

Abstract
Orofacial clefts are common birth defects of multifactorial etiology. Several novel approaches have recently been applied to investigate the causes of clefts. These include examining Mendelian forms of clefting to identify genes that might also be implicated in isolated clefting, analyzing chromosomal rearrangements in which clefting is part of the resultant phenotype, studying animal models in which clefts arise either spontaneously or as a result of mutagenesis experiments, exploring how expression patterns correlate with gene function and examining the effects of gene–environment interactions. Together, these complementary strategies are providing researchers with new clues as to what mechanisms underlie orofacial clefting.
Introduction
Orofacial clefts comprise a large fraction of all human birth defects and are notable for their significant lifelong morbidity and complex etiology. On the basis of anatomical, genetic and embryological findings, orofacial clefts are commonly subdivided into those affecting the lip and/or palate (CL/P) and those involving the palate only (CPO) [1]. Clefts can be further categorized into syndromic (see Glossary) and isolated forms, according to whether affected individuals have other physical and developmental anomalies. Because the great majority of clefts appear to be isolated (~70% CL/P and ~50% CPO) [2], understanding the causes of these forms of clefts has long been a focus of research.
Many aspects of clefting, including epidemiology, clinical care, and genetic and environmental risks, have been recently reviewed [3]. In this overview, we focus on recent developments in genetics [4], animal models [5] and gene–environment interactions [6].
Development of the lip and palate
After conception, a precisely coordinated cascade of developmental processes involving cell migration, growth, differentiation and apoptosis results in the development of craniofacial structures from the originating oropharyngeal membrane [7]. Early in the sixth week, the medial nasal prominences merge with each other and the bilateral maxillary processes to form the primary palate and the upper lip. The lower lip and jaw are produced by the mandibular prominences, which merge across the midline. The secondary palate begins to develop early in the sixth week from the two palatal shelves, which extend from internal aspects of the maxillary prominences. During weeks 7–8, apoptosis and epithelial–mesenchymal transformation (EMT) at the medial edges enable the palatal shelves to fuse after the shelves have ascended to an appropriate position above the tongue. Proteins such as integrins, matrix metalloproteinases, microtubules and actin cytoskeletons are involved in the EMT process [8].
The molecular events that underlie the formation of orofacial structures are under the strict control of an array of genes that includes the fibroblast growth factors (Fgfs), sonic hedgehog (Shh), bone morphogenetic proteins (Bmps), members of the transforming growth factor β (Tgf-β) superfamily, and transcription factors such as Dlx, Pitx, Hox, Gli and T-box families [2]. Hydration of extracellular matrix components (principally hyaluronan) in the shelf mesenchyme is thought to provide the necessary intrinsic force to cause shelf elevation [9]. However, contraction of elastic fibers and/or skeletal muscle fibers, and an increase in vascularity of the developing palate have also been proposed as alternative mechanisms underpinning shelf elevation. Palatal fusion itself appears to be driven by several cell adhesion molecules, including nectin 1, desmosomes and type IX collagen, and growth factors, such as TGFα/EGFR and TGF-β3 [8,9].
The search for candidate genes
A variety of genetic approaches have been used to identify candidate genes and loci responsible for clefting [4]. Compiled in Table 1 is a list of candidate genes derived from linkage and association studies, studies of the roles these genes play in animal development and the phenotypes they generate when disrupted in mouse knockouts [1,5]. Genome-wide linkage scans have also provided some important clues. To date, 13 genome-wide scans for nonsyndromic CL/P have been performed, and a meta-analysis (see Glossary) of these individual scans revealed significant heterogeneity LOD scores (see Glossary) on chromosomes 1p, 6p, 6q, 14q and 15q, and a particularly strong signal on 9q [10•].
Table 1.
Genes implicated in orofacial clefting based on evidence from animal models, expression analyses, and human linkage and/or association studies.
| Gene | Cytogenetic locationa | Gene function | Animal model phenotypeb | Expression data | Linkage/association | Known syndrome |
|---|---|---|---|---|---|---|
| IRF6 | 1q32 | TF | NA | + | + | Van der Woude |
| SKI1 | 1q32 | GF | + | + | + | |
| MTHFR | 1p36 | CS | NA | +/− | + | |
| TGFA | 2p13 | GF | − | +/− | + | |
| TP63 | 3q27 | TF | + | + | − | EEC |
| MSX1 | 4p16 | TF | + | + | + | Witkop |
| EDN1 | 6p24.1 | CAM | + | NA | + | |
| FGFR1 | 8p11.2–8p11.1 | GFR | + | + | + | Kallmann |
| PPP3CC | 8p21.3 | CS | NA | + | + | |
| FOXE1 | 9q22 | TF | + | + | + | Bamforth-Lazarus |
| PVRL1 | 11q23 | CAM | NA | +/− | + | Margarita Island |
| TGFB3 | 14q24 | GF | + | + | + | |
| GABRB3 | 15q11.2–15q12 | CS | + | − | + | |
| FOXC2 | 16q22–16q24 | TF | − | + | − | Lymphedema-distichiasis |
| RARA | 17q21 | CS | NA | + | + | |
| BCL3 | 19q13 | TF | − | − | + | |
| TBX22 | Xq21 | TF | NA | + | + | CP and ankyloglossia |
Abbreviations: CAM, cell adhesion molecule; CS, cell signaling; EEC, ectrodactyly ectodermal dysplasia; GF, growth factor; GFR, growth factor receptor; NA, not available TF, transcription factor; −, negative; +, positive; +/−, weak.
Cytogenetic location according to Entrez Gene (http://www.ncbi.nih.gov/entrez/query.fcgi?db=gene).
From the Mouse Genome Informatics database (MGI; http://www.informatics.jax.org/).
The past two years in particular have witnessed several exciting new advances in the mapping of genes for clefting. The latest data from mouse and human studies have helped identify several genes known to underlie Mendelian syndromic forms of CL/P as also playing a role in the etiology of isolated clefts. These include IRF6 [11••], MSX1 [12], PVRL1 [13], TBX22 [14] and FGFR1 [15] (Table 1).
Clues from Mendelian forms of clefts
Mendelian forms of clefting with phenotypes closely mimicking those of isolated clefts can greatly facilitate the mapping of genes underlying the isolated forms [2]. The autosomal dominant Van der Woude syndrome (VWS) is the best model studied to date. In addition to clefts, pits in the lower lip and hypodontia are the only additional features in VWS patients. Recently, mutations in the interferon regulatory factor 6 (IRF6) gene were reported to underlie VWS [16], and, subsequently, variants in IRF6 were found to be significantly associated with nonsyndromic clefting as well [11••,17]. In the mouse, Irf6 transcripts are highly expressed in the palatal medial edge epithelium (MEE) immediately before and during fusion of the palatal shelves [16] (Figure 1). It has been speculated that mutations in IRF6 might repress the TGF-β signaling pathway in a manner analogous to IRF1-mediated repression, leading to increased epithelial apoptosis before the bilateral processes have managed to fuse [8].
Figure 1.

Irf6 expression in the E14.5 prefusion mouse palate. The figure depicts an in situ frontal section through the posterior palate. Nuclei were stained with DAPI for background staining and the silver grains were pseudo-colored red in the merged image. (Photograph kindly provided by Alexandra Knight and Professor Michael J Dixon).
Knocking out a second gene, Msx1 (msh homeobox homolog 1), in mice results in clefting [18]. The Msx proteins are known to play key roles in epithelial–mesenchymal tissue interactions during craniofacial development [19]. In humans, MSX1 is deleted in cases of a 4p deletion syndrome that is frequently associated with clefting [20]. Moreover, a nonsense mutation in exon 1 of MSX1 caused tooth agenesis and various combinations of clefts in a Dutch family [21]. In a follow-up study of 1000 unrelated individuals with CL/P, complete sequencing of the gene showed that mutations in MSX1 alone could account for 2% of isolated CL/P [12,22].
A third gene, FGFR1 (fibroblast growth factor receptor-1), encodes a transmembrane receptor tyrosine kinase that transduces signals from secreted FGFs [23]. Loss-of-function mutations in FGFR1 cause the autosomal dominant form of Kallmann syndrome (KAL2), which is characterized by hypogonadism and anosmia, and clefting in around 5–10% of the cases [15]. The variable expression of FGFR1 variants results in some affected individuals presenting with isolated CL/P alone (JC Murray, unpublished).
Mutations in TP63 are implicated in five distinct human developmental disorders, characterized by limb abnormalities, ectodermal dysplasia and orofacial clefts [24]. Interestingly, the distribution of mutations over the different p63 protein domains shows a clear pattern of genotype–phenotype correlation. Other notable examples of clefting syndromes that might include phenocopies of isolated clefts are X-linked cleft palate with ankyloglossia, caused by mutations in TBX22 [14,25], cleft lip and palate-ectodermal dysplasia syndrome (PVRL1) [13,26], and lymphedema-distichiasis syndrome (FOXC2) [27,28]. Other genes underlying additional clefting syndromes that are also excellent candidates for investigating the causes of isolated clefts include FOXE1 in Bamforth-Lazarus syndrome [29] and FLNA in otopalatodigital syndromes types 1 and 2 [30].
Clues from genomic rearrangements
Genomic rearrangements can arise when interspersed repeat elements lying in tandem facilitate submicroscopic deletion and duplication events or translocations and/or inversions between or within chromosomes [31]. Genetic variants that result in a phenotype including clefting and that are found segregating with a genomic rearrangement in multiple members are best represented by the 22q deletion syndrome. Duplications of this same region have been associated with cleft palate [32], suggesting that genome-wide searches using comparative genomic hybridization (CGH; see Glossary), quantitative-PCR (see Glossary) or allele-loss might reveal additional clefting loci.
Recently, two relevant genes or gene clusters with balanced translocations and CL/P have been identified: the first gene at 19q13 [33] and the second gene at 2q32 [34•]. The candidate gene transected at 2q32 is SATB2. It is highly expressed in both the lip and the palate, making it an excellent candidate for isolated CL/P. Expression analyses in the mouse secondary palate reveal that the strongest expression of Satb2 occurs before palatal shelf fusion (E13.5), with a dramatic down-regulation after the shelves have fused (E14.5) (Figure 2). Additional examples of clefts arising from displaced genomic material are the Dancer mutation [35] and clefts in the 22q and 1p36 deletion syndromes [36,37].
Figure 2.
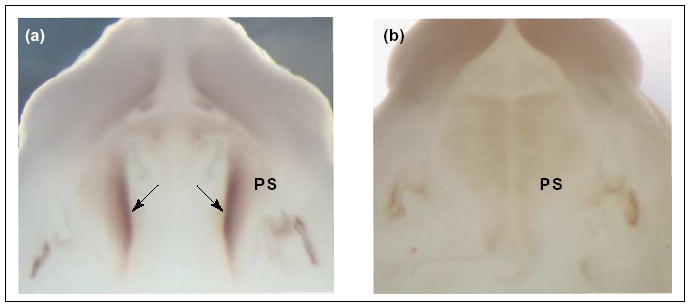
Satb2 expression during development of the murine secondary palate. Whole-mount in situ hybridization analysis of Satb2 expression in mouse embryos at (a) E13.5 and (b) E14.5. (a) At E13.5, the strongest expression of Satb2 is detected in the mesenchyme underlying the presumptive medial edge epithelia (arrows). (b) By the time of palatal shelf fusion at E14.5, the expression is dramatically down-regulated. Abbreviations: PS, palatal shelf. (Photographs kindly provided by Professor Michael J Dixon).
The transforming growth factors
Transforming growth factors are one of the most extensively studied gene families in relation to clefting. One member of this superfamily, transforming growth factor α (TGF-α), binds to the epidermal growth factor receptor (EGFR) and elicits responses similar to but more potent than EGF. The expression pattern of TGF-αin palatal tissues, especially in the midline seam and subjacent mesenchyme of the palatal shelves at the time of shelf fusion, supports a role for TGFA in clefting. Although inconclusive, data from association studies indicate that either TGFA itself or markers in its vicinity might play an important role in clefting [38].
Studies of expression patterns have shown that, although each Tgf-β is temporally and spatially expressed in the developing palate, only the Tgfb3−/− knockout inhibited normal palatal shelf fusion in mice [39]. Moreover, the mechanism by which Tgf-β3 affects palatal shelf fusion appears to be targeted and specific: the MEE in Tgfb3−/− mice fails to stop cellular proliferation [40], displays reduced apoptosis [41], fails to alter its morphology and adheres less well [42], fails to degrade the basement membrane and fails to undergo EMT [43]. Furthermore, exogenous TGF-β3 can induce palatal fusion in the chicken through a process that requires physical contact of the MEE and formation of the midline seam [39]. Thus, Tgfb3 signaling is unequivocally a key pathway in palate development in the mouse. It also appears to be involved in palatal development in humans, because association studies have provided some corroborative data [38].
Animal models and expression data
Molecular studies in the mouse and chick have been pivotal in the identification of genes that regulate the dynamic cellular changes in the MEE. Although chick palatal shelves grow towards one another above the tongue and make contact, they do not actually fuse. The chick, therefore, has the advantage of mirroring the pathology seen in cleft palate, whereas the mouse provides an excellent model to study palatal shelf fusion. Indeed, studies in these animal models have helped to identify a battery of genes essential for palatal formation: Tgfb3 [44]; Bmps [45]; Tbx22 [46]; Fgfrs [47]; Pdgfc [48]; RhoA [49]; the gene encoding PtdIns-3 kinase [43]; Gabrb3 [50]; Gad1 [51,52]; Cspg [53]; and Mmps and Timp2 [54].
Animal models with clefts arising spontaneously or as a result of mutagenesis experiments provide another exciting avenue for gene mapping [55]. The mouse is an excellent model for studying human clefting because the development of craniofacial structures in these two species is remarkably similar. Whereas cleft palate is a common phenotype in the mouse, cleft lip is rare. To date, four mutations have been reported with cleft lip and palate phenotypes in mice. These include two spontaneous mutations called Twirler and Dancer, a transgene insertion-induced deletion mutation called Legless, and a radiation-induced mutation called Brachyphalangy [56]. Both the Dancer and Twirler mutations are almost fully penetrant for CL/P in homozygotes. Furthermore, Dancer was shown to arise from a translocation of the p23 gene sequence into the Tbx10 locus, resulting in ectopic expression of Tbx10 under the influence of the p23 promoter [35].
In addition to these mutant strains, cleft lip also occurs spontaneously in around 5–30% of embryos and neonates in a well-studied family of inbred mouse strains (the ‘A’ strains) [56]. A genome-wide screen for cleft susceptibility loci in the A/WySn strain identified two epistatically interacting loci, clf1 and clf2, that contribute to the cleft lip phenotype [57]. The clf1 locus contains two Wnt genes, Wnt3 and Wnt9b, suggesting a potential role for the Wnt signaling pathway in orofacial development [58••].
As to expression analysis, the strongest candidate genes are likely to be those whose normal expressions encompass the critical time and tissue for lip and palate development. Three global approaches are currently available for gene expression analysis in craniofacial structures: (i) the ongoing studies of the Craniofacial and Oral Gene Expression Network (COGENE), which provides public web access to genome-wide expression analysis data of craniofacial tissues isolated from human embryos (http://humgen.wustl.edu/COGENE/); (ii) Optical Projection Tomography (OPT), which enables the visualization of the relative expression of genes both temporally and spatially [59]; and finally, (iii) the mouse N-ethyl-N-nitrosourea (ENU) mutagenesis projects [60], which in addition to helping identify potential candidates for craniofacial development also serve as a means of verifying whether the expression patterns of existing candidate genes are consistent with hypotheses about function. A recent study [61] of the effects of ENU mutagenesis on the offspring of male mice suggested that genes related to isolated cleft palate might be recessive in phenotype, whereas point mutations appeared to be more relevant to the pathogenesis of cleft lip and palate. See Box 1 for additional resources on the internet.
A role for environmental risk factors
Birth defects are likely to recur in families not only because of shared genetic factors but also as a result of shared environmental factors [62]. Cigarette smoking during pregnancy, with the attendant hypoxia, is associated with several adverse reproductive outcomes. The most recent meta-analysis on the effects of smoking indicates a moderately increased risk of orofacial clefts [63]. Specifically, estimates of relative risks were 1.34 (95% confidence interval (see Glossary) (CI); 1.25–1.44) for CL/P and 1.22 (95% CI; 1.10–1.35) for CPO.
Maternal nutrition during pregnancy also appears to play an important role. For example, low dietary intake of B-complex vitamins, in addition to exposure to deficient or excessive amounts of vitamin A, have been linked to increased risks of clefts [64,65]. Increased risks from exposures can suggest metabolic pathways whose disruption might trigger the development of clefts. Several studies have shown that folic acid and other B-complex vitamins might have a beneficial effect on reducing the risk of orofacial clefts [66–69].
The role of cholesterol-lowering drugs (e.g. statins) in prenatal development has been recently discussed [70]. Statins that reach the embryo through maternal intake of the drug might inhibit cholesterol biosynthesis and, consequently, affect the sterol-dependent Hedgehog family of morphogens, which is critical for the proper development of a range of structures, including the face. In a study of the adverse effects of gestational exposure to statins, two cases had cleft lip and two others had cleft palate among 31 adverse birth outcomes [71]. Other drugs, such as corticoids, have also received some attention, although the effects are modest in size [72].
Of particular importance to a complex trait such as clefts is the study of the likely impact of both genetic and environmental factors. Several studies have investigated interactions of a range of common environmental factors, such as cigarette smoking, alcohol intake, multivitamin/folic acid supplementation and the use of medication, with variant alleles in several genes that include TGFA, TGFB3, MSX1, BCL3, RARA, MTHFR, CYP1A1, NAT1, NAT2, GSTT1 and EPHX1. These have been reviewed elsewhere [1,73,74].
In assessing disease risk, most previous studies have typically focused on the affected child as the unit of analysis. Recent works in clefts, however, have started to focus on parental contributions too, particularly for the assessment of maternally mediated effects and the effects of imprinting [75,76,77•]. Another recent extension of the case–parent triad approach (see Glossary) consists of using information from grandparents to explore the joint effects of maternal and offspring genotypes and to provide a direct estimation of relative risks [78]. These new analytical approaches ensure improved power for the detection of an effect, if present, and the judicious use of all the available data from the families.
Conclusions
Great strides have been made in recent years in our understanding of how orofacial clefts arise at a molecular level. Contributions from the single genes IRF6, MSX1 and FGFR1 now seem to explain approximately 15% of isolated clefts. Cigarette smoking and, possibly, disruptions in the folate biosynthetic pathway represent potential environmental risks. Given the now large sample sets available for study, linkage scans will hopefully have sufficient power to identify gene–environment interactions. Coupled with new discoveries in gene expression and animal models, researchers are finally starting to unravel the causes of orofacial clefts and, hopefully, new opportunities for improvements in diagnosis and treatment of this complex genetic can soon be made available to cleft patients and their families.
Update
A genome-wide scan for loci involved in CPO was recently conducted in a group of Finnish multiplex families [79]. Finland has one of the highest rates of isolated CPO among Caucasian populations, and even more intriguing is the higher observed prevalence of CPO compared to CL/P. Finland is therefore especially attractive for the study of isolated cleft palate. This study reported suggestive linkage at 1p34, 2p24-p25, and 12q21. The authors also screened nine unrelated affected individuals for mutations in IRF6, but no mutation was found.
Lately, Loeys et al. [80] reported that mutations in TGFBR1 or TGFBR2 were the cause behind a novel syndrome that is characterized by altered cardiovascular, neurocognitive, skeletal and craniofacial development. Tissues from affected individuals showed increased TGF-β signaling, reflected by nuclear enrichment of phosphorylated Smad2. In a related paper, Cui and coworkers [81] demonstrated that over-expression of Smad2 rescue the cleft palate phenotype in Tgf-β3−/− could mutant mice. These reports provide further evidence that aberrant TGF-β signaling plays a prominent role in the pathogenesis of many common human malformations, including cleft palate.
Data from a recent study on Bmp-signaling in lip and palate fusion in mice uncovered a Bmp4–Bmpr1a genetic pathway involved in lip fusion, and revealed distinct roles of Bmp-signaling in lip and palate development [82]. Whereas Bmpr1a mutants had fully penetrant bilateral CL/P with tooth agenesis, most likely as a result of defective proliferation, Bmp4 mutants had isolated cleft lip, possibly caused by premature apoptosis in the medial nasal processes. This suggests that Bmp-signaling plays distinct roles in lip fusion and secondary palate development. Interestingly, signaling through Bmpr1a appeared to affect the expression of transcriptional regulators such as Barx1 and Pax9, but not of Msx1, Tbx22 or Osr2.
As to studies of gene–environment interactions in relation to clefts, a recent meta-analysis examined the association between maternal cigarette smoking and infant’s genotype at the TaqI site in TGFA [83]. Although maternal smoking was a consistent risk factor for both CL/P and CPO across all studies, the modest effects of interaction seemed to be restricted to cleft palate only.
Acknowledgments
We thank Dr Temis M Felix for reviewing this manuscript and apologize to all those authors whose work could not be cited because of lack of space. AJ is supported by a postdoctoral fellowship from the Research Council of Norway (NFR) and JCM by National Institutes of Health (NIH) grants DE08559 and DE16215. We would especially like to thank the many families and students who have contributed to our research over the years, and our colleagues Kaare Christensen, Mike Dixon, David Fitzpatrick, Andrew Lidral, Mary Marazita, Brian Shutte and Rolf Terje Lie for many helpful discussions.
Glossary
- Breakpoint mapping
Identification and delineation of chromosomal breakpoints caused by deletions, insertions, inversions or translocations. Breakpoints are mapped using high-resolution techniques such as fluorescence in situ hybridization (FISH) and comparative genomic hybridization (CGH; see below).
- Case-parent triad approach
A study design in which a triad made up of the mother, father and affected offspring is used as the unit of analysis. Parental alleles not transmitted to the offspring are used as ethnically matched genetic controls in statistical analyses.
- Comparative genomic hybridization (CGH)
CGH is a fluorescent molecular cytogenetic technique for detecting chromosomal imbalances. Two genomic DNA samples are simultaneously hybridized in situ to normal human metaphase spreads, and regions of increased or decreased copy number are located or mapped relative to the normal metaphase chromosomes.
- Confidence interval
This is an interval calculated from a given set of sample data that has a specified probability of containing the parameter being estimated. If samples of the same size are drawn repeatedly from a population and a confidence interval is calculated from each sample then 95% of these intervals should contain the population parameter.
- Haploinsufficiency
A locus shows haploinsufficiency if more gene product is required to produce a normal phenotype than the amount produced by a single copy. This situation might arise if an individual is heterozygous for a certain gene mutation or hemizygous at a particular locus owing to a deletion of the corresponding allele.
- LOD score
A measure to assess the strength of the evidence in favour of linkage. A LOD of 3 indicates 103 odds in favour of linkage compared to no linkage. HLOD is the LOD score corresponding to the likelihood ratio for linkage given heterogeneity.
- Meta-analysis
A statistical method that integrates the results of several existing independent studies in order to provide a larger sample size for evaluation, and to produce a stronger conclusion that can be provided by any single study.
- Quantitative-PCR
A sensitive method that enables the quantification of the amount of either DNA or RNA products generated during each cycle of the PCR.
- Syndromic
Refers to cleft cases with an accompanying physical and/or developmental anomaly.
Box 1 Additional resources on the internet
|
Center for Craniofacial Development and Disorders (CCDD)
http://www.hopkinsmedicine.org/craniofacial/Home/Index.cfm OMIM is a curated database of human genes and genetic disorders. It enables rapid and direct linking between disease, gene sequence and chromosomal locus. |
National Institute of Dental and Craniofacial Research (NIDCR)
http://www.nidcr.nih.gov/ NIDCR focuses on improving oral, dental and craniofacial health through research, research-training and the dissemination of health information. The site has numerous links to NIDCR clinical trials, funding opportunities for research and training, health information, and news and reports, among others. |
|
Craniofacial and Oral Gene Expression Network (COGENE)
http://humgen.wustl.edu/COGENE/ COGENE represents a consortium of investigators involved in describing human gene expression changes that occur during early stages of development, with particular emphasis on craniofacial development. |
N-ethyl-N-nitrosourea (ENU) mutagenesis projects
http://www.mouse-genome.bcm.tmc.edu/Home.asp The ENU mutagenesis projects aim at determining the function of genes on the mouse chromosome 11 by saturating the wild type chromosomes with point mutations using the chemical N-ethyl-N-nitrosourea (ENU). Many of the new mutants thus created might represent models of human diseases such as birth defects, patterning defects, etc. |
|
Developmental Genome Anatomy Project (DGAP)
http://www.bwhpathology.org/dgap/ DGAP looks for apparently balanced chromosomal rearrangements in patients with multiple congenital anomalies, and uses this information to map and identify genes that are disrupted or dysregulated at critical stages of human development. |
Online Mendelian Inheritance in Man (OMIM)
http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM OMIM is a curated database of human genes and genetic disorders. It enables rapid and direct linking between disease, gene sequence and chromosomal locus. |
|
Entrez Gene
http://www.ncbi.nih.gov/entrez/query.fcgi?db=gene Entrez Gene provides a unified query interface for gene- oriented searches. It provides information on official nomenclature, aliases, sequence accessions, phenotypes, homology, map locations, and related websites. |
Optical Projection Tomography (OPT)
http://genex.hgu.mrc.ac.uk/OPT_Microscopy/optwebsite/frontpage/index.htm This site provides extensive data on optical projection tomography microscopy, which is a new technique that enables 3D imaging of biological specimens. |
|
Mouse Genome Informatics (MGI)
http://www.informatics.jax.org/ MGI provides integrated access to data on the genetics, genomics and biology of the laboratory mouse. |
Single Nucleotide Polymorphism database (dbSNP)
http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=snp dbSNP is a central repository for a broad collection of simple genetic polymorphisms. |
|
Murray laboratory website
http://genetics.uiowa.edu/ This is JCM’s laboratory website. The web pages provide information on review protocols currently used in the lab, access to both published and unpublished data regarding genes and ongoing studies. Also included are extensive descriptions of each major project currently underway and options for obtaining additional information about them. |
Society of Craniofacial Genetics
http://www.craniofacialgenetics.org/ This is the official website of the Society of Craniofacial Genetics, developed to promote education, research and communication in normal and abnormal development of the craniofacies. |
Footnotes
This review comes from a themed issue on Genetics of disease
Edited by Veronica van Heyningen and David FitzPatrick
References
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Murray JC. Gene/environment causes of cleft lip and/or palate. Clin Genet. 2002;61:248–256. doi: 10.1034/j.1399-0004.2002.610402.x. [DOI] [PubMed] [Google Scholar]
- 2.Stanier P, Moore GE. Genetics of cleft lip and palate: syndromic genes contribute to the incidence of non-syndromic clefts. Hum Mol Genet. 2004;13 Spec No 1:R73–R81. doi: 10.1093/hmg/ddh052. [DOI] [PubMed] [Google Scholar]
- 3.Wyszynski DF. Cleft Lip and Palate: from Origin to Treatment. New York: Oxford University Press; 2002. [Google Scholar]
- 4.Lidral AC, Murray JC. Genetic approaches to identify disease genes for birth defects with cleft lip/palate as a model. Birth Defects Res A Clin Mol Teratol. 2004;70:893–901. doi: 10.1002/bdra.20096. [DOI] [PubMed] [Google Scholar]
- 5.Murray JC, Schutte BC. Cleft palate: players, pathways, and pursuits. J Clin Invest. 2004;113:1676–1678. doi: 10.1172/JCI22154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marazita ML, Mooney MP. Current concepts in the embryology and genetics of cleft lip and cleft palate. Clin Plast Surg. 2004;31 :125–140. doi: 10.1016/S0094-1298(03)00138-X. [DOI] [PubMed] [Google Scholar]
- 7.Sperber GH. Formation of the primary palate. In: Wyszynski DFE, editor. Cleft Lip and Palate: from Origin to Treatment. Oxford University Press: 2002; pp. 5–13. [Google Scholar]
- 8.Cox TC. Taking it to the max: the genetic and developmental mechanisms coordinating midfacial morphogenesis and dysmorphology. Clin Genet. 2004;65:163–176. doi: 10.1111/j.0009-9163.2004.00225.x. [DOI] [PubMed] [Google Scholar]
- 9.Moxham BJ. The development of the palate — a brief review. Eur J Anat. 2003;7:53–74. [Google Scholar]
- •10.Marazita ML, Murray JC, Lidral AC, Arcos-Burgos M, Cooper ME, Goldstein T, Maher BS, Daack-Hirsch S, Schultz R, Mansilla MA, et al. Meta-analysis of 13 genome scans reveals multiple cleft lip/palate genes with novel loci on 9q21 and 2q32–35. Am J Hum Genet. 2004;75:161–173. doi: 10.1086/422475. Marazita et al. combined the results of all previously performed genome-wide scans of isolated clefts with new data from their own analyses. More than 500 families are represented in this large dataset and one locus on 9q showed a heterogeneity LOD score of over 6.5, providing very strong support for a cleft locus or loci in this region. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••11.Zucchero TM, Cooper ME, Maher BS, Daack-Hirsch S, Nepomuceno B, Ribeiro L, Caprau D, Christensen K, Suzuki Y, Machida J, et al. Interferon regulatory factor 6 (IRF6) gene variants and the risk of isolated cleft lip or palate. N Engl J Med. 2004;351:769–780. doi: 10.1056/NEJMoa032909. Using the IRF6 gene, in which haploinsufficiency (see Glossary) results in the autosomal dominant Van der Woude’s syndrome, Zucchero et al. studied 36 single nucleotide polymorphisms from ten world-wide populations to demonstrate very strong transmission distortion of a particular risk allele of IRF6 in isolated clefts. Allelic variants of IRF6 had an attributable risk of approximately 12% for cleft lip and palate. This report represents the first time such a strong single-gene effect has been demonstrated in isolated clefting. [DOI] [PubMed] [Google Scholar]
- 12.Jezewski PA, Vieira AR, Nishimura C, Ludwig B, Johnson M, O’Brien SE, Daack-Hirsch S, Schultz RE, Weber A, Nepomucena B, et al. Complete sequencing shows a role for MSX1 in non-syndromic cleft lip and palate. J Med Genet. 2003;40 :399–407. doi: 10.1136/jmg.40.6.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sozen MA, Suzuki K, Tolarova MM, Bustos T, Fernandez Iglesias JE, Spritz RA. Mutation of PVRL1 is associated with sporadic, non-syndromic cleft lip/palate in northern Venezuela. Nat Genet. 2001;29:141–142. doi: 10.1038/ng740. [DOI] [PubMed] [Google Scholar]
- 14.Marcano AC, Doudney K, Braybrook C, Squires R, Patton MA, Lees MM, Richieri-Costa A, Lidral AC, Murray JC, Moore GE, et al. TBX22 mutations are a frequent cause of cleft palate. J Med Genet. 2004;41:68–74. doi: 10.1136/jmg.2003.010868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dode C, Levilliers J, Dupont JM, De Paepe A, Le Du N, Soussi-Yanicostas N, Coimbra RS, Delmaghani S, Compain-Nouaille S, Baverel F, et al. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet. 2003;33:463–465. doi: 10.1038/ng1122. [DOI] [PubMed] [Google Scholar]
- 16.Kondo S, Schutte BC, Richardson RJ, Bjork BC, Knight AS, Watanabe Y, Howard E, de Lima RL, Daack-Hirsch S, Sander A, et al. Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat Genet. 2002;32:285–289. doi: 10.1038/ng985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scapoli L, Palmieri A, Martinelli M, Pezzetti F, Carinci P, Tognon M, Carinci F. Strong evidence of linkage disequilibrium between polymorphisms at the IRF6 locus and nonsyndromic cleft lip with or without cleft palate, in an Italian population. Am J Hum Genet. 2004;76:180–183. doi: 10.1086/427344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Satokata I, Maas R. Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nat Genet. 1994;6:348–356. doi: 10.1038/ng0494-348. [DOI] [PubMed] [Google Scholar]
- 19.Alappat S, Zhang ZY, Chen YP. Msx homeobox gene family and craniofacial development. Cell Res. 2003;13:429–442. doi: 10.1038/sj.cr.7290185. [DOI] [PubMed] [Google Scholar]
- 20.van den Boogaard MJ. MSX1 and partial anodontia, orofacial clefting, and the Witkop syndrome. In: Epstein JC, Erickson RP, Wynshaw-Boris A, editors. Inborn Errors of Development. The Molecular Basis of Clinical Disorders of Morphogenesis; Oxford University Press: 2004. pp. 557–567. [Google Scholar]
- 21.van den Boogaard MJ, Dorland M, Beemer FA, van Amstel HK. MSX1 mutation is associated with orofacial clefting and tooth agenesis in humans. Nat Genet. 2000;24:342–343. doi: 10.1038/74155. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki Y, Jezewski PA, Machida J, Watanabe Y, Shi M, Cooper ME, Viet le T, Nguyen TD, Hai H, Natsume N, et al. In a Vietnamese population, MSX1 variants contribute to cleft lip and palate. Genet Med. 2004;6:117–125. doi: 10.1097/01.gim.0000127275.52925.05. [DOI] [PubMed] [Google Scholar]
- 23.Dode C, Hardelin JP. Kallmann syndrome: fibroblast growth factor signaling insufficiency? J Mol Med. 2004;82:725–734. doi: 10.1007/s00109-004-0571-y. [DOI] [PubMed] [Google Scholar]
- 24.van Bokhoven H, Brunner HG. Splitting p63. Am J Hum Genet. 2002;71:1–13. doi: 10.1086/341450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Braybrook C, Lisgo S, Doudney K, Henderson D, Marcano AC, Strachan T, Patton MA, Villard L, Moore GE, Stanier P, et al. Craniofacial expression of human and murine TBX22 correlates with the cleft palate and ankyloglossia phenotype observed in CPX patients. Hum Mol Genet. 2002;11:2793–2804. doi: 10.1093/hmg/11.22.2793. [DOI] [PubMed] [Google Scholar]
- 26.Suzuki K, Hu D, Bustos T, Zlotogora J, Richieri-Costa A, Helms JA, Spritz RA. Mutations of PVRL1, encoding a cell–cell adhesion molecule/herpesvirus receptor, in cleft lip/palate-ectodermal dysplasia. Nat Genet. 2000;25:427–430. doi: 10.1038/78119. [DOI] [PubMed] [Google Scholar]
- 27.Bahuau M, Houdayer C, Tredano M, Soupre V, Couderc R, Vazquez MP. FOXC2 truncating mutation in distichiasis, lymphedema, and cleft palate. Clin Genet. 2002;62:470–473. doi: 10.1034/j.1399-0004.2002.620608.x. [DOI] [PubMed] [Google Scholar]
- 28.Fang J, Dagenais SL, Erickson RP, Arlt MF, Glynn MW, Gorski JL, Seaver LH, Glover TW. Mutations in FOXC2 (MFH-1), a forkhead family transcription factor, are responsible for the hereditary lymphedema-distichiasis syndrome. Am J Hum Genet. 2000;67:1382–1388. doi: 10.1086/316915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Castanet M, Park SM, Smith A, Bost M, Leger J, Lyonnet S, Pelet A, Czernichow P, Chatterjee K, Polak M. A novel loss-of-function mutation in TTF-2 is associated with congenital hypothyroidism, thyroid agenesis and cleft palate. Hum Mol Genet. 2002;11:2051–2059. doi: 10.1093/hmg/11.17.2051. [DOI] [PubMed] [Google Scholar]
- 30.Robertson SP, Twigg SR, Sutherland-Smith AJ, Biancalana V, Gorlin RJ, Horn D, Kenwrick SJ, Kim CA, Morava E, Newbury-Ecob R, et al. Localized mutations in the gene encoding the cytoskeletal protein filamin A cause diverse malformations in humans. Nat Genet. 2003;33:487–491. doi: 10.1038/ng1119. [DOI] [PubMed] [Google Scholar]
- 31.Stankiewicz P, Inoue K, Bi W, Walz K, Park SS, Kurotaki N, Shaw CJ, Fonseca P, Yan J, Lee JA, et al. Genomic disorders: genome architecture results in susceptibility to DNA rearrangements causing common human traits. Cold Spring Harb Symp Quant Biol. 2003;68:445–454. doi: 10.1101/sqb.2003.68.445. [DOI] [PubMed] [Google Scholar]
- 32.Hassed SJ, Hopcus-Niccum D, Zhang L, Li S, Mulvihill JJ. A new genomic duplication syndrome complementary to the velocardiofacial (22q11 deletion) syndrome. Clin Genet. 2004;65:400–404. doi: 10.1111/j.0009-9163.2004.0212.x. [DOI] [PubMed] [Google Scholar]
- 33.Yoshiura K, Machida J, Daack-Hirsch S, Patil SR, Ashworth LK, Hecht JT, Murray JC. Characterization of a novel gene disrupted by a balanced chromosomal translocation t(2;19)(q11.2;q13.3) in a family with cleft lip and palate. Genomics. 1998;54:231–240. doi: 10.1006/geno.1998.5577. [DOI] [PubMed] [Google Scholar]
- •34.FitzPatrick DR, Carr IM, McLaren L, Leek JP, Wightman P, Williamson K, Gautier P, McGill N, Hayward C, Firth H, et al. Identification of SATB2 as the cleft palate gene on 2q32–q33. Hum Mol Genet. 2003;12:2491–2501. doi: 10.1093/hmg/ddg248. Fitzpatrick et al. used breakpoint mapping techniques (see Glossary), coupled with expression analyses, to identify the SATB2 gene located at a breakpoint in two different families with CL/P. SATB2 encodes a 733 amino acid DNA-binding protein of remarkable conservation between human and mouse. Although mutations were not identified in an initial screen of isolated cleft palate cases, the gene itself and its regulatory elements still remain strong candidates for isolated clefts of the lip and/or palate. [DOI] [PubMed] [Google Scholar]
- 35.Bush JO, Lan Y, Jiang R. The cleft lip and palate defects in Dancer mutant mice result from gain of function of the Tbx10 gene. Proc Natl Acad Sci USA. 2004;101:7022–7027. doi: 10.1073/pnas.0401025101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Colmenares C, Heilstedt HA, Shaffer LG, Schwartz S, Berk M, Murray JC, Stavnezer E. Loss of the SKI proto-oncogene in individuals affected with 1p36 deletion syndrome is predicted by strain-dependent defects in Ski−/− mice. Nat Genet. 2002;30:106–109. doi: 10.1038/ng770. [DOI] [PubMed] [Google Scholar]
- 37.Shaikh TH, Kurahashi H, Emanuel BS. Evolutionarily conserved low copy repeats (LCRs) in 22q11 mediate deletions, duplications, translocations, and genomic instability: an update and literature review. Genet Med. 2001;3:6–13. doi: 10.1097/00125817-200101000-00003. [DOI] [PubMed] [Google Scholar]
- 38.Marazita ML, Neiswanger K. Association studies. In: Wyszynski DFE, editor. Cleft Lip and Palate: from Origin to Treatment. Oxford University press; 2002. pp. 240–254. [Google Scholar]
- 39.Nawshad A, LaGamba D, Hay ED. Transforming growth factor β (TGFβ) signalling in palatal growth, apoptosis and epithelial mesenchymal transformation (EMT) Arch Oral Biol. 2004;49:675–689. doi: 10.1016/j.archoralbio.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 40.Cui XM, Chai Y, Chen J, Yamamoto T, Ito Y, Bringas P, Shuler CF. TGF-β3-dependent SMAD2 phosphorylation and inhibition of MEE proliferation during palatal fusion. Dev Dyn. 2003;227:387–394. doi: 10.1002/dvdy.10326. [DOI] [PubMed] [Google Scholar]
- 41.Martinez-Alvarez C, Tudela C, Perez-Miguelsanz J, O’Kane S, Puerta J, Ferguson MW. Medial edge epithelial cell fate during palatal fusion. Dev Biol. 2000;220:343–357. doi: 10.1006/dbio.2000.9644. [DOI] [PubMed] [Google Scholar]
- 42.Tudela C, Formoso MA, Martinez T, Perez R, Aparicio M, Maestro C, Del Rio A, Martinez E, Ferguson M, Martinez-Alvarez C. TGF-β3 is required for the adhesion and intercalation of medial edge epithelial cells during palate fusion. Int J Dev Biol. 2002;46:333–336. [PubMed] [Google Scholar]
- 43.Kang P, Svoboda KK. PI-3 kinase activity is required for epithelial-mesenchymal transformation during palate fusion. Dev Dyn. 2002;225:316–321. doi: 10.1002/dvdy.10161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brunet CL, Sharpe PM, Ferguson MW. Inhibition of TGF-β3 (but not TGF-β 1 or TGF-β 2) activity prevents normal mouse embryonic palate fusion. Int J Dev Biol. 1995;39:345–355. [PubMed] [Google Scholar]
- 45.Zhang Z, Song Y, Zhao X, Zhang X, Fermin C, Chen Y. Rescue of cleft palate in Msx1-deficient mice by transgenic Bmp4 reveals a network of BMP and Shh signaling in the regulation of mammalian palatogenesis. Development. 2002;129:4135–4146. doi: 10.1242/dev.129.17.4135. [DOI] [PubMed] [Google Scholar]
- 46.Herr A, Meunier D, Muller I, Rump A, Fundele R, Ropers HH, Nuber UA. Expression of mouse Tbx22 supports its role in palatogenesis and glossogenesis. Dev Dyn. 2003;226:579–586. doi: 10.1002/dvdy.10260. [DOI] [PubMed] [Google Scholar]
- 47.Rice R, Spencer-Dene B, Connor EC, Gritli-Linde A, McMahon AP, Dickson C, Thesleff I, Rice DP. Disruption of Fgf10/Fgfr2b-coordinated epithelial-mesenchymal interactions causes cleft palate. J Clin Invest. 2004;113:1692–1700. doi: 10.1172/JCI20384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ding H, Wu X, Bostrom H, Kim I, Wong N, Tsoi B, O’Rourke M, Koh GY, Soriano P, Betsholtz C, et al. A specific requirement for PDGF-C in palate formation and PDGFR-alpha signaling. Nat Genet. 2004;36:1111–1116. doi: 10.1038/ng1415. [DOI] [PubMed] [Google Scholar]
- 49.Kaartinen V, Haataja L, Nagy A, Heisterkamp N, Groffen J. TGFβ3-induced activation of RhoA/Rho-kinase pathway is necessary but not sufficient for epithelio-mesenchymal transdifferentiation: implications for palatogenesis. Int J Mol Med. 2002;9:563–570. [PubMed] [Google Scholar]
- 50.Hagiwara N, Katarova Z, Siracusa LD, Brilliant MH. Nonneuronal expression of the GABAA β3 subunit gene is required for normal palate development in mice. Dev Biol. 2003;254:93–101. doi: 10.1016/s0012-1606(02)00030-1. [DOI] [PubMed] [Google Scholar]
- 51.Asada H, Kawamura Y, Maruyama K, Kume H, Ding RG, Kanbara N, Kuzume H, Sanbo M, Yagi T, Obata K. Cleft palate and decreased brain gamma-aminobutyric acid in mice lacking the 67-kDa isoform of glutamic acid decarboxylase. Proc Natl Acad Sci USA. 1997;94:6496–6499. doi: 10.1073/pnas.94.12.6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Condie BG, Bain G, Gottlieb DI, Capecchi MR. Cleft palate in mice with a targeted mutation in the γ-aminobutyric acid-producing enzyme glutamic acid decarboxylase 67. Proc Natl Acad Sci USA. 1997;94:11451–11455. doi: 10.1073/pnas.94.21.11451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gato A, Martinez ML, Tudela C, Alonso I, Moro JA, Formoso MA, Ferguson MW, Martinez-Alvarez C. TGF-β3-induced chondroitin sulphate proteoglycan mediates palatal shelf adhesion. Dev Biol. 2002;250:393–405. [PubMed] [Google Scholar]
- 54.Blavier L, Lazaryev A, Groffen J, Heisterkamp N, DeClerck YA, Kaartinen V. TGF-beta3-induced palatogenesis requires matrix metalloproteinases. Mol Biol Cell. 2001;12:1457–1466. doi: 10.1091/mbc.12.5.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Herron BJ, Lu W, Rao C, Liu S, Peters H, Bronson RT, Justice MJ, McDonald JD, Beier DR. Efficient generation and mapping of recessive developmental mutations using ENU mutagenesis. Nat Genet. 2002;30:185–189. doi: 10.1038/ng812. [DOI] [PubMed] [Google Scholar]
- 56.Juriloff DM. Mapping studies in animal models. In: Wyszynski DFE, editor. Cleft Lip and Palate: from Origin to Treatment. Oxford University press; 2002. pp. 265–282. [Google Scholar]
- 57.Juriloff DM, Harris MJ, Dewell SL. A digenic cause of cleft lip in A-strain mice and definition of candidate genes for the two loci. Birth Defects Res A Clin Mol Teratol. 2004;70:509–518. doi: 10.1002/bdra.20041. [DOI] [PubMed] [Google Scholar]
- ••58.Juriloff DM, Harris JM, Dewell SL, Brown CJ, Mager DL, Gagnier L, Mah DG. Birth Defects Res A Clin Mol Teratol. Vol. 73. 2005. Investigations of the genomic region that contains the clf1 mutation, a causal gene in multifactorial cleft lip and palate in mice; pp. 103–113. Identification of an inserted element in the 3′ region of the Wnt9b gene in a well-studied strain of mice suggests that interfering with this important cell signaling pathway results in cleft lip. It opens the door for additional studies of this pathway in humans and provides an important animal model for a relatively isolated cleft phenotype. [DOI] [PubMed] [Google Scholar]
- 59.Sharpe J. Optical projection tomography as a new tool for studying embryo anatomy. J Anat. 2003;202:175–181. doi: 10.1046/j.1469-7580.2003.00155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nolan PM, Hugill A, Cox RD. ENU mutagenesis in the mouse: application to human genetic disease. Brief Funct Genomic Proteomic. 2002;1:278–289. doi: 10.1093/bfgp/1.3.278. [DOI] [PubMed] [Google Scholar]
- 61.Yamada T, Fujiwara K, Mishima K, Sugahara T. Effect of ENU (ethylnitrosourea) mutagenesis in cleft lip and/or palate pathogenesis in mice. Int J Oral Maxillofac Surg. 2005;34:74–77. doi: 10.1016/j.ijom.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 62.Hayes C. Environmental risk factors and oral clefts. In: Wyszynski DFE, editor. Cleft Lip and Palate: from Origin to Treatment. Oxford University press; 2002. pp. 159–169. [Google Scholar]
- 63.Little J, Cardy A, Munger RG. Tobacco smoking and oral clefts: a meta-analysis. Bull World Health Organ. 2004;82:213–218. [PMC free article] [PubMed] [Google Scholar]
- 64.Finnell RH, Shaw GM, Lammer EJ, Brandl KL, Carmichael SL, Rosenquist TH. Gene-nutrient interactions: importance of folates and retinoids during early embryogenesis. Toxicol Appl Pharmacol. 2004;198:75–85. doi: 10.1016/j.taap.2003.09.031. [DOI] [PubMed] [Google Scholar]
- 65.Munger RG. Maternal nutrition and oral clefts. In: Wyszynski DFE, editor. Cleft Lip and Palate: from Origin to Treatment. Oxford University press; 2002. pp. 170–192. [Google Scholar]
- 66.Botto LD, Olney RS, Erickson JD. Vitamin supplements and the risk for congenital anomalies other than neural tube defects. Am J Med Genet. 2004;125:12–21. doi: 10.1002/ajmg.c.30004. [DOI] [PubMed] [Google Scholar]
- 67.Krapels IP, van Rooij IA, Ocke MC, van Cleef BA, Kuijpers-Jagtman AM, Steegers-Theunissen RP. Maternal dietary B vitamin intake, other than folate, and the association with orofacial cleft in the offspring. Eur J Nutr. 2004;43:7–14. doi: 10.1007/s00394-004-0433-y. [DOI] [PubMed] [Google Scholar]
- 68.Krapels IP, van Rooij IA, Ocke MC, West CE, van der Horst CM, Steegers-Theunissen RP. Maternal nutritional status and the risk for orofacial cleft offspring in humans. J Nutr. 2004;134:3106–3113. doi: 10.1093/jn/134.11.3106. [DOI] [PubMed] [Google Scholar]
- 69.Munger RG, Sauberlich HE, Corcoran C, Nepomuceno B, Daack-Hirsch S, Solon FS. Maternal vitamin B-6 and folate status and risk of oral cleft birth defects in the Philippines. Birth Defects Res A Clin Mol Teratol. 2004;70:464–471. doi: 10.1002/bdra.20037. [DOI] [PubMed] [Google Scholar]
- 70.Edison RJ, Muenke M. Central nervous system and limb anomalies in case reports of first-trimester statin exposure. N Engl J Med. 2004;350:1579–1582. doi: 10.1056/NEJM200404083501524. [DOI] [PubMed] [Google Scholar]
- 71.Edison RJ, Muenke M. Mechanistic and epidemiologic considerations in the evaluation of adverse birth outcomes following gestational exposure to statins. Am J Med Genet. 2004;131A:287–298. doi: 10.1002/ajmg.a.30386. [DOI] [PubMed] [Google Scholar]
- 72.Kallen B. Maternal drug use and infant cleft lip/palate with special reference to corticoids. Cleft Palate Craniofac J. 2003;40:624–628. doi: 10.1597/02-077. [DOI] [PubMed] [Google Scholar]
- 73.Cummings AM, Kavlock RJ. Gene-environment interactions: a review of effects on reproduction and development. Crit Rev Toxicol. 2004;34:461–485. doi: 10.1080/10408440490519786. [DOI] [PubMed] [Google Scholar]
- 74.Zeiger JS, Beaty TH. Gene-environment interaction and risk to oral clefts. In: Wyszynski DFE, editor. Cleft Lip and Palate: from Origin to Treatment. Oxford University press; 2002. pp. 283–289. [Google Scholar]
- 75.Jugessur A, Lie RT, Wilcox AJ, Murray JC, Taylor JA, Saugstad OD, Vindenes HA, Abyholm F. Variants of developmental genes (TGFA, TGFB3, and MSX1) and their associations with orofacial clefts: a case–parent triad analysis. Genet Epidemiol. 2003;24:230–239. doi: 10.1002/gepi.10223. [DOI] [PubMed] [Google Scholar]
- 76.Jugessur A, Lie RT, Wilcox AJ, Murray JC, Taylor JA, Saugstad OD, Vindenes HA, Abyholm FE. Cleft palate, transforming growth factor alpha gene variants, and maternal exposures: assessing gene-environment interactions in case-parent triads. Genet Epidemiol. 2003;25:367–374. doi: 10.1002/gepi.10268. [DOI] [PubMed] [Google Scholar]
- •77.Jugessur A, Wilcox AJ, Lie RT, Murray JC, Taylor JA, Ulvik A, Drevon CA, Vindenes HA, Abyholm FE. Am J Epidemiol. Vol. 157. 2003. Exploring the effects of methylenetetrahydrofolate reductase gene variants C677T and A1298C on the risk of orofacial clefts in 261 Norwegian case-parent triads; pp. 1083–1091. Both fetal and maternal alleles might influence the outcome of pregnancy, particularly for genes that are involved in the metabolism of essential nutrients or the detoxification of harmful chemicals. A special strength of this study was that it assessed effects of the mother’s alleles separately from those of the fetal alleles at two MTHFR variants (C677T and A1298C) in a case–parent triad setting. There was no indication of an increased risk with the child’s genotypes at either C677T or A1298C, but mothers with either CT or TT at C677T appeared to lower the risk of CL/P in their children. In CPO, a dominant pattern of increased risk was observed with the child’s C677T genotypes. Haplotype analysis in the cleft palate category showed that, except for 677T/1298A, none of the other haplotypes were transmitted significantly in excess or in deficiency. [DOI] [PubMed] [Google Scholar]
- 78.Weinberg CR. Studying parents and grandparents to assess genetic contributions to early-onset disease. Am J Hum Genet. 2003;72:438–447. doi: 10.1086/346171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Koillinen H, Lahermo P, Rautio J, Hukki J, Peyrard-Janvid M, Kere J. A genome-wide scan of non-syndromic cleft palate only (CPO) in Finnish multiplex families. J Med Genet. 2005;42:177–184. doi: 10.1136/jmg.2004.019646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, Meyers J, Leitch CC, Katsanis N, Sharifi N, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 81.Cui XM, Shiomi N, Chen J, Saito T, Yamamoto T, Ito Y, Bringas P, Chai Y, Shuler CF. Overexpression of Smad2 in Tgf-β3-null mutant mice rescues cleft palate. Dev Biol. 2005;278:193–202. doi: 10.1016/j.ydbio.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 82.Liu W, Sun X, Braut A, Mishina Y, Behringer RR, Mina M, Martin JF. Distinct functions for Bmp signaling in lip and palate fusion in mice. Development. 2005;132:1453–1461. doi: 10.1242/dev.01676. [DOI] [PubMed] [Google Scholar]
- 83.Zeiger JS, Beaty TH, Liang KY. Oral clefts, maternal smoking, and TGFA: a meta-analysis of gene–environment interaction. Cleft Palate Craniofac J. 2005;42:58–63. doi: 10.1597/02-128.1. [DOI] [PubMed] [Google Scholar]