

Abstract
We previously showed that neuregulin-1 (NRG-1) protected neurons from death in vivo following focal ischemia. The goal of this study was to develop an in vitro rat ischemia model to examine the cellular and molecular mechanisms involved in the neuroprotective effects of NRG-1 on ischemia-induced neuronal death. Rat B-35 neuroblastoma cells differentiated by serum withdrawal, developed enhanced neuronal characteristics including, neurite extension and upregulation of neuronal markers of differentiation. When B35 neurons were subjected to oxygen glucose deprivation (OGD)/reoxygenation or glutamate, widespread neuronal death was seen after both treatments. Treatment with NRG-1 immediately after OGD significantly increased neuronal survival. NRG-1 administration also resulted in a significant decrease in annexin V, an early marker of apoptosis. However, the neurotoxic actions of glutamate were unaffected by NRG-1. The neuroprotective effects of NRG-1 were prevented by an inhibitor of the phosphatidylinositol-3-kinase/Akt pathway. These results provide a new model to gain insight into the mechanisms employed by NRG-1 to protect neurons from ischemic brain injury.
Keywords: neuregulin, acetylcholine receptor reducing activity, glutamate excitotoxicity, Glial growth factor, heregulin, neu differentiation factor, ischemia, apoptosis, oxygen glucose deprivation, neuronal cell culture
1. Introduction
Ischemic stroke results in cellular energy depletion, excessive neuronal depolarization and massive glutamate release from neurons [6,9,22,24]. The excess glutamate released overstimulates N-methyl-D-aspartate (NMDA)-type glutamate receptors, ultimately leading to glutamate-induced excitotoxicity and neuronal death. This excitotoxic neuronal injury also triggers inflammatory and oxidative stress responses that lead to a delayed apoptotic neuronal death. A number of recent reviews have described the delayed apoptotic and inflammatory mechanisms that occur in neuronal cells following ischemia [5,6,9,22,24,25,31].
The neuregulins are a family of multipotent growth factors that includes acetylcholine receptor inducing activities (ARIAs), glial growth factors (GGFs), heregulins and neu differentiation factors (NDFs) [13,19,20,27,42]. Neuregulin-1 (NRG- 1) has been shown to promote the survival and function of neuronal and non-neuronal cell types [1,3,11,15,38,39]. NRG-1 binds to erbB receptors and can signal through various pathways, including the phosphatidylinositol-3-kinase (PI3K)/Akt pathway [3,4,12]. Several recent reports from our laboratory and others demonstrated that NRG-1 is neuroprotective in vivo following ischemia [16,33,43–45]. The effects of NRG-1 were shown to be associated with an inhibition of mechanisms associated with inflammation and neuronal apoptosis [33,44,45].
In this study, we use a rat B35 neuronal cell line [32] to further elucidate the molecular mechanisms associated with the direct neuroprotective effects of NRG-1 in ischemia. Many neuronal cell lines require use of growth factors to differentiate into neurons. However, the use of growth factors for differentiation could complicate results seen when examining the effect of growth factors such as NRG-1. The advantage of B-35 neurons is the ability to differentiate simply upon serum withdrawal. They develop large growth cones and extend long neurites when grown in low serum in the absence of endogenous growth factors [30,32]. Here, we further characterize the differentiation of B35 neurons to determine their suitability to study ischemia in vitro. We also examine whether NRG-1 can directly prevent ischemia-induced neuronal death in vitro by activating the PI3K/Akt survival pathway.
2. Results
Characterization of B35 neurons upon serum withdrawal
Rat B35 neuroblastoma cells were derived from tumors of the neonatal rat central nervous system and display neuronal properties such as membrane excitability and expression of enzymes for neurotransmitter release, including acetylcholinesterase, and glutamic acid decarboxylase upon differentiation by serum withdrawal [32,41]. We examined the expression specific neuronal markers in undifferentiated B35 cells and upon serum withdrawal. As previously reported, B35 cells grown in serum are round, phase-bright cells with few, short processes (Fig. 1A). Upon serum withdrawal, cells begin to elaborate long processes within 2–3 days of culture which continues for several days in vitro (Fig. 1B). Undifferentiated B-35 cells expressed neuron specific enolase (NSE) as previously described [41], as well as neurofilament (NF200) and synaptophysin (SYN) when grown in the presence of serum (Fig. 2). These levels of these neuronal markers appeared virtually unchanged in differentiated cells after serum withdrawal. Consistent with previous studies [32], undifferentiated B-35 cells express choline acetyltransferase (ChAT); however, unlike the neuronal markers, serum withdrawal resulted in an increase in the levels of this enzyme (Fig. 3). B-35 cells did not synthesize tyrosine hydroxylase (TH), a marker for catecholaminergic neurons (Fig. 3), or serotonin (not shown) in either differentiation state as previously shown [32].
Figure 1.
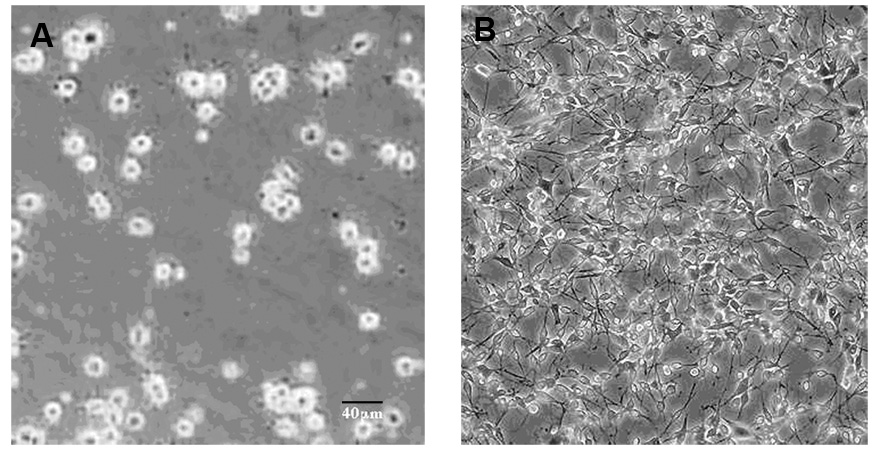
Differentiation of rat B-35 neuroblastoma cells. Rat B-35 neuroblastoma cells were grown in serum (A; undifferentiated) or serum-free medium (B; differentiated) for 3 days. Representative bright field images are shown. Upon serum withdrawal, cells begin to elaborate long processes within 2 days of culture which continues for several days in vitro. The scale bar is 40 µm.
Figure 2.
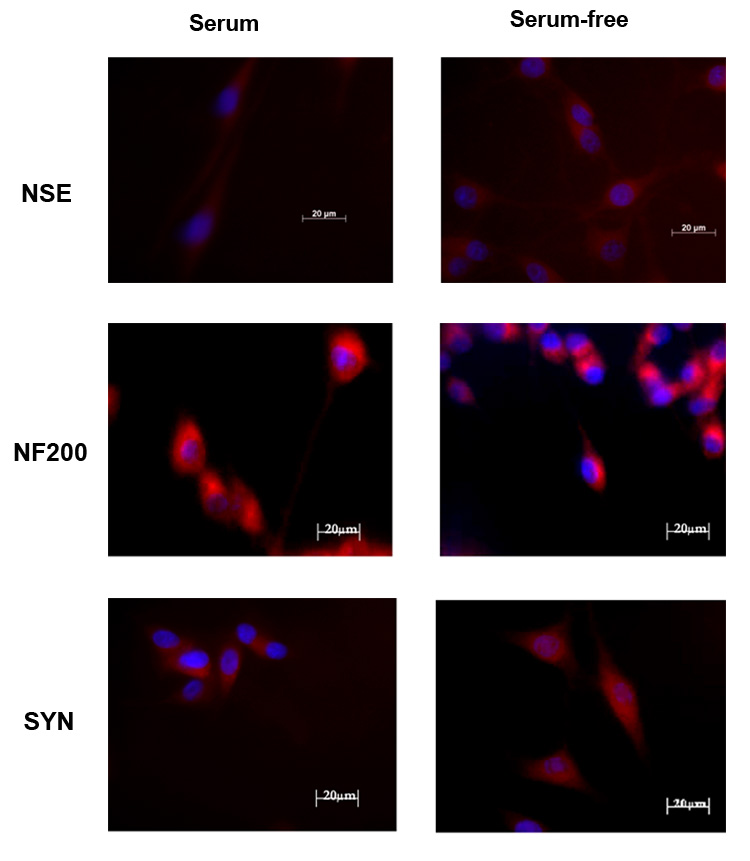
Rat B-35 neuroblastoma cells were grown in serum (left column) or serum-free medium (right column) for 3 days. Representative images are shown (n = 3). B35 cell express neuron specific enolase (NSE) as well as neurofilament 200 (NF200) and synaptophysin (SYN) when grown in the absence or presence of serum. These levels of these neuronal markers appeared virtually unchanged in differentiated cells after serum withdrawal. The scale bar is 20 µm.
Figure 3.
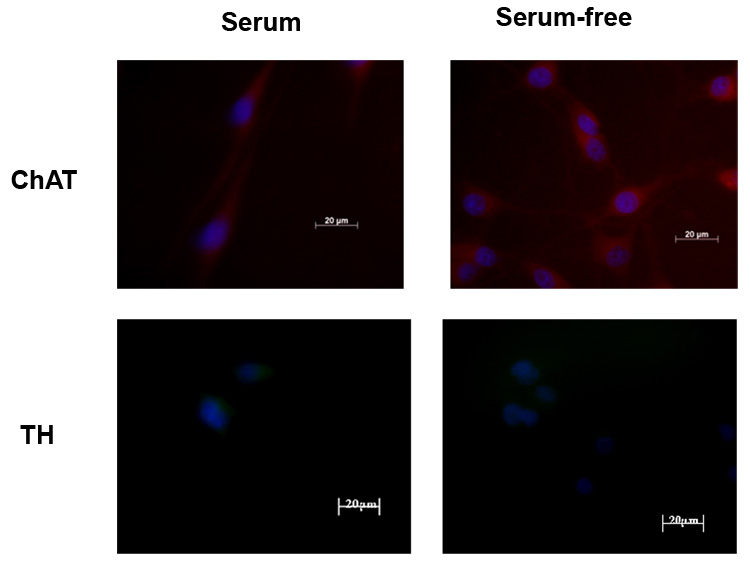
Rat B-35 neuroblastoma cells were grown in serum (left column) or serum-free medium (right column) for 3 days. Representative images are shown (n = 3). Consistent with previous studies [32], undifferentiated B-35 cells (left column) express choline acetyltransferase (ChAT). However, serum withdrawal (right column) resulted in an increase in the expression of ChAT. B-35 cells did not synthesize tyrosine hydroxylase (TH). The scale bar is 20 µm.
OGD and glutamate exposure decrease survival in differentiated B35 neurons
Glutamate excitotoxicity and oxygen glucose deprivation (OGD) are well-characterized neuronal cellular injury methods that mimic events that occur following an ischemic insult in vivo [2,14,17,29,36]. As an in vitro method of ischemia, B35 cells were subjected to OGD followed by reoxygenation for a total of 24 hours. We observed a time-dependent decrease in cell survival with increased OGD duration (Figs. 4A,B). To examine whether these cells exhibited glutamate excitotoxicity, B35 neurons were exposed to various concentrations of glutamate for 1 hour and allowed to recover for 23 hours. Quantitative analysis showed a dose-dependent decrease in neuronal survival after glutamate exposure (Figs. 4C,D).
Figure 4.
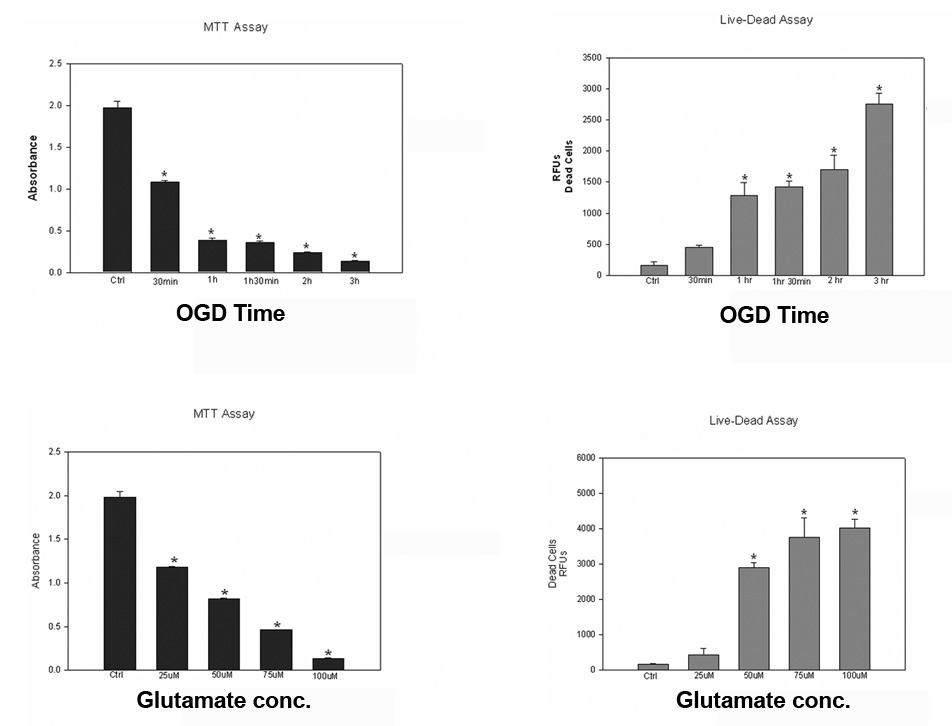
Effect of OGD and glutamate exposure time on B35 cell survival. B-35 cells were subjected to OGD at the indicated time followed by reoxygenation for a total of 24 hours. Results revealed a time-dependent decrease in cell death with increase in duration of OGD when using an MTT assay (upper left) or the Live-Dead cytotoxicity assay (upper right). B-35 cells were subjected to glutamate treatment for 1hr followed by 24 hours recovery and analyzed by the MTT (lower left) or the Live-Dead assay (lower right). Experiments were performed 3 or more times in triplicate. Values are represented as means ± SEM; * denotes significant difference from control.
NRG-1 increases neuronal survival after OGD, but not glutamate exposure
The capability of B35 cells to respond to NRG-1 was examined by determining the expression of the erbB receptors. Our results showed expression mRNA for the erbB2 and erbB3, but not erbB4 (Fig. 5). No change in receptor expression was seen following differentiation. To determine the neuroprotective effects of NRG-1 on cell survival after ischemia/reoxygenation in vitro, cells were exposed to OGD and treated with NRG-1 immediately after ischemia and prior to reoxygenation. Administration of NRG-1 immediately after OGD resulted in a 2-fold increase in cell viability compared to untreated cells (Fig. 6A). To examine whether NRG-1 could protect neurons from glutamate excitotoxicity, B-35 cells were incubated with 50 µM glutamate. After a 60 minute exposure, medium was removed and cells were re-incubated in conditioned medium for 24 hours. NRG-1 was added at the indicated concentrations one hour prior to glutamate exposure. Unlike OGD, when B35 cells were subjected glutamate treatment, no difference in survival was seen after NRG-1 treatment, even when up to 10-fold higher concentrations were used (Fig. 6B).
Figure 5.
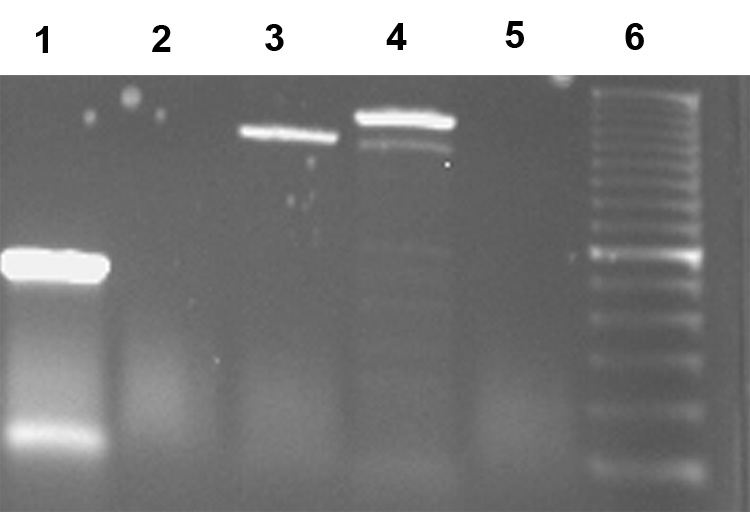
B35 cells express erbB receptor mRNA. RNA was isolated from B35 cells and erbB expression was examined by RT-PCR. GAPDH (lane 1), erbB1 (lane 2), erbB2 (lane 3), erbB3 (lane 4) and erbB4 (lane 5) PCR products along with DNA ladder (lane 6) were separated by agarose gel electrophoresis.
Figure 6.
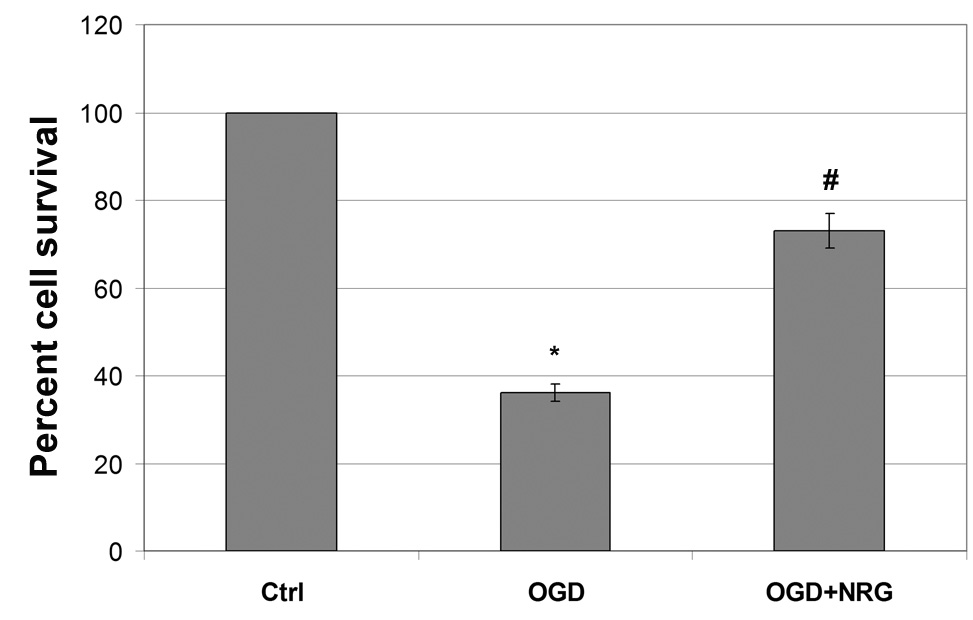
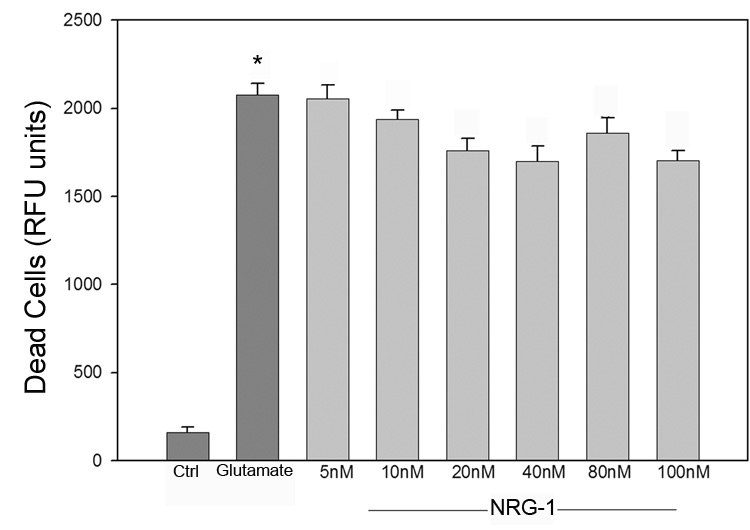
NRG-1 increases survival after OGD but not glutamate exposure. B-35 cells were subjected to 90 minutes of OGD 22.5 hours of reoxygenation. NRG-1 treatment resulted in a two-fold increase in cell viability (A). When B-35 cells were incubated with 50 µM glutamate, NRG-1 did not protect neurons from glutamate excitotoxicity. Values are represented as means ± SEM; * denotes difference from control, # denotes difference from OGD (n = 5; p<0.05).
To examine whether anti-apoptotic mechanisms could be involved in neuroprotection by NRG-1, we analyzed the labeling of cells with annexin V, an early marker of apoptosis that binds to phosphatidyl serine transposed in the outer membrane during the apoptotic process [34,37,40]. Control samples do not show annexin V binding, however, a significant increase in annexin V labeling was detected in cells following OGD (Fig. 7). Annexin V levels were significantly attenuated by 40% in cells treated with NRG-1 after OGD.
Figure 7.
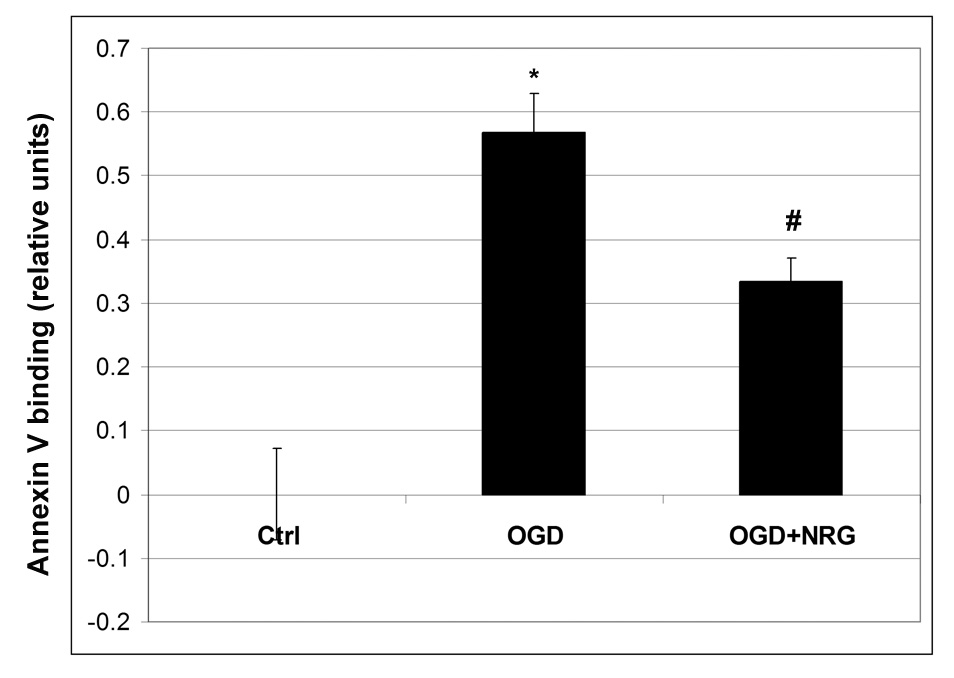
NRG-1 decreases annexin V labeling after OGD. OGD results in an increase in annexin V labeling of cells that is reduced by NRG-1. Values are represented as means ± SEM;* denotes difference from control, # denotes difference from OGD (n = 5; p<0.05)
Neuroprotection by NRG-1 in OGD involves the PI3K/Akt pathway
NRG-1 has been shown to stimulate the PI3K/Akt pathways, which has a well established role in cell survival [3,4,12]. Baseline pAkt levels are relatively low in control cultures, however, stimulation with NRG-1 results in a dramatic increase in pAkt levels while total Akt levels were unchanged following NRG-1 exposure (Fig. 8A). To determine whether PI3K/Akt signaling pathways was responsible for NRG-1’s neuroprotective capability, we applied wortmannin, an inhibitor of PI3K, to cultures prior to OGD. Wortmannin prevented NRG-1 from protecting neurons from OGD (Fig. 8B). Our data suggest that neuroprotective effect of NRG-1 in ischemia involves the activation of the PI3K/Akt pathway.
Figure 8.
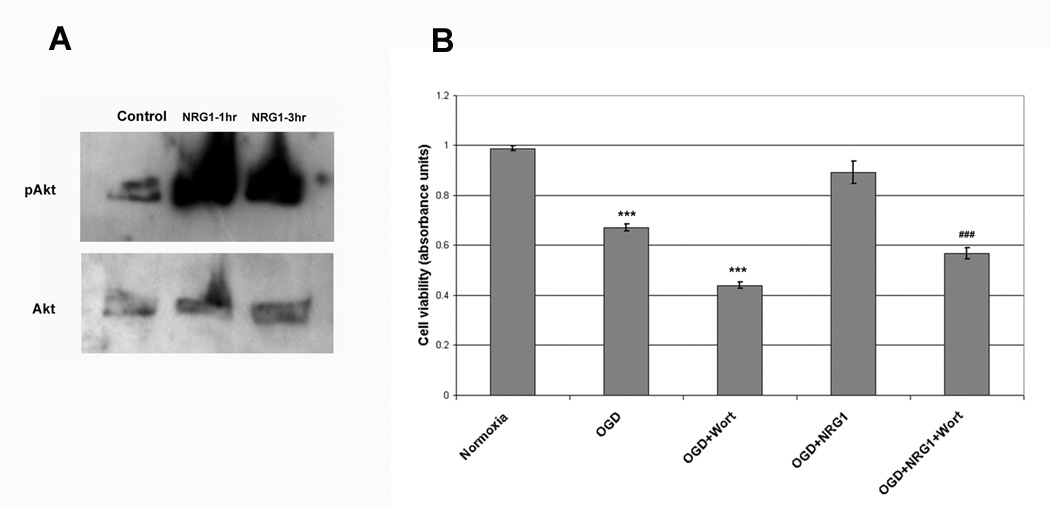
Neuroprotection by NRG-1 involved the PI3K/Akt pathway. Stimulation with NRG-1 resulted in a dramatic increase in pAkt levels while total Akt levels remained unchanged (A). Wortmannin, an inhibitor of PI3K, prevented NRG-1 from protecting neurons from OGD (B). Values are represented as means ± SEM; *** denotes difference from control, ### denotes difference from OGD (n = 3; p<0.001).
3. Discussion
Previous findings from our laboratory and others demonstrated that NRG-1 prevents neuronal death following ischemia in vivo [16,33,43–45]. However, the neuroprotection in the in vivo models could result either from direct effects on neurons or indirect effects on non-neuronal cells that facilitate ischemia-induced neuronal death. The results of this study clearly show the direct protective effects of NRG-1 on neurons following in vitro ischemia (OGD).
Cerebral ischemia results in an early glutamate-induced excitotoxicity [6,9,22,24]. Subsequently, delayed neuronal death occurs that is characterized by inflammation, oxidative stress and apoptosis. Glutamate excitotoxicity and OGD are two widely accepted methods for studying mechanisms associated with cerebral ischemia in vitro [17,29]. In this study, we examined the utility of B35 neurons as a model for ischemic neuronal injury. The advantage of B35 neurons is the ability to adopt neuronal characteristics upon serum withdrawal without the requirement of exogenously applied growth factors to induce neuronal differentiation [32]. The use of growth factors to stimulate differentiation as with many neuronal cells lines could confound the results seen when investigating the effects of growth factors like NRG-1 in neuronal cultures.
In this study, we showed that while NRG-1 protects neurons from OGD, it has no effect on glutamate neurotoxicity. This is consistent with our previous observations that NRG-1 protects neurons from delayed neuronal death and apoptosis in vivo [43–45]. The PI3K/Akt signaling pathway is a well described survival and anti-apoptotic factor in neurons and other cells during ischemia [18,21,23,26,46]. Binding of NRG-1 erbB receptors has been shown to activate the PI3K/Akt pathway [3,4,12]. NRG-1 strongly stimulated the phophorylation of Akt in B35 cells. Blockade of PI3K prevented NRG-1 from protecting neurons from OGD. This is consistent with previous findings in PC-12 cells demonstrating that NRG-1 induced the activation of PI3K and inhibition of the PI3K/Akt pathway activity prevented the NRG-1 mediate survival effect. [7,8,11,15]. The PI3K/Akt pathway has been shown to have direct effects on multiple apoptosis mechanisms [10,28,46]. For example, Akt has been shown to phosphorylate and inactivate the pro-apoptotic Bcl-2 related protein, BAD as well as other downstream effectors, including NF-κB, Forkhead and GSK-3 and MDM-2, which mediate the effects of Akt on protection from pro-apoptotic stimuli.
In conclusion, we showed that B35 cells are suitable in both glutamate excitotoxicity and OGD initiated in vitro ischemia models. Additionally, we demonstrated that NRG-1 can directly protect neurons from ischemic injury. These findings may aid in the development novel neuroprotective strategies for ischemia and other neurological disorders.
4. Experimental Procedure
Cell Cultures and Oxygen Glucose Deprivation
B35 rat neuroblastoma cells (ATCC, Manassas,VA) were cultured in Dulbecco modified eagle medium (DMEM), 10U/ml penicillin, 10mg/ml streptomycin, and 5% fetal bovine serum (FBS) at 37°C in a 5% CO2 incubator. Cells were differentiated by the changing to a serum-free culture medium 3–5 days prior to treatment. Cultures were subjected to oxygen glucose deprivation (OGD) by incubation in a balanced salt solution (BSS: 116 mM NaCl, 5.4mM KCl, 0.8mM MgSO4, 1.0mM NaH2PO4, 26.2mM NaHCO3, 1.8mM CaCl2, 0.01mM glycine, and 10mg/L phenol red) and placement in a hypoxic atmosphere of 1% O2, 5% CO2. Cultures were subjected to OGD for 90 minutes. After OGD, cultures were treated with 10nM NRG-1 (NRG-1β EGF-like domain, R&D Systems, Minneapolis, Minnesota dissolved in 1% BSA/PBS) or vehicle and returned to normoxic conditions (reoxygenation) for 24 hours. Control samples were placed in a balanced salt solution with 20mM D-glucose.
Glutamate Excitotoxicity
B-35 cells were washed in PBS solution and then incubated with 50 µM glutamate in Hanks BSS (116mM NaCl, 5.4mM KCl, 0.8mM MgSO4 , 1.0mM NaH2PO4, 26.2mM NaHCO3, 1.8mM CaCl2, 0.01mM glycine, and 20mM glucose). After a 60 minute exposure, medium was removed and cells washed in PBS and re-incubated in old conditioned medium for 24 hours. Response of experimental cultures to glutamate excitotoxicity was compared to control cultures in different wells of the same plate where glutamate was not added. NRG-1 was added at the indicated concentrations one hour prior to glutamate exposure.
Immunocytochemistry
Cells were fixed with 4% paraformaldehyde for 30 minutes at room temperature and washed three times in PBS buffer. Fixed cultures were blocked by incubation in PBS with 5% normal goat serum and 0.2% triton X-100 at room temperature for 1 hour. Cultures were subsequently processed for immunocytochemical staining using primary antibodies against neuron specific enolase (NSE), neurofilament 200 (NF200), synaptophysin (SYN), choline acetyltransferase (ChAT), glutamic acid decarboxylase (GAD67), tyrosine hydroxylase (TH) and serotonin. Antibodies were diluted in blocking buffer then cultures were incubated with primary antibody for either 2 hours at room temperature or overnight at 4 °C. Cells were washed four times with PBS, then incubated with fluorescent secondary antibodies for 1 hour at room temperature or overnight at 4 °C. Cultures were mounted with Vectasheild containing DAPI for counterstaining. Cells were visualized using fluorescence confocal microscopy.
Cell viability
Cell viability was measured using the Molecular Probes Live/Dead Cell Viability/Cytotoxicity Assay Kit (Invitrogen, Carlsbad,CA). Calcein (live cell activity) was measured in either a fluorescent plate reader at excitation /emission 485/530nm or via fluorescent microscopy using a fluorescein filter. EthD-1 (dead cell activity) was measured in either a fluorescent plate reader at excitation /emission 530/645nm or via fluorescent microscopy using a Texas red filter. Live and dead cell values are shown as the mean ± SEM of assay results from three separate experiments performed in triplicate. Statistical analysis was carried out using the student t-test.
Annexin V binding
Cultures were subjected to OGD/reoxygenation and analyzed using an Annexin V Assay Kit (BD Pharmingen, San Diego, CA). Cultures were washed twice with PBS and annexin V binding was measured by fluorescent spectroscopy at excitation /emission 485/530nm for annexin staining or by fluorescence microscopy. Relative fluorescent unit (RFU) values gathered by spectroscopy are presented as the mean ± SEM of assay results from three experiments. Statistical analysis was carried out using the student t-test.
RNA isolation and RT-PCR
Total RNA was isolated using the Ambion RNAqueous RNA isolation system (Austin, TX) according to the manufactures protocol. RT-PCR was used to amplify the isolated RNA in a Bio-Rad thermal cycler. To quantify mRNA expression primers for erbB2 (forward 5'-AGC TGG TGA CAC AGC TTA-3'; reverse 5'-TGG TTG GG ACT CTT GAC-3'); erbB3 (forward 5'-GAC CTA GAC CTA GAC TT-3'; reverse 5'-TCT GAT GAC TCT GAT GC-3'); erbB4 (forward 5'-CAT CTA CAC ATC CAG AAC A- 3'; reverse 5'-AAA CAT CTC AGC CGT TGC A-3'); 'and GAPDH (forward 5’-GAA GGG CTC ATG ACC ACA GTC C-3’; reverse 5’-TCC ACC ACC CTG TTG CTG TAG CC-3’) [35] were used for RT-PCR using the Qiagen (Valencia, CA) one-step kit (30min at 50° C, 15 min at 95° C, then 35 cycles of denaturation at 94° C for 30s, annealing at 60° C for 30s and extension at 72° C for 30s with a 10min final extension period at 72° C). PCR products were loaded on 2% agarose gel, quantified using Kodak digital imaging software and expressed as a ratio of the control gene (GAPDH). Intensity values are means ± SEM of PCR results from three experiments.
Protein Isolation and Western Analysis
Total protein was extracted from cells by lysing them with buffer (50mM Tris-HCl, pH 7.4, 150mM NaCl, 1mM EDTA, 0.5% Triton X-100, 0.5% Nonidet P-40, 1mM sodium orthovandate, 1mM phenyl methanesulfonyl fluoride, p 8.0 with 1:500 protease inhibitor cocktail) for 30 minutes at 4°C. Harvested lysates were denatured with loading buffer (final concentrations: 62.5 mM Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 50 mM DTT, 0.1% bromophenol blue), resolved in SDS/5% polyacrylamide gels and transferred to polyvinyldifluoride (PVDF) membranes (Bio-Rad). Membranes were blocked with 3% nonfat dry milk in phosphate buffered saline-0.05% Tween 20 (PBST) and exposed to primary antibodies Akt and phospho-Akt (Cell Signaling Technology, Danvers, MA) that were diluted in blocking buffer overnight at 4°C. Membranes were then incubated with secondary anti-rabbit HRP-linked antibodies and were visualized on x-ray film (Hyperfilm, Amersham Biosciences) using a chemiluminescence kit (New England Biolabs).
Acknowledgements
This work was supported by NIH grants R01 NS 34194; U54 NS34194 and the W.M. Keck Foundation. The investigation was conducted in a facility constructed with support from Research Facilities Improvement Grant #C06 RR-07571 from the National Center for Research Resources, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bermingham-McDonogh O, McCabe K, Reh T. Effects of GGF/neuregulins on neuronal survival and neurite outgrowth correlate with erbB2/neu expression in developing rat retina. Dev Suppl. 1996;122:1427–1438. doi: 10.1242/dev.122.5.1427. [DOI] [PubMed] [Google Scholar]
- 2.Bruno VM, Goldberg MP, Dugan LL, Giffard RG, Choi DW. Neuroprotective effect of hypothermia in cortical cultures exposed to oxygen-glucose deprivation or excitatory amino acids. J Neurochem. 1994;63:1398–1406. doi: 10.1046/j.1471-4159.1994.63041398.x. [DOI] [PubMed] [Google Scholar]
- 3.Buonanno A, Fischbach GD. Neuregulin and ErbB receptor signaling pathways in the nervous system. Curr Opin Neurobiol. 2001;11:287–296. doi: 10.1016/s0959-4388(00)00210-5. [DOI] [PubMed] [Google Scholar]
- 4.Burden S, Yarden Y. Neuregulins and their receptors: a versatile signaling module in organogenesis and oncogenesis. Neuron. 1997;18:847–855. doi: 10.1016/s0896-6273(00)80324-4. [DOI] [PubMed] [Google Scholar]
- 5.Chan PH. Mitochondria and neuronal death/survival signaling pathways in cerebral ischemia. Neurochem Res. 2004;29:1943–1949. doi: 10.1007/s11064-004-6869-x. [DOI] [PubMed] [Google Scholar]
- 6.del Zoppo G, Ginis I, Hallenbeck JM, Iadecola C, Wang X, Feuerstein GZ. Inflammation and stroke: putative role for cytokines, adhesion molecules and iNOS in brain response to ischemia. Brain Pathol. 2000;10:95–112. doi: 10.1111/j.1750-3639.2000.tb00247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di Segni A, Farin K, Pinkas-Kramarski R. ErbB4 Activation Inhibits MPP+-Induced Cell Death In PC12-ErbB4 Cells: Involvement of PI3K and Erk Signaling. J Mol Neurosci. 2006;29:257–268. doi: 10.1385/JMN:29:3:257. [DOI] [PubMed] [Google Scholar]
- 8.Di Segni A, Shaharabani E, Stein R, Pinkas-Kramarski R. Neuregulins rescue PC12-ErbB-4 cells from cell death induced by beta-amyloid peptide: involvement of PI3K and PKC. J Mol Neurosci. 2005;26:57–69. doi: 10.1385/JMN:26:1:057. [DOI] [PubMed] [Google Scholar]
- 9.Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- 10.Downward J. PI 3-kinase, Akt and cell survival. Semin Cell Dev Biol. 2004;15:177–182. doi: 10.1016/j.semcdb.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 11.Erlich S, Goldshmit Y, Lupowitz Z, Pinkas-Kramarski R. ErbB-4 activation inhibits apoptosis in PC12 cells. Neuroscience. 2001;107:353–362. doi: 10.1016/s0306-4522(01)00350-5. [DOI] [PubMed] [Google Scholar]
- 12.Falls DL. Neuregulins: functions, forms, and signaling strategies. Exp Cell Res. 2003;284:14–30. doi: 10.1016/s0014-4827(02)00102-7. [DOI] [PubMed] [Google Scholar]
- 13.Falls DL, Rosen KM, Corfas G, Lane WS, Fischbach GD. ARIA, a protein that stimulates acetylcholine receptor synthesis, is a member of the neu ligand family. Cell. 1993;72:801–815. doi: 10.1016/0092-8674(93)90407-h. [DOI] [PubMed] [Google Scholar]
- 14.Goldberg MP, Choi DW. Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calcium-independent mechanisms of neuronal injury. J Neurosci. 1993;13:3510–3524. doi: 10.1523/JNEUROSCI.13-08-03510.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldshmit Y, Erlich S, Pinkas-Kramarski R. Neuregulin rescues PC12-ErbB4 cells from cell death induced by H(2)O(2). Regulation of reactive oxygen species levels by phosphatidylinositol 3-kinase. J Biol Chem. 2001;276:46379–46385. doi: 10.1074/jbc.M105637200. [DOI] [PubMed] [Google Scholar]
- 16.Guo WP, Wang J, Li RX, Peng YW. Neuroprotective effects of neuregulin-1 in rat models of focal cerebral ischemia. Brain Res. 2006;1087:180–185. doi: 10.1016/j.brainres.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 17.Gwag BJ, Lobner D, Koh JY, Wie MB, Choi DW. Blockade of glutamate receptors unmasks neuronal apoptosis after oxygen-glucose deprivation in vitro. Neuroscience. 1995;68:615–619. doi: 10.1016/0306-4522(95)00232-8. [DOI] [PubMed] [Google Scholar]
- 18.Hillion JA, Li Y, Maric D, Takanohashi A, Klimanis D, Barker JL, Hallenbeck JM. Involvement of Akt in preconditioning-induced tolerance to ischemia in PC12 cells. J Cereb Blood Flow Metab. 2006;26:1323–1331. doi: 10.1038/sj.jcbfm.9600286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ho WH, Armanini MP, Nuijens A, Phillips HS, Osheroff PL. Sensory and motor neuron-derived factor. A novel heregulin variant highly expressed in sensory and motor neurons. J Biol Chem. 1995;270:26722. [PubMed] [Google Scholar]
- 20.Holmes WE, Sliwkowski MX, Akita RW, Henzel WJ, Lee J, Park JW, Yansura D, Abadi N, Raab H, Lewis GD, et al. Identification of heregulin, a specific activator of p185erbB2. Science. 1992;256:1205–1210. doi: 10.1126/science.256.5060.1205. [DOI] [PubMed] [Google Scholar]
- 21.Horn AP, Gerhardt D, Geyer AB, Valentim L, Cimarosti H, Tavares A, Horn F, Lenz G, Salbego C. Cellular death in hippocampus in response to PI3K pathway inhibition and oxygen and glucose deprivation. Neurochem Res. 2005;30:355–361. doi: 10.1007/s11064-005-2609-0. [DOI] [PubMed] [Google Scholar]
- 22.Iadecola C, Alexander M. Cerebral ischemia and inflammation. Curr Opin Neurol. 2001;14:89–94. doi: 10.1097/00019052-200102000-00014. [DOI] [PubMed] [Google Scholar]
- 23.Lawlor MA, Alessi DR. PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J Cell Sci. 2001;114:2903–2910. doi: 10.1242/jcs.114.16.2903. [DOI] [PubMed] [Google Scholar]
- 24.Lee JM, Grabb MC, Zipfel GJ, Choi DW. Brain tissue responses to ischemia. J Clin Invest. 2000;106:723–731. doi: 10.1172/JCI11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lo EH, Moskowitz MA, Jacobs TP. Exciting, radical, suicidal: how brain cells die after stroke. Stroke. 2005;36:189–192. doi: 10.1161/01.STR.0000153069.96296.fd. [DOI] [PubMed] [Google Scholar]
- 26.Malhotra S, Savitz SI, Ocava L, Rosenbaum DM. Ischemic preconditioning is mediated by erythropoietin through PI-3 kinase signaling in an animal model of transient ischemic attack. J Neurosci Res. 2006;83:19–27. doi: 10.1002/jnr.20705. [DOI] [PubMed] [Google Scholar]
- 27.Marchionni MA, Goodearl AD, Chen MS, Bermingham-McDonogh O, Kirk C, Hendricks M, Danehy F, Misumi D, Sudhalter J, Kobayashi K, et al. Glial growth factors are alternatively spliced erbB2 ligands expressed in the nervous system. Nature. 1993;362:312–318. doi: 10.1038/362312a0. [DOI] [PubMed] [Google Scholar]
- 28.Mitsiades CS, Mitsiades N, Koutsilieris M. The Akt pathway: molecular targets for anti-cancer drug development. Curr Cancer Drug Targets. 2004;4:235–256. doi: 10.2174/1568009043333032. [DOI] [PubMed] [Google Scholar]
- 29.Noraberg J, Poulsen FR, Blaabjerg M, Kristensen BW, Bonde C, Montero M, Meyer M, Gramsbergen JB, Zimmer J. Organotypic hippocampal slice cultures for studies of brain damage, neuroprotection and neurorepair. Curr Drug Targets CNS Neurol Disord. 2005;4:435–452. doi: 10.2174/1568007054546108. [DOI] [PubMed] [Google Scholar]
- 30.Otey CA, Boukhelifa M, Maness P. B35 neuroblastoma cells: an easily transfected, cultured cell model of central nervous system neurons. Methods Cell Biol. 2003;71:287–304. doi: 10.1016/s0091-679x(03)01013-6. [DOI] [PubMed] [Google Scholar]
- 31.Ouyang YB, Giffard RG. Cellular neuroprotective mechanisms in cerebral ischemia: Bcl-2 family proteins and protection of mitochondrial function. Cell Calcium. 2004;36:303–311. doi: 10.1016/j.ceca.2004.02.015. [DOI] [PubMed] [Google Scholar]
- 32.Schubert D, Heinemann S, Carlisle W, Tarikas H, Kimes B, Patrick J, Steinbach JH, Culp W, Brandt BL. Clonal cell lines from the rat central nervous system. Nature. 1974;249:224–227. doi: 10.1038/249224a0. [DOI] [PubMed] [Google Scholar]
- 33.Shyu WC, Lin SZ, Chiang MF, Yang HI, Thajeb P, Li H. Neuregulin-1 reduces ischemia-induced brain damage in rats. Neurobiol Aging. 2004;25:935–944. doi: 10.1016/j.neurobiolaging.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 34.Stadelmann C, Lassmann H. Detection of apoptosis in tissue sections. Cell Tissue Res. 2000;301:19–31. doi: 10.1007/s004410000203. [DOI] [PubMed] [Google Scholar]
- 35.Sundaresan S, Penuel E, Sliwkowski MX. The biology of human epidermal growth factor receptor 2. Curr Oncol Rep. 1999;1:16–22. doi: 10.1007/s11912-999-0005-7. [DOI] [PubMed] [Google Scholar]
- 36.Tymianski M, Charlton MP, Carlen PL, Tator CH. Source specificity of early calcium neurotoxicity in cultured embryonic spinal neurons. J Neurosci. 1993;13:2085–2104. doi: 10.1523/JNEUROSCI.13-05-02085.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van den Eijnde SM, Luijsterburg AJ, Boshart L, De Zeeuw CI, van Dierendonck JH, Reutelingsperger CP, Vermeij-Keers C. In situ detection of apoptosis during embryogenesis with annexin V: from whole mount to ultrastructure. Cytometry. 1997;29:313–320. doi: 10.1002/(sici)1097-0320(19971201)29:4<313::aid-cyto8>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 38.Vaskovsky A, Lupowitz Z, Erlich S, Pinkas-Kramarski R. ErbB-4 activation promotes neurite outgrowth in PC12 cells. J Neurochem. 2000;74:979–987. doi: 10.1046/j.1471-4159.2000.0740979.x. [DOI] [PubMed] [Google Scholar]
- 39.Verdi JM, Groves AK, Farinas I, Jones K, Marchionni MA, Reichardt LF, Anderson DJ. A reciprocal cell-cell interaction mediated by NT-3 and neuregulins controls the early survival and development of sympathetic neuroblasts. Neuron. 1996;16:515–527. doi: 10.1016/s0896-6273(00)80071-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vincent AM, Maiese K. Direct temporal analysis of apoptosis induction in living adherent neurons. J Histochem Cytochem. 1999;47:661–672. doi: 10.1177/002215549904700508. [DOI] [PubMed] [Google Scholar]
- 41.Vinores SA, Marangos PJ, Bonnin JM, Rubinstein LJ. Immunoradiometric and immunohistochemical demonstration of neuron-specific enolase in experimental rat gliomas. Cancer Res. 1984;44:2595–2599. [PubMed] [Google Scholar]
- 42.Wen D, Peles E, Cupples R, Suggs SV, Bacus SS, Luo Y, Trail G, Hu S, Silbiger SM, Levy RB, et al. Neu differentiation factor: a transmembrane glycoprotein containing an EGF domain and an immunoglobulin homology unit. Cell. 1992;69:559–572. doi: 10.1016/0092-8674(92)90456-m. [DOI] [PubMed] [Google Scholar]
- 43.Xu Z, Croslan DR, Harris AE, Ford GD, Ford BD. Extended therapeutic window and functional recovery after intraarterial administration of neuregulin-1 after focal ischemic stroke. J Cereb Blood Flow Metab. 2006;26:527–535. doi: 10.1038/sj.jcbfm.9600212. [DOI] [PubMed] [Google Scholar]
- 44.Xu Z, Ford GD, Croslan DR, Jiang J, Gates A, Allen R, Ford BD. Neuroprotection by neuregulin-1 following focal stroke is associated with the attenuation of ischemia-induced pro-inflammatory and stress gene expression. Neurobiol Dis. 2005;19:461–470. doi: 10.1016/j.nbd.2005.01.027. [DOI] [PubMed] [Google Scholar]
- 45.Xu Z, Jiang J, Ford G, Ford BD. Neuregulin-1 is neuroprotective and attenuates inflammatory responses induced by ischemic stroke. Biochem Biophys Res Commun. 2004;322:440–446. doi: 10.1016/j.bbrc.2004.07.149. [DOI] [PubMed] [Google Scholar]
- 46.Zhao H, Shimohata T, Wang JQ, Sun G, Schaal DW, Sapolsky RM, Steinberg GK. Akt contributes to neuroprotection by hypothermia against cerebral ischemia in rats. J Neurosci. 2005;25:9794–9806. doi: 10.1523/JNEUROSCI.3163-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]