

Abstract
In search of new selective antagonists and/or agonists for the human melanocortin receptor subtypes hMC1R to hMC5R to elucidate the specific biological roles of each GPCR, we modified the structures of the superagonist MT-II (Ac-Nle-c[Asp-His-D-Phe-Arg-Trp-Lys]-NH2) and the hMC3R/hMC4R antagonist SHU9119 (Ac-Nle-c[Asp-His-D-Nal(2′)-Arg-Trp-Lys]-NH2) by replacing the His-D-Phe and His-D-Nal(2′) fragments in MT-II and SHU9119, respectively, with Aba-Xxx (4-amino-1,2,4,5-tetrahydro-2-benzazepin-3-one-Xxx) dipeptidomimetics (Xxx = D-Phe/pCl-D-Phe/D-Nal(2′)). Employment of the Aba mimetic yielded novel selective high affinity hMC3R and hMC3R/hMC5R antagonists.
Keywords: Human melanocortin receptors; 4-Amino-1,2,4,5-tetrahydro-2-benzazepin-3-ones; Cyclic lactam analogues; Conformational restrictions; hMC3R/hMC5R antagonists
The α-melanocyte stimulating hormone (α-MSH, Ac-Ser-Tyr-Ser-Met-Glu-His-Phe-Arg-Trp-Gly-Lys-Pro-Val-NH2) plays a role in a wide range of biological responses like feeding behavior, pain modulation, learning behavior, pigmentation, sexual function, energy homeostasis, and thermoregulation.1 In particular, the human melanocortin 4 subtype receptor is an attractive drug target because of its role in regulation of feeding behavior.2,3 Design of selective hMC4R antagonists is considered to have great potential for the treatment of anorexia.4,5 The hMC3R on the other hand has been shown to play a role in the physiological process of energy partitioning and body weight.6 Controlled modulation of these receptors could lead to promising results in the field of feeding disorders.
The principal pharmacophore groups of α-MSH were found to be the side chains of the central tetrapeptide His6-Phe7-Arg8-Trp9.7,8 Molecular modeling as well as conformational analysis of a variety of cyclic analogues led to the discovery of a superpotent lactam analogue MT-II (Ac-Nle-c[Asp-His-D-Phe-Arg-Trp-Lys]-NH2).9 This cyclic peptide analogue, first introduced by Al-Obeidi et al.9 was a very potent, but non-selective, agonist for the human melanocortin receptor subtypes MC1R, MC3R, MC4R, and MC5R. It also showed a very high stability against all proteolytic enzymes and tissue homogenates.9 Because of the lack in selectivity, finding selective ligands for each of the four human melanocortin receptors (hMC1R, hMC3R to hMC5R) was consequently crucial for the determination of their individual physiological roles. Development of selective melanocortin ligands can help to ascribe specific biological functions for the corresponding receptor subtypes.
Several parameters of MT-II (e.g., ring size, introduction of hydrophobic groups) were modified to design potent and more selective peptide analogues.2,10,11 Recently, Grieco et al. reported the influence of replacing the His6 by Pro in MT-II, which resulted in retention of agonist potency for most melanocortin receptors.11 Upon substitution of His6 in SHU9119, a potent hMC3R/hMC4R antagonist (Ac-Nle-c[Asp-His-D-Nal(2′)-Arg-Trp-Lys]-NH2),12 by conformationally restricted amino acids, selective antagonists for the hMC3R and hMC4R were discovered.13 In particular, several conformationally constricted amino acids such as Tic, Oic, Aic, etc., were introduced and subsequently provided information about well-defined conformational spaces.13 Upon substitution of His6 by Tic a potent hMC3R (IC50 = 6.7 nM) and hMC4R (IC50 = 3.7 nM) antagonist was obtained. This work along with that of others14–16 provided clear evidence of the importance of position 6 for potency and melanocortin receptor selectivity in analogues of α-MSH.
The 4-amino-1,2,4,5-tetrahydro-2-benzazepin-3-one scaffold (Fig. 1) has proven to be an excellent tool for the design of novel peptide mimetics.17–22 Its qualities can be described as two folded: (1) the scaffold can be considered as a conformationally restricted Phe analogue where the aromatic side chain is anchored to the α-amine of the next residue by means of a methylene bridge (dotted line);17 (2) on the other hand, one can see the Aba template as a ‘privileged template’.23
Figure 1.
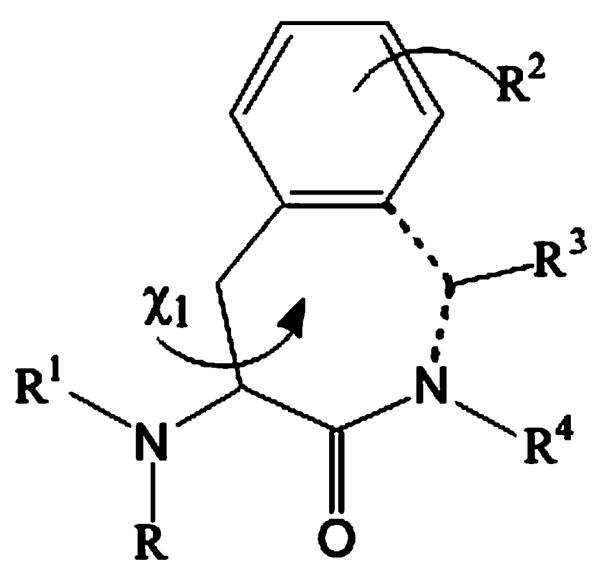
The 4-amino-1,2,4,5-tetrahydro-2-benzazepin-3-one (Aba) scaffold.
In scaffold 1 only the g(+) (χ1 = +60°) and trans (χ1 = 180°) staggered conformations are allowed for the Cα–Cβ bond. By applying this restraint bioactive conformations can be fixed.17,18 Different substitution patterns in 1 can induce the specific binding to certain subtypes of receptors or enzymes, for example ACE inhibitors,19 opioid receptors,20 B2 bradykinin receptors, 21 and farnesyl transferase inhibitors.22
Thus, replacement of the His6-D-Phe7 dipeptide in MT-II and His6-D-Nal(2′)7 in SHU9119 by Aba-D-Phe/Aba-pCl-D-Phe and Aba-D-Nal(2′), respectively, was expected to provide new structure–activity relationships in the search for selective and potent ligands for the hMC1–hMC5R.
All three α-amino protected dipeptomimetics Fmoc-Aba-D-Phe-OH 5a, Fmoc-Aba-pCl-D-Phe-OH 5b, and Fmoc-Aba-D-Nal(2′)-OH 5c were synthesized using a previously reported methodology based on an intramolecular benzazepinone ring formation (see Scheme 1).24 The depicted synthetic pathway starts from (S)-orthocyano-phenylalanine 1, obtained through an asymmetric phase transfer catalysis reaction.25 Using the phthaloylating agent MSB (methyl 2-[(succinimidooxy)carbonyl] benzoate), it was possible to efficiently protect amino acid 1.26 The conversion of the nitrile to the aldehyde 2 was realized in an acetic acid/water/pyridine mixture with Raney nickel as catalyst in good yield. This compound served as a precursor for the Aba-D-Xxx dipeptomimetics 5a–c. Reductive amination of this phthaloyl-protected o-formyl-Phe 2 with the corresponding amino acid benzyl esters resulted in the secondary amines 3a–c, which were subsequently ring closed with the activated carboxylic acids using DCC. Removal of the benzyl-protecting moiety, followed by phthaloyl-deprotection and final Fmoc-protection gave Fmoc-Aba blocks 5a–c. These were building blocks in the synthesis of the new MT-II and SHU9119 analogues using Fmoc solid-phase peptide synthesis.36
Scheme 1.
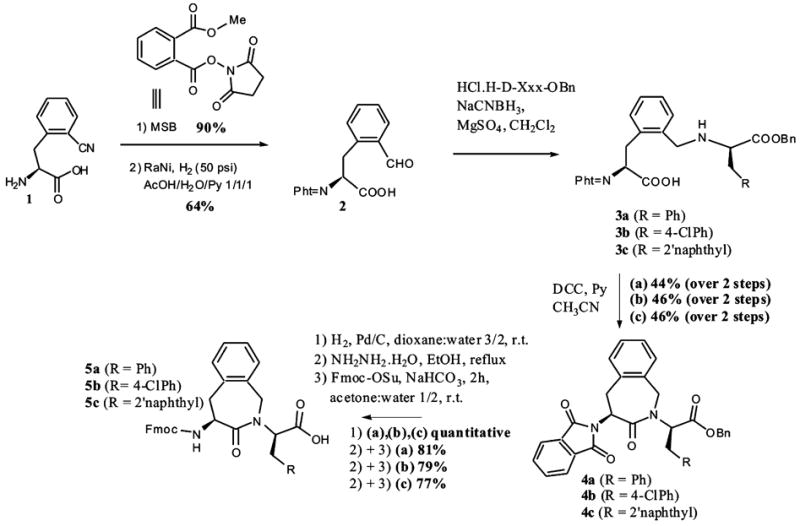
Synthetic pathway for the Aba-Xxx-dipeptomimetics 5a–c.
Molecular modeling proved to be very useful in the design of these hybrid peptides of MT-II and SHU9119. This study employed Macromodel 9.1, with the OPLS 2005 force field and a MCMM/LMCS (Monte Carlo Multiple Minima/Low Frequency Mode) conformational search method.27 We overlapped the NMR structure of the cyclic lactam α-MSH analogue MT-II28 with the global minima of Aba-2 (Ac-Nle-c-[Asp-Aba-D-Phe-Arg-Trp-Lys]-NH2), Aba-3 (Ac-Nle-c-[Asp-Aba-pCl-D-Phe-Arg-Trp-Lys]-NH2) and Aba-4 (Ac-Nle-c-[Asp-Aba-D-Nal(2′)-Arg-Trp-Lys]-NH2), and concluded that the backbone overlap was very good (RMSD = 1.53, 1.01, and 0.99 Å, respectively; non-hydrogen backbone pharmacophore atoms only) (see Fig. 2). It is evident that the backbone conformations of all three Aba peptides are remarkably similar. Moreover, the Aba-bearing peptidomimetics preserve the amphiphilic character for the message sequence (Aba-Xxx-Phe-Arg-Trp), a property occurring in most of the high affinity ligands for hMC1-, hMC3-, hMC4-, and hMC5R, needed for favorable interactions with the melanocortin receptors.28 The hydrophobic part, bearing the side chains of Aba, D-Xxx and Trp, is on one face, and the hydrophilic counterpart, consisting of the side chain of Arg, is oriented away from the aromatic moieties.
Figure 2.
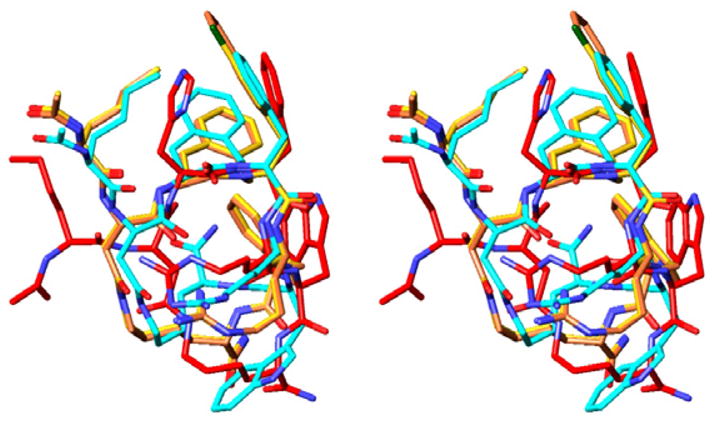
Overlap of Aba-2 (blue), Aba-3 (yellow), Aba-4 (orange), and MT-II (red).40
The calculated dihedral angles for the Aba-Xaa block loosely fit the criteria for a type IV β-turn, as defined by Scheraga and co-workers,29 although examination of the Cα(i)–Cα(i + 3) distances reveals that they are larger than the Cα Asp5-Arg8 distance in MT-II (Aba-2 = 6.54 Å, Aba-3 = 7.41 Å, Aba-4 = 7.39 Å, and MTII = 5.28 Å), and are outside the cut off value of 7 Åusually used to define a β-turn structure.29 The distances between the CO group of Asp5 and the NH group of Arg8 (Aba-2, 2.70 Å; Aba-3, 3.71 Å; Aba-4, 3.71 Å; MT-II, 3.58 Å) are also too large to expect a stable hydrogen bond between these groups. These observations are in accordance with our earlier reports on structural comparison of the β-turn-inducing properties of Aba with those of the so-called Freidinger lactams,30 as we found that only its spirocyclic derivative is capable of inducing a β-turn conformation in an Ac-spiro-Aba-Xxx-NHMe model25 or upon introduction in a biologically active peptide hormone like bradykinin.21 The introduction of this Aba-moiety also clearly avoids the typical stacking between the side chains of residues 6 (His) and 7 (D-Phe), which can be seen for MT-II (red) in Figure 2.
Nevertheless, introduction of the Aba block into the MT-II structure resulted in largely preserved pharmacophore topography (Fig. 3), which was expected to lead to good molecular recognition and high binding affinity to the hMCRs. Another aspect rising from these modeling studies was the fact that the g(+) conformation of the side chain of D-Phe was retained, analogously with MT-II.
Figure 3.
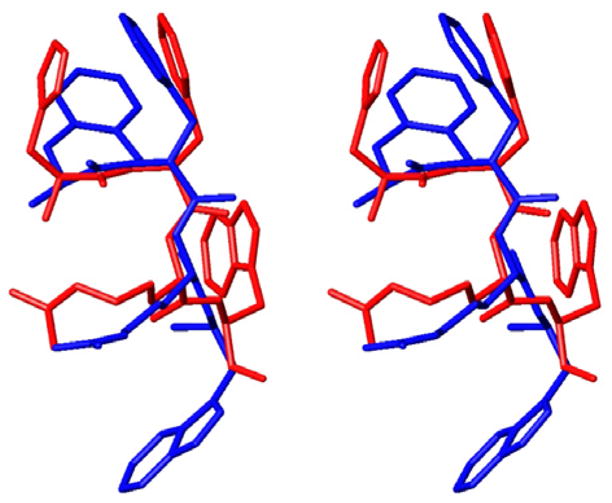
Overlap between the pharmacophore groups of MT-II and Aba-2.
The affinity for the Aba-containing linear and cyclic lactam analogues of MT-II Aba-1–Aba-3, as well as the cyclic SHU9119 analogue Aba-4 was evaluated by competition binding experiments carried out using HEK293 cells stably expressing the human MC1, MC3, MC4, and MC5 receptors. The binding affinities, expressed as IC50 values, are represented in Table 1.37–39
Table 1.
| Name | Sequence |
hMC1R
|
hMC3R
|
hMC4R
|
hMC5R
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (nM) | EC50 (nM) | Max effect (%) | IC50 (nM) | EC50 (nM) | Max effect (%) | IC50 (nM) | EC50 (nM) | Max effect (%) | IC50 (nM) | EC50 (nM) | Max effect (%) | ||
| Aba-1 | Ac-Nle-Asp-Aba-D-Phe-Arg-Trp-Lys-NH2 | >10,000 | >10,000 | 0 | >10,000 | >10,000 | 0 | >10,000 | >10,000 | 0 | >10,000 | >10,000 | 0 |
| Aba-2 | Ac-Nle-c[Asp-Aba-D-Phe-Arg-Trp-Lys]-NH2 | >10,000 | >10,000 | 0 | 50 ± 6 | >10,000 | 0 | >10,000 | >10,000 | 0 | 2,900 ± 300 | >10,000 | 0 |
| Aba-3 | Ac-Nle-c[Asp-Aba-p-Cl-D-Phe-Arg-Trp-Lys]-NH2 | >1,000 | 900 ± 100 | 60 | 29 ± 3 | >1,000 | 55 | 2,000 ± 200 | 3,200 | 33 | 123 ± 13 | 180 ± 20 | 45 |
| Aba-4 | Ac-Nle-c[Asp-Aba -D-Nal-Arg-Trp-Lys]-NH2 | 580 ± 70 | >2,500 | 60 | 43 ± 5 | >10,000 | 0 | 1700 ± 200 | >10,000 | 0 | 87 ± 10 | >10,000 | 0 |
| MT-II | Ac-Nle-c[Asp-His-D-Phe-Arg-Trp-Lys]-NH2 | 0.2 ± 0.01 | 0.3 ± 0.04 | 100 | 1.25 ± 0.2 | 1.85 ± 0.2 | 100 | 1.07 ± 0.3 | 2.87 ± 0.52 | 100 | 7.47 ± 0.23 | 3.3 ± 0.7 | 100 |
IC50, concentration of compound at 50% specific binding. Values are means of three experiments; standard deviation is given.37
EC50, effective concentration of compound that was able to generate 50% maximal intracellular cAMP accumulation. Compounds were tested at a range of concentrations from 10−10 to 10−5 M.38
Introduction of the Aba-D-Phe dipeptidomimetic in the linear sequence of MT-II (Aba-1) resulted in no binding to the melanotropin receptors up to a concentration of 10 μM. This observation suggests that the Aba mimetic alone cannot induce structural features necessary for binding, such as a β-turn, and needs to be used in conjunction with a global conformational constraint. The cyclic lactam analogue Aba-2 was found to show a good binding affinity for the hMC3R (IC50 = 50 nM) and a weak affinity for hMC5R (IC50 = 2.9 μM). No cAMP stimulation could be detected leading to the conclusion that these data are consistent with Aba-2 being a selective hMC3R antagonist (200-fold selective against the hMC4R, and about 60-fold selective against the hMC5R).
Halogenation of the para-position of D-Phe7 with F or Cl typically enhances agonist activity at the hMCRs,12,31 and some weak agonist activity was indeed observed at all receptor subtypes for Aba-3, which bears a pCl-D-Phe at position 7. In addition, this substitution resulted in somewhat improved binding affinities to all four receptor subtypes. The Aba-D-Nal(2′) analogue Aba-4 displayed high affinity hMC3R and hMC5R antagonist properties (IC50 = 43 and 87 nM, respectively), and a weak binding affinity to the hMC4R (IC50 = 1.7 μM), a weak partial agonist activity for the hMC1R (EC50 = 2.5 μM, 60% max cAMP).
The observed lack of agonist activity points to possible steric interference from Aba, analogous to the effect of replacing His6 residue in cyclic α-MSH with bulky Nle, recently presented by Mayorov et al.32 Their report also describes markedly similar biological profiles of Nle6 peptides, as the hMC3/4R agonist VJH085 (cyclo(5β→10ε)-[succinyl5-His6-D-Phe7-Arg8-Trp9-Lys10]-NH2) was thereby converted into a hMC3/4R antagonist (cyclo(5β→10ε)-[succinyl5-Nle6-D-Phe7-Arg8-Trp6-Lys10]-NH2), with a significant decrease in binding affinities (IC50 = 84 and 930 nM, respectively). The Nle6, D-Nal(2′)7 analogue (cyclo(5β →10ε)-[succinyl5-Nle6-D-Nal7-Arg8-Trp9-Lys10]-NH2) was also reported to display hMC3R/hMC5R antagonist properties (IC50 = 12 and 17 nM, respectively).32 Notably, the hMC3/5R antagonism was also observed in cyclic Nle4, D-Nal(2′)6-γ-MSH analogues and was hypothesized to be linked to steric hindrance of Arg7 binding space with Nle4.27 It seems plausible that the steric effects of Aba are responsible for the hMC3R and hMC3/5R antagonist properties of the Aba peptides described in this report. Alternatively, the structural deviations of the Aba-Xaa blocks from the type II β-turn structures found in potent melanocortin agonists28 may indicate the significance of type II β-turns for melanocortin agonist activity. The unique conformational features of Aba may also account for the enhanced receptor selectivity displayed by these peptides.
Molecular modeling experiments40 have suggested that the unique conformational properties of Aba mimetics can be used to design and obtain novel melanotropin peptides with significantly enhanced receptor selectivity. The peptide design was based on the MT-II/SHU9119 cyclic lactam template, where the His6-Xaa7 residues were replaced with Aba-Xaa block. The Fmoc-dipeptidomimetics 5a–c were prepared in facile manner using a previously reported method, which was based on a reductive amination/cyclization sequence. The cyclic lactam α-MSH analogues Aba-2–Aba-4 were synthesized by Nα-Fmoc solid-phase methodology. Competition binding experiments, combined with the adenylate cyclase assay, were used to evaluate the activities of these peptides at the human melanotropin receptors to reveal new highly selective high affinity hMC3R antagonist (Aba-2) and an hMC3R/hMC5R antagonist (Aba-4). These results, in conjunction with earlier SAR work on cyclic α- and γ-MSH analogues, suggest that the unique conformational and sterical attributes of the Aba mimetic may be responsible for the observed antagonist activities, and high hMC3R receptor selectivity against the hMC1R and hMC4R. The newly developed melanotropin peptides will be used to clarify the exact biological functions of the physiologically important melanocortin-3 receptor.
Acknowledgments
This work was supported by the Institute for the Promotion of Innovation through Science and Technology in Flanders (IWT Vlaanderen), and by grants from the U.S. Public Health Service, National Institutes of Health DK-17420 and DA-06284. The opinions expressed are those of the authors and not necessarily those of the USPHS.
References and notes
- 1.Cai M, Mayorov AV, Ying J, Stankova M, Trivedi D, Cabello C, Hruby VJ. Peptides. 2005;26:1481. doi: 10.1016/j.peptides.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 2.Hruby VJ, Cai M, Grieco P, Han G, Kavarana M, Trivedi D. Ann NY Acad Sci. 2003;994:12. doi: 10.1111/j.1749-6632.2003.tb03157.x. [DOI] [PubMed] [Google Scholar]
- 3.Holder JR, Haskell-Luevano C. Med Res Rev. 2004;24:325. doi: 10.1002/med.10064. [DOI] [PubMed] [Google Scholar]
- 4.Vergoni AV, Bertolini A. Eur J Pharmacol. 2000;405:25. doi: 10.1016/s0014-2999(00)00538-0. [DOI] [PubMed] [Google Scholar]
- 5.Harrold JA, Williams G, Widdowson PS. J Neurochem. 2000;74:1224. doi: 10.1046/j.1471-4159.2000.741224.x. [DOI] [PubMed] [Google Scholar]
- 6.Chen AS, Marsh DJ, Trumbauer ME, Frazier EG, Guan XM, Yu H, Rosenblum CI, Vongs A, Feng Y, Cao LH, Metzger JM, Strack AM, Camacho RE, Mellin TN, Nunes CN, Min W, Fisher J, Gopal-Truter S, MacIntyre DE, Chen HY, Van der Ploeg LHI. Nat Genet. 2000;26:97. doi: 10.1038/79254. [DOI] [PubMed] [Google Scholar]
- 7.de Lauro Castrucci A-M, Hadley ME, Sawyer TK, Wilkes BC, Al-Obeidi F, Staples DJ, de Vaux AE, Dym O, Hintz MF, Riehm JP, Rao KR, Hruby VJ. Gen Comp Endocrinol. 1989;73:157. doi: 10.1016/0016-6480(89)90066-x. [DOI] [PubMed] [Google Scholar]
- 8.Hruby VJ, Wilkes BC, Hadley ME, Al-Obeidi F, Sawyer TK, Staples DJ, et al. J Med Chem. 1987;30:2126. doi: 10.1021/jm00394a033. [DOI] [PubMed] [Google Scholar]
- 9.(a) Al-Obeidi F, de Lauro Castrucci A-M, Hadley ME, Hruby VJ. J Med Chem. 1989;32:2555. doi: 10.1021/jm00132a010. [DOI] [PubMed] [Google Scholar]; (b) Al-Obeidi F, Hadley ME, Pettitt BM, Hruby VJ. J Am Chem Soc. 1989;111:3413. [Google Scholar]
- 10.Kavarana MJ, Trivedi D, Cai M, Ying J, Hammer M, Cabello C, Grieco P, Han X, Hruby VJ. J Med Chem. 2002;45:2644. doi: 10.1021/jm020021z. [DOI] [PubMed] [Google Scholar]
- 11.(a) Bednarek MA, MacNeil T, Kalyani RN, Tang R, Van der Ploeg LHT, Weinberg DH. J Med Chem. 2001;44:3665. doi: 10.1021/jm010165y. [DOI] [PubMed] [Google Scholar]; (b) Grieco P, Han G, Hruby VJ. In: Peptides for the New Millenium. Fields GB, Tam JP, Barany G, editors. Kluwer Academic Publishers; The Netherlands: 2000. p. 541. [Google Scholar]
- 12.Hruby VJ, Lu D, Sharma SD, de Lauro Castrucci AM, Kesterson RA, Al-Obeidi FA, Hadley ME, Cone RD. J Med Chem. 1995;38:3454. doi: 10.1021/jm00018a005. [DOI] [PubMed] [Google Scholar]
- 13.Grieco P, Lavecchia A, Cai M, Trivedi D, Weinberg D, MacNeil T, Van der Ploeg LHT, Hruby VJ. J Med Chem. 2002;45:5287. doi: 10.1021/jm0202526. [DOI] [PubMed] [Google Scholar]
- 14.Cai M, Cai C, Mayorov AV, Xiong C, Cabello CM, Soloshonok VA, Swift JR, Trivedi D, Hruby VJ. J Pept Res. 2004;63:116. doi: 10.1111/j.1399-3011.2003.00105.x. [DOI] [PubMed] [Google Scholar]
- 15.Li SZ, Lee JH, Lee W, Yoon CJ, Baik JH, Lim SK. Eur J Biochem. 1999;265:430. doi: 10.1046/j.1432-1327.1999.00763.x. [DOI] [PubMed] [Google Scholar]
- 16.Nijenhuis WAJ, Kruijtzer JAW, Wanders N, Vrinten DH, Garner KM, Schaaper WMM, Meloen RH, Gispen WH, Liskamp RM, Adana RAH. Peptides. 2003;24:271. doi: 10.1016/s0196-9781(03)00032-9. [DOI] [PubMed] [Google Scholar]
- 17.Tourwé D, Verschueren K, Frycia A, Davis P, Porreca F, Hruby VJ, Toth G, Jaspers H, Verheyden P, Van Binst G. Biopolymers. 1995;38:1. doi: 10.1002/(sici)1097-0282(199601)38:1<1::aid-bip1>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 18.Ballet S, Frycia A, Piron J, Chung NN, Schiller PW, Kosson P, Lipkowski AW, Tourwé D. J Pept Res. 2005;66:222. doi: 10.1111/j.1399-3011.2005.00291.x. [DOI] [PubMed] [Google Scholar]
- 19.Flynn GA, Giroux L, Dage RC. J Am Chem Soc. 1987;109:7914. [Google Scholar]
- 20.Van den Eynde I, Laus G, Schiller PW, Kosson P, Chung NN, Lipkowski AW, Tourwe D. J Med Chem. 2005;48:3644. doi: 10.1021/jm0491795. [DOI] [PubMed] [Google Scholar]
- 21.Ballet S, De Wachter R, Van Rompaey K, Tömböly Cs, Feytens D, Töth G, Quartara L, Cucchi P, Meini S, Tourwé D. J Pept Sci. 2007;13 doi: 10.1002/psc.827. [DOI] [PubMed] [Google Scholar]
- 22.Le Diguarher T, Ortuno JC, Shanks D, Guilbaud N, Pierré E, Raimbaud E, Fauchère N, Hickman JA, Tucker GC, Casara PJ. Bioorg Med Chem Lett. 2004;14:767. doi: 10.1016/j.bmcl.2003.11.013. [DOI] [PubMed] [Google Scholar]
- 23.Evans BE, Rittle KE, Bock MG, DiPardo RM, Freidinger RM, Whitter WL, Lundell GF, Veber DF, Anderson PS, Chang RSL, Lotti VJ, Cerino DJ, Chen TB, Kling PJ, Kunkel KA, Springer JP, Hirshfield J. J Med Chem. 1988;31:2235. doi: 10.1021/jm00120a002. [DOI] [PubMed] [Google Scholar]
- 24.Van Rompaey K, Van den Eynde I, De Kimpe N, Tourwé D. Tetrahedron. 2003;59:4421. [Google Scholar]
- 25.Van Rompaey K, Ballet S, Tömböly Cs, De Wachter R, Vannommeslaeghe K, Biesemans M, Willem R, Tourwé D. Eur J Org Chem. 2006:2899. [Google Scholar]
- 26.Casimir JR, Guichard G, Briand JP. J Org Chem. 2002;67:3764. doi: 10.1021/jo016347h. [DOI] [PubMed] [Google Scholar]
- 27.Mayorov AV, Cai M, Chandler KB, Petrov RR, Van Scoy AR, Yu Z, Tanaka DK, Trivedi D, Hruby VJ. J Med Chem. 2006;49:1946. doi: 10.1021/jm0510326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ying J, Köver KE, Gu X, Trivedi DB, Kavarana MJ, Hruby VJ. Biopolymers. 2003;71:696. doi: 10.1002/bip.10596. [DOI] [PubMed] [Google Scholar]
- 29.Lewis PN, Momany FA, Scheraga HA. Biochim Biophys Acta. 1973;303:211. doi: 10.1016/0005-2795(73)90350-4. [DOI] [PubMed] [Google Scholar]
- 30.Freidinger RM, Veber DF, Perlow DS. Science. 1980;210:656. doi: 10.1126/science.7001627. [DOI] [PubMed] [Google Scholar]
- 31.Mayer JP, Hsiung HM, Flora DB, Edwards P, Smith DP, Zhang XY, Gadski RA, Heiman ML, Hertel JL, Emmerson PJ, Husain S, O’Brien TP, Kahl SD, Smiley DL, Zhang L, DiMarchi RD, Yan LZ. J Med Chem. 2005;48:3095. doi: 10.1021/jm0501432. [DOI] [PubMed] [Google Scholar]
- 32.Mayorov AV, Cai M, Han S-Y, Tan B, Van Scoy AR, Dedek M, Palmer ES, Trivedi D, Hruby VJ. Abstracts of Papers, 232nd ACS National Meeting; San Francisco, CA, United States. Sept. 10–14, 2006; MEDI-414. [Google Scholar]
- 33.Flora D, Mo H, Mayer JP, Khan MA, Yan LZ. Bioorg Med Chem Lett. 2005;15:1065. doi: 10.1016/j.bmcl.2004.12.025. [DOI] [PubMed] [Google Scholar]
- 34.Haskell-Luevano C, Miwa H, Dickinson C, Hruby VJ, Yamada T, Gantz I. Biochem Biophys Res Commun. 1994;204:1137. doi: 10.1006/bbrc.1994.2581. [DOI] [PubMed] [Google Scholar]
- 35.Gantz I, Miwa H, Konda Y, Shimoto Y, Tashiro T, Watson SJ, Delvalle J, Yamada T. J Biol Chem. 1993;268:15174. [PubMed] [Google Scholar]
- 36.All peptides in this study were synthesized manually, as described previously,27 by the Nα-Fmoc solid-phase methodology on Rink amide AM (w/Nle) resin using DIC and Cl–HOBt as the coupling reagents. Upon coupling the Asp residue, the orthogonal allylic protection for the side chains of Asp and Lys was removed using the well-established procedure,27,33 and the peptide cyclizations were found to proceed in facile manner with 6 equiv DIC, 6 equiv Cl–HOBt in THF (72 h), as determined by Kaiser ninhydrin test. Upon completion of cyclization the peptide sequence was finalized by coupling the Fmoc-Nle residue, removal of the N-terminal Fmoc group, and acetylation of the N-terminus. The strategy of performing the peptide cyclization prior to removal of the Nα-Fmoc protecting group of the Asp residue was chosen to minimize the competing aspartimide formation, as recently suggested by Flora et al.,33 and indeed was found to be superior to the previously reported procedure.13 The peptides were isolated and purified as described previously27 in 30–35% overall yield and were >95% pure as determined by analytical RP-HPLC. The structures of the pure peptides were confirmed by high-resolution electrospray ionization (ESI) mass-spectrometry.
- 37.Competition binding experiments were carried out using whole HEK293 cells stably expressing human MC1, MC3, MC4, and MC5 receptors. HEK293 cells transfected with hMCRs14,34,35 were seeded on 96-well plates 48 h before assay (50,000 cells/well). For the assay, the cell culture medium was aspirated and the cells were washed once with a freshly prepared MEM buffer containing 100% minimum essential medium with Earle’s salt (MEM, Gibco) and 25 mM sodium bicarbonate. Next, the cells were incubated for 40 min at 37 °C with different concentrations of unlabeled peptide and labeled [125I]-[Nle4, D-Phe7]-α-MSH (Perkin-Elmer Life Science, 20,000 cpm/well, 33.06 pM) diluted in a 125 μL of freshly prepared binding buffer containing 100% MEM, 25 mM Hepes (pH 7.4), 0.2% bovine serum albumin, 1 mM 1,10-phenanthroline, 0.5 mg/L leupeptin, 200 mg/L bacitracin. The assay medium was subsequently removed, the cells were washed once with basic medium, and then lysed by the addition of 100 μL of 0.1 M NaOH and 100 μL of 1% Triton X-100. The lysed cells were transferred to 12 × 75 mm borosilicate glass tubes, and the radioactivity was measured by a Wallac 1470 WIZARD Gamma Counter.
- 38.Adenylate Cyclase assay: HEK 293 cells transfected with human melanocortin receptors14 were grown to confluence in MEM Gibco) containing 10% fetal bovine serum, 100 units/mL penicillin and streptomycin, and 1 mM sodium pyruvate. The cells were seeded on 96-well plates 48 h before assay (50,000 cells/well). For the assay, the cell culture medium was removed and the cells were rinsed with 100 μL MEM buffer (Gibco). An aliquot (100 μL) of the Earle’s balanced salt solution with 5 nM isobutylmethylxanthine (IBMX) was placed in each well along for 1 min at 37 °C. Next, aliquots (25 μL) of melanotropin peptides of varying concentrations were added, and the cells were incubated for 3 min at 37 °C. The reaction was stopped by aspirating the assay buffer and adding 60 μL ice-cold Tris/EDTA buffer to each well, then placing the plates in a boiling water bath for 7 min. The cell lysates were then centrifuged for 10 min at 2300g. A 50μL aliquot of the supernatant was transferred to another 96-well plate and placed with 50 μL [3H]cAMP and 100 μL protein kinase A (PKA) buffer in an ice bath for 2–3 h. The PKA-buffer consisted of Tris/EDTA-buffer with 60 μg/mL PKA and 0.1% bovine serum albumin by weight. The incubation mixture was filtered through 1.0 μm glass fiber filters in MultiScreen™-FB 96-well plates (Millipore, Billerica, MA). The total [3H]cAMP was measured by a Wallac MicroBeta TriLux 1450 LSC and Luminescence Counter (Perkin-Elmer Life Science, Boston, MA). The cAMP accumulation data for each peptide analogue were determined with the help of a cAMP standard curve generated by the same method as described above.
- 39.IC50 and EC50 values represent the mean of two experiments performed in triplicate. IC50 and EC50 estimates and their associated standard errors were determined by fitting the data using a nonlinear least squares analysis, with the help of GraphPad Prism 4 (GraphPad Software, San Diego, CA).
- 40.Molecular modeling experiments employed Macromodel 9.1 equipped with the Maestro 7.5 graphical interface (Schrödinger, LLC, New York, NY, 2005) installed on a Linux Red Hat 9.0 system, and were performed as previously described using the OPLS 2005 force field.27 The superpositions of peptide structures were performed using the α-carbons of the core sequence Xaa-D-Phe/DNal(2′)-Arg-Trp.