

Abstract
Neuroimaging methods can be used to investigate whether sleep disorders are associated with specific changes in brain structure or regional activity. However, it is still unclear how these new data might improve our understanding of the pathophysiology underlying adult sleep disorders. Here we review functional brain imaging findings in major intrinsic sleep disorders (i.e., idiopathic insomnia, narcolepsy, and obstructive sleep apnea) and in abnormal motor behavior during sleep (i.e., periodic limb movement disorder and REM sleep behavior disorder). The studies reviewed include neuroanatomical assessments (voxel-based morphometry, magnetic resonance spectroscopy), metabolic/functional investigations (positron emission tomography, single photon emission computed tomography, functional magnetic resonance imaging), and ligand marker measurements.
Based on the current state of the research, we suggest that brain imaging is a useful approach to assess the structural and functional correlates of sleep impairments as well as better understand the cerebral consequences of various therapeutic approaches. Modern neuroimaging techniques therefore provide a valuable tool to gain insight into possible pathophysiological mechanisms of sleep disorders in adult humans.
Citation:
Desseilles M; Dang-Vu TD; Schabus M; Sterpenich V; Maquet P; Schwartz S. Neuroimaging insights into the pathophysiology of sleep disorders. SLEEP 2008;31(6):777–794.
Keywords: PET, SPECT, fMRI, insomnia, depression, narcolepsy, obstructive sleep apnea syndrome, restless legs syndrome, REM sleep behavior disorders
OVER THE LAST COUPLE OF DECADES, SEVERAL STUDIES HAVE USED FUNCTIONAL NEUROIMAGING TECHNIQUES TO INVESTIGATE THE CEREBRAL correlates and consequences of primary sleep disorders in adult humans. By revealing the regional patterns of activation associated with specific sleep disorders, the data from positron emission tomography (PET) and magnetic resonance imaging (MRI) techniques complement and extend previous findings mainly based on electroencephalography (EEG) and brain-damaged patients. Here we review recent functional neuroimaging data gained from adult patients having sleep disorders. Our goal is to assess how these new data might improve our knowledge of the neural mechanisms involved in the pathophysiology of some major sleep disorders. Critical EEG results are also considered, but a comprehensive integration of electrical neuroimaging with metabolic and hemodynamic findings is beyond the scope of the current review.
We first report functional imaging studies in intrinsic sleep disorders such as idiopathic insomnia, narcolepsy and obstructive sleep apnea. We then focus on neuroimaging findings in abnormal motor behavior during sleep (i.e., periodic limb movement disorder and REM sleep behavior disorder). We also consider brain functions in sleep disorders related to specific psychiatric disorders. Rare sleep disorders and case reports are not reviewed here.
Each sleep disorder section starts with an introduction to the disorder, including possible pathophysiological brain mechanisms, followed by a detailed review of the structural and functional neuroimaging findings, and ends with a summary of the main findings.
IDIOPATHIC INSOMNIA
Insomnia is characterized by complaints of difficulty in initiating or maintaining sleep or of nonrestorative sleep, which cause clinically significant distress or impairment in social, occupational, or other important areas of functioning.1 Insomnia therefore presents with subjective symptoms. Insomnia might arise directly from sleep/wake regulatory dysfunction or indirectly from comorbid behavioral, psychiatric, neurological, immune, or endocrine disorders, including disturbances secondary to the use of drugs. Insomnia appears to be a 24-h disorder because it is not restricted to sleep complaints alone but can affect several aspects of daytime functioning as well. Importantly, insomnia is a common disorder in our society, with 10% to 20% of the general population reporting insomnia complaints and related impairment of daytime functioning.2
Depression is often associated with insomnia.3 In this section, we also review the data pointing to some common underlying neurophysiological mechanisms for both sleep and mood regulation.
Hyperarousal Hypothesis in Insomnia
According to the International Classification of Sleep Disorders (ICSD-2), idiopathic insomnia “is a lifelong inability to obtain adequate sleep that is presumably due to an abnormality of the neurological control of the sleep-wake system.”4 Idiopathic insomnia is thought to reflect an imbalance between arousal and sleep promoting systems, which results in a global cortical hyperactivity as evidenced by EEG studies (see below). In line with the elevated arousal levels, several studies have reported increased alertness using the multiple sleep latency test as well as increased tension and anxiety during wakefulness, associated with a reduction of total sleep duration.5 Poor sleep may also have important consequences on daytime functioning such as altered mood and motivation, decreased attention and vigilance, low levels of energy and concentration, and increased daytime fatigue.5 In addition, insomnia increases the risk of major depression.3
Quantitative EEG recordings have confirmed an overall cortical hyperarousal in primary insomnia, characterized by an increase in beta/gamma activity at sleep onset and during NREM sleep.6 Insomnia would therefore result from a conditioned state of central nervous system (CNS) arousal, which enhances a variety of sensory and cognitive phenomena that are normally suppressed or at least diminished at sleep onset. Uncommon high-frequency activity associated with sleep onset might thus contribute to the frequent misperception of insomniacs of not being asleep while objective EEG parameters indicate otherwise.6
Functional Imaging in Insomnia
To our knowledge, only a few studies have assessed the functional neuroanatomy of idiopathic (or primary) insomnia disorder by recording brain activity during NREM sleep. Nofzinger et al. used 18fluorodeoxyglucose (18FDG) PET to measure regional brain metabolism (indexed by glucose consumption, CMRglu) in 7 patients with idiopathic insomnia and 20 healthy age-matched and gender-matched subjects during waking and NREM sleep.7 Insomnia patients showed increased global CMRglu during the transition from waking to sleep onset as compared to healthy subjects, suggesting that there is an overall cortical hyperarousal in insomnia. Moreover, insomniac patients exhibited less reduction of relative CMRglu from waking to NREM sleep in the ascending reticular activating system, hypothalamus, insular cortex, amygdala, hippocampus, anterior cingulate, and medial prefrontal cortices, as illustrated in Figure 1. Increased metabolism was also observed in the thalamus, which might reflect persistent sensory processing as well as subsequent shallower sleep. In contrast, during wakefulness decreased metabolism was found in subcortical (thalamus, hypothalamus, and brainstem reticular formation) as well as in cortical regions (prefrontal cortex bilaterally, left superior temporal, parietal, and occipital cortices). These findings suggest that insomnia might involve abnormally high regional brain activity during sleeping states, associated with reduced brain metabolism during waking. The observed reduction in prefrontal cortex activity during wakefulness is consistent with reduced attentional abilities and impaired cognitive flexibility resulting from inefficient sleep and is consistent with a chronic state of sleep deprivation.8–10
Figure 1.
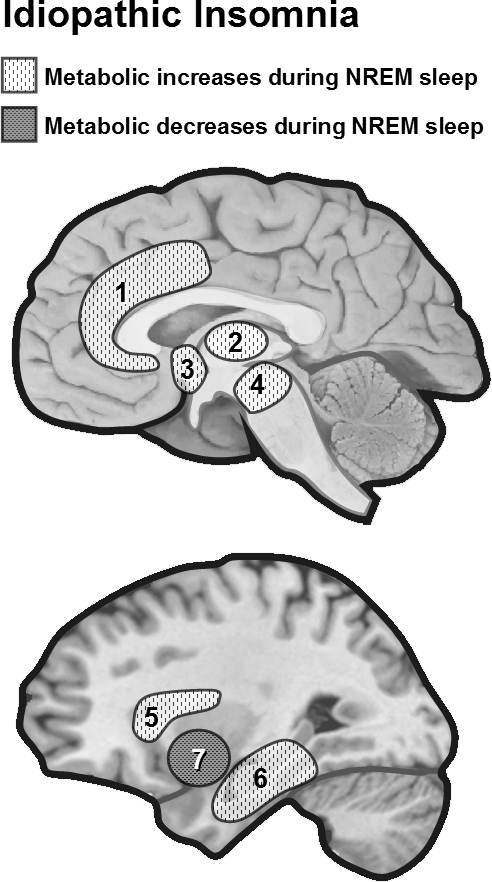
Regional cerebral metabolism during NREM sleep in idiopathic insomnia. Nofzinger et al. found increased regional metabolism (18FDG PET) from waking to NREM sleep in patients with idiopathic insomnia.7 Smith et al. found reduced regional cerebral blood flow (SPECT) in the basal ganglia in insomniacs.11 1 = anterior cingulate, 2 = thalamus, 3 = hypothalamus, 4 = ascending reticular activating system, 5 = insula, 6 = medial temporal, 7 = basal ganglia.
Another preliminary study by Smith et al.,11 which compared 5 insomniacs with 4 normal sleepers using single photon emission computed tomography (SPECT), found no significant regional increase during NREM sleep but reduced regional cerebral blood flow (rCBF) in frontal medial, occipital, and parietal cortices, as well as in the basal ganglia (Figure 1). Interestingly, in Nofzinger's study, decreases in activity in these same regions were also found in insomniacs, but during wakefulness. However, some of the methodological limitations in the Smith's study need to be considered. Firstly, the blood flow was only sampled during the first NREM cycle. Therefore, the observed decreased metabolism in insomniacs might reflect cortical hypoarousal during the initial phases of NREM sleep following sleep onset, while it remains possible that the patients were more aroused over later NREM sleep cycles, which would be more consistent with higher beta activity later at night (see above). Secondly, the blood flow was measured after a longer duration of NREM sleep in insomnia patients than in healthy subjects, leading to a possible underestimation of activity in the patients because blood flow decreases over long NREM periods. Because of such methodological limitations, these preliminary results cannot rule out the hyperarousal hypothesis of primary insomnia.
Four of the insomnia patients from the Smith's study were rescanned after they had been treated by cognitive behavioral therapy (which included sleep restriction and stimulus control12). After treatment, sleep latency was reduced by at least 43%, and there was a global 24% increase in CBF with significant increases in the basal ganglia. The authors proposed that such increase in brain activity might reflect the normalization of sleep homeostatic processes. These interesting initial results will inspire further investigation on the effects of psychotherapy on brain functioning in insomnia.
Depression and Insomnia
Depression is the most common primary diagnosis in patients suffering from insomnia.13 Of all psychiatric conditions associated with insomnia, depression (in particular unipolar depression) is the most frequently diagnosed one.3 Depressed patients frequently report increased daytime fatigue and tend to compensate with daytime napping. Patients with bipolar disorder, on the other hand, report insomnia while depressed, but also hypersomnia, with extended nocturnal sleep periods, difficulty in awakening, and excessive daytime sleepiness.13 Thus, sleep disturbances appear to vary even across depression subtypes. In addition, depression is associated with other sleep disorders like OSAS (see OSAS section).14 Here, we only focus on the links between depression and insomnia. Indications of hyperarousal in both conditions suggest shared neurophysiological mechanisms underlying both sleep and mood regulation.15
Hyperarousal Hypothesis in Depression
In depressed patients, modifications of the sleep architecture is characterized by reduced slow wave sleep (SWS), early onset of the first episode of REM sleep, and increased phasic REM sleep.16 Gillin et al.17 postulated that depression is closely linked to an abnormal increase in some aspects of physiological arousal. Consistent with this hypothesis, total scores on the Hamilton Depression Rating Scale (HDRS) as well as sleep disturbance in depression, a distinct symptom cluster included in the HDRS, have been found to correlate with increased metabolism and regional cerebral blood flow during wakefulness in a large set of cerebral areas including limbic structures, anterior cingulate, thalamus, and basal ganglia.18
Intriguingly, total sleep deprivation is the only known therapeutic intervention in depression that has proven antidepressant effects within 24 hours. Sleep deprivation has rapid beneficial effects, but unfortunately only for about half of the depressive population, with depressive symptoms reappearing after 1 night of recovery sleep.3 One hypothesis is that sleep deprivation can transiently counteract global hyperarousal in the responder population.19
Since hyperarousal has also been described in insomnia, this may be a common pathway underpinning the close relationship between sleep and mood disorders. Evidence for reciprocal relationship between sleep and depression is twofold: sleep disturbances often accompany depression whereas chronic insomnia is a risk factor for the development of depression.20 Subclinical sleep EEG alterations may persist in patients at risk for a depressive episode, thus offering further evidence of a close link between sleep and mood regulation.
Neuroimaging of Sleep in Depression
A pioneering study by Ho et al. examined NREM using PET in 10 patients with depression and 12 controls.21 The depressed patients showed higher CMRglu during NREM sleep in the pons, posterior cingulate, amygdala, hippocampus, and occipital and temporal cortices. There was a significant reduction of relative CMRglu in medial-orbital frontal and anterior cingulate cortices, caudate nucleus, and medial thalamus. These early findings support the hypothesis that hyperarousal in depression affects a network of limbic and posterior cortical regions, but also that the decreased medial frontal and striatal metabolism may be a hallmark of depression.22 More recent studies have confirmed that depressed patients have relatively persistent “elevated” activity measured by CMRglu across many brain regions during sleep compared to presleep wakefulness (REM: 24 depressed patients compared to 14 controls;23 NREM: 12 depressed patients compared to 13 controls,24 see Figure 2). Regions more activated during REM sleep included frontal, parietal, premotor, and sensorimotor cortices, as well as the insula, the ventral pallidum, and the midbrain reticular formation.23 Regions more activated during NREM sleep included the temporal and occipital cortices, as well as the insula, posterior cingulate, cerebellum, and thalamus.24 However, increased metabolism was also found in prefrontal cortex (unlike21). These results are again consistent with a general hyperactivation of arousal systems in depression that may underlie both sleep disturbances such as insomnia as well as nonrestorative sleep complaints in depressed patients.
Figure 2.
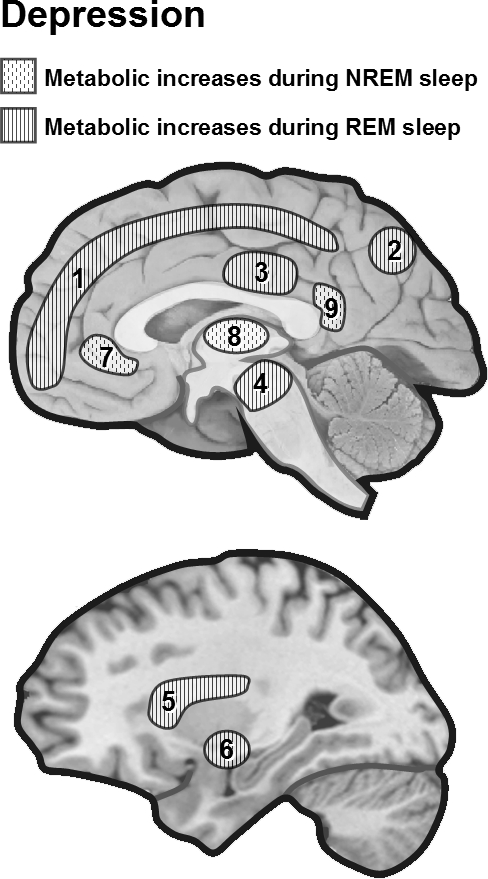
Metabolic changes during REM and NREM sleep in depression. During NREM23 and REM,24 depressed patients show “elevated” activity measured by CMRglu across several cortical and subcortical regions sleep compared to presleep wakefulness.23,24 1 = frontoparietal, 2 = posterior parietal, 3 = supplementary motor area, 4 = ascending reticular activating system, 5 = insula, 6 = ventral pallidum, 7 = medial prefrontal, 8 = thalamus, 9 = posterior cingulate.
Increased rapid eye movement density (number of REMs per minute of REM sleep) was found to correlate with depression severity and clinical outcomes.25 In humans, REMs bursts are classically thought to reflect ponto-geniculo-occipital (PGO) waves, possibly associated with orienting responses and arousal processes during sleep.26,27 An 18FDG PET study assessed cerebral glucose consumption in a group of 13 medication-free depressed patients during REM sleep.28 The average REM count (an automated analog of REM density) was found to positively correlate with the metabolism in a network of regions involved in emotional regulation and emotion-induced arousal (medial and ventrolateral prefrontal cortex) as well as in regions linking emotion and attention systems (striate cortex, precuneus, and posterior parietal cortex).29 Whether increased activity in these regions may drive hyperarousal during REM sleep remains unclear. These results might not be specific to depression because no control data were provided in that study and because the observed activation pattern overlapped with results of healthy controls from other studies.26,30
Summary
Because currently available data are limited and not perfectly consistent, any conclusion about the cerebral correlates of insomnia during NREM sleep has to remain tentative. Whilst there is some evidence for decreased activity in cortical areas during early NREM sleep as well as during wakefulness, several subcortical regions involved in sleep/wake regulation, including limbic and paralimbic regions, were found to be more active during the transition from waking to sleep states. The available data generally support the hyperarousal theory of insomnia with increased neuronal activity during NREM sleep being a possible key factor contributing to sleep misperception and disturbances in insomnia.
Depression is often associated with insomnia, as well as with hyperarousal characterized by persistent “elevated” activity across many brain regions during NREM sleep. Strong evidence for hyperarousal in both idiopathic insomnia and depression, together with persistent alterations in sleep architecture in remitted depression, corroborate common neurophysiological mechanisms underlying sleep and mood regulation.
Changes in brain functions after insomnia treatments have to be assessed more carefully in future neuroimaging studies. Indeed, functional imaging could be coupled with pharmacological or psychotherapeutic treatments in order to assess the neurophysiological response to such interventions, and thus allow a better understanding of the neural mechanisms underlying the recovery from primary insomnia.
NARCOLEPSY
Narcolepsy is a sleep/wake disorder characterized by the clinical tetrad of excessive daytime sleepiness, sudden loss of muscle tone (cataplexy), sleep paralysis, and hypnagogic hallucinations. Nocturnal sleep disruption is typical in narcolepsy. Almost all patients with narcolepsy (with cataplexy subgroup) are positive for the human leukocyte antigen (HLA) subtype DQB1*0602; this HLA subtype is much less frequent in the general population.31 Additional biological markers include sleep-onset REM periods (SOREMPs) on multiple sleep latency tests (MSLT) and low level of cerebrospinal fluid (CSF) hypocretin-1 (orexin A).
Lately, narcolepsy has been linked to a loss of hypothalamic neurons producing hypocretin (also called orexin), a neuropeptide implicated in arousal systems.32 Hypocretin neurons are localized in the lateral hypothalamus and have widespread projections throughout the brain. Hypocretin neurons receive inputs from excitatory (glutaminergic) and inhibitory (noradrenergic, serotonergic and GABAergic) neurons.32 Hypocretin neurons have been found to be implicated in maintaining wakefulness, as well as in the regulation of motor functions (locomotion, muscle tone), energy expenditure, and sympathetic activity.32 Postmortem autopsy studies showed a loss of hypocretin messenger ribonucleic acid (mRNA) and a reduction or loss of hypocretin peptides. Low CSF hypocretin-1 levels is an usual finding in narcolepsy with definite cataplexy.31 In contrast, in most patients with narcolepsy without cataplexy and in other primary sleep-wake disorders (such as insomnia or restless legs syndrome), CSF hypocretin-1 levels are normal. However, low CSF hypocretin-1 levels can also be found in several neurological disorders irrespective of sleep habits.
Over the past decade, brain imaging studies have provided major insights into the functional neuroanatomy of normal human waking state, REM sleep or SWS. Yet, only a few studies looked at how brain activity might be altered in narcoleptic patients. Moreover, the neural correlates of other characteristic symptoms in narcoleptic patients such as cataplexy, hypnopompic/hypnagogic hallucinations or sleep paralysis remain largely unknown.
Structural Abnormalities in Narcolepsy
Because it controls transitions between sleep states, the pontine tegmentum was first proposed as a possible main site of anatomical or functional impairments in narcolepsy. While Plazzi and coworkers33 had reported pontine tegmentum abnormalities (T2 hyperintensity) in 3 patients with narcolepsy using MRI, 2 other structural MRI studies34,35 found no pontine abnormalities in idiopathic narcolepsy (except in 2 out of 12 patients who had longstanding hypertension35). However, the MRI abnormalities found in the study of Plazzi and colleagues could reflect nonspecific age-related pontine vascular changes rather than a narcolepsy-related phenomenon, as they were indistinguishable from ischemic changes and were associated with similar anomalies in the hemispheres.
Differences in brain morphology that are not identifiable by routine inspection of individual structural MRI can be investigated using voxel-based morphometry (VBM). VBM allows between-group, statistical comparisons of tissue composition (gray and white matter) across all brain regions, based on high-resolution scans. To date, VBM studies reported equivocal results in narcoleptic patients. An early study found no structural change in brains of patients with hypocretin-deficient narcolepsy,36 suggesting that functional abnormalities of hypocretin neurons could either be associated with microscopic alterations undetectable by VBM or not be associated with any structural changes whatsoever. Two subsequent studies did find cortical gray matter reduction in patients with narcolepsy, predominantly in frontal brain regions37 as well as in inferior temporal regions38 (Figure 3). Relative global gray matter loss was independent of disease duration or medication history and there were no significant subcortical gray matter alterations.38 Significant gray matter concentration decreases were found in the hypothalamus, cerebellum (vermis), superior temporal gyrus and right nucleus accumbens in 29 narcoleptic patients relative to unaffected healthy controls.39 Given the major projection sites of hypocretin-1 (the hypothalamus among others) and hypocretin-2 (the nucleus accumbens among others), the decreases in gray matter could reflect secondary neuronal losses due to the destruction of specific hypocretin projections. A recent VBM study corroborated significant reduction in hypothalamic gray matter volume in 19 patients compared with 16 controls.40
Figure 3.

(a) Anatomical brain changes in narcoleptic patients assessed by VBM. In narcoleptic patients, cortical gray matter loss was found to affect frontal brain regions,37 temporal regions,38 as well as hypothalamus, cerebellum (vermis) and ventral striatum39 (see also40). However, hypothalamus damage is not systematically found in VBM studies.36 (b) Functional brain changes in narcolepsy. Narcoleptic patients show reduced baseline activity during wakefulness (18FDG PET, SPECT) in several regions including the hypothalamus and mediodorsal thalamic nuclei.45,46 During cataplexy, one SPECT study reported hyperperfusion in several brain regions including limbic area, brainstem and motor regions51 (but see also52). 1 = hypothalamus, 2 = nucleus accumbens (right), 3 = frontomesial, 4 = prefrontal (right), 5 = inferior temporal, 6 = inferior frontal, 7 = superior frontal, 8 = inferior parietal lobule, 9 = rectal/subcallosal gyrus, 10 = dorsal thalamus, 11 = hypothalamus, 12 = cingulate, 13 = post central/supramarginal, 14 = caudate, 15 = premotor and motor cortex, 16 = cingulate, 17 = thalamus, 18 = brainstem, 19 = insula (right), 20 = amygdala (right).
Proton magnetic resonance spectroscopy (1H-MRS) was also used to assess the N-acetylaspartate (NAA) and creatinine plus phosphocreatinine (Cr+PCr) content in the hypothalamus of narcoleptic patients. An analysis of spectral peak area ratios revealed a decrease in the NAA/Cr+PCr ratio in the hypothalamus of 23 narcoleptic patients compared with 10 control subjects.41 An earlier study found similar NAA/Cr+PCr ratios in the ventral pontine areas of 12 narcoleptic patients compared to 12 controls.42 Reduced NAA/Cr+PCr ratio indicates reduced neuronal function which could reflect neuronal loss (i.e., fewer neurons), but could also be due to reduced activity of existing neurons. Decreased NAA concentration is typically seen in neurodegenerative, inflammatory, or vascular disorders.43 The partial reversibility of NAA deficit during recovery from acute brain pathology44 suggests that reduced brain NAA may be not only related to neuronal loss, but also to neuronal dysfunction.
Several factors can explain inconsistencies across both VBM and spectroscopy studies such as inhomogeneous patient groups, history of treatment or, for VBM, pre-statistical image processing and limited sensitivity of this technique (which means that large sample sizes are needed to obtain reliable results). VBM studies with larger samples of drug-naive patients are required to identify reliably structural abnormalities in narcolepsy.
Functional Imaging in Narcolepsy
Baseline activity during wakefulness was assessed with 18FDG PET by measuring CMRGlu in 24 narcoleptic patients and 24 normal controls.45 Narcoleptic patients had reduced CMRGlu in bilateral precuneus, bilateral posterior hypothalami and mediodorsal thalamic nuclei45 (see Figure 3). A subsequent SPECT study showed hypoperfusion in bilateral anterior hypothalami.46 These two studies indicate lower waking baseline activity in narcolepsy.
Other studies have compared regional brain activity during wakefulness and sleep states. An early study using 133Xe inhalation showed that during wakefulness, brainstem and cerebellar blood flow was lower in narcoleptic patients than in normal subjects.47 In contrast, rCBF increased in all areas after sleep onset as compared to wakefulness, in particular in temporo-parietal regions, possibly related to visual dreaming or hypnagogic hallucinations. More recently, a 99mTC-HMPAO (technetium 99m-hexamethylpropyleneamine oxime) SPECT study in 6 narcoleptic patients found no difference between waking state and REM sleep suggesting similar overall cortical activity across these two states.48 However, the lack of control data significantly limits the interpretation of this result. Further studies are needed to confirm these findings on a larger narcoleptic population including systematic comparisons with matched controls.
Brain responses to visual and auditory stimuli were studied in 12 narcoleptic patients and 12 control subjects using functional MRI (fMRI).49 There was no group difference in spatial extent of cortical activation between control and narcoleptic subjects.
Finally, based on the clinical observation that cataplexy episodes are often triggered by positive emotions (e.g., hearing or telling jokes), a recent event-related fMRI study was performed on 12 narcoleptic patients and 12 controls while they watched sequences of humorous pictures. Both patients and controls were similar in humor appreciation and activated regions known to contribute to humor processing, including limbic and striatal regions. A direct statistical comparison between patients and controls revealed that humorous pictures elicited reduced hypothalamic response together with enhanced amygdala response in the patients. These results suggest that hypothalamic HCRT activity physiologically modulates the processing of emotional inputs within the amygdala, and that suprapontine mechanisms of cataplexy might involve a dysfunction of hypothalamic–amygdala interactions triggered by positive emotions.50
Neural Correlates of Cataplexy
There are very few data describing the neural correlates of cataplexy in narcoleptic patients. One study reported preliminary SPECT data on 2 patients during a cataplexy episode compared to REM sleep or baseline wakefulness.51 During cataplexy, perfusion increased in limbic areas (including amygdala, insula and cingulate gyri) and basal ganglia, thalami, premotor cortices, sensorimotor cortices and brainstem, whereas perfusion decreased in prefrontal cortex and occipital lobe (Figure 3). Increased cingulate and amygdala activity may relate to concomitant emotional processing that is usually reported as a powerful trigger of cataplexy. However, such hyperperfusion in the pons, thalami and amygdaloid complexes was not found in a recent single case report,52 which revealed increased activity in several cortical areas including cingulate cortex, orbitofrontal cortex and right putamen during an episode of cataplexy (here, during status cataplecticus). Future studies using well-designed fMRI protocols on larger samples of patients would be particularly suited to better characterize this complex symptom.50
Neurotransmission in Narcolepsy: Ligand Neuroimaging Studies
Given the role of acetylcholine as an important neurotransmitter in the generation of REM sleep, it was hypothesized that narcolepsy might also involve disturbances within the cholinergic system. However at present, this hypothesis is not supported by the available PET data which could not show any change in muscarinic cholinergic receptors in narcoleptic patients.53 Recently, the release of endogenous serotonin was measured during wakefulness and sleep in human brain using a serotonin antagonist as PET ligand (18FMPPF in 14 narcoleptic patients).54 Serotonin receptor availability increased in sleep compared to wakefulness in narcoleptic patients. Unfortunately, as there was no control group, these results can only support the fact that serotonin release promotes wakefulness and suppresses REM sleep, as suggested by previous animal data.55
Likewise, the dopamine system has been probed by PET in narcoleptic patients because increased dopamine D2 (dopamine receptor 2) binding was shown in the brain of deceased narcoleptic patients.56,57 One SPECT study found D2-receptor binding in the striatal dopaminergic system correlated with the frequency of cataplectic episodes and sleep attacks in 7 patients with narcolepsy.58 However, this finding was not confirmed by other PET59,60 or SPECT studies.61,62 Interestingly, although binding levels of IBZM (iodobenzamide, dopamine D2 receptor ligand) might be similar in narcoleptic patients and normal controls, treatment by stimulants and/or antidepressants for 3 months have been shown to significantly increase the ligand uptake in 4 out of 5 patients as compared to pretreatment scans.62 Therefore, a potential explanation for discrepancies with the postmortem studies might be related to the drug treatment of narcoleptic patients. Consistent with this hypothesis, Rinne et al.63 found no evidence of altered striatal dopamine transporter availability in 10 drug-free narcoleptics (without any treatment in the past) compared to 15 healthy controls with PET using a dopamine transporter ligand (11C-CFT). Collectively, these neuroimaging results suggest that the reported postmortem increase in dopamine binding might be due to long-term effects of prior treatment rather than intrinsic modifications due to the pathophysiology of narcolepsy.
Neural Consequences of Treatment in Narcolepsy
Madras et al. studied the neurochemical substrate of modafinil, a stimulant drug used in the treatment of narcolepsy, in vivo (in rhesus monkey) and in vitro (in human embryonic kidney cells).64 They found that modafinil occupies striatal dopamine transporter sites and thalamic norepinephrine transporter sites in vivo and modulates transporters of both catecholamines as well as serotonin in vitro. These results suggest that the therapeutic action of modafinil is mediated by the modulation of catecholamine receptors.
Moreover, the effects of stimulant drugs on cerebral function in narcoleptic patients were assessed by 2 fMRI studies. The first one tested the effect of modafinil on 8 narcoleptic patients and 8 control subjects while they were presented with multiplexed visual and auditory stimulation.49 Modafinil administration efficiently increased self-reported levels of alertness in 7 of 8 narcoleptic subjects but did not modify the average level of activation in either controls or narcoleptics. Another fMRI study assessed the effects of amphetamines in 2 patients with narcolepsy and 3 healthy controls.65 Whereas the extent of the brain response to auditory and visual stimulation decreased after amphetamine administration in controls, the reverse pattern was observed in narcoleptic patients, with increased response in primary and association sensory cortices. This latter finding suggests that the beneficial effects of amphetamine may be mediated by some enhancement of sensory processing in arousal-deficient subjects. These early neuroimaging results on the effects of stimulant drugs still need to be replicated over larger samples of patients.
One EEG study used advanced methods of distributed source localization (based on intracerebral current density estimates, LORETA66) to analyze waking EEG recordings in 15 narcoleptic patients before and after 3 weeks of modafinil or placebo.67 Cognitive performance (calculation task) was significantly better after modafinil and correlated with a decrease in prefrontal delta, theta, and alpha-1 power, suggesting that modafinil might influence medial prefrontal processes. Interestingly, Thomas and Kwong68 showed that modafinil can counteract the negative effects of a single night of sleep deprivation on working memory, but only when the difficulty of the task remains moderate (2-back task). This was associated with the recruitment of areas in the executive network including prefrontal and parietal regions.
Summary
Although human narcolepsy is associated with hypothalamic hypocretin/orexin dysfunction, no clear evidence for hypothalamic or pontine tegmentum abnormalities emerges from the structural imaging studies reviewed here, including MRI and spectroscopy data. By contrast, the few available functional imaging studies have consistently found hypoactivity in the hypothalamus. These findings suggest that narcolepsy is associated with abnormal hypothalamic function in the absence of consistent structural alterations detectable by current imaging methods. Higher-field MRI scanners with improved signal and spatial resolution might provide a more refined picture of the structural changes in narcolepsy. Neuroimaging data of narcolepsy during active tasks testing specific brain circuits,50 as well as during different sleep states are very promising.
One of the cardinal symptoms of narcolepsy, i.e., cataplexy, has been found to be associated with increased activity in areas involved in emotion and reward processing. These data collected on a total of 3 patients still need confirmation.
In addition to a hypocretin/orexin dysfunction, it has been suggested that the dopamine system might also be involved in pathophysiology of narcolepsy, based on postmortem studies showing an increase in striatal dopamine binding. However, the available neuroimaging results on living patients indicate that increases in dopamine activity might be due to long-term effects of prior treatment rather than intrinsic modifications due to the pathophysiology of narcolepsy.
OBSTRUCTIVE SLEEP APNEA SYNDROME (OSAS)
Obstructive sleep apnea syndrome (OSAS) is characterized by repetitive episodes of upper airway obstruction that occur during sleep and are usually associated with a reduction in blood oxygen saturation. These nocturnal respiratory disturbances result in brief arousals from sleep (i.e., sleep fragmentation) that considerably disturb sleep architecture and may lead to an almost complete deprivation of REM sleep and stages 3 and 4 of NREM sleep. Both sleep disturbances and hypoxemia contribute to excessive daytime sleepiness, a common symptom of the syndrome. OSAS is associated with significant morbidity, such as hypertension, cardiovascular disease, stroke and also motor vehicle accidents. Large epidemiologic studies revealed that OSAS affects 2% of women and 4% of men of the general adult population69 and up to 25% of the elderly (i.e., over 60 years).70
The pathophysiology of OSAS is complex and not yet completely understood. Several studies suggest that OSAS in all age groups is due to a combination of both anatomic airway narrowing and alterations in upper airway neuromuscular tone.71 The pathophysiology of OSAS also includes enhanced chemoreflex sensitivity and an exaggerated ventilatory response during hypoxemic episodes.72
Alterations of cognitive processes, behavior and interpersonal relations are commonly observed in OSAS patients. Both hypoxemia and fragmented sleep are proposed as the main factors leading to neurocognitive impairments during wakefulness. Several studies emphasized the deterioration of executive functions in OSAS patients, including the inability to initiate new mental processes, as well as deficits in working memory, selective attention and continuous attention.73 A recent meta-analysis showed that untreated patients with OSAS had negligible deficits in intellectual and verbal functioning but a substantial impairment of vigilance and executive functioning.74 ‘Cognitive reserve’ might protect against OSAS-related cognitive decline.75
It is still unclear whether the cognitive consequences of OSAS are reversible or not.76,77 Structural alterations may indicate irreversible cerebral changes that would underpin permanent cognitive impairments, although this proposal remains a matter of debate in the literature.78
Structural Changes in OSAS
Several studies used voxel-based morphometry (VBM) on high-resolution MRI scans to assess structural brain changes in patients with OSAS. An early study found regional gray matter loss in OSAS patients (n = 21) compared to healthy controls (n = 21) in regions involved in various cognitive functions and motor regulation of the upper airway, including frontal, temporal, and parietal regions, anterior cingulate, hippocampus, and cerebellum79 (see Figure 4). By contrast, another VBM study showed lower gray matter concentration limited to the left hippocampus in 7 OSAS patients compared to 7 controls, with no difference in total gray matter volume between the two groups.80 A more recent study on 25 OSAS patients and 23 controls found neither gray matter volume deficits nor focal structural changes in severe OSAS patients.81 Comparing both neuropathological and neuropsychological effects of hypoxia in patients with either carbon monoxide poisoning or OSAS, Gale and Hopkins82 reported hippocampal atrophy in both groups. Importantly, hippocampal volume correlated with performance on nonverbal tasks (Wechsler Adult Intelligence Scale–Revised Block Design) in both groups. There was no significant correlation between hippocampal volume and global memory performance but in the OSAS group only, there was a linear relationship between hippocampal volume and a subset of memory tests (e.g., delayed recall on the Rey-Osterrieth Complex Figure Design, Trial 6 of the Rey Auditory Verbal Learning Test). This suggests a link between hippocampus damage and some memory performance in OSAS.
Figure 4.

(a) Regional gray matter loss in OSAS patients. VBM results in OSAS patients revealed gray matter loss limited to the left hippocampus80 or extending to regions involved in cognitive functions and motor regulation of the upper airway.79 (b) Task-related activation in OSAS patients. Functional MRI in OSAS patients during a 2-back working memory task was associated with reduced dorsolateral prefrontal activity,87 while verbal learning was associated with increases in frontal cortex, thalamus and cerebellum.88 1 = left anterior cingulate cortex, 2 = posterior lateral parietal cortex, 3 = inferior temporal gyrus, 4 = parahippocampal gyrus, 5 = right quadrangular lobule, 6 = left hippocampus, 7 = dorsolateral prefrontal cortex, 8 = inferior/middle frontal, 9 = thalamus, 10 = cingulate gyrus, 11 = cerebellum.
Single voxel proton magnetic resonance spectroscopy (1H-MRS) has also been used to assess whether OSAS can induce axonal loss or dysfunction, or myelin metabolism impairment. An early study using 1H-MRS in 23 OSAS patients showed that the N-acetylaspartate/choline ratio (NAA/Cr) in cerebral white matter was significantly lower in patients with moderate to severe OSAS than in patients with mild OSAS and healthy subjects.83 In a more recent study, magnetic resonance spectra were obtained from prefrontal cortex, parieto-occipital, and frontal periventricular white matter. The NAA/Cr and choline/creatine (Cho/Cr) ratios as well as absolute concentrations of NAA and Cho were significantly lower in the frontal white matter of OSAS patients when compared to controls.84 These findings may explain some of the deficits in executive function associated with OSAS, but it is still unclear whether hypoxia or sleep fragmentation is the primary cause of such dysfunction.
Consistent with the VBM results above, decreases in absolute creatine-containing compounds in the left hippocampal area correlated with increased OSAS severity and worse neurocognitive performance.85 Interestingly, a recent study of Halbower et al.86 showed decreased NAA/Cho ratio in the left hippocampus and in the right frontal cortex using the same technique in a pediatric population with OSAS.
Taken together these VBM and spectroscopy studies point to an atrophy and/or dysfunction of hippocampal regions in OSAS.
Brain Imaging of Cognitive Functions in OSAS
Cognition in OSAS patients has been extensively studied, yet little is known about associated functional cerebral changes. Thomas et al.87 used fMRI to study 16 OSAS patients (8 of them were rescanned after treatment with positive airway pressure) and 16 healthy controls during a 2-back verbal working memory task. Both performance on the task and dorsolateral prefrontal activity were reduced in the patients' population, regardless of nocturnal hypoxia (Figure 4). After treatment, resolution of subjective sleepiness and the partial recovery of posterior parietal activation contrasted with persistent performance deficits and lack of prefrontal activation. Another fMRI study examined the cerebral correlates of learning and memory in 12 nontreated OSAS patients and 12 matched healthy controls.88 Verbal learning performance was similar for both groups, but OSAS patients showed increased brain activation in different regions, including bilateral inferior frontal and middle frontal gyri, cingulate gyrus, thalamus, and cerebellum. The recruitment of additional brain areas during the task in OSAS patients reflect an adaptive compensatory recruitment response.88 This hypothesis is consistent with increased brain activity seen after sleep deprivation in healthy subjects which has been thought to reflect dynamic, compensatory changes in cerebral activation during a task after sleep deprivation.89
Cognitive performance may improve with nasal continuous positive airway pressure (nCPAP) treatment, but evidence suggests that some cognitive impairments may be permanent. For instance, improved attention and vigilance is commonly reported after nCPAP treatment in OSAS patients, but no such improvement is found for constructional abilities or psychomotor functioning.76 Intrinsic neural dysfunction may explain the neuropsychological deterioration in OSAS patients.90 In addition, several studies have linked OSAS and depression.14 Overlaps of structural and functional deficits in the hippocampus, anterior cingulate, and frontal cortex (areas consistently showing abnormal structure or function in the depression literature91) provide several potential biological links between OSAS and mood disorders. Regardless of the mechanism, nCPAP therapy in OSAS patients can decrease depression scores and overall psychopathology, thus providing further evidence for a relationship between both these pathologies.92
Neural Correlates of Autonomic Dysfunction and Impaired Ventilatory Control
The apneas in OSAS patients have considerable hemodynamic consequences, involving a complex cascade of physiological events. Repetitive episodes of apnea trigger marked fluctuations in both blood pressure and heart rate and affect cardiovascular regulation. Several important regulatory mechanisms in cardiovascular homeostasis seem to be impaired in OSAS patients—for instance, the ventilatory response to carbon dioxide is elevated in OSAS patients.72 This may be associated with an altered autonomic balance and result in the subsequent development of cardiovascular diseases in patients with OSAS.
Several fMRI studies have been conducted to characterize the response to sympathetic challenge or respiratory stress in OSAS patients. Based on the observation that OSAS patients exhibit altered sympathetic outflow, Harper et al.93 used fMRI to assess changes in brain activity during a forehead cold pressor challenge, which typically elicits respiratory slowing, bradycardia, and enhanced sympathetic outflow. Compared with 16 control subjects, 10 OSAS patients exhibited signal increases in cingulate and cerebellar and frontal cortex; whereas fMRI signal decreased in medullary, midbrain areas, and cerebellar nuclei, as well as in ventral thalamus, hippocampus, and insula (with such signal modulation often paralleling changes in breathing and heart rate). In another study conducted in 8 drug-free OSAS patients (compared with 15 controls), the fMRI response to Valsalva maneuver revealed reduced brain response in parietal, temporal, and posterior insular cortex, as well as in the cerebellum and hippocampus; while activity was enhanced in the lateral precentral gyrus, anterior cingulate, and superior frontal cortex in OSAS.94 These findings suggest that OSAS impacts on cerebellar, limbic, and motor areas involved in the control of airway muscles that mediate a compensatory response to the Valsalva maneuver. Another fMRI study investigated brain activity changes during baseline and expiratory loading conditions in 9 OSAS and 16 controls.95 Both groups developed similar expiratory loading pressures, but OSAS patients failed to show the appropriate autonomic cerebral responses. Indeed, OSAS patients had reduced activation within the frontal cortex, anterior cingulate, cerebellar dentate nucleus, dorsal pons, anterior insula, and lentiform nuclei, together with increased activation in the ventral pons, midbrain, quadrangular cerebellar lobule, and hippocampus. Moreover, activity in the fastigial nuclei of the cerebellum and the amygdala showed substantial variability increase in OSAS subjects. A more recent fMRI study evaluated brain activity changes during baseline and inspiratory loading in 7 patients with OSAS and 11 controls.96 A number of cortical and subcortical areas mediating sensory, motor, and autonomic processes were affected in OSAS patients, with abnormal activation in primary sensory thalamus and sensory cortex, supplementary motor cortex, cerebellar cortex and deep nuclei, cingulate, medial temporal, and insular cortices, right hippocampus, and midbrain.96 Taken together, these fMRI results indicate abnormal brain responses to experimentally induced respiratory and cardiovascular stresses in OSAS, most frequently affecting the cerebellum, insula, cingulate, and motor cortices. This altered pattern of brain activity in OSAS patients during physiologic stress also suggests that similar brain dysfunctions may occur during pathological apneas in sleep, which could lead to permanent neural changes over time. Interestingly, an fMRI study showed significant signal increases in hippocampus, frontal cortex, precentral gyrus, frontal cortex, mediodorsal thalamus, and cerebellar cortex and decreases in the anterior cingulate cortex and postcentral gyrus, coincident with apnea during Cheyne-Stokes breathing (characterized by repeated episodes of apnea followed by increasing and declining respiratory efforts during sleep) in 2 patients.97
Neural Consequences of Treatment in OSAS
A few studies have assessed the long-term neural consequences of nasal continuous positive airway pressure (nCPAP) treatment in OSAS. In an early 99mTC-HMPAO SPECT study in 14 adult OSAS patients, tracer injections were performed between 02:00 and 04:00 during stage 2 sleep, when numerous episodes of obstructive apnea were observed.98 Reduced perfusion in the left parietal region was found, which was completely reversed under effective nCPAP therapy, suggesting that some deleterious effects of OSAS on brain activity might be reversible.87 In another study using 1H-MRS in 14 OSAS patients, NAA in the parietal-occipital cortex was significantly reduced more in OSAS patients than in controls but, unlike the SPECT study above, this reduction persisted after nCPAP therapy despite clinical, neuropsychological, and neurophysiological normalization.99
The effect of mandibular advancement (a frequent treatment of OSAS) was studied in 12 healthy subjects using fMRI during respiratory stress induced by resistive inspiratory loading.100 This treatment led to decreased fMRI response in the left cingulate gyrus and bilateral prefrontal cortices. Together with these objective results, the subjective effects of the treatment assessed by a visual analog scale confirmed the successful reduction of respiratory stress.
Based on the available data, it remains unresolved whether cerebral dysfunctions in OSAS can be alleviated by efficient treatment of nocturnal apnea.
Summary
The reviewed studies suggest that neuropsychological impairments in OSAS are attributable to functional alterations in prefrontal, anterior cingulate, hippocampal, and parietal cortices. Even if abnormal brain activations are sometimes reversible under nCPAP, persistent structural brain changes have been reported in OSAS patients. Consistent with such findings, several studies have suggested that not all neuropsychological impairments disappear after nCPAP treatment. Although the basic mechanisms underlying OSAS are not completely understood, a dysregulation in autonomic functions might contribute to the neural pathophysiology of OSAS. However, it is important to note that some of the deficits observed in OSAS patients may also be attributable to other concomitant factors such as age, elevated body mass index, or depression.
ABNORMAL MOTOR BEHAVIOR (1): PERIODIC LIMB MOVEMENTS AND RESTLESS LEGS SYNDROME
Periodic limb movements in sleep (PLMS) and restless legs syndrome (RLS) are distinct but overlapping syndromes. PLMS is characterized by periodic episodes of repetitive and highly stereotyped limb movements that occur during sleep (mainly NREM sleep).4,101 To date, the largest epidemiological study evaluating the simultaneous presence of PLMS and sleep complaints reported a 3.9% prevalence in 18,980 subjects from the general population between 15 and 100 years of age.102
The diagnosis of PLMS requires the presence of PLMS on polysomnography as well as an associated sleep complaint such as “unrefreshing sleep.” Partial arousal or even awakening frequently accompanies movements, but the patient is usually unaware of these movements or sleep disruption. Periodic limb movements are themselves nonspecific, occurring during sleep—(PLMS) with RLS and with other sleep disorders (e.g., narcolepsy, sleep apnea syndrome, REM sleep behavior disorder)—or during wakefulness (PLMW) and also in healthy subjects.103 Thus, the diagnosis of PLMS requires the exclusion of other potential causes for the associated sleep complaint.
Restless legs syndrome (RLS) is a disorder characterized by uncomfortable leg sensations, usually prior to sleep onset or during the night, which cause an almost irresistible urge to move the legs.4,101 The prevalence of RLS is estimated at 5% to 20%.101 The diagnosis of RLS, by contrast to PLMS, is essentially made on clinical grounds. In addition, most of the patients who suffer from RLS also have PLMS. Psychiatric illnesses such as depression and anxiety have been associated with chronic sleep loss20,104 and appear to be more prevalent in those with RLS and PLMS than with normal controls.105,106 Interestingly, several genetic variants associated with susceptibility to PLMS107 and RLS108 were discovered recently. As RLS and PLMS are not always distinguished in the literature, our review below will cover both RLS and PLMS together.
Structural Abnormalities
Recently structural cerebral abnormalities have been reported in patients with idiopathic RLS. High-resolution T1-weighted MRI of 51 patients compared with 51 controls using VBM revealed a bilateral gray matter increase in the pulvinar (Figure 5).109 The authors suggested that changes in thalamic structures could be directly involved in the pathogenesis of RLS or may instead reflect a consequence of dopaminergic therapy or of a chronic increase in afferent input due to sensory leg discomfort.
Figure 5.
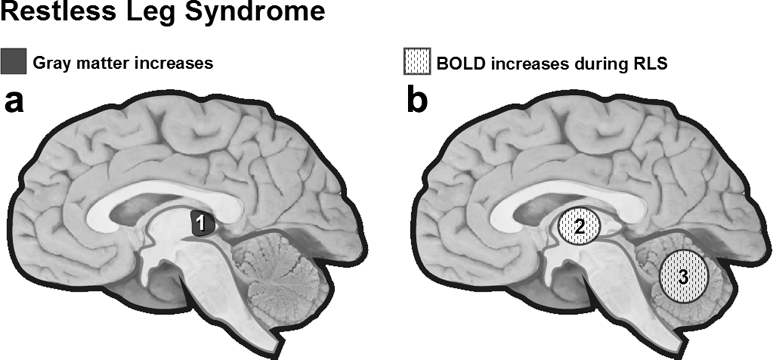
(a) Cortical gray matter changes in RLS. VBM in RLS patients revealed bilateral gray matter increase in the pulvinar.109 (b) BOLD increases during RLS. Cerebellum and thalamus are more activated (fMRI) when RLS patients experience leg discomfort.110 1 = pulvinar, 2 = thalamus, 3 = cerebellum.
Functional Imaging
Functional neuroimaging has also attempted to localize some cerebral generators of leg discomfort and periodic limb movements in RLS. An fMRI study (19 patients) performed during wakefulness showed bilateral activation of the cerebellum and contralateral activation of the thalamus when patients with RLS experienced leg discomfort.110 This is partially consistent with the VBM study reported above.109 During a second condition combining periodic limb movements and sensory leg discomfort, patients also showed activity in the cerebellum and thalamus, with additional activation in the red nuclei and brainstem close to the reticular formation. In neither condition was any cortical activation found. However, when subjects were asked to voluntarily imitate PLMS, there was no activation in the brainstem, but rather additional activation in the globus pallidus and motor cortex. These results are in agreement with those of an EEG study showing no cortical potentials prior to periodic leg movements during the daytime.111 Together, these results support an involuntary mechanism of induction and a subcortical origin for RLS, as also suggested by a transcranial magnetic stimulation study.112
Neurotransmission Abnormalities
A suprasegmental release of inhibition from descending inhibitory pathways implicating dopaminergic, adrenergic, and opiate systems is thought to be involved in RLS/PLMS pathogenesis.
Dopaminergic System
RLS becomes worse when dopamine antagonists are given, whereas dopaminergic drugs have been shown to relieve RLS.113 Studies using SPECT or PET examined both presynaptic DA transporter and postsynaptic D2-receptor binding in the striatum to better characterize the neurophysiological mechanisms underlying the deficit.
Presynaptic DA transporter reflects the number of DA neurons in the substantia nigra and was shown to be similar114–116 or moderately hypofunctional117,118 between patients with RLS (accompanied or not by PLMS) and controls. Two studies used a ligand marker binding dopamine transporter to evaluate striatal presynaptic DA status in RLS-PLMS patients and controls and found no difference between the 2 populations.115,116 In another study, presynaptic striatal dopamine activity correlated negatively with the number of PLMS in 11 patients with idiopathic Parkinson disease (PD) and periodic leg movements during sleep, suggesting that striatal dopaminergic nerve cell loss might cause PLMS in PD.119
For postsynaptic D2-receptor binding, the results are more equivocal. On the one hand, several SPECT studies114,120 comparing RLS (with or without PLMS) patients to age-matched controls failed to find any significant difference. In one of these studies,120 14 patients with idiopathic RLS and PLMS successfully treated by dopaminergic (e.g., ropinirole) and nondopaminergic (e.g., gabapentin) treatment were investigated while off medication and compared to 10 healthy controls. The patients presented sleep disturbances, severe PLMS, and severe RLS symptoms during scanning but did not show any significant differences in striatum/frontal D2 receptors binding ratio. These findings suggest that the dopaminergic system might be affected elsewhere, possibly in the diencephalo-spinal part of the dopaminergic system.
On the other hand, Staedt et al. have tested the hypothesis of a decrease in dopaminergic activity in PLMS patients across a series of SPECT studies121–123 and consistently reported decreased IBZM (postsynaptic) striatal uptake, indicating lower D2 receptor occupancy in PLMS patients. Treating patients with dopamine replacement therapy increased IBZM binding and improved the sleep quality in these patients.122 In line with these results, another SPECT study reported a small but statistically significant decrease in D2-receptor binding in 10 drug-naive patients suffering from both RLS and PLMS compared with 10 age-matched controls.116 Consistent with this observation, a PET study showed a decrease in striatal D2-receptor binding (postsynaptic) for raclopride (an in vivo marker of dopamine D2/D3 receptor binding) in 13 patients with RLS compared with controls.118 No relationship was observed, however, between D2-receptor binding and either RLS severity or PLMS indices.116,118
A recent study investigated both striatal and extrastriatal dopamine activity in 16 RLS patients naïive to dopaminergic drugs and 16 matched controls.124 The results confirmed augmented dopamine activity in the striatum, but also in the thalamus, insula, and anterior cingulate cortex. The latter is part of the medial nociceptive system, which is thought to regulate the affective and motivational component of pain. This pattern of results is thus consistent with the hypothesis of RLS as a disorder of somatosensory processing.
Overall, presynaptic DA transporter binding appears normal in patients with RLS, contrary to what is typically found in early Parkinson disease, suggesting that these two conditions do not share a common pathophysiology. However, postsynaptic D2-receptor binding may be decreased indicating a possible dysfunction of D2-receptors or down-regulation due to increased levels of site occupancy by endogenous DA resulting from an increase in DA release. In addition, extrastriatal (i.e., thalamus and anterior cingulate cortex) as well as striatal brain areas seem to be involved in DA dysfunctions and these extrastriatal areas may subtend a possible pathway for sensory symptoms of RLS.
Iron Metabolism Abnormalities
Some recent studies implicated the cerebral metabolism of iron in the physiopathology of RLS-PLMS.125 Importantly, iron and dopaminergic systems are linked since iron is an important cofactor for tyrosine hydroxylase, the step-limiting enzyme in DA synthesis, and also plays a major role in the functioning of post-synaptic D2 receptors.126
A neuropathologic study (7 RLS brain and 5 normal brains) showed a marked decrease of H-ferritin (ferritin heavy chain) and iron staining in RLS substantia nigra.127 Using a special MRI measurement (R2*), Allen et al.128 found decreased regional iron concentrations in the substantia nigra and in the putamen of 5 patients with RLS (compared to 5 controls), both in proportion to RLS severity, consistent with regional iron insufficiency in RLS patients. In a more recent study, the same team found diminished iron concentration across 10 brain regions in early-onset RLS (beginning of RLS symptoms before 45 years, n = 22), but not in late-onset RLS (n = 19) when compared to controls (n = 39).129 These convergent observations suggest that RLS may be associated with impaired iron metabolism (i.e., impaired regulation of transferring receptors), which might indirectly affect the dopamine system as well.
Opiate System
Opioid receptor agonists, acting predominantly on the pain system, have been found to significantly improve RLS symptoms.130 Nevertheless, available data suggest that this effect may be mediated by dopamine and may thus not be related to specific deficiencies of the endogenous opioid system.131 Using PET and 11C-diprenorphine (a nonselective opioid receptor radioligand), von Spiczak et al.132 found no difference in opioid receptor binding between 15 RLS patients and 12 controls. However, in this study, negative regional correlations between ligand binding and RLS severity was found in the pain system (medial thalamus, amygdala, caudate nucleus, anterior cingulate gyrus, insula, and orbitofrontal cortex). Moreover, pain scores correlated inversely with endogenous opioids binding in orbitofrontal cortex and anterior cingulate gyrus. Therefore, the most likely interpretation of decreased opioid binding (i.e., availability) may be an increase in endogenous opioid release consecutive to pain or dysesthesia.132
Summary
PMLS/RLS still appears as a complex and multifarious movement disorder which implicates many brain areas and most probably a variety of pathophysiological mechanisms. However, the recent findings from anatomical, functional, and ligand neuroimaging studies converge to suggest a critical involvement of mostly subcortical regions (brainstem, thalamus, cerebellum) and of the dopamine system in the control and generation of leg movements. However, because presynaptic dopamine function is normal in PMLS/RLS disorders, the underlying pathophysiological mechanisms must differ from Parkinson disease. In addition, several observations suggest that RLS may be associated with impaired iron metabolism that will indirectly affect the dopamine system. Finally, consistent with PMLS/RLS being related to major somatosensory disturbances, abnormal dopamine and opioid activity was found in regions belonging to the medial pain system (e.g., thalamus, anterior cingulate, insula) which mediate the unpleasant component of pain.133 It is noteworthy that there are no data available during sleep itself, probably given methodological difficulties such as the unpredictability of occurrence of leg movements, as well as scanning artifact due to movements.
ABNORMAL MOTOR BEHAVIOR (2): REM SLEEP BEHAVIOR DISORDER
REM sleep behavior disorder (RDB) is characterized by brisk movements of the body associated with dream mentation that usually disturb sleep continuity.134 This parasomnia has a prevalence estimated at 0.5% of the population,135 mainly affecting men older than 50 years of age. During the night, patients behave as if they were acting out their dreams. The disease may be idiopathic (up to 60%) or associated with other neurological disorders. A sizeable proportion of patients with RBD will develop extrapyramidal disorders,136,137 Lewy body dementia,138 or multiple system atrophy.139,140 Critically, signs of RBD may often precede the clinical onset of neurodegenerative diseases such as Parkinson disease or Lewy body dementia.136,138 Any evidence for RBD should thus be considered with great care as this may have major clinical implications.
Structural and Functional Abnormalities
An early experimental model of RBD in the cat showed that lesions in the mesopontine tegmentum can lead to the disappearance of muscle atonia during REM sleep and the appearance of dream-enactment behavior.141 A magnetic resonance imaging (MRI) study confirmed this hypothesis in man, showing that 3 of 6 patients with RDB had lesions affecting the dorsal mesopontine tegmentum (Figure 6).142 A study combining anatomical MRI and 123I-IMP SPECT measurements during REM sleep in 20 patients with RBD reported significantly decreased blood flow in the pons and superior frontal regions in patients with RBD, in comparison with 7 normal elderly subjects.143 Decreased blood flow in the frontal lobe of patients with RBD did not correlate with frontal lobe atrophy. Another SPECT study in 8 RBD patients during waking rest confirmed decreased activity in frontal and temporoparietal cortices and found increased activity in the pons, putamen, and right hippocampus.144 Similarly, “acting out” of oneiric behaviors was associated with significantly lower cerebral metabolic rate for glucose in a set of brain areas (parietal, temporal, and cingulate cortices) in both Alzheimer and dementia with Lewy body disease.145
Figure 6.

(a) Brain lesions in RDB. Patients with RDB have lesions affecting the dorsal mesopontine tegmentum.142 (b) Metabolic changes during REM sleep, patients with RBD show decreased blood flow (SPECT) in the pons and superior frontal regions,143 and increased activity in the pons, putamen and hippocampus during rest at wake.144 1 = mesopontine tegmentum, 2 = pons, 3 = superior frontal lobe, 4 = pons, 5 = right hippocampus.
Increased Cho/Cr ratio in the brainstem suggesting local neural dysfunction was revealed by proton magnetic resonance spectroscopy (1H-MRS) in a 69-year-old man with idiopathic RBD.146 However, another 1H-MRS study in a large group of patients with idiopathic RBD (n = 15) compared to matched controls (n = 15) did not reveal any difference in NAA/Cr, Cho/Cr, and myoinositol/Cr ratios in the pontine tegmentum and the midbrain.147 Mesopontine neuronal loss or 1H-MRS detectable metabolic disturbances in idiopathic RBD therefore remain hypothetical. Future 1H-MRS may provide a noninvasive metabolic evaluation of brainstem neuronal function in RBD and may usefully contribute to the differentiation of secondary RBD with neurodegenerative disorders from idiopathic disorders.148
Neurotransmission Abnormalities
Using SPECT, the binding of ligands of striatal presynaptic dopamine transporters in RBD patients (n = 5) during wakefulness was found to be lower than in normal controls but higher than in Parkinson patients (n = 14).149,150 This result suggests that the number of presynaptic dopamine transporters decreases in both Parkinson and RBD patients. Moreover, such findings provide evidence for a continuum of striatal presynaptic dopaminergic dysfunction in patients with subclinical RBD (i.e., individuals who have REM sleep without atonia but without behavioral manifestations), clinical RBD, and Parkinson disease.150 No such differences were found for a marker of postsynaptic D2 receptor density in RDB patients.149
The same conclusions were reached by another study that probed the density of striatal dopaminergic terminals using PET and 11C-dihydrotetrabenazine (11C-DTBZ, an in vivo marker for dopamine nerve terminals) in 6 elderly subjects with chronic idiopathic RBD compared to 19 age-matched controls.151 Significant reductions in striatal 11C-DTBZ binding were found in all striatal nuclei, with the greatest reduction in the posterior putamen. The striatal vesicular monoamine transporter density is actually considered to be a direct function of the number of DA neurons in the substantia nigra, therefore suggesting a loss of DA midbrain neurons in chronic RBD.
Recently, one SPECT study using a radiomarker of the presynaptic dopamine transporter in 11 RBD patients (mostly with narcolepsy) and controls revealed that 2 “idiopathic” RBD patients with severe olfactory dysfunction (anosmia) had degeneration of presynaptic nigrostriatal neurons, as determined by reduced dopamine transporter binding.152 The authors suggested that the discrepancies between studies may be related to RBD symptoms duration, since patients who had reduced (2 patients) or pathologically asymmetrical (1 patient with mild hyposmia) dopamine transporter binding had by far the longest duration of RBD symptoms (14, 16, and 38 years).
It remains to be shown whether these alterations (mainly dysfunction in mesopontine tegmentum and DA neurotransmission impairments) play a causal role in the pathophysiology of RBD, or reflect functional consequences or adaptations to the pathological condition. Although there is evidence that some Parkinson patients do show excessive nocturnal movements,119,153 only a small percentage of Parkinson patients develop full-blown RBD. RBD has been found to also occur in patients with voltage-gated potassium channel antibody-associated limbic encephalitis.154 These observations suggest that modifications involving other systems of neurotransmission and/or other regions (e.g., frontal lobe, limbic system) are probably necessary for full-blown RBD to occur.
Summary
Dream-enactment behavior during REM sleep, which characterizes RBD, may in some cases be idiopathic but is predominantly associated with neurodegenerative diseases. Results from proton magnetic resonance spectroscopy may usefully contribute to the differentiation of idiopathic versus secondary RBD associated with neurodegenerative disorders. Neuroimaging studies have revealed that RDB may affect several levels of cerebral organization, from neurotransmission (presynaptic striatal DA) to neuroanatomical integrity (lesions in mesopontine tegmentum) and brain function (frontal, temporoparietal and cingulate cortex dysfunctions). Whether these cerebral anomalies play a causal role in the pathophysiology of RBD or mainly reflect pharmacological consequences or adaptations to the pathological condition is still unclear.
CONCLUSIONS
Modern functional neuroimaging techniques provide unprecedented possibilities to explore brain functions during normal and pathological sleep. This review demonstrates that a functional brain imaging approach may address a wide range of issues pertaining to the treatment and underlying pathophysiology of sleep disorders.
One line of research aims to characterize the neural consequences of sleep disruption due to intrinsic sleep disorders or to extrinsic environmental or medical causes. Functional neuroimaging can also be used to assess the effects of hypnotic drugs on regional brain function. A second, more fundamental challenge for a neuroimaging approach is to determine to what extent cerebral dysfunctions at wake as well as during sleep contribute to the primary physiological mechanisms of sleep disorders.
Although this review shows that neuroimaging can be used to achieve these goals, the approach is hampered by several factors: (a) Scanning patients or controls during their sleep or during pathological manifestations requires specialized equipment (e.g., EEG) and many adjustments in the scanning parameters (especially for fMRI studies). In addition, it is never guaranteed that the participant will sleep during data acquisition. Clinical manifestations of sleep disorders are often unpredictable and transient (e.g., sleepwalking, RBD); thus one cannot predict if the pathological event will occur during the scanning period. Moreover, clinical manifestations are often associated with large body movements that may interfere with image acquisition and create artifacts. In this respect, SPECT is probably the most appropriate procedure because the radiotracer can be administered during the clinical events, well before the brain images are acquired. An excellent example of such a study pertains to sleepwalking.155 (b) Assessing short- or long-term effects of treatments remains complicated, since neuroimaging data collected at different time points cannot be straightforwardly compared because of unavoidable fluctuations in signal (e.g., fMRI), and more importantly because treatments may have nonspecific influences across a distributed network of areas (e.g., dopamine system). Moreover, as suggested by the present review, it is difficult to find large and homogeneous groups of patients. (c) Last but not least, the theoretical framework necessary for designing an efficient clinical neuroimaging protocol is only available for a few sleep disorders. One such example is narcolepsy; the discovery of the hypocretin system and its role in narcolepsy in the late 90s156 has indubitably changed the main goal and target systems (and thus the experimental designs) of neuroimaging studies in narcoleptic patients.39,50
Neuroimaging studies may usefully contribute to the establishment or refinement of a nosography of sleep disorders. For instance, neuroimaging could help classify different subtypes of insomnia in terms of their underlying characteristic patterns of regional brain activity, an approach that may prove complementary to standard clinical observation.
Although substantial methodological progress has been achieved, much effort is still needed to better characterize neurophysiological mechanisms involved in sleep disorders, especially in teasing apart those mechanisms which have a causal role in the pathophysiology from those which are secondary consequences. Future brain imaging will undoubtedly provide new and valuable information about the functional and structural consequences of long-term sleep disruption. Ultimately, neuroimaging will help with clinical diagnosis and prognosis of sleep pathologies at an individual level. These considerations argue for closer collaboration and partnership between basic and imaging neuroscientists, sleep researchers, and sleep clinicians for designing and conducting multimodal assessments of sleep disorders.
ACKNOWLEDGMENTS
This research was funded by the Swiss National Science Foundation (grants #310000-114008, #3200B0-104100 to S.S.). M.D., T.D., V.S. and P.M. are supported by the Fonds National de la Recherche Scientifique (FNRS) (Belgium). Additional support for the work presented here comes from the University of Liege, the Queen Elisabeth Medical Foundation and an Austrian Science Fund Erwin-Schrïodinger Fellowship J2470-B02 (to M. S.).
Footnotes
Disclosure Statement
This was not an industry supported study. The authors have indicated no financial conflicts of interest.
REFERENCES
- 1.Cortoos A, Verstraeten E, Cluydts R. Neurophysiological aspects of primary insomnia: implications for its treatment. Sleep Med Rev. 2006;10:255–66. doi: 10.1016/j.smrv.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 2.Ohayon MM. Epidemiology of insomnia: what we know and what we still need to learn. Sleep Med Rev. 2002;6:97–111. doi: 10.1053/smrv.2002.0186. [DOI] [PubMed] [Google Scholar]
- 3.Tsuno N, Besset A, Ritchie K. Sleep and depression. J Clin Psychiatry. 2005;66:1254–9. doi: 10.4088/jcp.v66n1008. [DOI] [PubMed] [Google Scholar]
- 4.American Academy of Sleep Medicine. diagnostic and coding manual. 2nd ed. Westchester, IL: American Academy of Sleep Medicine; 2005. International classification of sleep disorders. [Google Scholar]
- 5.Bonnet MH, Arand DL. Hyperarousal and insomnia. Sleep Med Rev. 1997;1:97–108. doi: 10.1016/s1087-0792(97)90012-5. [DOI] [PubMed] [Google Scholar]
- 6.Perlis ML, Merica H, Smith MT, Giles DE. Beta EEG activity and insomnia. Sleep Med Rev. 2001;5:365–76. doi: 10.1053/smrv.2001.0151. [DOI] [PubMed] [Google Scholar]
- 7.Nofzinger EA, Buysse DJ, Germain A, et al. Functional neuroimaging evidence for hyperarousal in insomnia. Am J Psychiatry. 2004;161:2126–8. doi: 10.1176/appi.ajp.161.11.2126. [DOI] [PubMed] [Google Scholar]
- 8.Drummond SP, Brown GG. The effects of total sleep deprivation on cerebral responses to cognitive performance. Neuropsychopharmacology. 2001;25:S68–73. doi: 10.1016/S0893-133X(01)00325-6. [DOI] [PubMed] [Google Scholar]
- 9.Durmer JS, Dinges DF. Neurocognitive consequences of sleep deprivation. Semin Neurol. 2005;25:117–29. doi: 10.1055/s-2005-867080. [DOI] [PubMed] [Google Scholar]
- 10.Thomas M, Sing H, Belenky G, et al. Neural basis of alertness and cognitive performance impairments during sleepiness. I. Effects of 24 h of sleep deprivation on waking human regional brain activity. J Sleep Res. 2000;9:335–52. doi: 10.1046/j.1365-2869.2000.00225.x. [DOI] [PubMed] [Google Scholar]
- 11.Smith MT, Perlis ML, Chengazi VU, et al. Neuroimaging of NREM sleep in primary insomnia: a Tc-99-HMPAO single photon emission computed tomography study. Sleep. 2002;25:325–35. [PubMed] [Google Scholar]
- 12.Smith MT, Perlis ML, Chengazi VU, et al. NREM sleep cerebral blood flow before and after behavior therapy for chronic primary insomnia: preliminary single photon emission computed tomography (SPECT) data. Sleep Med. 2005;6:93–4. doi: 10.1016/j.sleep.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 13.Benca RM. Mood disorders. In: Kryger MH, Roth T, Dement WC, editors. Principles and practice of sleep medicine. 4th ed. Elsevier Saunders; 2005. pp. 1311–26. [Google Scholar]
- 14.Schroder CM, O'Hara R. Depression and obstructive sleep apnea (OSA) Ann Gen Psychiatry. 2005;4:13–20. doi: 10.1186/1744-859X-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roth T, Roehrs T, Pies R. Insomnia: pathophysiology and implications for treatment. Sleep Med Rev. 2007;11:71–9. doi: 10.1016/j.smrv.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 16.Thase ME. Depression, sleep, and antidepressants. J Clin Psychiatry. 1998;59(Suppl 4):55–65. [PubMed] [Google Scholar]
- 17.Gillin JC, Buchsbaum M, Wu J, et al. Sleep deprivation as a model experimental antidepressant treatment: findings from functional brain imaging. Depress Anxiety. 2001;14:37–49. doi: 10.1002/da.1045. [DOI] [PubMed] [Google Scholar]
- 18.Milak MS, Parsey RV, Keilp J, et al. Neuroanatomic correlates of psychopathologic components of major depressive disorder. Arch Gen Psychiatry. 2005;62:397–408. doi: 10.1001/archpsyc.62.4.397. [DOI] [PubMed] [Google Scholar]
- 19.Clark CP, Brown GG, Archibald SL, et al. Does amygdalar perfusion correlate with antidepressant response to partial sleep deprivation in major depression? Psychiatry Res. 2006;146:43–51. doi: 10.1016/j.pscychresns.2005.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lustberg L, Reynolds CF. Depression and insomnia: questions of cause and effect. Sleep Med Rev. 2000;4:253–62. doi: 10.1053/smrv.1999.0075. [DOI] [PubMed] [Google Scholar]
- 21.Ho AP, Gillin JC, Buchsbaum MS, et al. Brain glucose metabolism during non-rapid eye movement sleep in major depression. A positron emission tomography study. Arch Gen Psychiatry. 1996;53:645–52. doi: 10.1001/archpsyc.1996.01830070095014. [DOI] [PubMed] [Google Scholar]
- 22.Drevets WC, Price JL, Simpson JR, Jr, et al. Subgenual prefrontal cortex abnormalities in mood disorders. Nature. 1997;386:824–7. doi: 10.1038/386824a0. [DOI] [PubMed] [Google Scholar]
- 23.Nofzinger EA, Buysse DJ, Germain A, et al. Increased activation of anterior paralimbic and executive cortex from waking to rapid eye movement sleep in depression. Arch Gen Psychiatry. 2004;61:695–702. doi: 10.1001/archpsyc.61.7.695. [DOI] [PubMed] [Google Scholar]
- 24.Germain A, Nofzinger EA, Kupfer DJ, Buysse DJ. Neurobiology of non-REM sleep in depression: further evidence for hypofrontality and thalamic dysregulation. Am J Psychiatry. 2004;161:1856–63. doi: 10.1176/ajp.161.10.1856. [DOI] [PubMed] [Google Scholar]
- 25.Buysse DJ, Tu XM, Cherry CR, et al. Pretreatment REM sleep and subjective sleep quality distinguish depressed psychotherapy remitters and nonremitters. Biol Psychiatry. 1999;45:205–13. doi: 10.1016/s0006-3223(98)00198-x. [DOI] [PubMed] [Google Scholar]
- 26.Peigneux P, Laureys S, Fuchs S, et al. Generation of rapid eye movements during paradoxical sleep in humans. Neuroimage. 2001;14:701–8. doi: 10.1006/nimg.2001.0874. [DOI] [PubMed] [Google Scholar]
- 27.Wehrle R, Czisch M, Kaufmann C, et al. Rapid eye movement-related brain activation in human sleep: a functional magnetic resonance imaging study. Neuroreport. 2005;16:853–7. doi: 10.1097/00001756-200505310-00015. [DOI] [PubMed] [Google Scholar]
- 28.Germain A, Buysse DJ, Wood A, Nofzinger E. Functional neuroanatomical correlates of eye movements during rapid eye movement sleep in depressed patients. Psychiatry Res. 2004;130:259–68. doi: 10.1016/j.pscychresns.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 29.Vuilleumier P, Driver J. Modulation of visual processing by attention and emotion: windows on causal interactions between human brain regions. Philos Trans R Soc Lond B Biol Sci. 2007;362:837–55. doi: 10.1098/rstb.2007.2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Braun AR, Balkin TJ, Wesensten NJ, et al. Dissociated pattern of activity in visual cortices and their projections during human rapid eye movement sleep. Science. 1998;279:91–5. doi: 10.1126/science.279.5347.91. [DOI] [PubMed] [Google Scholar]
- 31.Mignot E, Lammers GJ, Ripley B, et al. The role of cerebrospinal fluid hypocretin measurement in the diagnosis of narcolepsy and other hypersomnias. Arch Neurol. 2002;59:1553–62. doi: 10.1001/archneur.59.10.1553. [DOI] [PubMed] [Google Scholar]
- 32.Baumann CR, Bassetti CL. Hypocretins (orexins): clinical impact of the discovery of a neurotransmitter. Sleep Med Rev. 2005;9:253–68. doi: 10.1016/j.smrv.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 33.Plazzi G, Montagna P, Provini F, et al. Pontine lesions in idiopathic narcolepsy. Neurology. 1996;46:1250–4. doi: 10.1212/wnl.46.5.1250. [DOI] [PubMed] [Google Scholar]
- 34.Bassetti C, Aldrich MS, Quint DJ. MRI findings in narcolepsy. Sleep. 1997;20:630–1. doi: 10.1093/sleep/20.8.630. [DOI] [PubMed] [Google Scholar]
- 35.Frey JL, Heiserman JE. Absence of pontine lesions in narcolepsy. Neurology. 1997;48:1097–9. doi: 10.1212/wnl.48.4.1097. [DOI] [PubMed] [Google Scholar]
- 36.Overeem S, Steens SC, Good CD, et al. Voxel-based morphometry in hypocretin-deficient narcolepsy. Sleep. 2003;26:44–6. [PubMed] [Google Scholar]
- 37.Brenneis C, Brandauer E, Frauscher B, et al. Voxel-based morphometry in narcolepsy. Sleep Med. 2005;6:531–6. doi: 10.1016/j.sleep.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 38.Kaufmann C, Schuld A, Pollmacher T, Auer DP. Reduced cortical gray matter in narcolepsy: preliminary findings with voxel-based morphometry. Neurology. 2002;58:1852–5. doi: 10.1212/wnl.58.12.1852. [DOI] [PubMed] [Google Scholar]
- 39.Draganski B, Geisler P, Hajak G, et al. Hypothalamic gray matter changes in narcoleptic patients. Nat Med. 2002;8:1186–8. doi: 10.1038/nm1102-1186. [DOI] [PubMed] [Google Scholar]
- 40.Buskova J, Vaneckova M, Sonka K, et al. Reduced hypothalamic gray matter in narcolepsy with cataplexy. Neuro Endocrinol Lett. 2006;27:769–72. [PubMed] [Google Scholar]
- 41.Lodi R, Tonon C, Vignatelli L, et al. In vivo evidence of neuronal loss in the hypothalamus of narcoleptic patients. Neurology. 2004;63:1513–15. doi: 10.1212/01.wnl.0000142259.94107.4c. [DOI] [PubMed] [Google Scholar]
- 42.Ellis CM, Simmons A, Lemmens G, et al. Proton spectroscopy in the narcoleptic syndrome. Is there evidence of a brainstem lesion? Neurology. 1998;50:S23–6. doi: 10.1212/wnl.50.2_suppl_1.s23. [DOI] [PubMed] [Google Scholar]
- 43.Rudkin TM, Arnold DL. Proton magnetic resonance spectroscopy for the diagnosis and management of cerebral disorders. Arch Neurol. 1999;56:919–26. doi: 10.1001/archneur.56.8.919. [DOI] [PubMed] [Google Scholar]
- 44.De Stefano N, Matthews PM, Arnold DL. Reversible decreases in N-acetylaspartate after acute brain injury. Magn Reson Med. 1995;34:721–7. doi: 10.1002/mrm.1910340511. [DOI] [PubMed] [Google Scholar]
- 45.Joo EY, Tae WS, Kim JH, et al. Glucose hypometabolism of hypothalamus and thalamus in narcolepsy. Ann Neurol. 2004;56:437–40. doi: 10.1002/ana.20212. [DOI] [PubMed] [Google Scholar]
- 46.Joo EY, Hong SB, Tae WS, et al. Cerebral perfusion abnormality in narcolepsy with cataplexy. Neuroimage. 2005;28:410–16. doi: 10.1016/j.neuroimage.2005.06.019. [DOI] [PubMed] [Google Scholar]
- 47.Meyer JS, Sakai F, Karacan I, et al. Sleep apnea, narcolepsy, and dreaming: regional cerebral hemodynamics. Ann Neurol. 1980;7:479–85. doi: 10.1002/ana.410070515. [DOI] [PubMed] [Google Scholar]
- 48.Asenbaum S, Zeithofer J, Saletu B, et al. Technetium-99m-HMPAO SPECT imaging of cerebral blood flow during REM sleep in narcoleptics. J Nucl Med. 1995;36:1150–5. [PubMed] [Google Scholar]
- 49.Ellis CM, Monk C, Simmons A, et al. Functional magnetic resonance imaging neuroactivation studies in normal subjects and subjects with the narcoleptic syndrome. Actions of modafinil. J Sleep Res. 1999;8:85–93. doi: 10.1046/j.1365-2869.1999.00142.x. [DOI] [PubMed] [Google Scholar]
- 50.Schwartz S, Ponz A, Poryazova R, et al. Humor processing in narcoleptic patient assessed by functional MRI. Brain. 2008;131:514–22. doi: 10.1093/brain/awm292. [DOI] [PubMed] [Google Scholar]
- 51.Hong SB, Tae WS, Joo EY. Cerebral perfusion changes during cataplexy in narcolepsy patients. Neurology. 2006;66:1747–9. doi: 10.1212/01.wnl.0000218205.72668.ab. [DOI] [PubMed] [Google Scholar]
- 52.Chabas D, Habert MO, Maksud P, et al. Functional imaging of cataplexy during status cataplecticus. Sleep. 2007;30:153–6. doi: 10.1093/sleep/30.2.153. [DOI] [PubMed] [Google Scholar]
- 53.Sudo Y, Suhara T, Honda Y, et al. Muscarinic cholinergic receptors in human narcolepsy: a PET study. Neurology. 1998;51:1297–302. doi: 10.1212/wnl.51.5.1297. [DOI] [PubMed] [Google Scholar]
- 54.Derry C, Benjamin C, Bladin P, et al. Increased serotonin receptor availability in human sleep: evidence from an [18F]MPPF PET study in narcolepsy. Neuroimage. 2006;30:341–48. doi: 10.1016/j.neuroimage.2005.09.052. [DOI] [PubMed] [Google Scholar]
- 55.Portas CM, Bjorvatn B, Fagerland S, et al. On-line detection of extracellular levels of serotonin in dorsal raphe nucleus and frontal cortex over the sleep/wake cycle in the freely moving rat. Neuroscience. 1998;83:807–14. doi: 10.1016/s0306-4522(97)00438-7. [DOI] [PubMed] [Google Scholar]
- 56.Aldrich MS, Hollingsworth Z, Penney JB. Dopamine-receptor autoradiography of human narcoleptic brain. Neurology. 1992;42:410–15. doi: 10.1212/wnl.42.2.410. [DOI] [PubMed] [Google Scholar]
- 57.Kish SJ, Mamelak M, Slimovitch C, et al. Brain neurotransmitter changes in human narcolepsy. Neurology. 1992;42:229–34. doi: 10.1212/wnl.42.1.229. [DOI] [PubMed] [Google Scholar]
- 58.Eisensehr I, Linke R, Tatsch K, et al. Alteration of the striatal dopaminergic system in human narcolepsy. Neurology. 2003;60:1817–19. doi: 10.1212/01.wnl.0000069608.84542.46. [DOI] [PubMed] [Google Scholar]
- 59.MacFarlane JG, List SJ, Moldofsky H, et al. Dopamine D2 receptors quantified in vivo in human narcolepsy. Biol Psychiatry. 1997;41:305–10. doi: 10.1016/s0006-3223(96)00003-0. [DOI] [PubMed] [Google Scholar]
- 60.Rinne JO, Hublin C, Partinen M, et al. Positron emission tomography study of human narcolepsy: no increase in striatal dopamine D2 receptors. Neurology. 1995;45:1735–8. doi: 10.1212/wnl.45.9.1735. [DOI] [PubMed] [Google Scholar]
- 61.Hublin C, Launes J, Nikkinen P, Partinen M. Dopamine D2-receptors in human narcolepsy: a SPECT study with 123I-IBZM. Acta Neurol Scand. 1994;90:186–9. doi: 10.1111/j.1600-0404.1994.tb02703.x. [DOI] [PubMed] [Google Scholar]
- 62.Staedt J, Stoppe G, Kogler A, et al. [123I]IBZM SPET analysis of dopamine D2 receptor occupancy in narcoleptic patients in the course of treatment. Biol Psychiatry. 1996;39:107–11. doi: 10.1016/0006-3223(95)00087-9. [DOI] [PubMed] [Google Scholar]
- 63.Rinne JO, Hublin C, Nagren K, et al. Unchanged striatal dopamine transporter availability in narcolepsy: a PET study with [11C]-CFT. Acta Neurol Scand. 2004;109:52–5. doi: 10.1034/j.1600-0404.2003.00175.x. [DOI] [PubMed] [Google Scholar]
- 64.Madras BK, Xie Z, Lin Z, et al. Modafinil occupies dopamine and norepinephrine transporters in vivo and modulates the transporters and trace amine activity in vitro. J Pharmacol Exp Ther. 2006;319:561–9. doi: 10.1124/jpet.106.106583. [DOI] [PubMed] [Google Scholar]
- 65.Howard RJ, Ellis C, Bullmore ET, et al. Functional echoplanar brain imaging correlates of amphetamine administration to normal subjects and subjects with the narcoleptic syndrome. Magn Reson Imaging. 1996;14:1013–6. doi: 10.1016/s0730-725x(96)00238-x. [DOI] [PubMed] [Google Scholar]
- 66.Pascual-Marqui RD, Michel CM, Lehmann D. Low resolution electromagnetic tomography: a new method for localizing electrical activity in the brain. Int J Psychophysiol. 1994;18:49–65. doi: 10.1016/0167-8760(84)90014-x. [DOI] [PubMed] [Google Scholar]
- 67.Saletu M, Anderer P, Semlitsch HV, et al. Low-resolution brain electromagnetic tomography (LORETA) identifies brain regions linked to psychometric performance under modafinil in narcolepsy. Psychiatry Res. 2007;154:69–84. doi: 10.1016/j.pscychresns.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 68.Thomas RJ, Kwong K. Modafinil activates cortical and subcortical sites in the sleep-deprived state. Sleep. 2006;29:1471–81. doi: 10.1093/sleep/29.11.1471. [DOI] [PubMed] [Google Scholar]
- 69.Young T, Palta M, Dempsey J, et al. The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med. 1993;328:1230–5. doi: 10.1056/NEJM199304293281704. [DOI] [PubMed] [Google Scholar]
- 70.Duran J, Esnaola S, Rubio R, Iztueta A. Obstructive sleep apnea-hypopnea and related clinical features in a population-based sample of subjects aged 30 to 70 yr. Am J Respir Crit Care Med. 2001;163:685–9. doi: 10.1164/ajrccm.163.3.2005065. [DOI] [PubMed] [Google Scholar]
- 71.Arens R, Marcus CL. Pathophysiology of upper airway obstruction: a developmental perspective. Sleep. 2004;27:997–1019. doi: 10.1093/sleep/27.5.997. [DOI] [PubMed] [Google Scholar]
- 72.Mateika JH, Ellythy M. Chemoreflex control of ventilation is altered during wakefulness in humans with OSA. Respir Physiol Neurobiol. 2003;138:45–57. doi: 10.1016/s1569-9048(03)00174-5. [DOI] [PubMed] [Google Scholar]
- 73.Naegele B, Thouvard V, Pepin JL, et al. Deficits of cognitive executive functions in patients with sleep apnea syndrome. Sleep. 1995;18:43–52. [PubMed] [Google Scholar]
- 74.Beebe DW, Groesz L, Wells C, et al. The neuropsychological effects of obstructive sleep apnea: a meta-analysis of norm-referenced and case-controlled data. Sleep. 2003;26:298–307. doi: 10.1093/sleep/26.3.298. [DOI] [PubMed] [Google Scholar]
- 75.Alchanatis M, Zias N, Deligiorgis N, et al. Sleep apnea-related cognitive deficits and intelligence: an implication of cognitive reserve theory. J Sleep Res. 2005;14:69–75. doi: 10.1111/j.1365-2869.2004.00436.x. [DOI] [PubMed] [Google Scholar]
- 76.Aloia MS, Arnedt JT, Davis JD, et al. Neuropsychological sequelae of obstructive sleep apnea-hypopnea syndrome: a critical review. J Int Neuropsychol Soc. 2004;10:772–85. doi: 10.1017/S1355617704105134. [DOI] [PubMed] [Google Scholar]
- 77.Brown WD. The psychosocial aspects of obstructive sleep apnea. Semin Respir Crit Care Med. 2005;26:33–43. doi: 10.1055/s-2005-864199. [DOI] [PubMed] [Google Scholar]
- 78.Verstraeten E, Cluydts R, Pevernagie D, Hoffmann G. Executive function in sleep apnea: controlling for attentional capacity in assessing executive attention. Sleep. 2004;27:685–93. [PubMed] [Google Scholar]
- 79.Macey PM, Henderson LA, Macey KE, et al. Brain morphology associated with obstructive sleep apnea. Am J Respir Crit Care Med. 2002;166:1382–7. doi: 10.1164/rccm.200201-050OC. [DOI] [PubMed] [Google Scholar]
- 80.Morrell MJ, McRobbie DW, Quest RA, et al. Changes in brain morphology associated with obstructive sleep apnea. Sleep Med. 2003;4:451–4. doi: 10.1016/s1389-9457(03)00159-x. [DOI] [PubMed] [Google Scholar]
- 81.O'Donoghue FJ, Briellmann RS, Rochford PD, et al. Cerebral structural changes in severe obstructive sleep apnea. Am J Respir Crit Care Med. 2005;171:1185–90. doi: 10.1164/rccm.200406-738OC. [DOI] [PubMed] [Google Scholar]
- 82.Gale SD, Hopkins RO. Effects of hypoxia on the brain: neuroimaging and neuropsychological findings following carbon monoxide poisoning and obstructive sleep apnea. J Int Neuropsychol Soc. 2004;10:60–71. doi: 10.1017/S1355617704101082. [DOI] [PubMed] [Google Scholar]
- 83.Kamba M, Suto Y, Ohta Y, et al. Cerebral metabolism in sleep apnea. Evaluation by magnetic resonance spectroscopy. Am J Respir Crit Care Med. 1997;156:296–8. doi: 10.1164/ajrccm.156.1.9611063. [DOI] [PubMed] [Google Scholar]
- 84.Alchanatis M, Deligiorgis N, Zias N, et al. Frontal brain lobe impairment in obstructive sleep apnoea: a proton MR spectroscopy study. Eur Respir J. 2004;24:980–6. doi: 10.1183/09031936.04.00127603. [DOI] [PubMed] [Google Scholar]
- 85.Bartlett DJ, Rae C, Thompson CH, et al. Hippocampal area metabolites relate to severity and cognitive function in obstructive sleep apnea. Sleep Med. 2004;5:593–6. doi: 10.1016/j.sleep.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 86.Halbower AC, Degaonkar M, Barker PB, et al. Childhood obstructive sleep apnea associates with neuropsychological deficits and neuronal brain injury. PLoS Med. 2006;3:e301. doi: 10.1371/journal.pmed.0030301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Thomas RJ, Rosen BR, Stern CE, Weiss JW, Kwong KK. Functional imaging of working memory in obstructive sleep-disordered breathing. J Appl Physiol. 2005;98:2236–34. doi: 10.1152/japplphysiol.01225.2004. [DOI] [PubMed] [Google Scholar]
- 88.Ayalon L, Ancoli-Israel S, Klemfuss Z, Shalauta MD, Drummond SP. Increased brain activation during verbal learning in obstructive sleep apnea. Neuroimage. 2006;31:1817–25. doi: 10.1016/j.neuroimage.2006.02.042. [DOI] [PubMed] [Google Scholar]
- 89.Drummond SP, Brown GG, Gillin JC, et al. Altered brain response to verbal learning following sleep deprivation. Nature. 2000;403:655–7. doi: 10.1038/35001068. [DOI] [PubMed] [Google Scholar]
- 90.Beebe DW, Gozal D. Obstructive sleep apnea and the prefrontal cortex: towards a comprehensive model linking nocturnal upper airway obstruction to daytime cognitive and behavioral deficits. J Sleep Res. 2002;11:1–16. doi: 10.1046/j.1365-2869.2002.00289.x. [DOI] [PubMed] [Google Scholar]
- 91.Mayberg HS. Limbic-cortical dysregulation: a proposed model of depression. J Neuropsychiatry Clin Neurosci. 1997;9:471–81. doi: 10.1176/jnp.9.3.471. [DOI] [PubMed] [Google Scholar]
- 92.Engleman HM, Martin SE, Deary IJ, Douglas NJ. Effect of CPAP therapy on daytime function in patients with mild sleep apnoea/hypopnoea syndrome. Thorax. 1997;52:114–19. doi: 10.1136/thx.52.2.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Harper RM, Macey PM, Henderson LA, et al. fMRI responses to cold pressor challenges in control and obstructive sleep apnea subjects. J Appl Physiol. 2003;94:1583–95. doi: 10.1152/japplphysiol.00881.2002. [DOI] [PubMed] [Google Scholar]
- 94.Henderson LA, Woo MA, Macey PM, et al. Neural responses during Valsalva maneuvers in obstructive sleep apnea syndrome. J Appl Physiol. 2003;94:1063–74. doi: 10.1152/japplphysiol.00702.2002. [DOI] [PubMed] [Google Scholar]
- 95.Macey PM, Macey KE, Henderson LA, et al. Functional magnetic resonance imaging responses to expiratory loading in obstructive sleep apnea. Respir Physiol Neurobiol. 2003;138:275–90. doi: 10.1016/j.resp.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 96.Macey KE, Macey PM, Woo MA, et al. Inspiratory loading elicits aberrant fMRI signal changes in obstructive sleep apnea. Respir Physiol Neurobiol. 2006;151:44–60. doi: 10.1016/j.resp.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 97.Henderson LA, Macey KE, Macey PM, et al. Regional brain response patterns to Cheyne-Stokes breathing. Respir Physiol Neurobiol. 2006;150:87–93. doi: 10.1016/j.resp.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 98.Ficker JH, Feistel H, Moller C, et al. [Changes in regional CNS perfusion in obstructive sleep apnea syndrome: initial SPECT studies with injected nocturnal 99mTc-HMPAO] Pneumologie. 1997;51:926–30. [PubMed] [Google Scholar]
- 99.Tonon C, Vetrugno R, Lodi R, et al. Proton magnetic resonance spectroscopy study of brain metabolism in obstructive sleep apnoea syndrome before and after continuous positive airway pressure treatment. Sleep. 2007;30:305–11. doi: 10.1093/sleep/30.3.305. [DOI] [PubMed] [Google Scholar]
- 100.Hashimoto K, Ono T, Honda E, et al. Effects of mandibular advancement on brain activation during inspiratory loading in healthy subjects: a functional magnetic resonance imaging study. J Appl Physiol. 2006;100:579–86. doi: 10.1152/japplphysiol.00169.2005. [DOI] [PubMed] [Google Scholar]
- 101.Allen RP, Picchietti D, Hening WA, et al. Restless legs syndrome: diagnostic criteria, special considerations, and epidemiology. A report from the restless legs syndrome diagnosis and epidemiology workshop at the National Institutes of Health. Sleep Med. 2003;4:101–19. doi: 10.1016/s1389-9457(03)00010-8. [DOI] [PubMed] [Google Scholar]
- 102.Ohayon MM, Roth T. Prevalence of restless legs syndrome and periodic limb movement disorder in the general population. J Psychosom Res. 2002;53:547–54. doi: 10.1016/s0022-3999(02)00443-9. [DOI] [PubMed] [Google Scholar]
- 103.Pennestri MH, Whittom S, Adam B, et al. PLMS and PLMW in healthy subjects as a function of age: prevalence and interval distribution. Sleep. 2006;29:1183–7. doi: 10.1093/sleep/29.9.1183. [DOI] [PubMed] [Google Scholar]
- 104.Neckelmann D, Mykletun A, Dahl AA. Chronic insomnia as a risk factor for developing anxiety and depression. Sleep. 2007;30:873–80. doi: 10.1093/sleep/30.7.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Saletu B, Anderer P, Saletu M, et al. EEG mapping, psychometric, and polysomnographic studies in restless legs syndrome (RLS) and periodic limb movement disorder (PLMD) patients as compared with normal controls. Sleep Med. 2002;3(Suppl):S35–42. doi: 10.1016/s1389-9457(02)00147-8. [DOI] [PubMed] [Google Scholar]
- 106.Picchietti D, Winkelman JW. Restless legs syndrome, periodic limb movements in sleep, and depression. Sleep. 2005;28:891–8. [PubMed] [Google Scholar]
- 107.Stefansson H, Rye DB, Hicks A, et al. A genetic risk factor for periodic limb movements in sleep. N Engl J Med. 2007;357:639–47. doi: 10.1056/NEJMoa072743. [DOI] [PubMed] [Google Scholar]
- 108.Winkelmann J, Schormair B, Lichtner P, et al. Genome-wide association study of restless legs syndrome identifies common variants in three genomic regions. Nat Genet. 2007;39:1000–6. doi: 10.1038/ng2099. [DOI] [PubMed] [Google Scholar]
- 109.Etgen T, Draganski B, Ilg C, et al. Bilateral thalamic gray matter changes in patients with restless legs syndrome. Neuroimage. 2005;24:1242–7. doi: 10.1016/j.neuroimage.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 110.Bucher SF, Seelos KC, Oertel WH, et al. Cerebral generators involved in the pathogenesis of the restless legs syndrome. Ann Neurol. 1997;41:639–45. doi: 10.1002/ana.410410513. [DOI] [PubMed] [Google Scholar]
- 111.Trenkwalder C, Bucher SF, Oertel WH, et al. Bereitschaftspotential in idiopathic and symptomatic restless legs syndrome. Electroencephalogr Clin Neurophysiol. 1993;89:95–103. doi: 10.1016/0168-5597(93)90090-c. [DOI] [PubMed] [Google Scholar]
- 112.Tergau F, Wischer S, Paulus W. Motor system excitability in patients with restless legs syndrome. Neurology. 1999;52:1060–3. doi: 10.1212/wnl.52.5.1060. [DOI] [PubMed] [Google Scholar]
- 113.Stiasny K, Oertel WH, Trenkwalder C. Clinical symptomatology and treatment of restless legs syndrome and periodic limb movement disorder. Sleep Med Rev. 2002;6:253–65. doi: 10.1053/smrv.2001.0193. [DOI] [PubMed] [Google Scholar]
- 114.Eisensehr I, Wetter TC, Linke R, et al. Normal IPT and IBZM SPECT in drug-naive and levodopa-treated idiopathic restless legs syndrome. Neurology. 2001;57:1307–9. doi: 10.1212/wnl.57.7.1307. [DOI] [PubMed] [Google Scholar]
- 115.Linke R, Eisensehr I, Wetter TC, et al. Presynaptic dopaminergic function in patients with restless legs syndrome: are there common features with early Parkinson's disease? Mov Disord. 2004;19:1158–62. doi: 10.1002/mds.20226. [DOI] [PubMed] [Google Scholar]
- 116.Michaud M, Soucy JP, Chabli A, et al. SPECT imaging of striatal pre- and postsynaptic dopaminergic status in restless legs syndrome with periodic leg movements in sleep. J Neurol. 2002;249:164–70. doi: 10.1007/pl00007859. [DOI] [PubMed] [Google Scholar]
- 117.Ruottinen HM, Partinen M, Hublin C, et al. An FDOPA PET study in patients with periodic limb movement disorder and restless legs syndrome. Neurology. 2000;54:502–4. doi: 10.1212/wnl.54.2.502. [DOI] [PubMed] [Google Scholar]
- 118.Turjanski N, Lees AJ, Brooks DJ. Striatal dopaminergic function in restless legs syndrome: 18F-dopa and 11C-raclopride PET studies. Neurology. 1999;52:932–7. doi: 10.1212/wnl.52.5.932. [DOI] [PubMed] [Google Scholar]
- 119.Happe S, Pirker W, Klosch G, et al. Periodic leg movements in patients with Parkinson's disease are associated with reduced striatal dopamine transporter binding. J Neurol. 2003;250:83–6. doi: 10.1007/s00415-003-0957-8. [DOI] [PubMed] [Google Scholar]
- 120.Tribl GG, Asenbaum S, Happe S, et al. Normal striatal D2 receptor binding in idiopathic restless legs syndrome with periodic leg movements in sleep. Nucl Med Commun. 2004;25:55–60. doi: 10.1097/00006231-200401000-00008. [DOI] [PubMed] [Google Scholar]
- 121.Staedt J, Stoppe G, Kogler A, et al. Dopamine D2 receptor alteration in patients with periodic movements in sleep (nocturnal myoclonus) J Neural Transm Gen Sect. 1993;93:71–4. doi: 10.1007/BF01244940. [DOI] [PubMed] [Google Scholar]
- 122.Staedt J, Stoppe G, Kogler A, et al. Single photon emission tomography (SPET) imaging of dopamine D2 receptors in the course of dopamine replacement therapy in patients with nocturnal myoclonus syndrome (NMS) J Neural Transm Gen Sect. 1995;99:187–93. doi: 10.1007/BF01271478. [DOI] [PubMed] [Google Scholar]
- 123.Staedt J, Stoppe G, Kogler A, et al. Nocturnal myoclonus syndrome (periodic movements in sleep) related to central dopamine D2-receptor alteration. Eur Arch Psychiatry Clin Neurosci. 1995;245:8–10. doi: 10.1007/BF02191538. [DOI] [PubMed] [Google Scholar]
- 124.Cervenka S, Palhagen SE, Comley RA, et al. Support for dopaminergic hypoactivity in restless legs syndrome: a PET study on D2-receptor binding. Brain. 2006;129:2017–28. doi: 10.1093/brain/awl163. [DOI] [PubMed] [Google Scholar]
- 125.Allen R. Dopamine and iron in the pathophysiology of restless legs syndrome (RLS) Sleep Med. 2004;5:385–91. doi: 10.1016/j.sleep.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 126.Kryger MH, Roth T, Dement WC. Principles and practice of sleep medicine. Fourth ed. W.B. Saunders Company; 2005. p. 1517. [Google Scholar]
- 127.Connor JR, Boyer PJ, Menzies SL, et al. Neuropathological examination suggests impaired brain iron acquisition in restless legs syndrome. Neurology. 2003;61:304–9. doi: 10.1212/01.wnl.0000078887.16593.12. [DOI] [PubMed] [Google Scholar]
- 128.Allen RP, Barker PB, Wehrl F, et al. MRI measurement of brain iron in patients with restless legs syndrome. Neurology. 2001;56:263–5. doi: 10.1212/wnl.56.2.263. [DOI] [PubMed] [Google Scholar]
- 129.Earley CJ, P BB, Horska A, Allen RP. MRI-determined regional brain iron concentrations in early- and late-onset restless legs syndrome. Sleep Med. 2006;7:458–61. doi: 10.1016/j.sleep.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 130.Walters AS. Review of receptor agonist and antagonist studies relevant to the opiate system in restless legs syndrome. Sleep Med. 2002;3:301–4. doi: 10.1016/s1389-9457(02)00011-4. [DOI] [PubMed] [Google Scholar]
- 131.Barriere G, Cazalets JR, Bioulac B, et al. The restless legs syndrome. Prog Neurobiol. 2005;77:139–65. doi: 10.1016/j.pneurobio.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 132.von Spiczak S, Whone AL, Hammers A, et al. The role of opioids in restless legs syndrome: an [11C]diprenorphine PET study. Brain. 2005;128:906–17. doi: 10.1093/brain/awh441. [DOI] [PubMed] [Google Scholar]
- 133.Sewards TV, Sewards MA. The medial pain system: neural representations of the motivational aspect of pain. Brain Res Bull. 2002;59:163–80. doi: 10.1016/s0361-9230(02)00864-x. [DOI] [PubMed] [Google Scholar]
- 134.Schenck CH, Bundlie SR, Ettinger MG, Mahowald MW. Chronic behavioral disorders of human REM sleep: a new category of parasomnia. Sleep. 1986;9:293–308. doi: 10.1093/sleep/9.2.293. [DOI] [PubMed] [Google Scholar]
- 135.Ohayon MM. Prevalence of DSM-IV diagnostic criteria of insomnia: distinguishing insomnia related to mental disorders from sleep disorders. J Psychiatr Res. 1997;31:333–46. doi: 10.1016/s0022-3956(97)00002-2. [DOI] [PubMed] [Google Scholar]
- 136.Schenck CH, Bundlie SR, Mahowald MW. Delayed emergence of a parkinsonian disorder in 38% of 29 older men initially diagnosed with idiopathic rapid eye movement sleep behaviour disorder. Neurology. 1996;46:388–93. doi: 10.1212/wnl.46.2.388. [DOI] [PubMed] [Google Scholar]
- 137.Gagnon JF, Bedard MA, Fantini ML, et al. REM sleep behavior disorder and REM sleep without atonia in Parkinson's disease. Neurology. 2002;59:585–9. doi: 10.1212/wnl.59.4.585. [DOI] [PubMed] [Google Scholar]
- 138.Fantini ML, Ferini-Strambi L, Montplaisir J. Idiopathic REM sleep behavior disorder: toward a better nosologic definition. Neurology. 2005;64:780–6. doi: 10.1212/01.WNL.0000152878.79429.00. [DOI] [PubMed] [Google Scholar]
- 139.Plazzi G, Corsini R, Provini F, et al. REM sleep behavior disorders in multiple system atrophy. Neurology. 1997;48:1094–7. doi: 10.1212/wnl.48.4.1094. [DOI] [PubMed] [Google Scholar]
- 140.Gilman S, Koeppe RA, Chervin RD, et al. REM sleep behavior disorder is related to striatal monoaminergic deficit in MSA. Neurology. 2003;61:29–34. doi: 10.1212/01.wnl.0000073745.68744.94. [DOI] [PubMed] [Google Scholar]
- 141.Sakai K, Sastre JP, Salvert D, et al. Tegmentoreticular projections with special reference to the muscular atonia during paradoxical sleep in the cat: an HRP study. Brain Res. 1979;176:233–54. doi: 10.1016/0006-8993(79)90981-8. [DOI] [PubMed] [Google Scholar]
- 142.Culebras A, Moore JT. Magnetic resonance findings in REM sleep behavior disorder. Neurology. 1989;39:1519–23. doi: 10.1212/wnl.39.11.1519. [DOI] [PubMed] [Google Scholar]
- 143.Shirakawa S, Takeuchi N, Uchimura N, et al. Study of image findings in rapid eye movement sleep behavioural disorder. Psychiatry Clin Neurosci. 2002;56:291–2. doi: 10.1046/j.1440-1819.2002.00961.x. [DOI] [PubMed] [Google Scholar]
- 144.Mazza S, Soucy JP, Gravel P, et al. Assessing whole brain perfusion changes in patients with REM sleep behavior disorder. Neurology. 2006;67:1618–22. doi: 10.1212/01.wnl.0000242879.39415.49. [DOI] [PubMed] [Google Scholar]
- 145.Caselli RJ, Chen K, Bandy D, et al. A preliminary fluorodeoxyglucose positron emission tomography study in healthy adults reporting dream-enactment behavior. Sleep. 2006;29:927–33. doi: 10.1093/sleep/29.7.927. [DOI] [PubMed] [Google Scholar]
- 146.Miyamoto M, Miyamoto T, Kubo J, et al. Brainstem function in rapid eye movement sleep behavior disorder: the evaluation of brainstem function by proton MR spectroscopy (1H-MRS) Psychiatry Clin Neurosci. 2000;54:350–1. doi: 10.1046/j.1440-1819.2000.00710.x. [DOI] [PubMed] [Google Scholar]
- 147.Iranzo A, Santamaria J, Pujol J, et al. Brainstem proton magnetic resonance spectroscopy in idopathic REM sleep behavior disorder. Sleep. 2002;25:867–70. [PubMed] [Google Scholar]
- 148.Vion-Dury J, Meyerhoff DJ, Cozzone PJ, Weiner MW. What might be the impact on neurology of the analysis of brain metabolism by in vivo magnetic resonance spectroscopy? J Neurol. 1994;241:354–71. doi: 10.1007/BF02033352. [DOI] [PubMed] [Google Scholar]
- 149.Eisensehr I, Linke R, Noachtar S, et al. Reduced striatal dopamine transporters in idiopathic rapid eye movement sleep behaviour disorder. Comparison with Parkinson's disease and controls. Brain. 2000;123((Pt 6)):1155–60. doi: 10.1093/brain/123.6.1155. [DOI] [PubMed] [Google Scholar]
- 150.Eisensehr I, Linke R, Tatsch K, et al. Increased muscle activity during rapid eye movement sleep correlates with decrease of striatal presynaptic dopamine transporters. IPT and IBZM SPECT imaging in subclinical and clinically manifest idiopathic REM sleep behavior disorder, Parkinson's disease, and controls. Sleep. 2003;26:507–12. doi: 10.1093/sleep/26.5.507. [DOI] [PubMed] [Google Scholar]
- 151.Albin RL, Koeppe RA, Chervin RD, et al. Decreased striatal dopaminergic innervation in REM sleep behavior disorder. Neurology. 2000;55:1410–12. doi: 10.1212/wnl.55.9.1410. [DOI] [PubMed] [Google Scholar]
- 152.Stiasny-Kolster K, Doerr Y, Moller JC, et al. Combination of, idiopathic' REM sleep behaviour disorder and olfactory dysfunction as possible indicator for alpha-synucleinopathy demonstrated by dopamine transporter FP-CIT-SPECT. Brain. 2005;128:126–37. doi: 10.1093/brain/awh322. [DOI] [PubMed] [Google Scholar]
- 153.Trenkwalder C. Sleep dysfunction in Parkinson's disease. Clin Neurosci. 1998;5:107–14. [PubMed] [Google Scholar]
- 154.Iranzo A, Graus F, Clover L, et al. Rapid eye movement sleep behavior disorder and potassium channel antibody-associated limbic encephalitis. Ann Neurol. 2006;59:178–81. doi: 10.1002/ana.20693. [DOI] [PubMed] [Google Scholar]
- 155.Bassetti C, Vella S, Donati F, et al. SPECT during sleepwalking. Lancet. 2000;356:484–5. doi: 10.1016/S0140-6736(00)02561-7. [DOI] [PubMed] [Google Scholar]
- 156.Lin L, Faraco J, Li R, et al. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell. 1999;98:365–76. doi: 10.1016/s0092-8674(00)81965-0. [DOI] [PubMed] [Google Scholar]