

Abstract
Placental growth factor (PGF, previously known as PlGF) is prominently expressed by trophoblasts in human placenta, whereas most nontrophoblast cells express low levels of PGF mRNA under normal physiological conditions. We have shown that hypoxia decreases PGF expression in the trophoblast, but little is known about transcriptional regulation of PGF gene expression. We sought to determine promoter regions of the human PGF gene that contribute to its restricted high constitutive expression in the trophoblast. Overlapping putative promoter regions of human PGF gene encompassing −1.5 kb were cloned into reporter vectors and co-transfected into trophoblast and nontrophoblast cell lines. Promoter activity generated by a −1.5-kb clone was significantly higher in trophoblasts than in nontrophoblasts. Selective deletion mutants showed that a clone encompassing the PGF (−828/+34) region generated promoter activity similar to the −1.5-kb region in the trophoblast. However, deletion of another 131 bp from this subclone (−698/+34) resulted in significantly less promoter activity in the trophoblast. The (−828/−698) region significantly enhanced activity of a minimal promoter construct in trophoblast but not in nontrophoblast cells, suggesting that this region contributes to regulating PGF transcription in the trophoblast. Site-directed mutagenesis of a glial cell missing 1 (GCM1) motif in the 131-bp region significantly decreased enhancer activity in the trophoblast. Furthermore, overexpression of GCM1 significantly increased PGF −1.5-kb promoter activity and PGF mRNA expression in trophoblast and nontrophoblast cells. Forced overexpression of GCM1 restored PGF expression in the hypoxic trophoblast. These data support a functional role for GCM1 contributing to constitutively high trophoblast PGF expression and is the first direct evidence of an oxygen-responsive, trophoblast-specific transcription factor contributing to the regulation of PGF expression.
Keywords: gene regulation, growth factors, oxygen tension, placenta, placenta growth factor, placental growth factor, PlGF, pregnancy, transcription, trophoblast
Introduction
Placental growth factor protein (PGF, previously known as PlGF) is a member of the vascular endothelial growth factor (VEGF) family of proangiogenic factors [1] and is highly expressed by trophoblasts [2, 3] and decidual natural killer cells [4] within the human maternal-fetal interface. PGF exists as different protein isoforms due to alternative splicing of the mRNA from a single copy gene [5, 6], and four isoforms are expressed by trophoblasts [7]. PGF is chemotactic, mitogenic, and angiogenic for cultured endothelial cells and induces angiogenesis in vivo [8]. PGF overexpression produces abundant angiogenesis in normal mouse tissues [9] and stimulates collateral vessel growth in ischemic tissues [10]. Although PGF expression is normally low in most other organs/tissues, expression is inducible and required in tissues undergoing pathological angiogenesis [11], including vascularized tumors [12]. In addition to its known angiogenic activity, PGF promotes extravillous trophoblast proliferation [13], while data from our laboratory [14] and others [15] have shown that PGF can also function as a survival factor for trophoblasts. Furthermore, PGF induces relaxation of human placental vessels, which likely contributes to the reduction in blood flow impedance within the fetoplacental circulation [16]. Thus, PGF may contribute to trophoblast function as well as vascular development and function in the placenta.
We have shown that serum titers of PGF protein increase dramatically early in the second trimester of normal human pregnancy, whereas titers are significantly reduced in women diagnosed with preeclampsia [17]. Subsequently, it was found that decreased blood [18, 19] and urinary [20] levels of PGF protein early in pregnancy are promising predictors of the subsequent development of preeclampsia. Quantitative studies have confirmed that preeclamptic trophoblasts express less PGF mRNA and protein than normal trophoblasts [21, 22], and reduced bioavailability of PGF and VEGF produces a preeclampsia-like syndrome including hypertension, proteinuria, and glomerular endotheliosis in a pregnant rodent model [23]. Aberrations in trophoblast and/or leukocyte PGF production may compromise endothelial cell, trophoblast, and vascular function during gestation and contribute to the pathophysiologies commonly noted in perfusion-compromised pregnancies [24]. Although low oxygen tensions are known to decrease PGF expression in trophoblasts [3, 21], the intrinsic molecular and cellular mechanisms that function to regulate PGF gene expression in human trophoblasts are not known. In this study, we describe for the first time the unique functional transcriptional regulatory regions of the human PGF gene and demonstrate that the transcription factor glial cell missing 1 (GCM1) contributes to the constitutive expression of PGF in the human trophoblast under different oxygen tensions.
Materials and Methods
Computational Analysis of the PGF 5′UTR for Putative Promoter Regions
As a reference to begin to determine the functional promoter region of human PGF, a 2500-bp upstream sequence of the human PGF gene was analyzed using two independent search engines: WWW Promoter Scan from the National Institutes of Health (Bethesda, MD; http://www-bimas.cit.nih.gov/molbio/proscan/) [25] and Genomatix PromoterInspector (Munich, Germany; http://genomatix.de). These promoter scan programs are designed to locate regions of DNA that contain significant numbers and types of known transcription factor binding sites associated with eukaryotic polymerase II promoter sequences.
Cloning of 5′ PGF UTR
Human yeast artificial chromosome hYAC 964_e_2 was used as the template for generating PGF upstream regions. Different lengths of the 5′ untranslated upstream region of PGF were generated by PCR amplification using primer sequences given in Table 1. The PCR cycling conditions were: 95°C for 3 min, 30 cycles of 95°C for 1 min, 63°C for 1 min, and 70°C for 2 min. Resultant PCR amplicons of the appropriate predicted sizes were cloned into the beta-galactosidase reporter vector pBlue-TOPO-TA (Invitrogen, Carlsbad, CA), according to the manufacturer's instructions and transformed into chemically competent DH5-α E.coli cells, and plasmid DNA was isolated by QIAprep Miniprep kit (Qiagen, Valencia, CA). All clones were sequenced in both directions (University of Illinois Biotechnology Center, Urbana, IL), and the validity of the sequences was verified against the published human genome sequence.
Table 1.
Primer sequences used to generate the different human PGF promoter clones.
| Primer name (sequence location) | Sequence |
|---|---|
| 4 Antisense (+34 bp) | 5′-GCAGGAAGCAAGGGAACAGCC-3′ |
| 8 Sense (−305 bp) | 5′-AGCTACGGGAGGACCTGGAGT-3′ |
| 10 Sense (−698 bp) | 5′-CCTGCAGAGGCGGTTAGATCA-3′ |
| 11 Sense (−828 bp) | 5′-CCATTCGACATATGCAGGCAG-3′ |
| 5 Antisense (−284 bp) | 5′-ACTCCAGGTCCTCCCGTAGCT-3′ |
| 6 Antisense (−650 bp) | 5′-GTCCAGCGTCAGCCGTCAAGG-3′ |
| 9 Sense (−1042 bp) | 5′-CTTCCCAAGGAGGCAACAGGC-3′ |
| 4 Sense (−1521 bp) | 5′-CAGAAGGCAGTGAGAGGAGTGG-3′ |
Cell Culture
Human choriocarcinoma cell lines JEG-3 and JAR and nontrophoblast cell lines hEK-293 (embryonic kidney; all from ATCC, Manassas, VA), HeLa (cervical carcinoma, gift from Dr. Mary McAsey, Southern Illinois University School of Medicine), and MCF-7 (breast carcinoma, gift from Dr. Jay Wimalasena, University of Tennessee) were grown in Dulbecco modified Eagle medium (DMEM; Fisher Scientific, Hanover Park, IL) supplemented with 10% fetal bovine serum (Sigma, St. Louis, MO) and antibiotics (penicillin G [50 U/ml] and streptomycin [50 μg/ml]; Fisher Scientific). Cells were cultured under 21% O2 or 1% O2 oxygen tensions as previously described [22].
Transient Transfection Assays
Transfections of the cell lines were performed using ExGen-500 (MBI Fermentas, Hanover, MD) according to the manufacturer's instructions. In experiments testing PGF promoter responses, 1.5 μg of the various PGF reporter constructs were co-transfected with 0.5 μg of pRSVLuc vector, and cells were incubated at 37°C. For GCM1 overexpression experiments, 0.75 μg of PGF −1.5-kb clone was cotransfected with either 1.0 μg of pHA-GCMa [26] (gift from Dr. Hungwen Chen, Institute of Biological Chemistry, Academia Sinica, Taiwan) or 1.0 μg of its corresponding empty vector pEF1/MycHis. Transfection efficiencies in these experiments were controlled with 0.25 μg of pRSVLuc. For GCM1 overexpression under hypoxic conditions, JEG-3 and JAR cells were transfected with 2 μg of pHA-GCMa or pEF1-HisMyc and cultured under 21% O2 or 1% O2 conditions for 24 h. For all experiments, the transfection medium was changed and fresh medium (DMEM + 10% fetal calf serum) was added 3 h after transfection. After an additional 48 h, cells were lysed and chemiluminescence detected using the Dual Light Kit (Applied Biosystems, Foster City, CA). All transfections were done in duplicate, and each individual lysate was tested in duplicate. Results were quantified using a Beckman Coulter LD400 Luminometer with dual injectors (Fullerton, CA). Luciferase values were used to normalize beta-galactosidase values to control for transfection efficiency. Luciferase and beta-galactosidase activities expressed by untransfected cell lysates were used as background and subtracted from all transfected cells. Luciferase readings were averaged across a replicate group, and each luciferase reading was corrected by dividing the group average luciferase reading by the individual luciferase reading. This corrected luciferase reading was then multiplied to the average beta-galactosidase reading to give a corrected beta-galactosidase activity. Relative changes in expression levels were calculated as fold differences in beta-galactosidase activity from a control clone in which the region from −1521 to −650 was inserted in an opposite orientation (−1521/−650opp). This control clone produced no significant beta-galactosidase activity in any of the cell lines (P = 0.19, paired Student t-test versus nontransfected cells). Therefore, basal beta-galactosidase values of the control (−1521/−650opp) clone were set to equal 1 to calculate relative fold changes in promoter activity for each transfection. Statistical analyses were performed between groups with the Student t-test, and significance was set as P < 0.05. All experiments were repeated at least three times, and all data is presented as mean ± SEM.
Functional Analysis of the PGF (−828/−698) Region
The clone encompassing (−1521/+34) was used as the template for PCR amplification of the (−828/−698) region with forward and reverse primers (5′-GAATTCGTCCATTCGACATATGCAGG-3′ and 5′-GAATTCTAACCGCCTCTGCAGGAG-3′, respectively). PCR reactions were carried out using puReTaq Ready-To-Go PCR Beads (Amersham Biosciences, Piscataway, NJ), and the cycling conditions were 94°C for 5 min followed by 31 cycles of 94°C for 30 sec, 56.6°C for 30 sec, and 72°C for 30 sec. An additional 10 min at 72°C was included at the end of 31 cycles. PCR amplicons were analyzed on 1.0% agarose gels to verify expected length, cloned into the pBlue-TOPO-TA, and subsequently subcloned into pMLuc-2 (Novagen, San Diego, CA). Clones were identified by bidirectional sequencing to verify sequence and orientation of the insets. Clones with sense or antisense orientation of (−828/−698) region were named pE173-MlucS and pE173-MlucAS, respectively. Transient transfection assays were performed as above except that chemiluminescence was detected with Dual-Luciferase Reporter Assay system (Promega, Madison, WI). Relative changes in expression levels were calculated as fold differences in corrected Renilla luciferase activity from empty pMLuc-2 vector.
Site-Directed Mutagenesis
GCM1 site mutation (indicated by lowercase nucleotides) was introduced with mutagenic primers (GCM-MS 5′-CCGAGAGCACCCCTACCTtgATATGTCGAATGGAC-3′ and GCM-MAS 5′-GTCCATTCAACATATcaAGGCAGGGGTGCTCTCAA-3′) using the (−844/−678) clone pE173-MlucS as template. These mutagenic primers were extended during PCR cycling with Pfu Turbo DNA polymerase (Quik Change XL Site-Directed Mutagenesis kit; Stratagene, LaJolla, CA). Dpn1 digestion was carried out afterwards to digest the potential DNA template and to select for the mutants. Mutants were confirmed by bidirectional sequencing and verified against pE173-MlucS and named pE173-GCM. Transient transfection assays were performed as above. GCM1 function was determined as fold differences in Renilla luciferase activity between pE173-GCM and pE173-MlucS.
Real-Time RT-PCR
Cells were lysed 48 h after transfection. RNA was extracted and 200 ng of total RNA converted to cDNA with iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA), and 1/10th of the cDNA was subjected to quantitative PCR using primers: (F): 5′-AGAAGATGCCGGTCATGAG-3′ and (R): 5′-ACACTTCCTGGAAGGGTAC-3′. Ribosomal protein (RPL32) was used as the normalization control gene and was amplified with RPL32(F): 5′-CCCAAGATCGTCAAAAAGA-3′ and RPL32(R): 5′-TCAATGCCTCTGGGTTT-3′ [27]. PCR reactions were performed in the iQ5 Real Time PCR Detection System with iQ SYBR Green Supermix (Bio-Rad). The cycling conditions for PGF and RPL32 were: 95°C for 4.5 min, 40 cycles of 95°C for 30 sec, 62°C (PGF) or 53°C (RPL32) for 30 sec, and 72°C for 30 sec. PCR reaction efficiencies were monitored and found to be 93.7% (PGF) and 95.1% (RPL32). PGF expression was normalized to RPL32, and relative change of expression between treatment and control was calculated by the 2−ΔΔCT formula [28].
Immunoblots for HA-GCM1 Overexpression
Cells were lysed 48 h after transfection in lysis buffer (20 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1% Triton X-100, 10 mM PMSF, 1 mM Na3VO4, and 10 nM NaF) supplemented with complete protease inhibitor cocktail (Calbiochem, San Diego, CA), and protein concentrations were determined using the DC protein Assay kit (Bio-Rad). Ten micrograms of each cell lysate was separated on 12% SDS-PAGE and transferred to a nitrocellulose membrane. Membranes were blocked with 5% BSA in Tris-buffered saline containing 0.1 % Tween-20 (TBST) for 1 h at room temperature and incubated with rabbit anti-HA antibody (1:2000; Bethyl, Montgomery, TX) overnight at 4°C. Membranes were washed with TBST 3 times, incubated with anti-rabbit horseradish peroxidase-conjugated secondary antibody (1:10 000; Santa Cruz Biotechnology, Santa Cruz, CA) for 1 h at room temperature, then washed 3 times with TBST and once with TBS. Immunoreactivty was detected by chemiluminesence as described (Amersham Biosciences) for 1 min. Membranes were stripped (0.2 M glycine [pH 2.2], 0.1% SDS, and 10% Tween 20) for 20 min at room temperature, then reprobed for human β-actin (Sigma) to control for loading consistency.
GCM1 Binding Assay
GCM1 binding to the potential binding site contained within the PGF promoter was performed as described [29] with minor modifications. Briefly, equimolar concentrations (10 pmol) of a biotinylated 42-mer oligonucleotide containing triplicate repeats of the putative GCM1 sequence (5′ATGCAGGC3′) present in the −828/−698 region of PGF (5′ CATATGCAGGCAGGCATATGCAGGCAGGCATATGCATATGCAGGCAGG 3′) or a non-sense control sequence (5′ AAGCTTTAAATTCCGCCCAAGTTTTAAATTCCG CCCAAGCTT 3′) were annealed. Each biotinylated double-stranded oligonucleotide was bound to avidin beads (VECTREX Avidin-D matrix; Vector Laboratories, Burlingame, CA), washed with 1 ml of TBST, and preincubated with 20 μg of nuclear extract from hEK-293 cells to reduce non-specific binding. Both the GCM1-nucleotide-bead matrix and the control nucleotide-bead matrix (24 μl) were combined with 20 μg of nuclear extract from hEK-293 cells, previously transfected with pHA-GCMa, in binding buffer (100 mM Tris [pH 7.5], 50 mM KCl, 1 mM MgCl2, 1 mM EDTA, 5.5 mM dithiothreitol, 10% Glycerol, 0.03% NP-40, 200 mM Na3VO4, 100 mM PMSF) containing 0.1 μg of sheared salmon sperm DNA and allowed to incubate for 30 min with end-over-end rotation at room temperature. The matrix was pelleted by brief centrifugation, unbound proteins in the supernatant were removed, and the beads were washed twice with 1 ml of binding buffer. The beads were resuspended in 30 μl of 4×Laemmli sample buffer (Sigma), boiled for 5 min, and centrifuged to pellet the matrix. Proteins in the supernatant were separated on a 10% SDS-PAGE gel, transferred to nitrocellulose membrane, and immunoblotted for HA using conditions described above.
Results
Generation of PGF Reporter Constructs
To determine functional promoter regions responsible for high constitutive trophoblast-specific expression and to identify regions that may regulate trophoblast restricted transcription of PGF, selective reporter clones of the PGF 5′ UTR were generated (Fig. 1). Two independent promoter analysis programs predicted putative, overlapping, basal promoter regions between −538 bp and −346 bp and −666 bp and −416 bp upstream of the first ATG codon. These predicted promoter sequences overlap a common 122 bases and lie within 20–96 bp, respectively, of the single transcription start site of the hPGF gene (−320 bp) [1, 30]. Three clones—PGF (−305/+34), PGF (−698/+34), and PGF (−828/+34)—contain the first ATG codon of the translation start site of hPGF and extend to −828 bp upstream. Clone PGF (−698/−284) contains the predicted promoter sites from the −346 bp to −666 bp region. Clones PGF (−1042/−650) and PGF (−1521/−650) contain regions upstream of the predicted promoter sites. Clone PGF (−1521/+34) spans 1.5 kb upstream of the PGF gene.
Fig. 1.
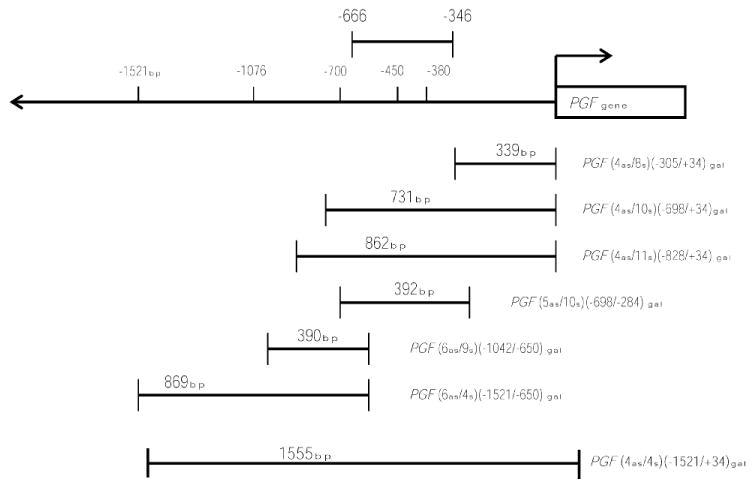
Schematic representation of PGF promoter clones. Putative PGF promoter clones were generated by ligating upstream regions of human PGF gene amplicons into a beta-galactosidase reporter vector. Clones of different sizes spanning a 1.5-kb upstream region of the PGF gene are depicted. Computational analyses of the upstream DNA sequences by two independent programs predicted that regions between −666 and −346 contained DNA binding motifs able to provide promoter activity.
Functional Analysis of hPGF Promoter Constructs in Trophoblast Cell Lines
The promoter constructs were transfected in two choriocarcinoma cell lines, JEG-3 and JAR, to determine functional activities. Both of these trophoblast cell lines have been shown to express high levels of PGF mRNA under standard culture conditions [3, 25]. The three clones that expressed significant promoter activity were: −1.5-kb clone (−1521/+34), (−828/+34), and (−698/+34) (Fig. 2A). In both trophoblast cell lines, promoter activity of the −1.5-kb clone (1521/+34) was significantly higher than that of cells transfected with the (−1521/−650opp) control clone. JEG-3 cells showed a 14.3 ± 1.1 (P < 0.001)-fold increase in beta-galactosidase activity, while JAR cells showed 10.5 ± 2.1 (P < 0.005)-fold increase in beta-galactosidase activity. There was no significant difference (P = 0.63) in the expression levels of the −1.5-kb clone between the two trophoblast cell lines. These results confirm the ability of the −1.5-kb region of the PGF 5′UTR to drive reporter gene expression in the trophoblast.
Fig. 2.
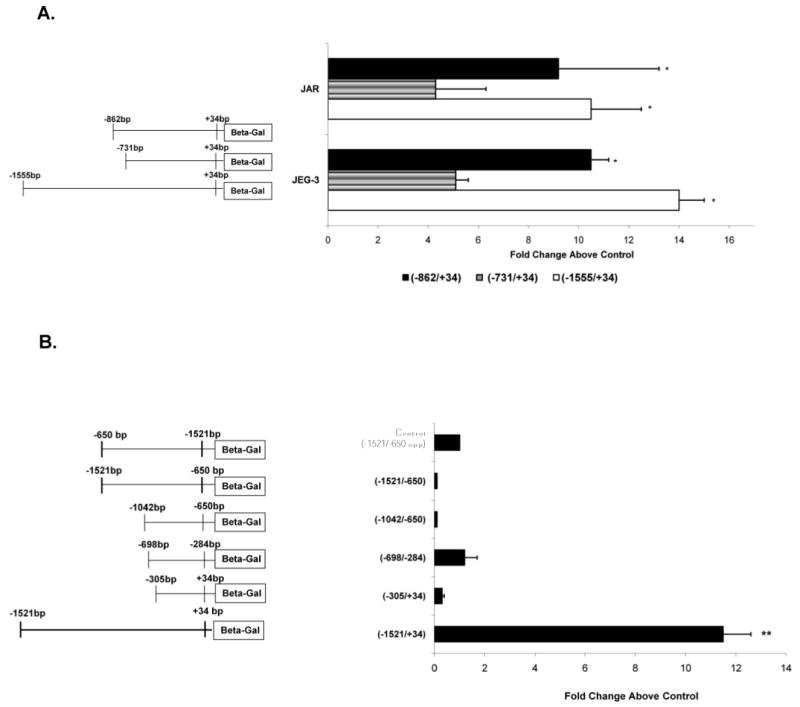
Functional promoter activity within 1.5 kb upstream of the PGF gene. A) JEG-3 and JAR cells were transfected individually with 1.5 μg of clone (−828/+34), clone (−698/+34), or the −1.5-kb clone (−1521/+34) and 0.5 μg of pRSVLuc. After 48 h, cell lysates were prepared and chemiluminesence measured as described in Materials and Methods. The relative fold increase in activity above levels produced by the transfection control clone (−1521/−650opp) for each of the clones in JEG-3 and JAR cells are shown. All activities shown are significantly higher than those produced by the control clone (set = 1). *, P < 0.05 versus activity produced by the (−698/+34) clone. B) Promoter activity produced by clones (−1521/+34), (−305/+34), (−698/−284), (−1042/−650), and (−1521/−650) in JEG-3 cells. **, P < 0.05 between activities of the promoter clones versus transfection control clone (−1521/−650opp) (set = 1).
Trophoblast cells transfected with clone (−698/+34), which contains both the predicted promoter region as well as the gene proximal 284-bp region, produced significantly (P < 0.05) higher promoter activity in JEG-3 cells (5.2 ± 0.5-fold) and in JAR cells (4.3 ± 1.6-fold) as compared to promoter activity produced by the control clone. However, the promoter activity exhibited by this clone was significantly lower than promoter activity produced by the −1.5-kb clone in JEG-3 cells (P < 0.001) and in JAR cells (P < 0.05; Fig. 2A).
Interestingly, clone (−828/+34), which contains an additional 131 bp upstream of the (−698/+34) region, produced 10.5 ± 0.7–fold promoter activity in JEG-3 cells and 9.2 ± 3.3 (P < 0.05)-fold promoter activity in JAR cells. There was no significant difference (P > 0.07) observed in the promoter activity of clone (−828/+34) and that of clone (−1.5 kb/+34) in JEG-3 or JAR cells (Fig. 2A). This clone produced a 2-fold increase in promoter activity compared to the (−698/+34) clone, suggesting that the region between −828 bp and −698 bp contains elements that can augment high trophoblast-specific promoter activity.
Subsequent analyses of various deletion mutant clones of the −1.5-kb region showed that clones encompassing (−305/+34), (−698/−284), (−1042/−650), and (−1521/−650) produce little to no promoter activity in JEG-3 cells (Fig. 2B). Identical results were obtained with all of these deletion clones in JAR cells (data not shown). In particular, clone (−698/−284), which encompasses both of the predicted putative promoter regions, produced low promoter activity that was not significantly above control levels (P = 0.7) in either JEG-3 or JAR cells. Thus, the predicted promoter region between −698 bp and −284 bp is not sufficient alone to produce promoter activity in trophoblast cells and requires the involvement of accessory elements for maximal activity.
Functional Analyses of hPGF Promoter Constructs in Nontrophoblast Cell Lines
To determine regions in the PGF promoter responsible for directing tissue-specific expression, we compared the promoter activity of the clones in the two trophoblast cell lines with that generated in three different nontrophoblast cell lines: hEK-293, HeLa, and MCF-7 cells. Promoter activities produced by the −1.5-kb clone in hEK-293 (4.2 ± 1.6–fold) and MCF-7 cells (3.6 ± 1–fold) were significantly higher than compared to the control clone (−1521/−650)opp (Fig. 3A). Clone −1.5 kb failed to produce promoter activity above background in HeLa cells. Although activity in the hEK-293 and MCF-7 cells was significantly higher than control clone activity, this intermediate level of activity was significantly (P < 0.05) lower compared to the mean activity of this region in the trophoblast cells. To validate these reporter clone studies, we determined the relative expression levels of endogenous PGF mRNA in the cells by real time RT-PCR. Expression of PGF mRNA in the HeLa and hEK-293 cell lines under standard culture conditions was found to be much lower than that seen in the trophoblast cell lines (Fig. 3B). Endogenous PGF mRNA was almost undetectable in HeLa cells, which is in agreement with a previous report [31] and the promoter clone data above. Similarly, the intermediate activity of the 1.5-kb clone in MCF-7 and hEK-293 cells corresponds well to the relative levels of PGF mRNA found in hEK-293 cells and previously reported by us in MCF-7 cells [32]. Collectively, these findings show that the promoter activity produced by the −1.5-kb clone in trophoblast and nontrophoblast cells accurately reflects PGF mRNA expression levels by the different cell lines and further highlights cell type-specific regulation of PGF expression. Similar to that seen in trophoblast cells, undetectable levels of promoter activity were produced by clones (−305/+34), (−698/−284), (−1042/−650), and (−1521/−650) in all three nontrophoblast cells (data not shown). The ability of the (−1.5 kb/+34) clone, but not clone (−305/+34), to produce significant activity in some, but not all, nontrophoblast cells suggests that regions between −1.5 and −305 may be important for regulating promoter activity in these cells in a similar manner to trophoblast cells.
Fig. 3.
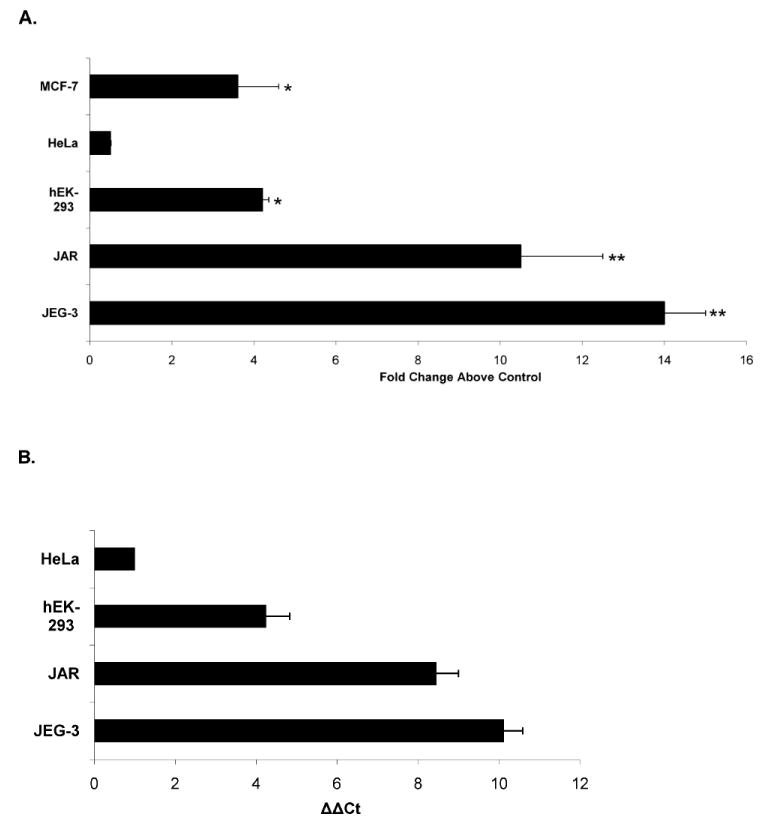
PGF promoter activity and mRNA expression are significantly lower in nontrophoblast cells than trophoblast cells. A) MCF-7, HeLa, hEK-293, JEG-3, and JAR cells were transfected with 1.5 μg of clone (−1521/+34) or 1.5 μg of control plasmid (−1521/−650opp) and 0.5 μg of pRSVLuc. Cells were processed and relative fold increase in activity above levels produced by the transfection control clone (−1521/−650opp) (set to = 1) in each cell type are shown. *, P < 0.05 comparing promoter activities produced by the −1.5-kb clone and (−1521/−650opp) control clone in each cell type. **, P < 0.005 comparison of promoter activity of the −1.5-kb clone in trophoblast cells to that produced in each nontrophoblast cell type. B) Relative PGF mRNA expression in trophoblast and nontrophoblast cell lines. Total RNA (200 ng) from each cell line was reverse transcribed into cDNA and subjected to qPCR for both PGF and RPL32 detection. ΔCT of HeLa (CTPgf-CTRPL32)-1 is used as the normalization control, and ΔΔCT of each cell type was plotted to show PGF mRNA expression level relative to that in HeLa cells. Data represents mean values (± SEM) of at least three independent measurements.
Functional Analyses of Tissue-Specific Regulatory Regions of Human PGF
To further characterize potential tissue-specific regulatory regions of the PGF 5′ UTR, promoter activities of clones (−698/+34) and (−828/+34) were determined in nontrophoblast cells (Fig. 4). Clone (−698/+34) produced approximately 3-fold more promoter activity in HeLa and hEK-293 cells than the control clone (−1521/−650opp). In contrast, addition of the 131-bp sequences (clone −828/+34) produced approximately 6-fold less activity versus that produced by the (−698/+34) clone in the same cells. There was little difference in mean promoter activity of these clones in the MCF-7 breast cancer cells. Collectively, this pattern of promoter activity is opposite to that seen in trophoblast cells by these clones, where inclusion of the 131-bp region between −828 and −698 resulted in an approximate 2-fold increase in promoter activity (Fig. 4). Thus, the presence of this 131-bp region in clone (−828/+34) compared to clone (−698/+34) decreases promoter activity in most nontrophoblast cells, but increases promoter activity in trophoblast cells.
Fig. 4.
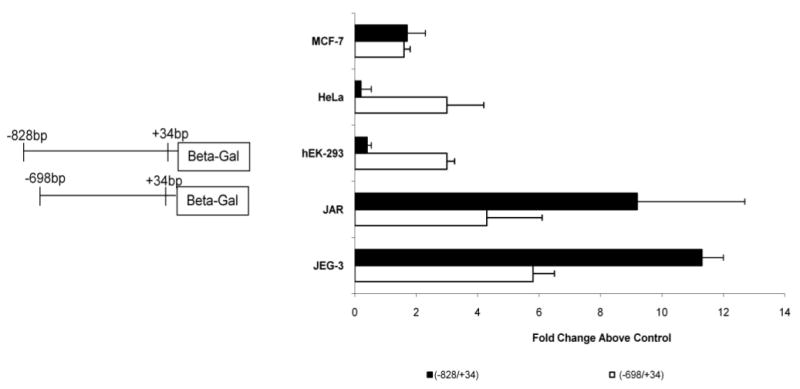
Tissue-specific PGF expression is mediated by the region between −828 and −698 bp upstream of the PGF translation start site. JEG-3, JAR, hEK-293, HeLa, and MCF-7 cells were transfected with 1.5 μg of clone (−698/+34) or clone (−828/+34) and 0.5 μg of pRSVLuc. Cells were processed and fold activity above background plotted as in Figure 2. Clone (−828/+34) produced high promoter activity in trophoblast cells JEG-3 and JAR, but not in the nontrophoblast cells hEK-293 and HeLa. Clone (−698/+34) produced relatively similar activity in the various cell lines.
GCM1 Functions to Regulate PGF Promoter Activity and Gene Expression
To specifically confirm the functional ability of this 131-bp PGF promoter region to promote transcriptional activity in trophoblasts, the PGF region encompassing (−828/−698) was cloned downstream of a minimal thymidine kinase (TK)-driven Renilla luciferase cassette in both sense and antisense orientations. In trophoblasts, both the sense- and antisense-oriented 131-bp element generated a significant increase in basal TK promoter activity (Fig. 5A). In contrast, the 131-bp element in either orientation did not significantly influence TK promoter activity in the HeLa or hEK-293 cells. Differences in functional activity of these clones in the cells suggest the presence of trophoblast-specific transactivating factors that recognize unique DNA elements within the region.
Fig. 5.
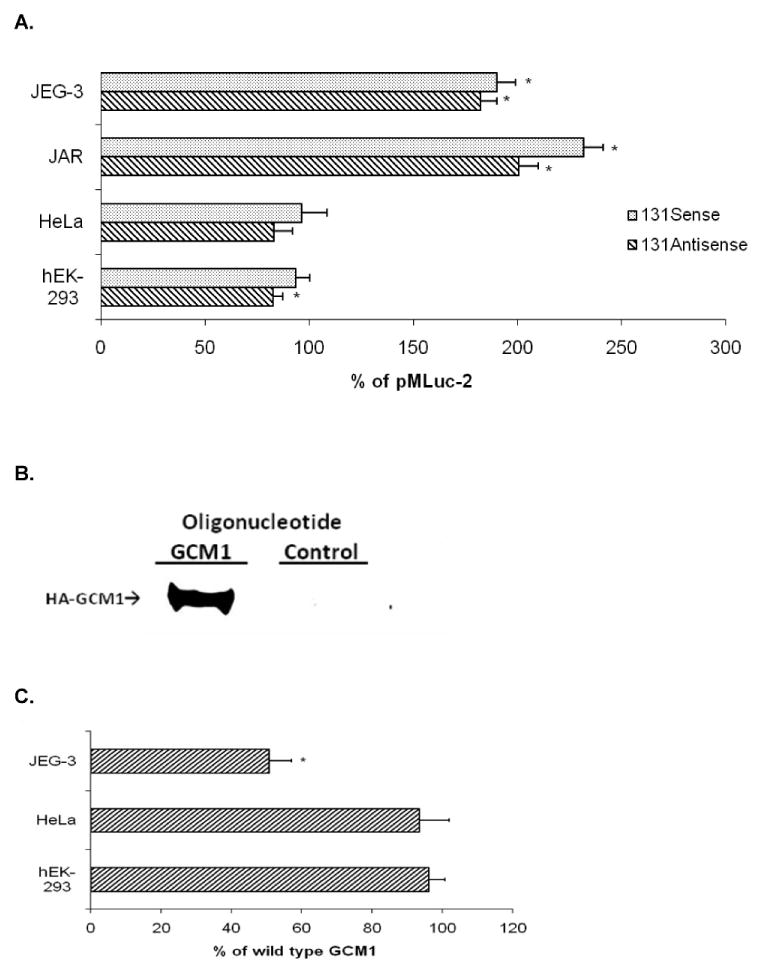
PGF (−828/−698) region enhances basal promoter activity in trophoblast but not nontrophoblast cells. A) JEG-3, JAR, HeLa, and hEK-293 cells were transfected with 1.5 μg of pE173-MlucS or pE173-MlucAS clones and 0.5 μg of pRSVLuc. Statistical analyses compared promoter activity of each individual clone with its parental control clone pMLuc-2 (set to 100%) in each cell type (*, P < 0.005). B) Nuclear extracts from hEK-293 cells previously transfected with pHA-GCM1 were incubated with bead-conjugated oligonucleotides containing either three copies of the putative GCM1 motif from human PGF (GCM1) or corresponding scrambled sequences (control). Western blots for HA-tagged GCM1 (HA-GCM1) were carried out to examine the binding ability of the GCM1 motif. C) GCM1 element of PGF (−828/−698) region in pE173-MLucS was mutated and transfected into JEG-3, HeLa, and hEK-293 cells. Activity of mutant GCM1 element relative to wild-type activity in each cell type was plotted (*, P < 0.005).
To elucidate functional elements that may be responsible for enhancing PGF gene expression in the trophoblast, we analyzed the (−844/−678) region electronically and identified a motif at position −818 to −810 with 87.5% sequence homology with consensus GCM1 motif (A/G)CCCGCAT [33]. In vitro binding experiments were used to investigate the functional ability of this motif to bind GCM1 protein (Fig. 5B). These studies established that the putative GCM1 motif in PGF is capable of directly binding exogenously expressed GCM1 protein in nuclear extracts of transfected hEK-293 cells (Fig. 5B). These results are in agreement with previous findings showing that a sequence with a similar mismatch maintains high binding capacity for Drosophila and murine GCM1 [34]. GCM1 protein is expressed in trophoblast cells [35] and has been shown to specifically regulate expression of certain genes in these cells [36]. To verify that the putative GCM1 element functions to regulate PGF enhancer activity, we carried out site-directed mutagenesis of the GCM1 motif in the (−844/−678) region. Transient expression experiments of the promoter constructs were performed in trophoblast and nontrophoblast cells to compare functional activity of wild-type GCM1 to mutated GCM1 motif (Fig. 5C). Mutation of the GCM1 motif resulted in significantly less promoter activity compared to wild-type GCM1 in the trophoblast. However, there was no significant difference in activity between mutated and wild-type GCM1 constructs in nontrophoblast cells. These data provided the initial evidence suggesting that the GCM1 motif within the (−844/−678) region of PGF contributes to transcriptional activity in the trophoblast.
To confirm the role of GCM1 in regulating PGF transcriptional activity, we overexpressed exogenous GCM1 in trophoblast and nontrophoblast cells in the presence of the −1.5-kb reporter construct (Fig. 6A). Real time RT-PCR analyses indicated that relative expression levels of endogenous GCM1 mRNA were similar between the two trophoblast cell lines and that they expressed approximately 32-fold more GCM1 mRNA than did the two nontrophoblast cells (data not shown). GCM1 overexpression induced significant PGF promoter activity in JEG-3 (3.1-fold), JAR (11.0-fold), HeLa (3.4-fold), and hEK-293 (72-fold) cells (Fig. 6B). To confirm the ability of GCM1 to specifically induce PGF gene expression, we overexpressed GCM1 protein in HeLa and hEK-293 cells and monitored endogenous PGF mRNA expression (Fig. 6C). The significant increase in PGF promoter activity following overexpression of exogenous GCM1 in these cells (Fig. 6B) was reflected in a significant increase in expression of PGF mRNA in the cells (Fig. 6C). Overexpression of GCM1 increased PGF mRNA in HeLa cells, with the highest induction of ∼108-fold with 1.0 μg of pHA-GCM1 as compared to transfections with backbone vector pEF1-MycHis. Similarly, in hEK-293 cells, as little as 0.5 μg pHA-GCM1 increased PGF mRNA expression 19-fold, and the induced PGF mRNA expression levels were similar up to 2.0 μg of pHA-GCM1. Transfection of the GCM1 expression vector into trophoblast cells also increased endogenous PGF mRNA levels ∼30% in the already highly expressing JEG-3 cells and approximately 2.5- to 4.0-fold in the JAR cells (data not shown).
Fig. 6.
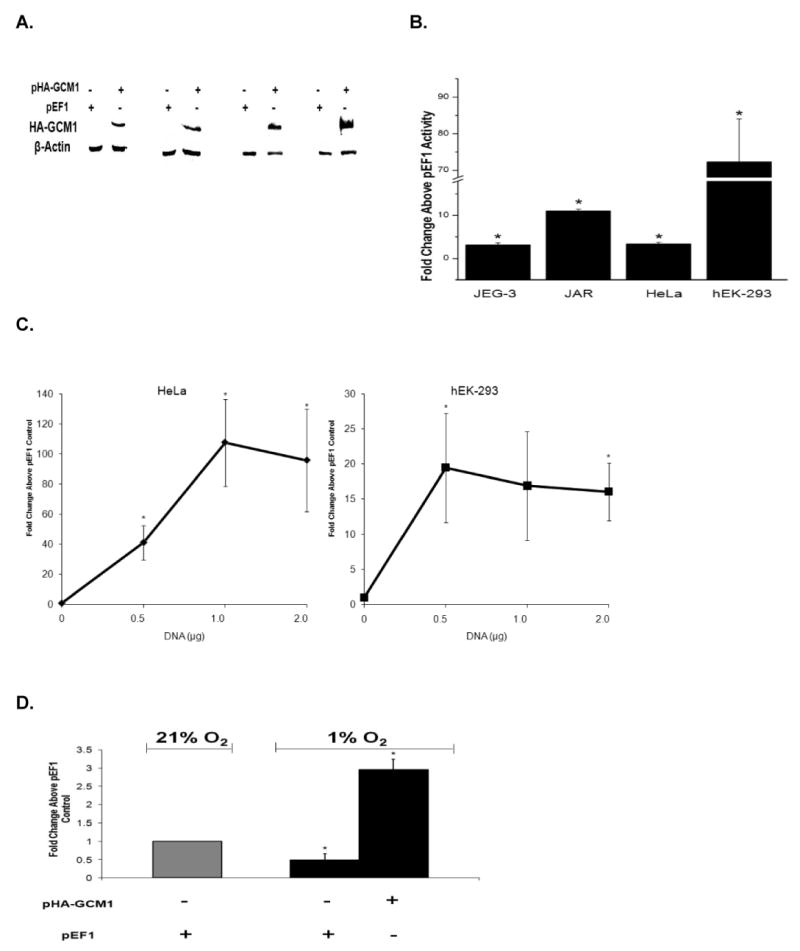
GCM1 regulates PGF promoter activity and mRNA expression. A) Expression of exogenous GCM1 was confirmed by Western blot for HA-tagged GCM1. Cells were transfected with pHA-GCM1 or a control empty vector (pEF1/MycHis) and immunoblotted for anti-HA and β-actin. Paired lanes correspond to JEG-3, JAR, HeLa, and hEK-293, respectively. B) GCM1 increased transcriptional activity of the −1.5-kb clone in both trophoblasts and nontrophoblasts. Cells were cotransfected with pHA-GCM1 or pEF1/MycHis and (−1521/+34) PGF reporter construct. PGF reporter activity was determined 48 h later and fold activity above cells transfected with pEF1/MycHis for each cell type plotted. GCM1 overexpression significantly increased PlGF −1.5-kb promoter activity in both trophoblast and nontrophoblast cells (*, P < 0.05). C) GCM1 overexpression increased (*, P < 0.05) PGF mRNA expression in nontrophoblasts. GCM1 overexpression was accomplished with increasing concentrations of pHA-GCM1, and qRT-PCR was performed 48 h later to determine PGF and RPL32 mRNA expression. PGF mRNA expression was normalized to RPL32 expression, and relative changes in PGF mRNA expression were compared to control transfections with the pEF1/MycHis empty vector (set = 1). D) GCM1 overexpression restores PGF mRNA expression in hypoxic trophoblasts. JEG-3 cells were transfected with GCM1 overexpression plasmid (pHA-GCM1) or pEF1/MycHis and cultured under either 21% O2 or 1% O2 for 24 h. Quantitative RT-PCR was performed to determine PGF and RPL32 mRNA expression. PGF mRNA expression was normalized to RPL32 expression and relative changes in PGF mRNA expression were compared to the result of pEF1/MycHis transfected cells cultured at 21% O2 (set = 1). *, P < 0.05.
Low oxygen tension has been shown to specifically decrease PGF mRNA expression in trophoblasts [3, 37]. We sought to determine if GCM1 mediates this effect since others have shown that low oxygen tension decreases GCM1 mRNA expression in BeWo cells [38]. We performed qRT-PCR and confirmed that GCM1 mRNA expression was significantly decreased (>80%) when JEG-3 cells were cultured in 1% O2 (data not shown). To determine if maintenance of GCM1 expression could rescue decreased PGF mRNA expression in hypoxic trophoblasts, we overexpressed GCM1 in hypoxic JEG-3 cells (Fig. 6D). As we have previously shown, exposure of JEG-3 cells to 1% O2 significantly decreased PGF expression by 24 h. However, overexpression of exogenous GCM1 in the hypoxic trophoblast increased endogenous PGF mRNA levels about 6-fold above those in hypoxic cells lacking GCM1 and about 3-fold above levels noted in normoxic cells. Similar results were observed in JAR trophoblast cells (data not shown).
Discussion
At the maternal-fetal interface, expression of PGF mRNA is normally high in trophoblasts [22] and uterine lymphocytes [39]. In contrast, most other cell types show little to no PGF expression under normal physiologic conditions [40]. The mechanisms underlying the differential high PGF expression in trophoblasts as compared to other cell types are not known. Our study focused on defining the regions of the PGF gene responsible for regulating its uniquely high and constitutive transcription in trophoblast cells. High PGF promoter activity was produced in two different trophoblast cell lines by a 1.5-kb clone that spans bases −1521 to +34. However, a significantly lower level of activity was observed when this clone was transfected in three different nontrophoblast cell lines. The promoter activity correlated directly with the endogenous levels of PGF mRNA reported in these cell lines by us [3] and others [6, 31]. These data suggest that the −1521-bp region proximal to the PGF gene is capable of producing constitutive and trophoblast-specific promoter activity.
Transfection studies in both the trophoblast and nontrophoblast cell lines with various reporter gene constructs showed that the region between −828 bp and +34 bp of the PGF gene maintained a high level of activity in trophoblast cells and little activity in nontrophoblasts. We also determined that the region −698 bp to +34 bp can function as a basal promoter with reduced activity in trophoblasts and yet still be permissible for increased constitutive activity in certain nontrophoblast cells. However, this region did not seem to significantly regulate the expression of PGF in the MCF-7 breast cancer line. This is in line with our previous findings showing that these cells express low but detectable levels of PGF mRNA [22].
We subsequently sought to determine what DNA elements might be present in the 131-bp region between −698 and −828 that mediate the increased transcriptional activity in trophoblasts. The 131-bp region between −828 bp and −698 bp significantly augmented minimal TK promoter activity independent of orientation in trophoblast, but not nontrophoblast, cells. These findings, which are suggestive of an enhancer-like effect, are similar to fold inductions noted for other placenta-specific enhancers [41, 42]. In contrast, the same region did not show a significant effect on promoter activity in nontrophoblasts. Collectively, these results suggest the presence of trophoblast-specific elements within this 131-bp region.
A number of placenta- and trophoblast-specific elements have been identified that can regulate placenta and trophoblast expression of different genes. Placenta-specific elements include cAMP responsive elements, upstream regulatory element (URE), trophoblast specific element and URE1 of human gonadotropin α subunit hormone [31, 33], chorionic somatomammotropin enhancer factor of the human chorionic somatomammotropin-B gene [34], and placental leptin elements (PLE)-1 and PLE-3 [30]. However, computer analyses of the 131-bp sequence between −698 bp and −828 bp and that of the entire 1.5-kb upstream region of the PGF gene did not reveal sequence homologies at 80% stringency for any of these sites.
Our in silico analyses did highlight several putative transcription factor binding elements within this 131-bp region, including Oct-1, SP-1, ETS, and transcription enhancing factor (TEF-1). Some of these putative elements have been shown to function at least in part to regulate transcription of various genes in the trophoblast [41, 43–45]. However, most notable was the presence of a motif that shared high sequence homology (87.5%) with GCM1 binding sites [46]. Based on the trophoblast-specific expression of GCM1 [35], its ability to transcriptionally regulate other trophoblast-specific genes including syncytin [47] and aromatase [36], and its decreased expression in preeclampsia [48] and hypoxic trophoblasts [38], we focused on the corresponding DNA element for GCM1 as a regulator for PGF gene expression. This putative GCM1 element in human PGF specifically bound GCM1 proteins in direct binding assays, confirming its functional protein-binding capacity. Site-directed mutagenesis of the single GCM1 binding motif in the 131-bp region significantly inhibited the enhancing effect of this region in the trophoblast. However, the mutated GCM1 motif had no effect on the basal promoter activity of the reporter clone in nontrophoblast cells. To confirm the ability of GCM1 to regulate PGF promoter activity, we overexpressed GCM1 protein in both trophoblast and nontrophoblast cells. GCM1 overexpression increased activity of the full-length PGF 5′UTR (−1.5-kb region) in both cell types, but to differing degrees. Trophoblasts express high levels of GCM1 (this report and [35]), whereas the nontrophoblast cells we tested (HeLa, hEK-293) express almost undetectable levels of GCM1 mRNA. Forced overexpression of GCM1 significantly increased transcriptional activity of the −1.5-kb PGF reporter clone and resulted in significant increases (20- to 100-fold) in endogenous PGF mRNA expression in the nontrophoblast. In addition, increased expression of GCM1 increased endogenous PGF mRNA expression in the trophoblast. These data indicate that GCM1 is able to regulate PGF promoter activity and that GCM1 cell type-specific expression might be partially responsible for high PGF promoter activity in trophoblasts, but lower activity in most nontrophoblasts. Whether the other identified putative transcription factor binding elements contribute to trophoblast-specific PGF expression is currently under investigation.
Our data supports that GCM1 alone can positively regulate PGF promoter activity by direct interaction with PGF DNA; however, it is possible that other transcription factors may cooperate with GCM1 to regulate its functional activity in a cell type-specific manner. For instance, coexpression of GCM1 and PITX1 or PITX2 synergistically increased transcriptional activity of a synthetic GCM1 reporter construct in trophoblast (JEG-3 cells), but repressed GCM1 activity in hEK-293 cells [49]. Similarly, functional activity of GCM1 in neuronal cell types has been shown to be negatively regulated by repressor proteins [50].
The ability of GCM1 to regulate PGF gene activity correlates well with their mutual expression patterns in trophoblasts in vitro and in normal and preeclamptic placentae. Sustained GCM1 expression is restricted to cytotrophoblasts and syncytiotrophoblasts in human placentae [35, 48, 51]. Furthermore, expression of GCM1 mRNA and protein is significantly lower in trophoblasts of late third trimester placentae than in midgestation trophoblasts [48]. Expression of PGF protein is temporally similar in that maternal systemic levels steadily decrease from a peak at approximately 26–30 wk gestation to term [17], and placental PGF mRNA expression is lower at term than in midgestation [22]. Placentae from near-term preeclamptic pregnancies express less GCM1 protein and mRNA and there are fewer GCM1 expressing trophoblast cells than are evident in gestational-age matched normal placentae [48]. These findings correlate well with the decreased PGF protein [17–20, 52, 53] and mRNA [22] expression noted in preeclamptic pregnancies. Furthermore, expression of GCM1 is significantly decreased in choriocarcinoma cells and primary term trophoblasts exposed to low oxygen tension [38]. Low oxygen tension is known to specifically decrease PGF expression in trophoblasts [3, 37], which is mediated at least in part by decreased transcription (R.M. Gobble, D.S. Torry, et al. unpublished results). We confirmed that low oxygen tension decreased GCM1 and PGF mRNA expression in trophoblast cells. Importantly, forced overexpression of GCM1 in hypoxic trophoblasts maintained high levels of PGF mRNA expression in the cells. Collectively, these results suggest that GCM1 plays a critical role in regulating PGF transcription in trophoblasts and that decreased trophoblast GCM1 expression noted in preeclampsia may contribute to the decreased expression levels of PGF.
It is increasingly evident that adequate growth and maintenance of the vasculature is an important component of successful placentation [54]. Furthermore, discrete threshold differences in growth factor expression may provide abnormal vascular growth and placentation [55]. Clearly, studies designed to determine tissue and cell type-specific regulatory regions of angiogenic growth factors like PGF in the placenta are needed. In summary, our studies have localized promoter regions contributing to constitutive expression of the PGF gene in human trophoblast. We have identified a 131-bp region located between −828 bp and −698 bp of the PGF gene that contributes to increased promoter activity in trophoblast cells and have identified GCM1 as a mediator of this enhancing effect. Furthermore, oxygen regulation of PGF gene expression in trophoblasts is reflected, at least in part, by the effects of oxygen on this trophoblast-specific transcription factor. Given the important role of PGF in regulating pathological angiogenesis [11], our identification of the putative transcription factor binding sites of the PGF gene may also highlight molecular mechanisms responsible for the increased regulation of PGF expression in well-vascularized tumors and wound healing.
Acknowledgments
We sincerely appreciate the generous gift of the GCMa (synonymous with GCM1 in this manuscript) expression clone from Dr. Hungwen Chen, Institute of Biological Chemistry, Academia Sinica, Taiwan.
Footnotes
Supported in part by NIH 5RO1HD36830, HL72802, and an Excellence in Academic Medicine Award from Southern Illinois University School of Medicine.
References
- 1.Maglione D, Guerriero V, Viglietto G, Delli-Bovi P, Persico MG. Isolation of a human placenta cDNA coding for a protein related to the vascular permeability factor. Proc Natl Acad Sci U S A. 1991;88:9267–9271. doi: 10.1073/pnas.88.20.9267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khaliq A, Li XF, Shams M, Sisi P, Acevedo CA, Whittle MJ, Weich H, Ahmed A. Localisation of placenta growth factor (PGF) in human term placenta. Growth Factors. 1996;13:243–250. doi: 10.3109/08977199609003225. [DOI] [PubMed] [Google Scholar]
- 3.Shore VH, Wang TH, Wang CL, Torry RJ, Caudle MR, Torry DS. Vascular endothelial growth factor, placenta growth factor and their receptors in isolated human trophoblast. Placenta. 1997;18:657–665. doi: 10.1016/s0143-4004(97)90007-2. [DOI] [PubMed] [Google Scholar]
- 4.Hanna J, Goldman-Wohl D, Hamani Y, Avraham I, Greenfield C, Natanson-Yaron S, Prus D, Cohen-Daniel L, Arnon TI, Manaster I, Gazit R, Yutkin V, et al. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat Med. 2006;12:1065–1074. doi: 10.1038/nm1452. [DOI] [PubMed] [Google Scholar]
- 5.Cao Y, Ji WR, Qi P, Rosin A. Placenta growth factor: identification and characterization of a novel isoform generated by RNA alternative splicing. Biochem Biophys Res Commun. 1997;235:493–498. doi: 10.1006/bbrc.1997.6813. [DOI] [PubMed] [Google Scholar]
- 6.Maglione D, Guerriero V, Viglietto G, Ferraro MG, Aprelikova O, Alitalo K, Del Vecchio S, Lei KJ, Chou JY, Persico MG. Two alternative mRNAs coding for the angiogenic factor, placenta growth factor (PlGF), are transcribed from a single gene of chromosome 14. Oncogene. 1993;8:925–931. [PubMed] [Google Scholar]
- 7.Yang W, Ahn H, Hinrichs M, Torry RJ, Torry DS. Evidence of a novel isoform of placenta growth factor (PlGF-4) expressed in human trophoblast and endothelial cells. J Reprod Immunol. 2003;60:53–60. doi: 10.1016/s0165-0378(03)00082-2. [DOI] [PubMed] [Google Scholar]
- 8.Ziche M, Maglione D, Ribatti D, Morbidelli L, Lago CT, Battisti M, Paoletti I, Barra A, Tucci M, Parise G, Vincenti V, Granger HJ, et al. Placenta growth factor-1 is chemotactic, mitogenic, and angiogenic. Lab Invest. 1997;76:517–531. [PubMed] [Google Scholar]
- 9.Odorisio T, Cianfarani F, Failla CM, Zambruno G. The placenta growth factor in skin angiogenesis. J Dermatol Sci. 2006;41:11–19. doi: 10.1016/j.jdermsci.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 10.Luttun A, Tjwa M, Moons L, Wu Y, Angelillo-Scherrer A, Liao F, Nagy JA, Hooper A, Priller J, De Klerck B, Compernolle V, Daci E, et al. Revascularization of ischemic tissues by PlGF treatment, and inhibition of tumor angiogenesis, arthritis and atherosclerosis by anti-Flt1. Nat Med. 2002;8:831–840. doi: 10.1038/nm731. [DOI] [PubMed] [Google Scholar]
- 11.Carmeliet P, Moons L, Luttun A, Vincenti V, Compernolle V, De Mol M, Wu Y, Bono F, Devy L, Beck H, Scholz D, Acker T, et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat Med. 2001;7:575–583. doi: 10.1038/87904. [DOI] [PubMed] [Google Scholar]
- 12.Roy H, Bhardwaj S, Yla-Herttuala S. Biology of vascular endothelial growth factors. FEBS Lett. 2006;580:2879–2887. doi: 10.1016/j.febslet.2006.03.087. [DOI] [PubMed] [Google Scholar]
- 13.Athanassiades A, Lala PK. Role of placenta growth factor (PlGF) in human extravillous trophoblast proliferation, migration and invasiveness. Placenta. 1998;19:465–473. doi: 10.1016/s0143-4004(98)91039-6. [DOI] [PubMed] [Google Scholar]
- 14.Desai J, Holt-Shore V, Torry RJ, Caudle MR, Torry DS. Signal transduction and biological function of placenta growth factor in primary human trophoblast. Biol Reprod. 1999;60:887–892. doi: 10.1095/biolreprod60.4.887. [DOI] [PubMed] [Google Scholar]
- 15.Zhou Y, McMaster M, Woo K, Janatpour M, Perry J, Karpanen T, Alitalo K, Damsky C, Fisher SJ. Vascular endothelial growth factor ligands and receptors that regulate human cytotrophoblast survival are dysregulated in severe preeclampsia and hemolysis, elevated liver enzymes, and low platelets syndrome. Am J Pathol. 2002;160:1405–1423. doi: 10.1016/S0002-9440(10)62567-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szukiewicz D, Szewczyk G, Watroba M, Kurowska E, Maslinski S. Isolated placental vessel response to vascular endothelial growth factor and placenta growth factor in normal and growth-restricted pregnancy. Gynecol Obstet Invest. 2004;59:102–107. doi: 10.1159/000082622. [DOI] [PubMed] [Google Scholar]
- 17.Torry DS, Wang HS, Wang TH, Caudle MR, Torry RJ. Preeclampsia is associated with reduced serum levels of placenta growth factor. Am J Obstet Gynecol. 1998;179:1539–1544. doi: 10.1016/s0002-9378(98)70021-3. [DOI] [PubMed] [Google Scholar]
- 18.Levine RJ, Maynard SE, Qian C, Lim KH, England LJ, Yu KF, Schisterman EF, Thadhani R, Sachs BP, Epstein FH, Sibai BM, Sukhatme VP, Karumanchi SA. Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med. 2004;350:672–683. doi: 10.1056/NEJMoa031884. [DOI] [PubMed] [Google Scholar]
- 19.Tidwell SC, Ho HN, Chiu WH, Torry RJ, Torry DS. Low maternal serum levels of placenta growth factor as an antecedent of clinical preeclampsia. Am J Obstet Gynecol. 2001;184:1267–1272. doi: 10.1067/mob.2001.113129. [DOI] [PubMed] [Google Scholar]
- 20.Levine RJ, Thadhani R, Qian C, Lam C, Lim KH, Yu KF, Blink AL, Sachs BP, Epstein FH, Sibai BM, Sukhatme VP, Karumanchi SA. Urinary placental growth factor and risk of preeclampsia. JAMA. 2005;293:77–85. doi: 10.1001/jama.293.1.77. [DOI] [PubMed] [Google Scholar]
- 21.Ahmed A, Dunk C, Ahmad S, Khaliq A. Regulation of placental vascular endothelial growth factor (VEGF) and placenta growth factor (PlGF) and soluble Flt-1 by oxygen—a review. Placenta. 2000;21:S16–S24. doi: 10.1053/plac.1999.0524. [DOI] [PubMed] [Google Scholar]
- 22.Torry DS, Hinrichs M, Torry RJ. Determinants of placental vascularity. Am J Reprod Immunol. 2004;51:257–268. doi: 10.1111/j.1600-0897.2004.00154.x. [DOI] [PubMed] [Google Scholar]
- 23.Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, Libermann TA, Morgan JP, Sellke FW, Stillman IE, Epstein FH, Sukhatme VP, Karumanchi SA. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003;111:649–658. doi: 10.1172/JCI17189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Torry DS, Mukherjea D, Arroyo J, Torry RJ. Expression and function of placenta growth factor: implications for abnormal placentation. J Soc Gynecol Investig. 2003;10:178–188. doi: 10.1016/s1071-5576(03)00048-0. [DOI] [PubMed] [Google Scholar]
- 25.Prestridge DS. Predicting Pol II promoter sequences using transcription factor binding sites. J Mol Biol. 1995;249:923–932. doi: 10.1006/jmbi.1995.0349. [DOI] [PubMed] [Google Scholar]
- 26.Chang CW, Chuang HC, Yu C, Yao TP, Chen H. Stimulation of GCMa transcriptional activity by cyclic AMP/protein kinase A signaling is attributed to CBP-mediated acetylation of GCMa. Mol Cell Biol. 2005;25:8401–8414. doi: 10.1128/MCB.25.19.8401-8414.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coulet F, Nadaud S, Agrapart M, Soubrier F. Identification of hypoxia response element in the human endothelial nitric oxide synthase gene promoter. J Biol Chem. 2003;47:46230–46240. doi: 10.1074/jbc.M305420200. [DOI] [PubMed] [Google Scholar]
- 28.Hashimoto JG, Beadles-Bohling AS, Wiren KM. Comparison of Ribo-Green and 18S rRNA quantitation for normalizing real-time RT-PCR expression analysis. Biotechniques. 2004;36:54–56. doi: 10.2144/04361BM06. [DOI] [PubMed] [Google Scholar]
- 29.Ebert BL, Bunn HF. Regulation of transcription by hypoxia requires a multiprotein complex that includes hypoxia-inducible factor 1, an adjacent transcription factor, and p300/CREB binding protein. Mol Cell Biol. 1998;18:4089–4096. doi: 10.1128/mcb.18.7.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang H, Palmer R, Gao X, Kreidberg J, Gerald W, Hsiao L, Jensen RV, Gullans SR, Haber DA. Transcriptional activation of placental growth factor by the forkhead/winged helix transcription factor FoxD1. Curr Biol. 2003;13:1625–1629. doi: 10.1016/j.cub.2003.08.054. [DOI] [PubMed] [Google Scholar]
- 31.Cao Y, Chen H, Zhou L, Chiang MK, Anand-Apte B, Weatherbee JA, Wang Y, Fang F, Flanagan JG, Tsang ML. Heterodimers of placenta growth factor/vascular endothelial growth factor. Endothelial activity, tumor cell expression, and high affinity binding to Flk-1/KDR. J Biol Chem. 1996;271:3154–3162. doi: 10.1074/jbc.271.6.3154. [DOI] [PubMed] [Google Scholar]
- 32.Torry DS, Ahn H, Barnes EL, Torry RJ. Placenta growth factor: potential role in pregnancy. Am J Reprod Immunol. 1999;41:79–85. doi: 10.1111/j.1600-0897.1999.tb00078.x. [DOI] [PubMed] [Google Scholar]
- 33.Akiyama Y, Hosoya T, Poole AM, Hotta Y. The gcm-motif: a novel DNA-binding motif conserved in Drosophila and mammals. Proc Natl Acad Sci U S A. 1996;93:14912–14916. doi: 10.1073/pnas.93.25.14912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schreiber J, Enderich J, Wegner M. Structural requirements for DNA binding of GCM proteins. Nucleic Acids Res. 1998;26:2337–2343. doi: 10.1093/nar/26.10.2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nait-Oumesmar B, Copperman AB, Lazzarini RA. Placental expression and chromosomal localization of the human Gcm 1 gene. J Histochem Cytochem. 2000;48:915–922. doi: 10.1177/002215540004800704. [DOI] [PubMed] [Google Scholar]
- 36.Yamada K, Ogawa H, Honda S, Harada N, Okazaki T. A GCM motif protein is involved in placenta-specific expression of human aromatase gene. J Biol Chem. 1999;274:32279–32286. doi: 10.1074/jbc.274.45.32279. [DOI] [PubMed] [Google Scholar]
- 37.Khaliq A, Dunk C, Jiang J, Shams M, Li XF, Acevedo C, Weich H, Whittle M, Ahmed A. Hypoxia down-regulates placenta growth factor, whereas fetal growth restriction up-regulates placenta growth factor expression: molecular evidence for “placental hyperoxia” in intrauterine growth restriction. Lab Invest. 1999;79:151–170. [PubMed] [Google Scholar]
- 38.Knerr I, Schubert SW, Wich C, Amann K, Aigner T, Vogler T, Jung R, Dotsch J, Rascher W, Hashemolhosseini S. Stimulation of GCMa and syncytin via cAMP mediated PKA signaling in human trophoblastic cells under normoxic and hypoxic conditions. FEBS Lett. 2005;579:3991–3998. doi: 10.1016/j.febslet.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 39.Tayade C, Fang Y, Hilchie D, Croy BA. Lymphocyte contributions to altered endometrial angiogenesis during early and midgestation fetal loss. J Leukoc Biol. 2007;82:877–886. doi: 10.1189/jlb.0507330. [DOI] [PubMed] [Google Scholar]
- 40.Viglietto G, Maglione D, Rambaldi M, Cerutti J, Romano A, Trapasso F, Fedele M, Ippolito P, Chiappetta G, Botti G, et al. Upregulation of vascular endothelial growth factor (VEGF) and downregulation of placenta growth factor (PlGF) associated with malignancy in human thyroid tumors and cell lines. Oncogene. 1995;11:1569–1579. [PubMed] [Google Scholar]
- 41.Cheng YH, Handwerger S. Identification of an enhancer of the human activating protein-2alpha gene that contains a critical Ets1 binding site. J Clin Endocrinol Metab. 2003;88:3305–3311. doi: 10.1210/jc.2002-021831. [DOI] [PubMed] [Google Scholar]
- 42.Peng L, Payne AH. AP-2 gamma and the homeodomain protein distal-less 3 are required for placental-specific expression of the murine 3 beta-hydroxysteroid dehydrogenase VI gene, Hsd3b6. J Biol Chem. 2002;277:7945–7954. doi: 10.1074/jbc.M106765200. [DOI] [PubMed] [Google Scholar]
- 43.Cheng YH, Richardson BD, Hubert MA, Handwerger S. Isolation and characterization of the human syncytin gene promoter. Biol Reprod. 2004;70:694–701. doi: 10.1095/biolreprod.103.023473. [DOI] [PubMed] [Google Scholar]
- 44.Cheng YH, Handwerger S. A placenta-specific enhancer of the human syncytin gene. Biol Reprod. 2005;73:500–509. doi: 10.1095/biolreprod.105.039941. [DOI] [PubMed] [Google Scholar]
- 45.Peng L, Huang Y, Jin F, Jiang SW, Payne AH. Transcription enhancer factor-5 and a GATA-like protein determine placental-specific expression of the Type I human 3beta-hydroxysteroid dehydrogenase gene, HSD3B1. Mol Endocrinol. 2004;18:2049–2060. doi: 10.1210/me.2004-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heaton JH, Nebes VL, O'Dell LG, Morris SM, Jr, Gelehrter TD. Glucocorticoid and cyclic nucleotide regulation of plasminogen activator and plasminogen activator-inhibitor gene expression in primary cultures of rat hepatocytes. Mol Endocrinol. 1989;3:185–192. doi: 10.1210/mend-3-1-185. [DOI] [PubMed] [Google Scholar]
- 47.Yu C, Shen K, Lin M, Chen P, Lin C, Chang GD, Chen H. GCMa regulates the syncytin-mediated trophoblastic fusion. J Biol Chem. 2002;277:50062–50068. doi: 10.1074/jbc.M209316200. [DOI] [PubMed] [Google Scholar]
- 48.Chen CP, Chen CY, Yang YC, Su TH, Chen H. Decreased placental GCM1 (glial cells missing) gene expression in pre-eclampsia. Placenta. 2004;25:413–421. doi: 10.1016/j.placenta.2003.10.014. [DOI] [PubMed] [Google Scholar]
- 49.Schubert SW, Kardash E, Khan MA, Cheusova T, Kilian K, Wegner M, Hashemolhosseini S. Interaction, cooperative promoter modulation, and renal colocalization of GCMa and Pitx2. J Biol Chem. 2004;279:50358–50365. doi: 10.1074/jbc.M404587200. [DOI] [PubMed] [Google Scholar]
- 50.Mondal S, Ivanchuk SM, Rutka JT, Boulianne GL. Sloppy paired 1/2 regulate glial cell fates by inhibiting Gcm function. Glia. 2007;55:282–293. doi: 10.1002/glia.20456. [DOI] [PubMed] [Google Scholar]
- 51.Baczyk D, Satkunaratnam A, Nait-Oumesmar B, Huppertz B, Cross JC, Kingdom JC. Complex patterns of GCM1 mRNA and protein in villous and extravillous trophoblast cells of the human placenta. Placenta. 2004;25:553–559. doi: 10.1016/j.placenta.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 52.Thadhani R, Mutter WP, Wolf M, Levine RJ, Taylor RN, Sukhatme VP, Ecker J, Karumanchi SA. First trimester placental growth factor and soluble fms-like tyrosine kinase 1 and risk for preeclampsia. J Clin Endocrinol Metab. 2004;89:770–775. doi: 10.1210/jc.2003-031244. [DOI] [PubMed] [Google Scholar]
- 53.Staff AC, Braekke K, Harsem NK, Lyberg T, Holthe MR. Circulating concentrations of sFlt1 (soluble fms-like tyrosine kinase 1) in fetal and maternal serum during pre-eclampsia. Eur J Obstet Gynecol Reprod Biol. 2005;1:33–39. doi: 10.1016/j.ejogrb.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 54.Huppertz B, Peeters LL. Vascular biology in implantation and placentation. Angiogenesis. 2005;8:157–167. doi: 10.1007/s10456-005-9007-8. [DOI] [PubMed] [Google Scholar]
- 55.Torry DS, Leavenworth J, Chang M, Maheshwari V, Groesch K, Ball ER, Torry RJ. Angiogenesis in implantation. J Assist Reprod Genet. 2007;24:303–315. doi: 10.1007/s10815-007-9152-7. [DOI] [PMC free article] [PubMed] [Google Scholar]