

Abstract
tRNA pseudouridine synthase I (ΨSI) catalyzes the conversion of uridine to Ψ at positions 38, 39, and/or 40 in the anticodon loop of tRNAs. ΨSI forms a covalent adduct with 5-fluorouracil (FUra)-tRNA (tRNAPhe containing FUra in place of Ura) to form a putative analog of a steady-state intermediate in the normal reaction pathway. Previously, we proposed that a conserved aspartate of the enzyme serves as a nucleophilic catalyst in both the normal enzyme reaction and in the formation of a covalent complex with FUra-tRNA. The covalent adduct between FUra-tRNA and ΨSI was isolated and disrupted by hydrolysis and the FUra-tRNA was recovered. The target FU39 of the recovered FUra-tRNA was modified by the addition of water across the 5,6-double bond of the pyrimidine base to form 5,6-dihydro-6-hydroxy-5-fluorouridine. We deduced that the conserved aspartate of the enzyme adds to the 6-position of the target FUra to form a stable covalent adduct, which can undergo O-acyl hydrolytic cleavage to form the observed product. Assuming that an analogous covalent complex is formed in the normal reaction, we have deduced a complete mechanism for ΨS.
Keywords: enzyme mechanism, 5-fluorouridine hydrate
Pseudouridine synthase (ΨS) catalyzes the conversion of uridine residues in RNA to pseudouridine (Ψ), an unusual nucleoside with a carbon-carbon glycoside bond (1–3). The minimal mechanism for the reaction involves cleavage of the N-glycosidic bond of the target Urd, movement of the cleaved uracil to juxtapose C5 of the pyrimidine and C1′ of the ribosyl moiety of RNA, and formation of the C1′-C5 carbon-carbon bond.
The earliest proposed mechanism for ΨS was based on chemical considerations and analogies with enzymes such as thymidylate synthase (4). This involved nucleophilic attack of the thiol of a Cys residue of the enzyme at C6 of the pyrimidine to form a 5,6-dihydropyrimidine (as in Scheme S1, R = H, except that Cys replaces the Asp residue). The formation of a 5,6-dihydropyrimidine adduct could facilitate all of the reactions necessary for the subsequent conversion to products. It would (i) enhance the lability of the N-glycosidic bond (5), (ii) provide an axis for a 180° rotation of the pyrimidine ring to juxtapose the C1′ and C5 positions, and (iii) activate the 5-carbon of the pyrimidine toward electrophilic attack by C1′ and facilitate subsequent β-elimination to give product and unchanged enzyme (6).
Scheme 1.

Proposed mechanism I for ΨSI.
Evidence for an essential Cys in ΨS was provided by the observation that enzyme activity was inhibited by sulfhydryl reagents (7). By analogy with thymidylate synthase, m5U-tRNA methyl transferase (MTase) and m5C-DNA MTase, such covalent adducts may form via initial attachment of a Cys thiol to the 6-position of the target 5-fluorouracil (FUra) (6, 8). Further, the involvement of an enzyme nucleophile was indicated by isolation of a covalent adduct between ΨSI and FUra-tRNA (tRNAPhe containing FUra in place of Ura) (9). However, sequence comparison of putative and proven ΨSs did not reveal a conserved Cys residue that could serve as the essential nucleophilic catalyst (9). Moreover, mutation of any or all of the Cys residues of ΨSI to Ala did not destroy the catalytic activity of the enzyme (10). Thus, the proposal of an essential Cys residue in the mechanism of ΨS was abandoned.
Recently, sequence comparisons of ΨSs revealed a completely conserved Asp residue (D60 in Escherichia coli ΨSI) which when mutated destroyed both catalytic activity and covalent adduct formation with FUra-tRNA (9). To account for all available data, we proposed that catalysis by ΨSs proceeded by one of two possible mechanisms. The first is that shown in Scheme S1 (R = H) where the conserved Asp-60 serves as the nucleophile attacking C6 of the target Urd residue. The second (Scheme S2, R = H), was born by analogy to glycosidases that use Asp as a nucleophile to displace a leaving group (Ura, in the case of ΨS) from the C1′ position of sugars and form covalent intermediates between the catalytic Asp residue and C1′ of the sugar (9).
Scheme 2.

Proposed mechanism II for ΨSI.
In the present work, we show that the ΨSI-FUra-tRNA covalent complex can be hydrolyzed to release a modified FUra-tRNA, and describe analyses of the FUra-tRNA product. Assuming that covalent adduct formation is mechanistically analogous to the normal catalytic reaction, we are able to distinguish between the two aforementioned pathways and propose a mechanism for ΨS that is consistent with all available data.
Materials and Methods
Materials.
Recombinant tRNA ΨSI (9) and T7 RNA polymerase (11) were as described. Plasmid p67YF0 used for preparation of yeast tRNAPhe was a gift from O.C. Uhlenbeck (University of Colorado, Boulder). [5′-α-32P]CTP and [5′-32P]pCp (3,000 Ci/mmol) were from Amersham Pharmacia. [6-3H]FUTP (2 Ci/mmol) was from Moravek Biochemicals (Brea, CA), and [6-3H]-5-fluorouridine (FUrd) was prepared by alkaline phosphatase digestion of [6-3H]FUTP. Other materials were the purest grade available.
In Vitro tRNA Synthesis.
Yeast FUra-tRNAPhe was synthesized by using BstN I-linearized p67Y0 as template, NTPs (2 mM each of ATP, CTP, GTP, and FUTP) and T7 RNA polymerase (12, 13). [5′-32P]CMP-FUra-tRNA (FUra-tRNA made with [5′-α-32P]CTP) and [6-3H]FUra-tRNA were prepared similarly by using 0.2 mM (0.5 Ci/mmol) of the appropriate labeled NTP. tRNAs were purified by using 7 M urea-15% PAGE. The tRNA-containing band was located under a UV lamp or by autoradiography and excised. tRNA was extracted with 0.5 M NaOAc, 20 mM EDTA, 0.01% SDS and precipitated with ethanol. The concentration of tRNA was calculated by assuming 1,600 pmol per A260 (14).
Isolation of ΨSI-Modified FUra-tRNA.
Typically, a reaction mixture (250 μl) containing 40 μM FUra-tRNA and 80 μM ΨSI in TNE buffer (20 mM Tris⋅HCl, pH 8.0/100 mM NH4Cl/2 mM DTT) (7) was incubated at room temperature for 3 hr and subjected to SDS-12% PAGE. The slower migrating band containing the ΨSI-FUra-tRNA complex was located under a UV lamp, excised, and extracted overnight with water. The extract was filtered through glass wool in a 1-ml pipette tip and incubated at 90°C for 5 min to disrupt the covalent complex. After phenol extraction and addition of 0.1 vol of 3 M NH4OAc, pH 6, the ΨSI-modified FUra-tRNA was precipitated with ethanol, dried, and dissolved in 20 μl of H2O (≈125 μM, ≈25% yield).
Mass Spectrometric Analysis.
FUra-tRNA and ΨSI-modified FUra-tRNA were purified by using 7 M urea-12% PAGE, with 50 mM Tris-borate, pH 8.4, as running buffer. The tRNA-containing bands were excised, extracted with 0.5 M NH4OAc (15), precipitated with ethanol, and dissolved in H2O. tRNAs (4 μl, ≈1 nmol/μl) were incubated with 1 μl of RNase A (1 μg/μl) at room temperature for 1 hr and stored at 4°C. UV-matrix-assisted laser desorption ionization-MS was performed with 3-hydroxypicolinic acid as the matrix. The molecular weights of mononucleotides were as reported (16). MS was performed by the University of California, San Francisco MS Facility or the Protein and Carbohydrate Structure Facility of the University of Michigan, Ann Arbor.
3′-End Labeling of tRNA.
FUra-tRNA and ΨSI-modified FUra-tRNA were labeled at the 3′ end with [5′-32P]pCp using T4 RNA ligase (17) and the resulting FUra-tRNA-[32P]pCp derivatives were purified by 7 M urea-10% PAGE.
Sequence Analysis of ΨSI-Modified FUra-tRNA.
Partial digestion of 3′-32P-labeled unmodified or ΨSI-modified FUra-tRNA-[32P]pCp with RNase A (0.1 ng) was performed in 0.1 M Tris⋅HCl, pH 7.5, 10 mM EDTA at 0°C for 20 min; reactions were terminated by heating at 90°C for 5 min, and reaction mixtures were subjected to 7 M urea-12% PAGE. The reference RNase T1 and T2 sequence ladders were generated as described (18).
RNase A Digestion and 3′-End Group Analysis.
Unmodified and ΨSI-modified [5′-32P]CMP-FUra-tRNA (50 pmol, ≈6 × 105 cpm) were digested with RNase A (5 μg) in 6 μl of 50 mM Tris⋅HCl, pH 8.0, at 37°C for 30 min, and digests were separated by 7 M urea-25% PAGE. The five largest oligoribonucleotides were excised, eluted with RNA extraction buffer, precipitated with ethanol in the presence of E. coli tRNA (10 μg/ml), and dissolved in 10 μl of H2O. Aliquots (≈ 104 cpm) were digested to completion with 1 μg of nuclease P1 in 10 μl of 30 mM NaOAc, pH 5.3, at 37°C for 2 h, or with 10 units of RNase T2 in 10 μl of 50 mM NaOAc, pH 4.5, and 2 mM EDTA at 37°C for 2 h. The resulting [5′-32P]-NMPs or [3′-32P]NMPs were subjected to two-dimensional TLC on cellulose plates (20 × 20 cm), using the mobile phases (first dimension) isobutyric acid-concentrated NH4OH-H2O (66:1:33, vol/vol/vol), (second dimension) isopropanol-concentrated HCl-H2O (70:15:14, vol/vol/vol) (18). The nucleotides were detected by autoradiography.
RNase A Digestion of the ΨSI-[5′-32P]CMP-[6-3H]FUra-tRNA Complex.
A reaction mixture (40 μl) containing 3.2 μM [5′-32P]CMP-[6-3H]FUra-tRNA (1.9 × 106 dpm 3H, 3.2 × 106 cpm 32P) and 12.5 μM ΨSI in NTE buffer was incubated at 37°C for 3 h, digested with 5 μl of RNase A (10 μg/μl) at room temperature for 30 min and subjected to SDS/12% PAGE. The band containing the ΨSI-ribonucleotide complex was excised, extracted overnight with water, and incubated at 90°C for 5 min. After phenol extraction, addition of 5 μg of carrier E. coli tRNA and 0.1 vol of 3 M NH4OAc, pH 6, oligoribonucleotides were precipitated with ethanol, dried, dissolved in 10 μl of H2O, and purified on 7 M urea-20% PAGE. The major radioactive band was excised, extracted overnight with 0.5 ml of RNA extraction buffer, and counted.
RNase A Digestion of ΨSI-Modified [5′-32P]CMP-[6-3H]FUra-tRNA.
ΨSI-modified [5′-32P]CMP-[6-3H]FUra-tRNA (20 pmol, 3 × 105 dpm 3H, 4 × 105 cpm 32P) and unmodified [5′-32P]CMP-[6-3H]FUra-tRNA (32 pmol, 5 × 105 dpm 3H, 7 × 105 cpm 32P) were digested with RNase A (1 μg) in 15 μl of 50 mM Tris⋅HCl, pH 8.0, at 37°C for 30 min; the digests were separated by 7 M urea-25% PAGE. For each reaction, the three largest oligonucleotides were excised, eluted with 0.5 ml of RNA extraction buffer overnight, and counted for 32P and 3H.
Preparation of 5,6-Dihydro-6-Hydroxy-FUrds (FUrd-H2Os).
Twenty milliliters of aqueous FUrd (0.2 mg/ml), 3.3 ml in each of six 1-cm quartz cuvettes, was irradiated at 254 nm with a Bleit 155 lamp (Spectronic, Westbury, NY) at 0.5 cm until the A260 decreased by over 90% (15 min). The samples were concentrated in vacuo to 2 ml, and the product was purified by HPLC using a Ranin Dynamax-300A C8 column (21.4 × 250 mm) and 10 ml/min water as eluent. The two major peaks with absorbance at 220 nm were concentrated in a speed-vac, and the purity of each was verified by HPLC using a Ranin Microsorb-MV C18 column (4.6 × 250 mm) with 0.5 ml/min water as eluent. Mass spectral analysis showed that both products had a molecular mass of 280 Da, in agreement with that for FUrd-H2O. 1H NMR (300 MHz, D2O) of photoproduct 1 (retention time, 7.4 min): δ 5.68 (dd, 1H, J = 5.8, 1.5 Hz, 1′-H), 5.59 (dd, 1H, J = 3.9, 3.6 Hz, 6-H), 5.44 (dd, 1H, J = 45, 3.6 Hz, 5-H), 4.19 (m, 1H, 2′-H), 4.11 (m, 1H, 3′-H), 3.96 (m, 1H, 4′-H), 3.73 (m, 2H, 5′-H); 1H NMR (300 MHz, D2O) of photoproduct 2 (retention time, 9.1 min) δ 5.69 (d, 1H, J = 5.7 Hz, 1′-H), 5.60 (dd, 1H, J = 3.9, 3.9 Hz, 6-H), 5.39 (dd, 1H, J = 46, 3.9 Hz, 5-H), 4.32 (m, 1H, 2′-H), 4.13 (m, 1H, 3′-H), 3.96 (m, 1H, 4′-H), 3.72 (m, 2H, 5′-H). The NMR spectra of photoproducts 1 and 2 were essentially identical to those reported for the cis-5S,6R and cis-6R,5S diastereoisomers, respectively, of FUrd-H2O designated as the North and South diastereoisomers (19).
[6-3H]FUrd-H2O was prepared by irradiation of a 3-ml aqueous solution of 0.6 mg [6-3H]FUrd (5 μCi) in a 1-cm quartz cuvette with a short wave UV lamp as described above, concentrated in vacuo to 0.1 ml, and purified on a Rainin Microsorb-MV C18 column (5 μm, 4.6 × 250 mm) using water as eluent at 0.5 ml/min. The eluent was monitored at 220 nm and radioactivity. The two major radioactive peaks, photoproducts 1 and 2, were individually collected and concentrated to dryness in a speed vacuum.
Nucleoside Analysis of ΨSI-Modified [6-3H]FUra-tRNA.
Reaction mixtures (40 μl) containing 0.8 μM of [6-3H]FUra-tRNA (6.4 × 105 dpm), or 0.02 μM [5′-32P]CMP-FUra-tRNA (1.8 × 104 cpm) and 12.5 μM ΨSI in NTE buffer were incubated at room temperature for 3 h and subjected to SDS/12% PAGE. The band containing the ΨSI-[6-3H]FUra-tRNA complex was localized by reference to the band for the ΨSI-[5′-32P]CMP-FUra-tRNA complex in an adjacent lane. The 3H-labeled band was excised and extracted overnight with water. The extract was filtered through glass wool in a 1-ml pipette tip and incubated at 90°C for 5 min to disrupt the complex. After phenol extraction and addition of 0.1 vol of 3 M NH4OAc, pH 6, the ΨSI-modified [6-3H]FUra-tRNA was precipitated with ethanol, dried, and dissolved in 20 μl of H2O. Aliquots (10 μl, ≈105 dpm) were digested with nuclease P1, followed by alkaline phosphatase (20). Digests were supplemented with 10 μl each of 1 μg/μl 5-FUrd photoproducts 1 and 2 and 5-FUrd. Analytical HPLC was performed on a Ranin Microsorb-MV C18 column (4.6 × 250 mm) by using 0.5 ml/min water as eluent. The eluent was monitored at 220 nm, and 0.2-ml fractions were collected and counted. Retention times were: photoproduct 1 (FUrd-H2O, cis, North; ref. 19), 7.4 min; photoproduct 2 (FUrd- H2O, cis, South), 9.1 min; 5-FUrd, 20.6 min.
ΨSI-modified [6-3H]FUra-tRNA (5 μl, 2 × 105 dpm) was digested with nuclease P1, followed by alkaline phosphatase. The digest was chromatographed on a cellulose TLC plate by using isobutyric acid-concentrated NH4OH-H2O (66:1:33, vol/vol/vol) as mobile phase. The plate was briefly wetted with Amplify (Amersham Pharmacia), dried, and analyzed by autoradiography.
Results
ΨSI-Modified FUra-tRNA.
The ΨSI-FUra-tRNA covalent complex was prepared by treatment of FUra-tRNA with excess ΨSI, followed by purification on and isolation from SDS/PAGE without prior heating in SDS loading buffer. Fig. 1A shows the SDS/PAGE of free FUra-tRNA (lane 1) and the ΨSI-FUra-tRNA complex (lane 2). The covalent complex was disrupted by heating an aqueous solution at 90°C for 5 min, and the released protein was removed by phenol extraction to provide the ΨSI-modified FUra-tRNA in about 25% overall yield.
Figure 1.

Denaturing PAGE of [32P]FUra-tRNAs and complexes with ΨSI. (A) SDS/PAGE of FUra-tRNA (lane 1), ΨSI-FUra-tRNA covalent complex (lane 2), and RNase A digest of ΨSI-FUra-tRNA covalent complex from lane 2 (lane 3). (B) 7 M urea-15% PAGE of unmodified (lane 1), ΨSI-modified FUra-tRNA (lane 2), and the oligo containing FU39 after hydrolysis of the RNase A-digested ΨSI-FUra-tRNA complex from A, lane 3.
As indicated above, the ΨSI-FUra-tRNA covalent complex was not heated in loading buffer before SDS/PAGE lest it undergo hydrolysis. Because some proteins require heating in SDS for denaturation, this limitation raises the question as to whether the complex isolated in SDS is indeed covalent. However, the complex also can be isolated by 7 M urea-PAGE (data not shown), which does not require heat for denaturation of proteins. Moreover, digestion of the complex with RNase A provides a ΨSI 7-mer oligonucleotide complex that is stable to SDS/PAGE (Fig. 1 A and B, lanes 3), which is highly unlikely to be associated by noncovalent interactions.
Sequence Ladder of ΨSI-Modified FUra-tRNA.
FUra-tRNA and ΨSI-modified FUra-tRNA were labeled at the 3′ end with [5′-32P]pCp, purified on 7 M urea-12% PAGE and subjected to partial digestion with RNase A to generate sequence ladders. The ladder generated upon digestion of unmodified FUra-tRNA (Fig. 2, lane 2) showed the band resulting from cleavage 3′ to FU39, whereas this band was not seen with ΨSI-modified FUra-tRNA (Fig. 2, lane 1). Thus, ΨSI modification of FUra-tRNA induced resistance of FU39 toward RNase A digestion.
Figure 2.
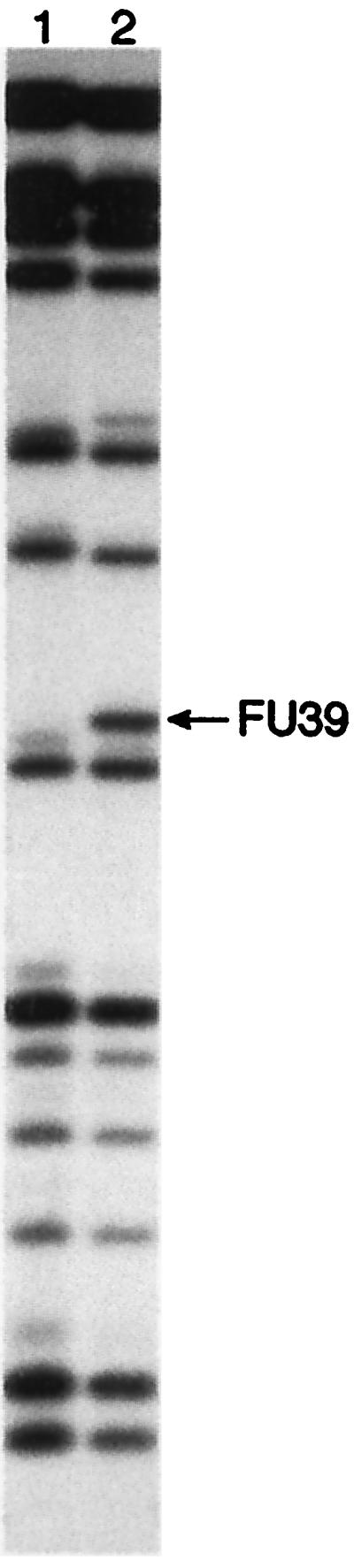
7 M urea-PAGE of partial RNase A digests of unmodified (lane 2) and ΨSI-modified FUra-tRNA (lane 1). FUra-tRNA was labeled with [32P]pCp, and the location of the oligo containing modified FU39 (arrow) was determined by comparison to sequence ladders from RNase T1 and T2 digests (data not shown).
3′-End Group Analysis of RNase A-Digested ΨSI-Modified FUra-tRNA.
[5′-32P]CMP-FUra-tRNA was prepared by using [5′-α-32P]CTP to label phosphate groups 5′ to C residues, thus introducing a 32P label 5′ to C40 and 3′ to the ΨSI target FU39. Complete RNase A digestion of the control unmodified [5′-32P]CMP-FUra-tRNA gave the expected bands on 7 M urea-25% PAGE for cleavage 3′ to all pyrimidine nucleotides (Fig. 3A, lane 1). The five largest fragments, including the hexanucleotide containing FU39 (Fig. 3A, lane 1, band 3), were subjected to 3′-end group analysis. In accord with the expected sequence, digestion of band 3 (GpApApGpApFU32p) to 3′NMPs with RNase T2 gave [32P]3′-FUMP, whereas cleavage with nuclease P1 to 5′NMPs gave 32Pi but no 32P nucleotides (data not shown). When ΨSI-modified [5′-32P]CMP-FUra-tRNA was completely digested with RNase A, 7 M urea-25% PAGE of the digest showed a shift of the GpApApGpApFU32p band 3 of unmodified FUra-tRNA to one that migrates between the first and second bands of unmodified [5′-32P]CMP-FUra-tRNA (Fig. 3A, lane 2, band 3). Digestion of the shifted band 3 with nuclease P1 to provide 5′NMPs gave 5′-32P-CMP, indicating a 3′ terminal C residue (Fig. 3B); cleavage to 3′ nucleotides with RNase T2 gave an unknown [32P]3′NMP (Fig. 3C), which was presumed to be a modified residue located 5′ to the terminal CMP. RNase T2 digestion of ΨSI modified-[5′-32P]CMP-FUra-tRNA showed the same modified residue (Fig. 3D). These results indicate that this fragment of ΨS-modified FUra-tRNA is the heptanucleotide GpApApGpApXpCp, where X indicates a modified FUrd.
Figure 3.

3′ End-group analysis of RNase A-digested ΨSI-modified FUra-tRNA. (A) 7 M urea-25% PAGE after complete RNase A digests of [5′-32P]CMP-FUra-tRNA (lane 1) and ΨSI-modified [5′-32P]CMP-FUra-tRNA (lane 2). (B) Two-dimensional TLC of band 3, lane 2 of A after nuclease P1 digestion to 5′-NMPs. (C) Two-dimensional TLC of band 3, lane 2 of A after RNase T2 digestion to 3′-NMPs. (D) Two-dimensional TLC of RNase T2 digest of ΨSI-modified [5′-32P]CMP-FUra-tRNA. 32P bands and spots were located by autoradiography.
MS of RNase A Digests of Unmodified and ΨSI-Modified FUra-tRNA.
Unmodified and ΨSI-modified FUra-tRNA were completely digested with RNase A, and the digests were analyzed by UV-matrix-assisted laser desorption ionization-MS. Only one peak in the mass spectrum of RNase A-digested ΨSI-modified FUra-tRNA was shifted from RNase A-digested FUra-tRNA (the others were as predicted after cleavage 3′ to the phosphate of pyrimidines). The peak, containing FU39, shifted from mass of ca. 2,022 Da in unmodified FUra-tRNA (34GpApApGpApFUp) to 2,344 Da in ΨSI-modified FUra-tRNA. The 322.8-Da increase in molecular mass of the fragment corresponds to the molecular mass of a CMP residue (305.2 Da) plus H2O (18.0 Da). One explanation for this result is that FU39 is modified by the addition of H2O and thus is rendered resistant to RNase A; consequently, RNase A cleaves at C40 to produce GpApApGpApXpCp, where X is a modified FUrd.
FUra 39 Is Retained in ΨSI-Modified FUra-tRNA.
[5′-32P]CMP-[6-3H]FUra-tRNA was prepared by using [5′-α-32P]CTP and [6-3H]FUTP to introduce a 32P label 3′ to FU39 and 3H at position 6 of FU residues. ΨSI-modified [5′-32P]CMP-[6-3H]FUra-tRNA was prepared, purified, and completely digested with RNase A; the control was [5′-32P]CMP-[6-3H]FUra-tRNA treated with RNase A. If the FUra pyrimidine of FU39 was covalently attached to the protein in the ΨSI-FUra-tRNA complex, as in Scheme S1 (1B), the tritium would be retained in the oligonucleotide containing FU39; if the FUra was not covalently attached, as in Scheme S2 (5B), the tritium would be lost. The 3H/32P ratios of three largest fragments (6–8 nt), including the hexa- or heptanucleotide containing FU39 obtained from complete RNase A digestion of [5′-32P]CMP-[6-3H]FUra-tRNA or ΨSI-modified [5′-32P]CMP-[6-3H]FUra-tRNA, respectively, were determined. For band 1 (Fig. 3A, lane 1) (18-GpGpGpApGpApG32pCp), which did not contain FUra, the 3H/32P ratio was < 0.05 (i.e., 3H undetectable), whereas for band 2 (42-GpGpApGpGpFU32p), which contained FUra at position 47, the ratio was 0.83. For band 3, which contained the ΨSI target FU39, for both unmodified (Fig. 3A, lane 1, GpApApGpApFU32p) and ΨSI-modified (Fig. 3A, lane 2, GpApApGpApX32pCp) [5′-32P]CMP-[6-3H]FUra-tRNA, the 3H/32P ratio was 0.77, demonstrating retention of FUra in both oligonucleotides.
In a second experiment, the target oligonucleotide containing FU39 was prepared by complete RNase A digestion of the ΨSI-[5′-32P]CMP-[6-3H]FUra-tRNA covalent complex, hydrolysis, and isolation. The 3H/32P ratio of the resulting target oligonucleotide (Fig. 1B, band 3) was 0.9, showing again that the target FUra at position 39 was not lost from the oligonucleotide.
Identification of Photoproducts of 5-FUrd.
We prepared two isomers of FUrd-H2O by photohydration of FUrd. The individual isomers were isolated by preparative HPLC and shown by mass spectral and NMR analysis to be identical to the two cis diastereoisomers (5S,6R and 5R,6S) of FUrd-H2O prepared by chemical synthesis (19).
Nucleoside Analysis of Modified FUra-tRNA.
ΨSI-modified [6-3H]FUra-tRNA was digested with nuclease P1 and phosphatase, and the products were subjected to HPLC. HPLC analysis showed [6-3H]FUrd [retention time (RT) = 23 min] and an unknown radioactive peak (RT = 7.4 min) at a ratio of 14:1 (theoretical 16:1). The latter peak coeluted on HPLC with authentic cis-5S,6R FUrd-H2O photoproduct 1 (North isomer, terminology of Visser et al.; ref. 19) and was separable from the cis-5R,6S FUrd-H2O isomer (RT = 10.5 min) (Fig. 4). On TLC, the HPLC-purified product moved as did photoproducts 1 or 2; likewise, the total digest of ΨSI-modified FUra-tRNA showed [6-3H]FUrd (Rf = 0.76) and the unknown (Rf = 0.63) that moved with the unresolved photoproducts 1 or 2 in a ratio of 12:1 (Fig. 5, lane 5). As control, nuclease P1 and phosphatase digestion of unmodified [6-3H]FUra-tRNA (Fig. 5, lane 6) gave only [6-3H]FUrd.
Figure 4.

HPLC analysis of modified FUrd. The nuclease P1 and phosphatase digest of ΨSI-modified [6-3H]FUra-tRNA was cochromatographed with FUrd and its photoproducts 1 and 2. The eluent was monitored by absorbance at 220 nm (line trace) and 3H counts (bar graph).
Figure 5.

TLC of standards and radioactive nucleosides derived from digestion of ΨSI-modified [6-3H]FUra-tRNA. (Lane 1) [6-3H]5-FUrd; (lane 2) photoproducts of [6-3H]5-FUrd; (lane 3) HPLC-purified photoproduct 1 of [6-3H]5-FUrd; (lane 4) HPLC-purified photoproduct 2 of [6-3H]5-FUrd; (lane 5) nuclease P1 and alkaline phosphatase digests of ΨSI-modified [6-3H]FUra-tRNA; (lane 6) nuclease P1 and alkaline phosphatase digests of [6-3H]FUra-tRNA. Lanes 5 and 6 migrate aberrantly because of the high salt concentration; for clarity, the components of each lane are segregated by vertical lines.
Discussion
The present work sought to determine the structure of the ΨSI-FUra-tRNA covalent complex (9). We assumed that the mechanism of interaction of ΨSI with FUra-tRNA was analogous to the normal enzymic reaction and that the structure of the complex would reveal information from which the mechanism of ΨSI could be deduced.
Our initial experiments were aimed at distinguishing whether the site of covalent attachment of ΨSI to FUra-tRNA involved the FUra base or the polynucleotide backbone (Schemes S1 and S2, R = F). In the mechanism depicted in Scheme S1, the putative Asp nucleophile is attached to the 6-position of the target FUra, and the intermediate contains an intact N-glycosidic bond linking the modified base to the polynucleotide chain (1B). In Scheme S2, the putative Asp nucleophile is covalently attached to the 1′-C of the sugar, and the N-glycosidic bond to FUra has been disrupted (5B). If the latter mechanism is correct, the FUra base must be tightly, albeit reversibly, bound to the enzyme, because it does not undergo exchange with Ura present in the medium (9).
The properties of the ΨSI-FUra-tRNA complex that enabled the present work are as follows. First, the covalent complex can readily be separated from the unbound components by SDS/PAGE or urea/PAGE. Upon protein denaturation, 1B (Scheme S1) would yield a complex containing the FUra base connected to the polynucleotide chain, whereas 5B (Scheme S2) would lose FUra from the protein-RNA complex. Second, the ΨSI-FUra-tRNA complex can be disrupted to its protein and nucleic acid components by heating in aqueous solution. Here, 1B could undergo β-elimination to regenerate unmodified FUra-tRNA or undergo ester hydrolysis to form 6-hydroxy-5,6-dihydro-FUMP (8B, Scheme S3) contained within the tRNA; 5B or 6B (Scheme S2) could undergo only hydrolysis to release FUra and provide an apyrimidinic tRNA. As described below, heat disruption of the ΨSI-FUra-tRNA complex results in a “modified” form of FUra-tRNA, the identification of which was key to the conclusions of this work.
Scheme 3.

Hydrolysis of the ester linkage from ΨSI-FUra-tRNA complex.
The sequence ladder generated by partial RNase A digestion of FUra-tRNA showed that FUra was cleaved 3′ to its 3′ phosphate. However, the sequence ladder generated by RNase A digestion of ΨSI-modified FUra-tRNA demonstrated that the target FU39 residue was resistant to digestion. We performed end-group analysis of the oligonucleotide-containing residue 39 (Fig. 3A, lane 2, shifted band 3) obtained from RNase digestion of ΨSI-modified FUra-tRNA. We identified CMP as the 3′ terminal nucleotide and found that the penultimate residue generated by complete RNase T2 digestion to 3′ nucleotides was not 3′FUMP. Thus, upon treatment of FUra-tRNA with ΨSI and heat disruption of the complex, FU39 was modified to a RNase A-resistant derivative, and cleavage occurred 3′ to the adjacent C40 to provide the heptanucleotide GpApApGpApXpCp.
To determine whether the N-glycoside bond to FUra was intact in the covalent complex, we asked whether FUra was retained in the covalent ΨSI-FUra-tRNA complex. We prepared the covalent complex from ΨSI and FUra-tRNA uniformly labeled with [6-3H]FUra and [5′-32P]CMP; the latter introduced a 32P label 3′ to FU39. We purified the complex on SDS/PAGE, heat-dissociated it, and then completely digested it with RNase A to give the heptanucleotide containing the target residue 39 and a single [5′-32P]CMP. Isotopic analysis showed that the [6-3H]FUra and [5′-32P]CMP were present in approximately equimolar amounts. This experiment showed that the N-glycoside bond to FUra was intact in the isolated oligonucleotide and hence must have been intact in the covalent complex. Confirming this conclusion, a similar result was obtained when the covalent complex was directly digested with RNase A and the complex was analyzed after SDS/PAGE. These data rule out the mechanism depicted in Scheme S2, R = F, and indicate that the of modification of residue 39 is the FUra base.
Mass spectral analysis of complete RNase A digests of unmodified FUra-tRNA showed the expected hexanucleotide GpApApGpApFUp, resulting from cleavage 3′ to pyrimidine nucleotides. In contrast, mass spectral analysis of RNase A digests of ΨSI-modified FUra-tRNA showed the absence of this hexanucleotide, and the appearance of a new peak with an increase of 323 mass units corresponded to the addition of CMP and H2O to the hexanucleotide. The results are consistent with the addition of water to the pyrimidine base of FU39 in ΨSI-modified FUra-tRNA, because the modified FUra residue is resistant to RNase A; consequently, RNase A cleavage occurs at the adjacent C40 residue.
From chemical considerations and the data presented above, the most plausible structure for the ΨSI-FUra-tRNA covalent complex is that in which Asp-60 is attached to the 6-position of FU39 (1B; Scheme S1); the rearranged 4B also was initially consistent with the data. A relevant chemical model for the covalent ΨSI-FUra-tRNA complex is 5-flouro-6-acetoxy-5,6-dihydro-Urd (FUrd-AcOH) (19). The two cis diastereoisomers (5S,6R and 5R,6S) of FUrd-AcOH are formed by AcOF addition to the 5,6-double bond of FUrd. As with the covalent ΨSI-FUra-tRNA complex, FUrd-AcOH is fairly stable in water at room temperature, but is converted to FUrd-H2O upon heating. Further, because the separated pure diastereoisomers of FUrd-AcOH hydrolyze with complete retention of configuration at C-6, it may be concluded that hydrolysis occurs by O-acyl rather than N-1-assisted O-alkyl scission; the latter would destroy symmetry at C-6 and give both configurations of FUrd-H2O.
Definitive evidence for the structure of the modified FU39 in the covalent complex was obtained by nucleoside analysis of ΨSI-modified FUra-tRNA. We digested [6-3H]FUra-tRNA and ΨSI-modified [6-3H]FUra-tRNA to component nucleosides and analyzed the products by HPLC (Fig. 4) and TLC (Fig. 5). The [6-3H]FUra-tRNA digest showed only [6-3H]FUrd, whereas the ΨSI-modified [6-3H]FUra-tRNA digest showed [6-3H]FUrd and a radioactive product (≈14:1) that cochromatographed with an authentic sample of cis-5S,6R FUrd-H2O. From chemical considerations, and the aforementioned properties of the model FUrd-AcOH, it was concluded that the FUrd-H2O resulted from hydrolysis of the ester bond between the Asp residue of the enzyme and the 6-position of FU39 in the ΨSI-FUra-tRNA complex (Scheme S3). Further, because the isolated FUrd-H2O chromatographed as the cis-5S,6R diastereoisomer and hydrolysis of the model FUrd-AcOH stereoisomers proceeds with retention of configuration at C6, it was deduced that the covalent complex also has the cis-5S,6R configuration (8B, Scheme S3). Thus, Asp-60 of ΨSI attacks FU39 of tRNA on the 6R face of the pyrimidine, and a proton adds to the 5-carbon from the opposite face (trans) to give the cis-5S,6R configuration. The stereochemical course of the ΨSI reaction is similar to that observed with m5C-DNA methyl transferase (MTase) and thymidylate synthase, both of which proceed via trans addition (6, 21), but opposite to that observed in RNA 54U5 MTase, which adds cis to provide a cis-5R,6S adduct (22).
It is not obvious why the Asp carboxylate is used as a catalytic nucleophile by ΨS rather than the Cys thiol found in the mechanistically related thymidylate synthase (6). Presumably, the catalytic nucleophiles of these enzymes evolved for functional reasons because substitution of the catalytic Cys of thymidylate synthase by Asp (23) or the catalytic Asp of ΨS by Cys (24) lead to inactive enzymes. This finding is not entirely unexpected because the functional groups of these residues have different chemical properties. For example, the thiolate of Cys is a much more nucleophilic than the carboxylate of Asp and is expected to provide more efficient covalent bond formation with the target pyrimidine; in contrast, the carboxylate of Asp provides a much more efficient leaving group than thiolate. Although certain steps in the catalytic pathways of ΨS and thymidylate synthase are similar and could be facilitated by a thiol, others are quite different and could be facilitated by an Asp. We speculate that the nucleophilic catalysts in these enzymes were evolved to represent a balance that serves to optimize the combined requirements of all steps of the pathways.
Conclusions
Together with reported data (9), the present work shows that the covalent complex formed between ΨSI and FUra-tRNA involves Michael adduct formation between Asp-60 of ΨSI and the 6-carbon of FU39 of tRNA to form a covalent ΨSI-FUra-tRNA complex. If we assume that an analogous covalent complex is formed in the normal catalytic reaction, and that this complex is on the reaction pathway, the mechanism for ΨS can be proposed to be as depicted in Scheme S1. Recently, the crystal structure of ΨSI has been determined (25), and Asp-60 lies at the base of a cavity believed to be the active site of the enzyme. The structure of a ΨSI-tRNA complex should be solved in the near future and will confirm or refute the mechanism proposed here.
Acknowledgments
This work was supported by U.S. Public Health Service Grant GM 51232.
Abbreviations
- Ψ
pseudouridine
- ΨSI
pseudouridine synthase I
- FUra
5-fluorouracil
- FUrd
5-fluorouridine
- FUrd-H2O
5,6-dihydro-6-hydroxy-FUrd
- FUra-tRNA
tRNAPhe containing FUra in place of Ura
- [5′-32P]CMP-FUra-tRNA
FUra-tRNA made with [5′-α-32P]CTP
- FUra-tRNA-[32P]pCp
FUra-tRNA 3′-end labeled with [5′-32P]pCp
- FUrd-AcOH
5-flouro-6-acetoxy-5,6-dihydro-Urd
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Björk G R, Ericson J U, Gustafsson C E, Hagervall T G, Jönsson Y H, Wikström P M. Annu Rev Biochem. 1987;56:263–287. doi: 10.1146/annurev.bi.56.070187.001403. [DOI] [PubMed] [Google Scholar]
- 2.Bakin A, Ofengand J. Biochemistry. 1993;32:9754–9762. doi: 10.1021/bi00088a030. [DOI] [PubMed] [Google Scholar]
- 3.Patton J R. Biochem J. 1993;290:595–600. doi: 10.1042/bj2900595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Santi D V, Wataya Y, Matsuda A. In: Proceedings of the International Symposium on Substrate-Induced Irreversible Inhibition of Enzymes. Seiler N, Jung M J, Koch-Weser J, editors. Strasbourg, France: Elsevier; 1978. pp. 291–303. [Google Scholar]
- 5.Prior J J, Maley J, Santi D V. J Biol Chem. 1984;259:2422–2428. [PubMed] [Google Scholar]
- 6.Carreras C W, Santi D V. Annu Rev Biochem. 1995;64:721–762. doi: 10.1146/annurev.bi.64.070195.003445. [DOI] [PubMed] [Google Scholar]
- 7.Kammen H O, Marvel C C, Hardy L, Penhoet E E. J Biol Chem. 1988;263:2255–2263. [PubMed] [Google Scholar]
- 8.Ivanetich K M, Santi D V. Prog Nucleic Acid Res Mol Biol. 1992;42:127–156. doi: 10.1016/s0079-6603(08)60575-9. [DOI] [PubMed] [Google Scholar]
- 9.Huang L, Pookanjanatavip M, Gu X, Santi D V. Biochemistry. 1998;37:344–351. doi: 10.1021/bi971874+. [DOI] [PubMed] [Google Scholar]
- 10.Zhao X M, Horne D A. J Biol Chem. 1997;272:1950–1955. doi: 10.1074/jbc.272.3.1950. [DOI] [PubMed] [Google Scholar]
- 11.Grodberg J, Dann J J. J Bacteriol. 1988;170:1245–1253. doi: 10.1128/jb.170.3.1245-1253.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sampson J R, Uhlenbeck O C. Proc Natl Acad Sci USA. 1988;85:1033–1037. doi: 10.1073/pnas.85.4.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gu X, Santi D V. Biochemistry. 1992;31:10295–10302. doi: 10.1021/bi00157a017. [DOI] [PubMed] [Google Scholar]
- 14.Stanley W M., Jr Methods Enzymol. 1974;29:530–547. doi: 10.1016/0076-6879(74)29049-9. [DOI] [PubMed] [Google Scholar]
- 15.Nordhoff E, Kirpekar F, Karas M, Cramer R, Hahner S, Hilenkamp F, Kristiansen K, Roepstorff P, Lezius A. Nucleic Acids Res. 1994;22:2460–2465. doi: 10.1093/nar/22.13.2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pomerantz S C, Kowalak J A, Mccloskey J A. J Am Soc Mass Spectrometry. 1993;4:204–209. doi: 10.1016/1044-0305(93)85082-9. [DOI] [PubMed] [Google Scholar]
- 17.England T E, Bruce A G, Uhlenbeck O C. Methods Enzymol. 1980;65:65–74. doi: 10.1016/s0076-6879(80)65011-3. [DOI] [PubMed] [Google Scholar]
- 18.Silberklang M, Gillum A M, RajBhandary U L. Methods Enzymol. 1979;59:58–109. doi: 10.1016/0076-6879(79)59072-7. [DOI] [PubMed] [Google Scholar]
- 19.Visser G W M, Herder R E, Noordhuis P, Zwaagatra O, Herscheid J D M. J. Chem. Soc. Perkin Trans. 1. 1988. 2547–2554. [Google Scholar]
- 20.Nègre D, Weitzmann C, Ofengand J. Proc Natl Acad Sci USA. 1989;86:4902–4906. doi: 10.1073/pnas.86.13.4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reinisch K M, Chen L, Verdine G L, Lipscomb W N. Cell. 1995;82:143–153. doi: 10.1016/0092-8674(95)90060-8. [DOI] [PubMed] [Google Scholar]
- 22.Kealey J T, Lee S, Floss H G, Santi D V. Nucleic Acids Res. 1991;19:6465–6468. doi: 10.1093/nar/19.23.6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Climie S, Ruiz-Perez L, Gonzalez-Pacanowska D, Prapunwattana P, Cho S-W, Stroud R, Santi D V. J Biol Chem. 1990;265:18776–18779. [PubMed] [Google Scholar]
- 24.Ramamurthy V, Swann S L, Paulson J L, Spedaliere C J, Mueller E G. J Biol Chem. 1999;274:22225–22230. doi: 10.1074/jbc.274.32.22225. [DOI] [PubMed] [Google Scholar]
- 25.Foster, P. G., Huang, L., Santi, D. V. & Stroud, R. M. (1999) Nat. Struct. Biol., in press. [DOI] [PubMed]