its expression is easily visualized.
Table 1.
Summary of RSF containing lacI plasmids.
| Name | Size (bp) | No. lacI | No. lacIq | No. lac o |
|---|---|---|---|---|
| R0 | 2600 | 0 | 0 | 0 |
| R1 | 3656 | 1 | 0 | 2 |
| R2 | 3244 | 1 | 0 | 0 |
| R3 | 3847 | 0 | 1 | 2 |
| R4 | 3435 | 0 | 1 | 0 |
| R5 | 4906 | 0 | 2 | 1 |
| R6 | 4706 | 0 | 2 | 0 |
| R7 | 5937 | 0 | 3 | 0 |
As an initial test, plasmids R0 to R7 were co-transformed with pCR4-TOPO-Blunt-EGFP and visualized on plates supplemented with 1% charcoal (to enhance fluorescent visualization) and antibiotics in the absence or presence of IPTG (Fig 2). As expected, constitutive expression of EGFP was observed in the absence of lac repressor (R0). Very low but detectable levels of expression were observed with lacI-containing plasmids (R1–R7). Even greater suppression of basal expression was observed by increasing the concentration of chloramphenicol. Practically all of the co-transformations displayed inducible gene expression when IPTG was added to the plate. This test demonstrates that, in principle, inducible gene expression is possible from a high-copy plasmid. Unexpectedly, the level of induction was not correlated with the amount of lacI repressor using this plate assay. This may be due to the different growth characteristics acquired by the colonies when they are co-transformed and growing on a solid surface, and/or the rate of diffusion of IPTG through the colony mass.
Figure 2. Baseline levels of lac-driven EGFP expression from a high-copy plasmid can be minimized by co-transformation of plasmids R1–R7.
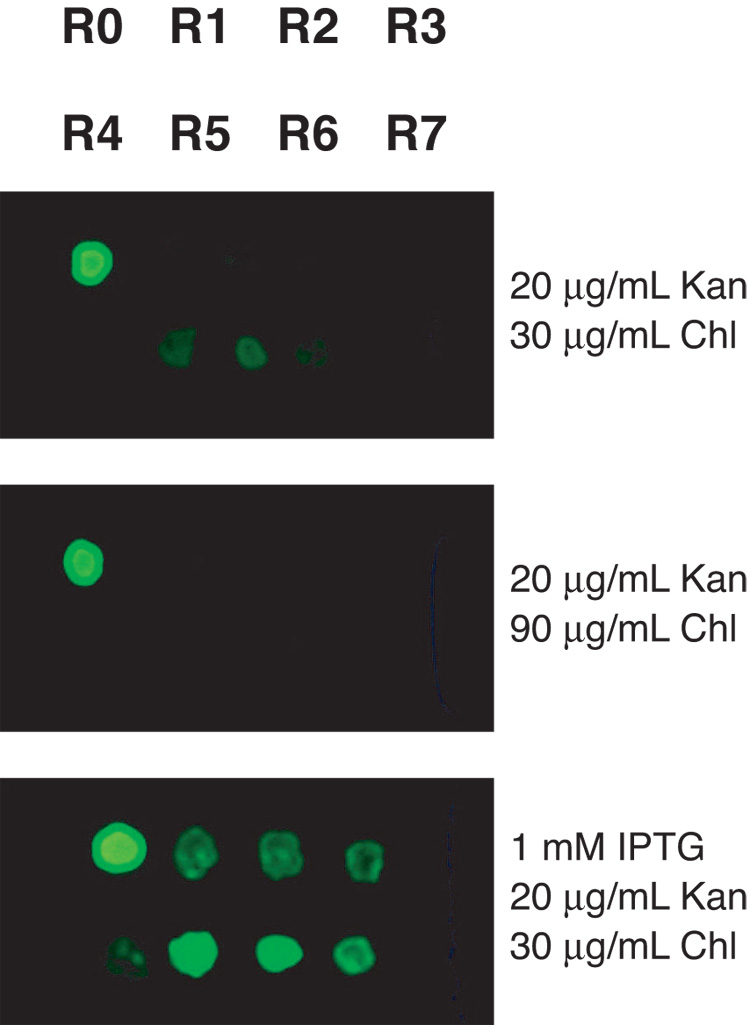
To determine if plasmids R0 to R7 were capable of suppressing basal gene expression from the lac promoter in a high-copy plasmid, these plasmids were cotransformed with pCR4-TOPO-Blunt-EGFP as a reporter. In this manner, fluorescence from colonies serves as a visual indicator of the degree of regulation. Bacterial colonies were grown on charcoal plates supplemented with the indicated antibiotics. Using this assay, expression of EGFP was substantially upregulated in the presence of IPTG.
Baseline and Induced Levels of Gene Expression
To obtain a better idea of how well the lacI-containing plasmids suppress gene expression, inductions using IPTG were carried out for 3 hours in liquid culture. Under these conditions, basal levels of EGFP were very low (Fig. 3A). In fact, basal levels of EGFP are lower than that observed when EGFP is expressed from a pET vector in BL21 (Fig. 3A and B). As expected, there was more LacI protein produced with co-transformation of plasmids with greater copies of the lacIq gene (Fig. 3B).
Figure 3. Comparable baseline and induced expression of the EGFP reporter is observed in the Dual Vector and pET systems.
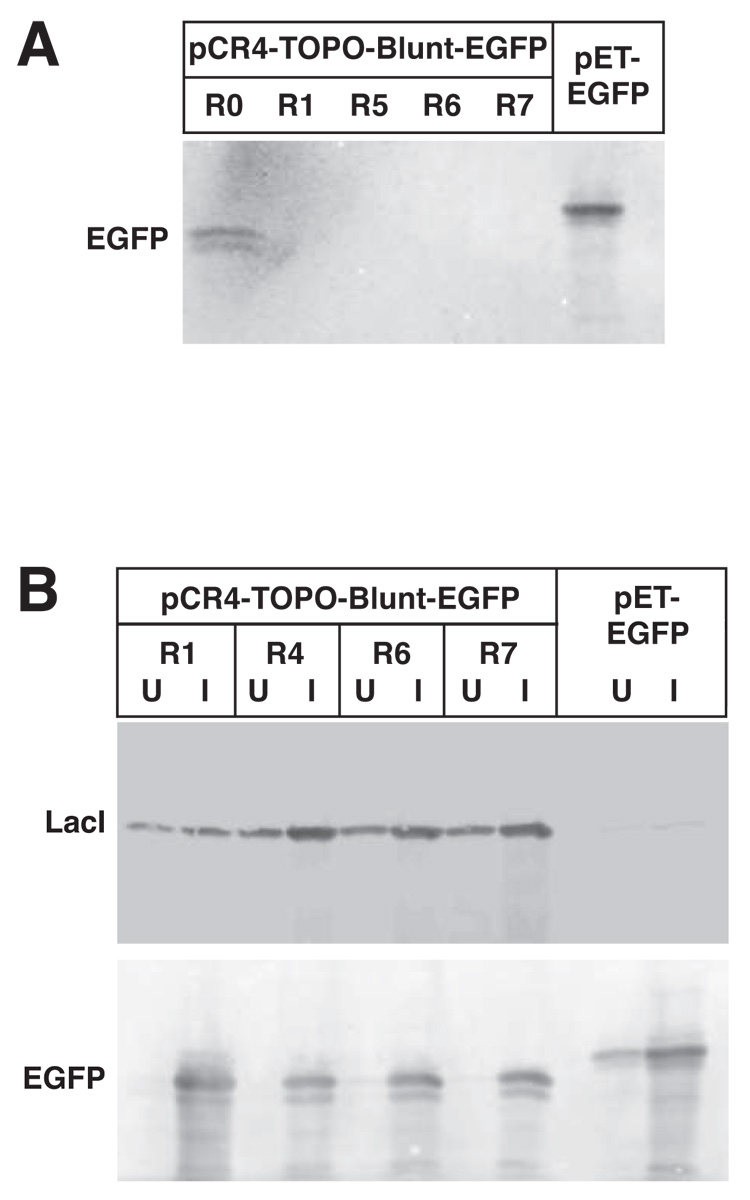
A. Baseline levels of expression observed using EGFP as a reporter. Protein extracts were made from overnight cultures of pCR4-TOPO-Blunt-EGFP co-transformed with the indicated RSF plasmid in TOP10 cells, or pET-EGFP in BL21 cells. Extracts were immunoblotted against a polyclonal antibody directed towards GFP. B. pCR4-TOPO-Blunt-EGFP was co-expressed with the indicated RSF plasmid in TOP10 cells until log phase. Similarly, pET-EGFP in BL21 was grown till log phase. Cells were then incubated for a further 3 hours without additives (U) or induced with 1 mM IPTG for 3 hours (I), pelleted, and crude protein extracts immunoblotted with the indicated antibodies.
To determine the basal level of expression and induction of gene expression, co-transformed E. coli were induced for 6 hours in liquid culture under a variety of conditions (Fig. 4). In general, inducible gene expression was highly robust in this system. However, there was some “leakiness” observed when pCR4-TOPO-Blunt-EGFP was co-transformed with R1, which contains a single lacI gene. Attempts to manipulate the copy number of the lacI-containing plasmid by increasing chloramphenicol concentration failed to alleviate this leakiness using the R1 plasmid. The R6 and R7 plasmids, which contain 2 and 3 lacIq genes respectively, were far more effective in suppressing basal expression of EGFP. Increasing chloramphenicol concentration resulted in a decrease in the amount of expressed EGFP protein, and R7 co-transformed cells had lower levels of EGFP expression than R6 co-transformed cells. Increasing the amount of IPTG did not appear to have an effect on induction. Thus, high levels of lac repressor can suppress basal expression of recombinant protein (i.e. improve “leakiness”), but can simultaneously decrease maximal levels of induction.
Figure 4. Robust induction of lac-driven EGFP using the Dual Vector method for regulating gene expression from a high-copy plasmid.
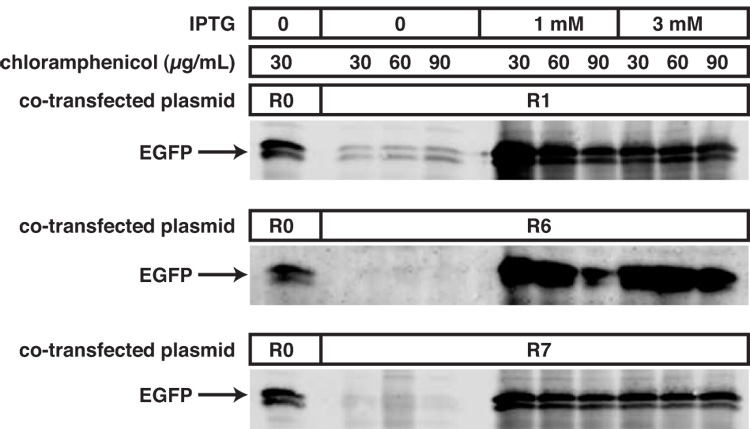
pCR4-TOPO-Blunt-EGFP was co-expressed with the indicated RSF plasmid in TOP10 cells until log phase, in the presence of the indicated amount of chloramphenical. Cells were then incubated for a further 6 hours with the indicated amount of IPTG, pelleted, and crude protein extracts immunoblotted with an antibody directed towards GFP.
High-Copy Expression of the Lethal ccdB Gene
To demonstrate that it is possible to attain stringent gene regulation using a high-copy plasmid, we attempted to control expression of the lethal ccdB gene in E. coli. To express the gene in E. coli, we ligated the high-copy vector pCR-blunt (Invitrogen) in the absence of insert DNA to provide read-through transcription of the ccdB gene. As expected, transformation of pCR-blunt alone yielded very few colonies. By contrast, co-transformation of pCR-blunt with the plasmid R5 yielded hundreds of colonies (data not shown). Of 6 colonies selected, no variation in DNA sequence of pCR-blunt was observed, suggesting that the plasmid is capable of read-through transcription of the ccdB gene, yet was able to be propagated because ccdB transcription was suppressed by sufficient amounts of lacI repressor.
As there are no known sources of antibodies directed toward the ccdB protein, we constructed a short in-frame epitope that is recognized by Flag monoclonal antibodies (Materials and Methods), to generate a method of monitoring the expression of the Flag-ccdB protein. Oligonucleotides corresponding to this epitope were ligated into pCR4-TOPO-Blunt; cloning in either orientation will lead to in-frame readthrough of 8 amino acids. As expected, pCR4-TOPO-Blunt ligated to the oligonucleotides resulted in very few E. coli colonies following transformation. However, hundreds of colonies were observed when the same ligation mixture was co-transformed with either plasmid R5 or R7. Sequencing of several colonies revealed that about half contained the correct orientation of Flag epitope.
We next induced expression of Flag-ccdB during the late log growth phase (Fig. 5). We observed that expression of Flag-ccdB from pCR4-TOPO-Blunt (co-transformed with plasmid R7) can be robustly activated by addition of IPTG into the bacterial media (Fig. 5). Since even minimal residual expression of this gene would prove fatal to E. coli, these results demonstrate that gene expression can be tightly regulated by this dual vector system.
Figure 5. Western blot of bacterial lysates expressing Flag-ccdB in the presence of plasmid R7.
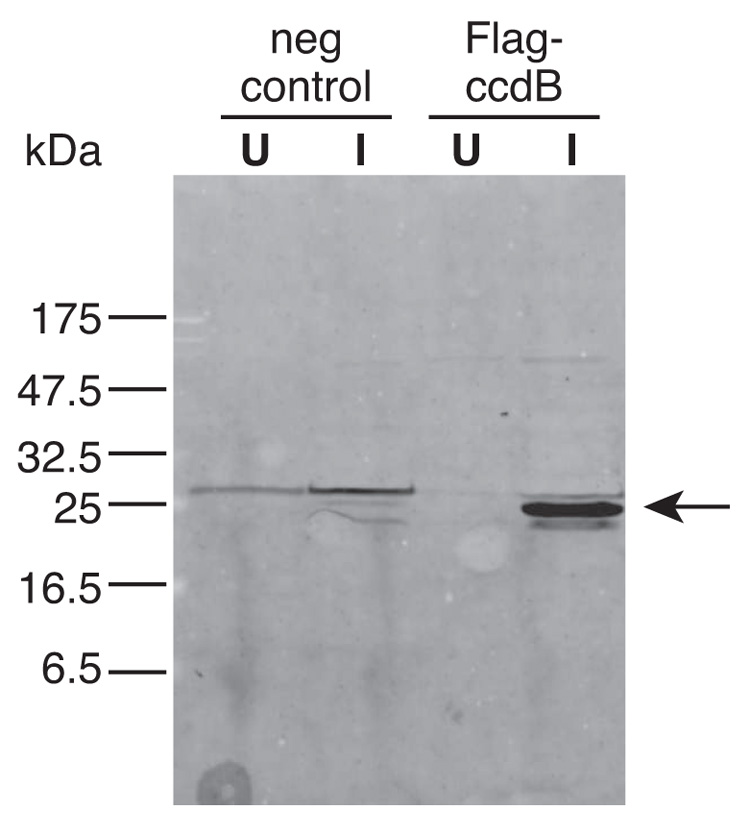
The blot was probed with a monoclonal antibody directed towards the Flag epitope. The negative control represents lysate from pCR4-TOPO-Blunt with an irrelevant insert. Lysates were derived from uninduced (U) or induced (I) bacteria that were incubated in the presence of IPTG for four hours. Arrow points to the position of Flag-ccdB, which is expected to migrate at 25 kDa. The ~28 kDa band visible in the negative control represents non-specific binding to the Flag antibody.
DISCUSSION
The expression of recombinant proteins from a plasmid in E. coli is now a routine procedure in laboratories worldwide. Since many recombinant proteins are poorly tolerated in E. coli, tight regulation of gene expression is desirable. For example, strict gene expression is necessary to evaluate protein folding kinetics and metabolic rates, as well as to propagate proteins that are toxic to E. coli. To optimize protein production, transcription is typically suppressed until cells have reached a high cell density (late log phase), before activating gene expression. Thus, it is important to maintain a low basal level of gene expression for these types of application.
The lac promoter and its variants are among the most popular means of regulating gene expression in E. coli, but gene expression using the lac promoter is notoriously “leaky” due to insufficient suppression. Transcription from the lac operator is regulated by the lac repressor, which binds to the lac operator located downstream of the promoter. The “leakiness” of the lac promoter in high-copy plasmids is most likely due to insufficient titration of the lac repressor to the lac operator. This can be especially problematic if the residual gene is toxic to E. coli, since expression hinders cell replication and decreases protein production.
To address this problem, strategies to lower basal gene expression, such as using a medium- or low-copy plasmid, have been developed. For example, the widely used pET vector system utilizes a medium-copy plasmid in which gene expression is driven by the T7 promoter, which itself is regulated by a T7 RNA polymerase gene that must be activated before gene expression can occur [10]. Thus, the pET vector system utilizes two levels of gene regulation in order to exert tight control over the expression of recombinant protein.
Dual plasmid systems for regulating gene expression in E. coli have been described previously. One system employed the trp repressor and trp promoter to achieve regulation of human interferon-β in E. coli [4]. Another system involved regulation of medium-copy T7-based expression plasmids with compatible ColE1-derived plasmids [3].
The dual vector system described here offers a number of advantages. The system utilizes plasmids that are commercially available and a series of lacIq-containing plasmids for titration of repression. We have demonstrated that it effectively converts a popular high-copy plasmid, pCR4-TOPO-Blunt, into a regulable expression plasmid, thus avoiding the process of subcloning DNA fragments into low-copy plasmids. One advantage of this method is that it can be applied to the expression of membrane proteins and toxic or poorly tolerated proteins to E. coli. This system can also be applied to any plasmid containing the popular pUC replication of origin, which confers high-copy number and includes many commercially available sequencing vectors that also carry the lac promoter. The activation of gene expression from this system is highly robust, as it is initiated at high gene dosage, yet basal levels of gene expression are comparable to low expression systems such as pET. Overall, this method offers an easier means of obtaining rigid control over gene expression.
ACKNOWLEDGEMENTS
This research was supported by NIH GM070348. We thank Shelby Sturgis for his excellent technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Baneyx F. Recombinant protein expression in Escherichia coli. Curr. Opin. Biotechnol. 1999;10:411–421. doi: 10.1016/s0958-1669(99)00003-8. [DOI] [PubMed] [Google Scholar]
- 2.Makrides SC. Strategies for achieving high-level expression of genes in Escherichia coli. Microbiol. Rev. 1996;60:512–538. doi: 10.1128/mr.60.3.512-538.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Munson M, Predki PF, Regan L. ColE1-compatible vectors for high-level expression of cloned DNAs from the T7 promoter. Gene. 1994;144:59–62. doi: 10.1016/0378-1119(94)90203-8. [DOI] [PubMed] [Google Scholar]
- 4.Warne SR, Thomas CM, Nugent ME, Tacon WC. Use of a modified Escherichia coli trpR gene to obtain tight regulation of high-copy-number expression vectors. Gene. 1986;46:103–112. doi: 10.1016/0378-1119(86)90172-1. [DOI] [PubMed] [Google Scholar]
- 5.Lee CL, Ow DS, Oh SK. Quantitative real-time polymerase chain reaction for determination of plasmid copy number in bacteria. J. Microbiol. Methods. 2006;65:258–267. doi: 10.1016/j.mimet.2005.07.019. [DOI] [PubMed] [Google Scholar]
- 6.Bernard P, Couturier M. Cell killing by the F plasmid CcdB protein involves poisoning of DNA-topoisomerase II complexes. J. Mol. Biol. 1992;226:735–745. doi: 10.1016/0022-2836(92)90629-x. [DOI] [PubMed] [Google Scholar]
- 7.Maki S, Takiguchi S, Miki T, Horiuchi T. Modulation of DNA supercoiling activity of Escherichia coli DNA gyrase by F plasmid proteins. Antagonistic actions of LetA (CcdA) and LetD (CcdB) proteins. J. Biol. Chem. 267;1992:12244–12251. [PubMed] [Google Scholar]
- 8.Phillips GJ, Park SK, Huber D. High copy number plasmids compatible with commonly used cloning vectors. Biotechniques. 2000;28:400–402. 404, 406 passim. doi: 10.2144/00283bm02. [DOI] [PubMed] [Google Scholar]
- 9.Müller-Hill B, Crapo L, Gilbert W. Mutants that make more lac repressor. Proc. Natl. Acad. Sci. U. S. A. 1968;59:1259–1264. doi: 10.1073/pnas.59.4.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dubendorff JW, Studier FW. Controlling basal expression in an inducible T7 expression system by blocking the target T7 promoter with lac repressor. J. Mol. Biol. 1991;219:45–59. doi: 10.1016/0022-2836(91)90856-2. [DOI] [PubMed] [Google Scholar]